


Abstract
The yeast gene BFR1 was originally isolated from a genetic screen for high-copy suppressors of brefeldin A-induced lethality in Saccharomyces cerevisiae. While this result suggested a possible role for the encoded protein, Bfr1p, in the secretory pathway, subsequent data have not fully supported this conclusion. Alternatively, Bfr1p has also been found by yeast two-hybrid analysis to interact with Bbp1p, a component of the spindle pole body. Finally, we have reported that Bfr1p associates with cytoplasmic mRNP complexes containing Scp160p, raising the possibility that Bfr1p may function in mRNA metabolism. Here, we have explored this possibility further. We report that Bfr1p associates with yeast polyribosomes and mRNP complexes even in the absence of Scp160p, and that its interaction with Scp160p-containing mRNP complexes is RNA-dependent. Furthermore, we have determined by fluorescence microscopy and subcellular fractionation that Bfr1p and Scp160p demonstrate similar cytoplasmic localization with enrichment around the nuclear envelope/endoplasmic reticulum. Finally, we report that loss of Bfr1p disrupts the interaction of Scp160p with polyribosomes, thereby demonstrating that the relationship between these two proteins is functional as well as physical. Considered together, these data raise the intriguing possibility that Bfr1p may provide a link between mRNA metabolism, the chromosomal segregation machinery and perhaps secretion in yeast.
INTRODUCTION
Bfr1p is a 55 kDa protein of unknown function in Saccharomyces cerevisiae. The gene, BFR1, was cloned in 1994 from a genetic screen for high-copy suppressors of brefeldin-A (BFA) induced lethality (1). Since BFA is known to block secretion in eukaryotic cells (2,3), Bfr1p was originally hypothesized to function in the yeast secretory pathway (1). In contrast to this hypothesis, however, bfr1-null cells did not exhibit a secretion defect (1). Indeed, the only additional data supporting a role for Bfr1p in secretion were the observations that BFR1 over-expression partially suppressed defects in yeast sec17 mutants, and that BFR1 deletion mildly accentuated the phenotype of sec21 mutants (1).
Other studies have implicated Bfr1p for a role in nuclear segregation during mitosis and meiosis (4). bfr1 null cells display a phenotype that includes increased ploidy and abnormal cell shape and size (1,4). 4′,6-Diamino-2-phenylindole (DAPI) staining of fixed bfr1 null cultures shows a sub-population of cells containing either enlarged nuclei or, alternatively, no detectable nuclei at all (1,4). Moreover, diploid strains lacking both BFR1 alleles are reported to be partially defective in meiosis (4). In some cells, a unique asci-within-asci phenotype was observed following sporulation (4). A potential role for Bfr1p in nuclear segregation was further supported by an observed yeast two-hybrid interaction with Bbp1p, an essential component of the yeast spindle pole apparatus (4–6). While BBP1 deletion is lethal in yeast, moderate over-expression leads to a phenotype similar to that of BFR1 deletion: increased ploidy, and the formation of asci-within-asci during sporulation of diploids (4).
We reported recently that Bfr1p associates with complexes containing Scp160p (7). Scp160p is a 1222 amino acid protein in yeast that includes 14 copies of the hnRNP K homology (KH) domain, a highly conserved motif found in many RNA-binding proteins, including the fragile-X mental retardation protein, Fmrp (8–10). Scp160p demonstrates significant similarity to a class of multiple KH-domain proteins collectively known as vigilins. First identified in chicken (11), vigilin homologs have now been found in human (12), Xenopus laevis (13), Drosophila melanogaster (14), Caenorhabditis elegans (8), Schizosaccharomyces pombe (GenPept #7493335) and Neurospora crassa (GenPept #7899383).
While all of the vigilin proteins studied to date are reported to bind nucleic acid, both the type of nucleic acid bound and the functional significance of these interactions remain unclear. In the case of Scp160p, we have demonstrated that the protein exists primarily associated with polyribosomes, and that it is released by EDTA treatment as a component of large mRNP complexes that also contain poly(A) binding protein (Pab1p), Bfr1p, and additional unidentified proteins and polyadenylated RNAs (7). Deletion of SCP160 results in a phenotype similar to that observed in bfr1 null cells, including increased ploidy and abnormal cell size and shape (15).
We report here further characterization of the mRNP and polyribosome associations of Bfr1p in both the presence and absence of Scp160p. Our data support the hypothesis that Bfr1p functions in mRNA metabolism in yeast, and suggest that the observed phenotypes of BFR1 deletion and over-expression may not demonstrate a direct role of the protein in secretion or nuclear segregation, but rather may reflect downstream effects resulting from the aberrant expression of other yeast genes.
MATERIALS AND METHODS
Plasmids, yeast strains and culture conditions
The N-terminally HA-tagged allele of BFR1 was generated by PCR-amplification of the BFR1 locus from wild-type (W303) yeast genomic DNA using the primers BFR1HAF1 (5′-CCGCGGATCCATGTACCCATACGACGTCCCAGACTACGCTATGTCCTCCCAACAACACAA-3′) and BFR1HINDR1 (5′-CCGCAAGCTTGTCGACTATTTCATATGCCACAGGAAACAG-3′), and subcloned into YIPlac211 (16). The BFR1 promoter region was PCR-amplified in a similar manner using the primers BFR1SACF1 (5′-CCGCGAGCTCAGCATTAAGCATTCACGAGC-3′) and BFR1BAMR1 (5′-CCGCGGATCCGGCAATGGCTGTGTTGTTAGA-3′) and subcloned into the appropriate position upstream of the HA-Bfr1p open reading frame in the plasmid backbone. The entire open reading frame was confirmed by dideoxy sequencing. Finally, the HA-BFR1 allele was substituted into the BFR1 genomic locus with linearization at the SphI site followed by standard two-step gene replacement techniques (17). The N-terminally GFP-tagged version of Bfr1p was created by PCR amplification of the GFP coding sequence, with primers GFPBFRF1 (5′-CCTCCTTTTATCAACGTAATAGCATATTTTCTAACAACACAGCCATTGCCGTCGACATGGCTAGCAAAGGAGAAGAA-3′) and GFPBFRR1 (5′-TTTCTTGTCTCTAACGGAGACATCTGGGCGCTTGAACTTGTGTTGTTGGGAGGAACCACCGCAGCCGGATCCTTTGTATAG-3′). This PCR product was then subcloned into the HA-tagged BFR1 construct using a BamHI site. A genomic disruption of the BFR1 locus was achieved by one-step gene replacement (17) using a cassette consisting of the BFR1 gene with an internal PstI fragment representing the majority of the coding sequence removed and replaced by a kanamycin resistance cassette [BamHI–SacI fragment derived from the plasmid pFA6a-KanMX6 (18)]. Genomic disruption was confirmed by PCR amplification of the locus. The HIS3 disrupted allele of SCP160 was created by first cutting the SCP160 sequence with ApaI and KpnI, to remove the majority of the coding sequence, and ligating in a fragment containing the yeast HIS3 gene. All yeast transformations and culture manipulations were performed according to standard protocols as described elsewhere (17).
Confirmation of genomic integrations
All genomic integrations, including deletions and introduction of epitope tags, were confirmed by PCR amplifications from yeast genomic DNA using primers that flanked the engineered regions. The presence of epitope tags was further confirmed by western blot analyses using the appropriate anti-tag primary antibodies.
Polyribosome isolation
Polyribosomes were isolated as described previously (7,19). For EDTA controls, lysis buffer containing 5 mM MgCl2 was used, and 30 mM EDTA was added to the sample before loading onto the gradient. For RNase controls, 50 U/ml of RNase I (Promega) were added prior to loading the sample onto the gradient.
Gel filtration chromatography
Gel filtration chromatography was performed as described previously (7), using a 120 ml Hi-Prep S-300 Sephacryl column (Pharmacia) with a cut-off of 1300 kDa, attached to an FPLC system (Pharmacia). Fractions (2.0 ml) were collected, from which 12 µl were combined with sample buffer (2% SDS, 10% glycerol, 100 mM dithiothreitol, 60 mM Tris pH 6.8, 0.001% bromophenol blue), size-fractionated by SDS–PAGE, and analyzed by western blot using the indicated antibodies.
α-FLAG affinity chromatography
For most experiments, 1 l yeast cultures were grown to early log phase and harvested by centrifugation. Cells were washed twice in T75 buffer (25 mM Tris pH 7.5, 75 mM NaCl) and then lysed by vortex agitation with an equal volume of glass beads in 4 ml T75 buffer containing 30 mM EDTA. Each lysate was transferred to a clean microfuge tube, and centrifuged at 3000 g for 10 min at 4°C. Each supernatant was again transferred to a clean microfuge tube and centrifuged at 12 000 g for 15 min at 4°C, and finally passed through a 0.2 µm syringe filter. The filtrate was then size-fractionated by running over an S-300 gel-filtration column in T75 buffer, with pooling of the void volume fractions (∼10 ml total). A low concentration (<10 µg/ml) of FLAG peptide (N-DYKDDDDK-C) was added to the sample to reduce background. This void material was then loaded onto a 1 ml M2 α-FLAG column (Sigma); column flow-through was passed over the column a second time. The column was then washed extensively with 75 ml T75 buffer; material bound to the column was eluted with 5 ml T75 containing 184 mg/ml FLAG peptide. The peptide solution was allowed to incubate on the column for 20 min prior to collection of the first 1 ml fraction; subsequent fractions were collected every 10 min. Samples of crude material, S-300 void, α-FLAG flow-through, first and last wash, and eluate fractions were analyzed by western blot using the indicated antibodies.
α-HA affinity chromatography
Soluble lysates and void volume fractions were prepared from yeast expressing either HA-Bfr1p and FLAG-Scp160p (JFy3535) or HA-Bfr1p in the absence of Scp160p (JFy3537), as described above. Samples representing 36–48 ml elution volume (Fig. 5A) were then pooled and incubated at 4°C overnight with 250 µl of anti-HA affinity matrix prepared by cross-linking 12CA5 monoclonal antibody to protein A–Sepharose beads (Amersham-Pharmacia), as described previously (20). The slurry was then centrifuged for 5 min at low speed in a Clay-Adams II Compact Centrifuge to pellet the affinity matrix, and the supernatant was removed and stored (‘flow-through’ in Fig. 5B). The pelleted beads were then washed once with 1 ml PBS (pH 7.4) and re-centrifuged as before; this supernatant was considered the ‘first wash’ (Fig. 5B). The washing step was then repeated an additional six times; the supernatant from the last step was considered the ‘last wash’ (Fig. 5B). Finally, bound material was released from the affinity matrix by rapid washing of the pellet with 1 ml of 0.1 M glycine (pH 3.5). This eluate was concentrated for further analysis as described below, and the affinity matrix was neutralized and washed (3 × 10 ml) using PBS, and then stored at 4°C.
Figure 5.

Bfr1p associates with mRNP complexes in the absence of Scp160p. (A) S300 gel filtration chromatography followed by western blot analysis of EDTA-treated lysates from yeast expressing HA-Bfr1p in the presence (top panel) or absence (lower panel) of Scp160p. (B) α-HA purification and western blot analyses of S300 fractions (pooled 36–48 ml elution volume) derived from the same ΔSCP160 strain used in (A), probed to reveal both the HA-Bfr1p signal (top panel) and the Pab1p signal (lower panel). FT, flow-through fraction; E, elution fractions.
Fluorescence microscopy
Indirect immunofluorescence and DAPI staining were performed as described previously (21). GFP-tagged proteins were detected using a rabbit polyclonal α-GFP antiserum (22), (a generous gift from Dr Anita Corbett, Emory University, Atlanta, GA) at a 1:10 000 dilution, and a Texas Red-conjugated α-rabbit secondary antibody (Jackson Research Labs) at a 1:2000 dilution. Cells were visualized through a Texas Red optimized filter (Chroma Technology) using an Olympus BX60 microscope equipped with a DAGE/MTI video camera.
Cell fractionation
Subcellular fractionation was accomplished using a protocol modified from that of Stoltenburg et al. (23). In brief, cells were grown, harvested, washed and lysed exactly as described above for polyribosome isolation. The cell lysate was then clarified by centrifuging at 1000 g for 5 min. An aliquot of 100 µl of supernatant was then transferred to a fresh microfuge tube, and centrifuged at 10 000 g for 8 min. The supernatant was removed and stored. The pellet containing the membranous fraction was washed twice with 100 µl polyribosome lysis buffer and centrifuged again at 10 000 g. The second wash was performed either in the presence or absence of 50 U/ml RNase I (Promega) as indicated, and the tubes were incubated at room temperature for 10 min. Samples were then centrifuged a final time at 10 000 g for 8 min. The supernatant was removed and stored, and the pellet was resuspended in 100 µl 1× sample buffer.
Concentration of samples
Where indicated, 400 µl protein samples were concentrated using the method of Traub et. al. (24), prior to polyacrylamide gel electrophoresis.
Western blot analysis
Western blot analyses were performed as described previously (25). FLAG-Scp160p fusion protein was detected using the mouse M2 anti-FLAG monoclonal antibody (10 µg/ml final concentration), followed by HRP-conjugated sheep anti-mouse secondary antibody (Amersham), diluted 1:5000, and ECL reagent (Amersham), as per the manufacturer’s instructions. HA-tagged Scp160p and HA-tagged Bfr1p were detected using the 12CA5 mAb (Boehringer-Mannheim) at a final concentration of 0.8 µg/ml, and Pab1p was detected using 1G1 mAb, a generous gift from Maurice Swanson (University of Florida), at 1:5000 dilution (26,27).
RESULTS
Bfr1p associates with polyribosomes
To facilitate detection of Bfr1p in yeast lysates, an N-terminally HA-tagged allele of BFR1 was created and integrated into the yeast genome in place of the native sequence (7). By cell morphology, these cells were indistinguishable from their wild-type counterparts, indicating that the tagged Bfr1p protein remained functional in vivo (7).
To test the hypothesis that Bfr1p associates with polyribosomes, we used sucrose gradient ultracentrifugation to size-fractionate the subcellular components of soluble lysates prepared from yeast expressing both HA-tagged Bfr1p protein, and an N-terminally FLAG-tagged Scp160p protein (7). Western blot analyses of resultant gradient fractions with the α-HA antibody 12CA5 revealed a 55 kDa band (HA-Bfr1p) that was most abundant in the denser fractions, consistent with the location of polyribosomes (>80S) (Fig. 1A). The lower panel of Figure 1A illustrates the distribution of FLAG-Scp160p in this same gradient. Although the Bfr1p and Scp160p signals clearly overlap, there is a notable difference in their distributions; Bfr1p appears only in the heaviest polyribosome fractions, while Scp160p is detectable in all fractions greater than about 40S. The significance of this difference is discussed below.
Figure 1.

Association of Bfr1p with polyribosomes. (A) The top panel shows the OD254 profile of a 15–45% sucrose gradient. Arrows indicate peaks representing small (40S) and large (60S) ribosomal subunits, and single ribosomes (80S). The center panel shows an α-HA western blot of gradient fractions concentrated and run on a 10% SDS–PAGE gel, to demonstrate the distribution of HA-Bfr1p in the gradient. The bottom panel shows an α-FLAG western blot of the same gradient fractions, demonstrating the distribution of FLAG-Scp160p. (B) Gradient profile and western blot analyses of a yeast lysate pre-treated with 30 mM EDTA. (C) Gradient profile and western blot analyses of a lysate pretreated with 50 U/ml RNase I.
To test further whether the migration pattern of Bfr1p in sucrose gradients reflected association with yeast polyribosomes, lysates were pre-treated with either 30 mM EDTA, or 50 U/ml RNase I (Promega) immediately prior to fractionation. EDTA chelates Mg2+ cations, resulting in dissociation of the small and large ribosomal subunits, which is reflected in gradient profiles (OD254) by the disappearance of 80S monosomes and polyribosomes, with a concomitant increase in the abundance of free ribosomal subunits (Fig. 2B). Under these conditions, the greatest intensity of HA-Bfr1p signal was seen only in the upper-most fractions of the gradient (Fig. 1B). Alternatively, pre-treatment of lysates with RNase, which results in inter-ribosomal severing of translating messages, gave rise to a large pool of single 80S monosomes (Fig. 1C). Again, HA-Bfr1p was shifted to the uppermost fractions of these gradients. As a positive control, α-FLAG western blot analyses were also performed on all samples, confirming the expected shift of FLAG-Scp160p to the upper fractions of these gradients. Together, these data demonstrate that like Scp160p, Bfr1p associates with yeast polyribosomes.
Figure 2.

RNA-dependent association of Bfr1p with Scp160p-containing mRNP complexes. (A) S300 gel filtration chromatography followed by western blot analysis of EDTA-treated lysates from yeast co-expressing FLAG-Scp160p and HA-Bfr1p. (B) α-FLAG purification and western blot analyses of S300 void fractions derived from the same strain as in (A), with and without RNase pre-treatment. An α-Pab1p western blot of the same samples is shown as a control. FT, flow-through fraction; E, elution fractions.
Association of Bfr1p and Scp160p is RNase sensitive
Previously, we have reported that Scp160p in yeast lysates remains in large complexes following treatment with EDTA, despite its dissociation from polyribosomes (7). To address whether Bfr1p would show a similar distribution, we performed gel-filtration chromatography on EDTA-treated lysates of cells expressing both FLAG-Scp160p and HA-Bfr1p. Western blot analyses of the resulting fractions demonstrated that Bfr1p, like Scp160p, remained exclusively in large complexes (Fig. 2A).
To determine whether the association of Bfr1p with EDTA-resistant Scp160p-containing complexes was dependent on RNA, void material from the gel-filtration column was treated with RNase immediately prior to loading onto a 1 ml α-FLAG affinity column. Figure 2B illustrates that, following this treatment, Bfr1p no longer co-purified with FLAG-Scp160p, demonstrating that this interaction was RNA-dependent. As a control, all fractions were also probed with a polyclonal antiserum against Pab1p, confirming the previously observed RNase-sensitive association of this protein with Scp160p in these samples (7).
Subcellular localization of Bfr1p
Scp160p has previously been observed by immunofluorescence microscopy to localize to the yeast nuclear envelope and rough endoplasmic reticulum (ER), where it ostensibly associates with membrane-bound polyribosomes (15). To determine whether Bfr1p displays a similar localization in cells, an N-terminal GFP tag was engineered onto the Bfr1p coding sequence, and the modified allele integrated into the genomic BFR1 locus in place of the native sequence. Cells remained morphologically indistinguishable from their wild-type counterparts, demonstrating functionality of the fusion protein. As shown in Figure 3A, GFP-Bfr1p, like GFP-Scp160p, localized predominantly to the cytoplasm with a concentration around the nuclear periphery.
Figure 3.
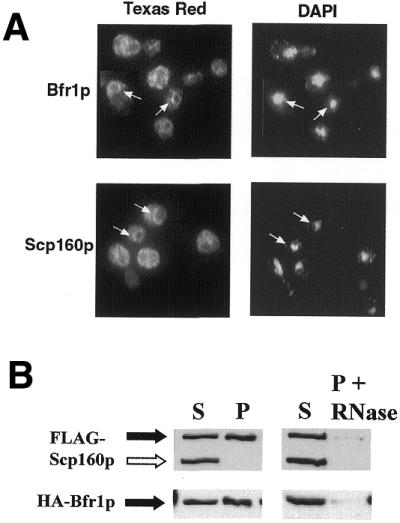
Cytoplasmic localization and membrane association of Bfr1p. (A) Anti-GFP immunofluorescence microscopy of fixed cells expressing either GFP-Bfr1 or Scp160-GFP. Bfr1p (top panel) shows an almost identical distribution to Scp160p (bottom panel), appearing cytoplasmic with an enrichment around the nuclear envelope/ER. Panels to the right demonstrate DAPI staining of nuclear and mitochondrial DNA in the same cells. Arrows indicate clear examples of perinuclear staining. (B) Western blot analyses following subcellular fractionation of lysates from yeast expressing HA-Bfr1p and FLAG-Scp160p. Pellets were washed in the presence (right panels) or absence (left panels) of 50 U/ml RNase I. The open arrow indicates the location of an endogenous α-FLAG cross-reacting protein which does not associate with membrane pellets in this assay.
To investigate further the potential membrane association of Bfr1p, we used differential centrifugation to separate crude cell lysates into a soluble cytoplasmic supernatant and a membrane pellet, as described previously (23). As shown in Figure 3B, the majority of Bfr1p associated with the membrane pellet in these assays, and could be released from the pellet by treatment with RNase. Consistent with published reports (8), a large percentage of Scp160p also associated with the membrane pellet, and was also released by RNase treatment. An endogenous yeast protein of unknown identity that cross-reacts with the α-FLAG antibody (Fig. 3B, open arrow) served as a convenient negative control for non-specific pellet association in this experiment.
Deletion of BFR1 results in loss of Scp160p-polyribosome association
To address the possibility of a functional link between Bfr1p and Scp160p, we created a bfr1 null strain that expressed FLAG-Scp160p (see Materials and Methods). These cells were grown to early log phase and subjected to sucrose gradient subcellular fractionation, as described above. α-FLAG western blot analyses of the resultant fractions revealed that in these cells Scp160p no longer migrated in the heaviest fractions of the gradient, indicating that the protein was no longer associated with polyribosomes (Fig. 4A). Gel-filtration of these lysates followed by α-FLAG chromatography demonstrated, however, that Scp160p nonetheless remained in large complexes containing Pab1p (Fig. 4C and D). These data support the conclusion that polyribosome association of Scp160p is a Bfr1p-dependent process, but that mRNP-association of Scp160p is not. In contrast, HA-Bfr1p expressed in scp160 null cells (Fig. 4B) demonstrated a sucrose gradient fractionation profile that was indistinguishable from that seen in SCP160 wild-type cells (Fig. 1A).
Figure 4.

Deletion of BFR1 disrupts the association of Scp160p with cytosolic polyribosomes. (A) Sucrose gradient analysis of bfr1 null yeast expressing FLAG-Scp160p. (B) Distribution of HA-Bfr1p in an scp160 null strain. (C) Gel filtration chromatography followed by western blot analysis of an EDTA-treated soluble lysate from bfr1 null yeast. The solid arrow indicates FLAG-Scp160p. The open arrow indicates an ∼100 kDa cross-reacting endogenous yeast protein. (D) α-FLAG purification of FLAG-Scp160p from a bfr1 null strain, demonstrating continued association with Pab1p. (E) Subcellular fractionation of lysates from bfr1 null yeast demonstrating that a small amount of FLAG-Scp160p remains associated with the membrane pellet (solid arrow). As in (C), the open arrow indicates an endogenous yeast cross-reacting protein. (F) Subcellular fractionation of lysates from scp160 null yeast demonstrating that the membrane association of Bfr1p is apparently unaffected by loss of Scp160p.
Finally, as an approach to test whether BFR1 deletion affected the membrane association of Scp160p, we fractionated cell lysates by differential centrifugation, as described above. In contrast to wild-type cells, which demonstrated a majority of Scp160p in the pellet (Fig. 3B), preparations from cells lacking Bfr1p demonstrated only a small fraction of the total Scp160p signal in the membrane pellet (Fig. 4E). Nonetheless, this signal remained sensitive to RNase treatment (Fig. 4E). Consistent with the sucrose fractionation data, Bfr1p also remained associated with membrane pellets in lysates derived from cells lacking scp160 (Fig. 4F).
Bfr1p associates with mRNP complexes even in the absence of Scp160p
To address the question of whether Bfr1p association with mRNP complexes was Scp160p-dependent, we prepared soluble lysates (see Materials and Methods) from both wild-type and scp160 null yeast expressing HA-Bfr1p, and size-fractionated these samples by gel-filtration column chromatography, as described above. Western blot analyses of the resulting fractions with 12CA5 antibody (Fig. 5A) demonstrated that the HA-Bfr1p protein remained predominantly in large complexes, regardless of whether Scp160p was present or not. In these experiments, we also observed a variable proportion of HA-Bfr1p signal that ran as an apparent monomer through the column. Although the significance of this apparently monomeric pool remains unclear, it may represent free HA-Bfr1p released from larger complexes that were disrupted in sample preparation.
Following column chromatography, fractions representing the large HA-Bfr1p-containing complexes (36–48 ml elution volume; Fig. 5A) were pooled and subjected to anti-HA affinity isolation, as described in Materials and Methods. Representative fractions from the different steps of this procedure were then subjected to PAGE followed by western blot analysis with both the 12CA5 and anti-Pab1p antibodies (Fig. 5B). The results clearly demonstrated that HA-Bfr1p remained in Pab1p-containing complexes despite the absence of Scp160p.
Yeast lacking both Scp160p and Bfr1p are viable
As a final test of the functional interaction of Bfr1p and Scp160p, we asked whether deletion of both genes would be synthetically lethal, by creating a diploid strain doubly heterozygous for both null alleles, sporulating and dissecting tetrads (17). In particular, we used a diploid strain of W303 yeast in which the coding region of one SCP160 allele was replaced by the HIS3 marker, and one BFR1 allele was disrupted with the KANR marker. Although the double null genotype was seen in lower frequency than would have been expected given random assortment and equal spore viability, we did obtain viable colonies that contained both disruption markers, confirmed at the molecular level by PCR of the appropriate loci. When these cells were examined microscopically, although they were clearly abnormal, they did not display any obvious morphological phenotypes beyond those seen in the single null strains. Further experiments will be required to define in quantitative terms any potential synergy of phenotypic outcome resulting from these two gene deletions.
DISCUSSION
Although Bfr1p was originally hypothesized to function in secretion and/or in spindle function in yeast, we recently reported an association of the protein with Scp160p-containing mRNP complexes, implicating a potential role for Bfr1p in cytoplasmic mRNA metabolism (7). The experiments reported here were designed to explore further the association of Bfr1p with mRNP complexes and with polyribosomes, as well as to probe whether the association between Bfr1p and Scp160p is not only physical but also functional. In particular, we have addressed four specific questions. (i) Does Bfr1p associate with polyribosomes? (2) Is the Bfr1p–Scp160p interaction RNA-dependent? (iii) Does loss of either Bfr1p or Scp160p alter the subcellular distribution of the other protein (in terms of association with mRNPs and polysomes)? (iv) Are cells deleted for both BFR1 and SCP160 viable?
To address the first question, we used sucrose gradient ultracentrifugation to demonstrate that Bfr1p, like Scp160p, co-sediments with large polyribosomes. Furthermore, following disruption of these polyribosomes with EDTA, gel filtration chromatography revealed that Bfr1p nonetheless remained predominantly in large complexes, as did Scp160p (7). However, while the sucrose gradient distribution profiles of Bfr1p and Scp160p clearly overlapped, they were not identical. Bfr1p appeared to be more concentrated in the fractions containing the heaviest polyribosomes, whereas Scp160p was more evenly distributed in fractions >40S. This result suggests that although they can interact, both proteins may also form complexes that are independent of one another. Additional data presented here further support this hypothesis.
To test whether the association of Scp160p and Bfr1p is RNA-dependent, we used affinity isolation of Scp160p-containing complexes in the presence and absence of RNase, followed by western blot analysis of the preparation. Bfr1p co-purified with Scp160p only in the absence of RNase, demonstrating that the association between these two proteins was RNA-dependent. In addition, we explored the sub-cellular localization of Bfr1p using both fluorescence microscopy and biochemical fractionation. As would be expected for a polyribosome-associated protein, Bfr1p appeared predominantly cytoplasmic with a concentration around the nuclear envelope, presumably where it is in contact with membrane-bound polyribosomes of the yeast rough ER. Differential centrifugation further demonstrated that a significant proportion of both Bfr1p and Scp160p associated with the membrane pellet, and were released by treatment with RNase. This observation supports the conclusion that both proteins are associated with membrane-bound polyribosomes. As mentioned in the Introduction, previous yeast two-hybrid studies have implicated an association of Bfr1p with the yeast spindle pole body (4–6); our results neither confirmed nor disproved this association.
The third question, regarding the possibility of a functional relationship between Bfr1p and Scp160p, was addressed biochemically. Using sucrose gradient fractionation, we found that in bfr1 null cells, Scp160p failed to associate with polyribosomes, thereby demonstrating a functional interaction between the two proteins. Although the exact nature of this interaction remains unclear, one candidate scenario would involve a role for Bfr1p in helping to bring Scp160p-containing mRNPs to polyribosomes for translation. Once specific mRNA binding targets for Scp160p have been identified, this will be a testable hypothesis. It is also important to recognize that deletion of BFR1 did not fully eliminate the association of Scp160p with the membrane pellet. There are two possible explanations for this result. One is that BFR1 deletion may have affected only the association of Scp160p with a sub-set of polyribosomes, leaving some of the membrane-bound Scp160p–polyribosome complexes intact. Since sucrose gradients were run using soluble lysates only, these membrane-bound Scp160p–containing complexes would not have been detected by this assay (Fig. 4A). Alternatively, Scp160p may contribute to RNase-sensitive membrane associated complexes other than, or in addition to, polyribosomes, that are bfr1-independent. In this light it is particularly interesting to note that although Bfr1p loss had a major impact on Scp160p subcellular distribution, the converse was not seen. Indeed, deletion of SCP160 did not appreciably affect the subcellular localization of Bfr1p, either in terms of polyribosome association (Fig. 4), or in terms of association with Pab1p-containing mRNP complexes (Fig. 5). Studies are currently underway to explore the significance of this observation.
Finally, we have shown by standard genetic techniques that deletion of both SCP160 and BFR1 in the same cell is not a lethal event. Clearly, further studies will be required to define in more quantitative terms the phenotype of the double-null strain in comparison with both single-null strains and wild-type cells. Nonetheless, viability of the double-null strain provides an opportunity for further analyses of the structures and pathways ostensibly impacted by these two proteins.
Considered together, our data support the hypothesis that Bfr1p functions in mRNA metabolism in yeast, and suggest that the observed phenotypes of BFR1 deletion and over-expression may reflect indirect rather than direct effects, resulting from the aberrant expression of other yeast genes. A preliminary examination of the Bfr1p sequence suggests that three regions of the protein are consistent with the formation of coiled coils (SwissProt), structural motifs implicated in mediating a variety of protein–protein interactions (28). Clearly, the presence of these motifs is consistent with the proposed function of Bfr1p as a component of a large complex. The data presented here further raise the intriguing possibility that Bfr1p may provide a link between mRNA metabolism, the chromosomal segregation machinery and perhaps also secretion in yeast. Indeed, there is precedence for this scenario. For example, the ribonuclease Xrn1p is believed to be the major cytoplasmic mRNase in yeast; however, deletion of the gene causes increased sensitivity to the microtubule destabilizing drug benomyl (29). Additionally, mutations in either of two genes encoding the translation release factors eRF3 and eRF1 also cause increased sensitivity to benomyl, as well as impaired chromosome stability (30). With regard to Bfr1p, future studies will be required to test this hypothesis.
Acknowledgments
ACKNOWLEDGEMENTS
We gratefully acknowledge the assistance of the following people: Maurice Swanson for the anti-Pab1p monoclonal antibody, Yue Feng and Devin Absher for assistance in polyribosome analysis, Keith Wilkinson for materials and assistance in α-FLAG purification, David Pallas and Carlos Moreno for materials and assistance in α-HA purification, Kristen Riehman and Alice Watson for excellent technical assistance, and Charles Buck, Anita Corbett, Steve Warren, and Keith Wilkinson for many helpful discussions. This research was supported in part by NIH grant 1P01HD35576-010006 (to J.L.F.K.), and in part by an Emory University Research Committee Grant (to J.L.F.K.).
References
- 1.Jackson C.L. and Kepes,F. (1994) BFR1, a multicopy suppressor of brefeldin A-induced lethality, is implicated in secretion and nuclear segregation in Saccharomyces cerevisiae. Genetics, 137, 423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klausner R.D., Donaldson,J.G. and Lippincott-Schwartz,J. (1992) Brefeldin A: insights into the control of membrane traffic and organelle structure. J. Cell Biol., 116, 1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lippincott-Schwartz J., Donaldson,J.G., Schweizer,A., Berger,E.G., Hauri,H.P., Yuan,L.C. and Klausner,R.D. (1990) Microtubule-dependent retrograde transport of proteins into the ER in the presence of brefeldin A suggests an ER recycling pathway. Cell, 60, 821–836. [DOI] [PubMed] [Google Scholar]
- 4.Xue Z., Shan,X., Sinelnikov,A. and Melese,T. (1996) Yeast mutants that produce a novel type of ascus containing asci instead of spores. Genetics, 144, 979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wigge P.A., Jensen,O.N., Holmes,S., Soues,S., Mann,M. and Kilmartin,J.V. (1998) Analysis of the Saccharomyces spindle pole by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry. J. Cell Biol., 141, 967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schramm C., Elliott,S., Shevchenko,A. and Schiebel,E. (2000) The Bbp1p–Mps2p complex connects the SPB to the nuclear envelope and is essential for SPB duplication. EMBO J., 19, 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lang B.D. and Fridovich-Keil,J.L. (2000) Scp160p, a multiple KH-domain protein, is a component of mRNP complexes in yeast. Nucleic Acids Res., 28, 1576–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weber V., Wernitznig,A., Hager,G., Harata,M., Frank,P. and Wintersberger,U. (1997) Purification and nucleic-acid-binding properties of a Saccharomyces cerevisiae protein involved in the control of ploidy. Eur. J. Biochem., 249, 309–317. [DOI] [PubMed] [Google Scholar]
- 9.Ashley C.T.,Jr, Wilkinson,K.D., Reines,D. and Warren,S.T. (1993) FMR1 protein: conserved RNP family domains and selective RNA binding. Science, 262, 563–566. [DOI] [PubMed] [Google Scholar]
- 10.Siomi H., Matunis,M.J., Michael,W.M. and Dreyfuss,G. (1993) The pre-mRNA binding K protein contains a novel evolutionarily conserved motif. Nucleic Acids Res., 21, 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidt C., Henkel,B., Poschl,E., Zorbas,H., Purschke,W.G., Gloe,T.R. and Muller,P.K. (1992) Complete cDNA sequence of chicken vigilin, a novel protein with amplified and evolutionary conserved domains. Eur. J. Biochem., 206, 625–634. [DOI] [PubMed] [Google Scholar]
- 12.Plenz G., Kugler,S., Schnittger,S., Rieder,H., Fonatsch,C. and Muller,P.K. (1994) The human vigilin gene: identification, chromosomal localization and expression pattern. Hum. Genet., 93, 575–582. [DOI] [PubMed] [Google Scholar]
- 13.Dodson R.E. and Shapiro,D.J. (1997) Vigilin, a ubiquitous protein with 14 K homology domains, is the estrogen-inducible vitellogenin mRNA 3′-untranslated region-binding protein. J. Biol. Chem., 272, 12249–12252. [DOI] [PubMed] [Google Scholar]
- 14.Cortes A., Huertas,D., Fanti,L., Pimpinelli,S., Marsellach,F.X., Pina,B. and Azorin,F. (1999) DDP1, a single-stranded nucleic acid-binding protein of Drosophila, associates with pericentric heterochromatin and is functionally homologous to the yeast Scp160p, which is involved in the control of cell ploidy. EMBO J., 18, 3820–3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wintersberger U., Kuhne,C. and Karwan,A. (1995) Scp160p, a new yeast protein associated with the nuclear membrane and the endoplasmic reticulum, is necessary for maintenance of exact ploidy. Yeast, 11, 929–944. [DOI] [PubMed] [Google Scholar]
- 16.Gietz R.D. and Sugino,A. (1988) New yeast–Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene, 74, 527–534. [DOI] [PubMed] [Google Scholar]
- 17.Guthrie C. and Fink,G.R. (1991) Guide to yeast genetics and molecular biology. Methods Enzymol., 194. [PubMed] [Google Scholar]
- 18.Longtine M.S., McKenzie,A.,III, Demarini,D.J., Shah,N.G., Wach,A., Brachat,A., Philippsen,P. and Pringle,J.R. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast, 14, 953–961. [DOI] [PubMed] [Google Scholar]
- 19.Stansfield I., Grant,G.M., Akhmaloka and Tuite,M.F. (1992) Ribosomal association of the yeast SAL4 (SUP45) gene product: implications for its role in translation fidelity and termination. Mol. Microbiol., 6, 3469–3478. [DOI] [PubMed] [Google Scholar]
- 20.Schneider C., Newman,R.A., Sutherland,D.R., Asser,U. and Greaves,M.F. (1982) A one-step purification of membrane proteins using a high efficiency immunomatrix. J. Biol. Chem., 257, 10766–10769. [PubMed] [Google Scholar]
- 21.Koepp D.M., Corbett,A.H., Kahana,J. and Silver,P.A. (1997) Indirect immunofluorescence in Saccharomyces cerevisiae. In Spector,D. Goldman,R. and Leiwand,L. (eds), Cells: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
- 22.Kahana J.A., Schlenstedt,G., Evanchuk,D.M., Geiser,J.R., Hoyt,M.A. and Silver,P.A. (1998) The yeast dynactin complex is involved in partitioning the mitotic spindle between mother and daughter cells during anaphase B. Mol. Biol. Cell, 9, 1741–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stoltenburg R., Wartmann,T., Kunze,I. and Kunze,G. (1995) Reliable method to prepare RNA from free and membrane-bound polysomes from different yeast species. Biotechniques, 18, 564–566, 568. [PubMed] [Google Scholar]
- 24.Traub L.M., Kornfeld,S. and Ungewickell,E. (1995) Different domains of the AP-1 adaptor complex are required for Golgi membrane binding and clathrin recruitment. J. Biol. Chem., 270, 4933–4942. [DOI] [PubMed] [Google Scholar]
- 25.Fridovich-Keil J.L., Quimby,B.B., Wells,L., Mazur,L.A. and Elsevier,J.P. (1995) Characterization of the N314D allele of human galactose-1-phosphate uridylyltransferase using a yeast expression system. Biochem. Mol. Med., 56, 121–130. [DOI] [PubMed] [Google Scholar]
- 26.Anderson J.T., Paddy,M.R. and Swanson,M.S. (1993) PUB1 is a major nuclear and cytoplasmic polyadenylated RNA-binding protein in Saccharomyces cerevisiae. Mol. Cell Biol., 13, 6102–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson J.T., Wilson,S.M., Datar,K.V. and Swanson,M.S. (1993) NAB2: a yeast nuclear polyadenylated RNA-binding protein essential for cell viability. Mol. Cell Biol., 13, 2730–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lupas A. (1997) Predicting coiled-coil regions in proteins. Curr. Opin. Struct. Biol., 7, 388–393. [DOI] [PubMed] [Google Scholar]
- 29.Johnson A.W. (1997) Rat1p and Xrn1p are functionally interchangeable exoribonucleases that are restricted to and required in the nucleus and cytoplasm, respectively. Mol. Cell. Biol., 17, 6122–6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borchsenius A.S., Tchourikova,A.A. and Inge-Vechtomov,S.G. (2000) Recessive mutations in SUP35 and SUP45 genes coding for translation release factors affect chromosome stability in Saccharomyces cerevisiae. Curr. Genet., 37, 285–291. [DOI] [PubMed] [Google Scholar]