

Abstract
Ferroptosis is a recently identified iron‐dependent form of nonapoptotic cell death implicated in brain, kidney, and heart pathology. However, the biological roles of iron and iron metabolism in ferroptosis remain poorly understood. Here, we studied the functional role of iron and iron metabolism in the pathogenesis of ferroptosis. We found that ferric citrate potently induces ferroptosis in murine primary hepatocytes and bone marrow–derived macrophages. Next, we screened for ferroptosis in mice fed a high‐iron diet and in mouse models of hereditary hemochromatosis with iron overload. We found that ferroptosis occurred in mice fed a high‐iron diet and in two knockout mouse lines that develop severe iron overload (Hjv–/– and Smad4Alb/Alb mice) but not in a third line that develops only mild iron overload (Hfe –/– mice). Moreover, we found that iron overload–induced liver damage was rescued by the ferroptosis inhibitor ferrostatin‐1. To identify the genes involved in iron‐induced ferroptosis, we performed microarray analyses of iron‐treated bone marrow–derived macrophages. Interestingly, solute carrier family 7, member 11 (Slc7a11), a known ferroptosis‐related gene, was significantly up‐regulated in iron‐treated cells compared with untreated cells. However, genetically deleting Slc7a11 expression was not sufficient to induce ferroptosis in mice. Next, we studied iron‐treated hepatocytes and bone marrow–derived macrophages isolated from Slc7a11–/– mice fed a high‐iron diet. Conclusion: We found that iron treatment induced ferroptosis in Slc7a11–/– cells, indicating that deleting Slc7a11 facilitates the onset of ferroptosis specifically under high‐iron conditions; these results provide compelling evidence that iron plays a key role in triggering Slc7a11‐mediated ferroptosis and suggest that ferroptosis may be a promising target for treating hemochromatosis‐related tissue damage. (Hepatology 2017;66:449–465).
Abbreviations
- ALT
alanine aminotransferase
- ANOVA
analysis of variance
- ARE
antioxidant response element
- BMDM
bone marrow–derived macrophage
- ER
endoplasmic reticulum
- ERK
extracellular signal–regulated kinase
- FAC
ferric citrate
- Ferr‐1
ferrostatin‐1
- Fpn1
ferroportin‐1
- Gpx4
glutathione peroxidase 4
- GSH
glutathione
- HFE
hemochromatosis protein
- HH
hereditary hemochromatosis
- HID
high‐iron diet
- HJV
hemojuvelin
- JNK
c‐Jun NH2‐terminal kinase
- LID
low‐iron diet
- MAPK
mitogen‐activated protein kinase
- MDA
malondialdehyde
- NADPH
nicotinamide adenine dinucleotide phosphate, reduced form
- Nrf2
nuclear erythroid 2 p45‐related factor 2
- p‐
phosphorylated
- ROS
reactive oxygen species
- SLC7A11
solute carrier family 7, member 11
Iron is an essential element for maintaining health in virtually all organisms. Although free iron is highly reactive and toxic, iron is required for the proper function of many proteins, including enzymes that regulate the respiratory complex, oxygen transport, and DNA synthesis. In mammals, the uptake, transport, use, and storage of iron are tightly coordinated by various proteins and pathways in order to maintain iron homeostasis at both the cellular and systemic levels.1 Thus, iron disorders—including both iron deficiency and iron overload—can disrupt normal cellular function, leading to disease.2
Hereditary hemochromatosis (HH) is an iron‐overload disease caused by mutations in genes whose protein products limit iron absorption, including hemochromatosis protein (HFE), hemojuvelin (HJV), transferrin receptor 2, SLC40A1 (Ferroportin‐1, Fpn1), and hepcidin antimicrobial peptide. In HH, iron accumulates in the parenchymal cells of various organs. This excess iron generates reactive oxygen species (ROS) through the Fenton reaction, thereby inducing cell death and global oxidative damage, ultimately leading to severe chronic complications, including hepatic cirrhosis, diabetes, and heart disease.1, 3, 4 Given its genomics‐based etiology, patients with HH currently have relatively few therapeutic options. Therefore, finding new therapeutic targets of iron‐related pathological processes is essential for treating HH‐associated complications.
Ferroptosis is a recently identified form of cell death that was first observed in Ras mutant tumor cells treated with oncogenic Ras‐selective lethal small molecules called ferroptosis inducers, which include erastin and RSL3.5 Ferroptosis is morphologically, biochemically, and genetically distinct from other forms of cell death, including apoptosis, necrosis, and autophagy.6, 7 Ferroptosis is dependent upon intracellular iron and can be specifically rescued using an iron chelator. Currently, three biomarkers can be used to detect ferroptosis: lipid peroxidation, increased PTGS2 expression, and decreased content of the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH).8, 9 Interestingly, ferroptosis can be inhibited by the specific inhibitor ferrostatin‐1 (Ferr‐1) but not by inhibitors of other forms of cell death.10 Recent studies indicate that ferroptosis contributes to pathological process in a variety of diseases and conditions, including acute organ failure secondary to ischemia/reperfusion, Huntington disease, and other neurodegenerative diseases.10 Thus, inhibiting ferroptosis may represent a promising new approach for treating cell death–related diseases.11 However, the precise roles of iron and iron metabolism in ferroptosis are currently unknown.
Although the regulatory mechanisms that underlie ferroptosis are poorly understood, several molecules that play a role in iron and redox metabolism have been implicated in ferroptosis. For example, glutathione peroxidase 4 (GPX4), nuclear erythroid 2 p45‐related factor 2 (Nrf2), and metallothionein‐1G are negative regulators of ferroptosis,8, 12, 13 whereas transferrin receptor 1, heme oxygenase 1 (HO1), and glutaminase 2 (GLS2) appear to promote ferroptosis.14, 15 Moreover, inhibiting GPX4 using an RSL3 analog can induce ferroptosis and suppress tumor growth.8 Therefore, identifying novel regulators of ferroptosis is important for understanding ferroptosis and for developing therapies for ferroptosis‐related diseases.
System xc –, a heterodimer composed of solute carrier family 7, member 11 (SLC7A11) and SLC3A2, is a cystine/glutamate antiporter that mediates the efflux of cellular glutamate and the influx of cystine at a 1:1 molar ratio.16 Upon entering the cell, cystine is reduced to form cysteine, the limiting amino acid in the synthesis of glutathione (GSH).17 GSH is an ROS scavenger and the most abundant cellular antioxidant, and reducing GSH levels by deleting the enzyme glutamate cysteine ligase induces ferroptosis.18 Erastin can deplete intracellular GSH by targeting system xc –, suggesting that system xc – and/or its constituent components plays a role in regulating ferroptosis.5 The SLC7A11 subunit of system xc – contains 12 transmembrane domains and is the pore‐forming subunit19; moreover, recent in vitro data indicate that inhibiting SLC7A11 can induce ferroptosis. For example, pharmacological inhibition of SLC7A11 by either erastin or sulfasalazine induces ferroptosis.20 Moreover, the tumor suppressor protein p53 can induce ferroptosis by suppressing transcription of the SLC7A11 gene.21, 22 Despite these compelling in vitro findings, however, the role of SLC7A11 in regulating ferroptosis has not been studied in vivo.
Here, we studied the role of iron homeostasis in Slc7a11‐mediated ferroptosis and found that iron overload is sufficient to trigger ferroptosis both in vitro and in vivo. We also found that Slc7a11 expression is up‐regulated by iron through the ROS–Nrf2–antioxidant response element (ARE) axis. In addition, using Slc7a11 knockout mice, we found that the absence of Slc7a11 is not sufficient to induce ferroptosis under basal conditions but facilitates iron overload–induced ferroptosis due to impaired cystine uptake and increased ROS production, suggesting that Slc7a11 confers protection against ferroptosis during iron overload. Finally, we found that iron‐induced ferroptosis is not mediated by endoplasmic reticulum (ER) stress or the mitogen‐activated protein kinase (MAPK) pathway. Thus, our results suggest that ferroptosis is a potential therapeutic target for treating iron overload–associated diseases, including hemochromatosis.
Materials and Methods
MICE
Hfe–/–, Hjv–/–, and Fpn1flox/flox mice (129/SvEvTac background) were provided by Dr. Nancy C. Andrews.23, 24, 25 Smad4flox/flox and Smad4Alb/Alb mice (129/SvEvTac background) were provided by Dr. Chu‐Xia Deng.26 Fpn1LysM/LysM mice were have been described.27, 28 Slc7a11–/– mice (C57BL/6J background) were provided by Dr. Hideyo Sato.29 Unless stated otherwise, the mice were fed a standard AIN‐76A diet containing 50 mg iron/kg (Research Diets, Inc., New Brunswick, NJ). Both the low‐iron diet (LID; 0.9 mg iron/kg) and the high‐iron diet (HID; 8.3 g carbonyl iron/kg) were egg white–based AIN‐76A diets (Research Diets, Inc.). All experimental protocols were approved by the Institutional Animal Care and Use Committee of the Laboratory Animal Center, Zhejiang University.
IRON PARAMETERS
Serum iron and tissue non‐heme iron were measured as described.27, 28
LIVER DAMAGE AND FIBROSIS
Serum alanine aminotransferase (ALT) was measured using an enzymatic assay kit (ShenSuoYouFu Diagnostics, Shanghai, China). Liver and spleen sections were stained with sirius red, and stained sections were analyzed using a positive pixel‐count algorithm in the software program ImageJ.30
ISOLATION AND CULTURE OF PRIMARY HEPATOCYTES AND BONE MARROW–DERIVED MACROPHAGES
Hepatocytes and bone marrow–derived macrophages (BMDMs) were isolated and cultured as described.27, 28 Notably, 50 μM β‐mercaptoethanol was included in the conditioned medium, while Slc7a11–/– BMDMs were being differentiated.31 No differences in differentiation or viability were observed between wild‐type and Slc7a11–/– BMDMs (data not shown).
IN VITRO DRUG TREATMENT
Ferric citrate (FAC) was used at the indicated concentrations. The other inhibitors were used at the following concentrations: Ferr‐1, 2 μM; necrostatin‐1, 10 μg/mL; Z‐VAD‐FMK 10 μg/mL; SP600125, 10 μM; SB202190, 10 μM; PD98059, 10 μM; β‐mercaptoethanol, 100 μM; trigonelline, 100 μM; and 3‐methylademine, 2 mM. Except where indicated otherwise, all drugs were purchased from Sigma‐Aldrich.
IN VIVO TREATMENT WITH Ferr‐1
Mice were given daily intraperitoneal injections of either phosphate‐buffered saline (control) or Ferr‐1 (2.5 μmol/kg body weight) for 3 weeks and then sacrificed.
CELL VIABILITY, ROS, LIPID PEROXIDATION MEASUREMENTS, AND TRANSMISSION ELECTRON MICROSCOPY
Cell viability was measured using the Cell Counting Kit‐8 viability assay (Sigma‐Aldrich). ROS and lipid peroxidation were measured using fluorescence‐activated cell sorting with H2DCFDA (Sigma‐Aldrich) or C11‐BODIPY (581/591) (Invitrogen) staining.5 Transmission electron microscopy was performed using a Tecnai 10 microscope (FEI, Hillsboro, OR) at the Electron Microscopy Core Facility, Zhejiang University.
MEASUREMENT OF MALONDIALDEHYDE, NADPH, AND GSH CONTENT
Hepatic malondialdehyde (MDA) content was measured using a kit (Beyotime, Haimen, China). NADPH content was measured using a fluorometric nicotinamide adenine dinucleotide phosphate/NADPH assay (Abcam). GSH content was measured using the GSH‐Glo Glutathione Assay (Promega).
REAL‐TIME PCR ANALYSIS
Total RNA was isolated using TRIzol (Invitrogen), then reverse‐transcribed into complementary DNA using the PrimeScript RT kit (Takara). The sequences of the primers are provided in Supporting Table S1. Real‐time PCR was performed using the two‐step quantitative RT‐PCR method (Bio‐Rad).
MICROARRAY ANALYSIS
Fpn1flox/flox and Fpn1LysM/LysM BMDMs were cultured for 12 hours in the presence or absence of 100 μM FAC.28 RNA was extracted and reverse‐transcribed into complementary DNA as described above. The complementary DNA was then processed for expression microarray analysis in accordance with Affymetrix's instructions. The fold change in expression between FAC‐treated and untreated cells was calculated for each gene, and genes that were up‐regulated >2‐fold in both the Fpn1flox/flox and Fpn1LysM/LysM BMDMs are listed in Supporting Table S2.
NUCLEAR PROTEIN ISOLATION AND WESTERN BLOT ANALYSIS
Nuclear proteins were extracted using the Nuclear and Cytoplasmic Protein Extraction Kit (Thermo‐Fisher). The following primary antibodies were used for western blot analysis: anti–phosphorylated c‐Jun NH2‐terminal kinase (P‐JNK; no. 9255; Cell Signaling Technology), anti‐JNK (no. 9525; Cell Signaling Technology), anti–phosphorylated extracellular signal–regulated kinase (P‐Erk) 1/2 (no. 4376; Cell Signaling Technology), anti‐Erk1/2 (no. 5013; Cell Signaling Technology), anti‐P‐p38 (no. 9216; Cell Signaling Technology), anti‐p38 (no. 9212; Cell Signaling Technology), anti‐LC3I/II (no. 4108; Cell Signaling Technology), anti‐Nrf2 (no. 137550; Abcam), anti‐p62 (no. 109012; Abcam), and anti‐Slc7a11 (no. 37185; Abcam).
STATISTICAL ANALYSES
All summary data are presented as the mean ± standard error of the mean. Groups were compared using the Student t test or one‐way analysis of variance (ANOVA) with Tukey's post hoc test, where appropriate. Differences with P < 0.05 were considered statistically significant. All statistical analyses were performed using the R software package.
Results
IRON OVERLOAD INDUCES FERROPTOSIS BOTH IN VITRO AND IN VIVO
Previous studies found that iron overload causes several forms of cell death, including apoptosis and necrosis32, 33; however, whether iron overload also causes ferroptosis is currently unknown. Hepatocytes and macrophages play key roles in iron metabolism by mediating iron storage and recycling.1 Therefore, to investigate whether iron overload causes ferroptosis, mouse primary hepatocytes and BMDMs were treated with FAC, and ferroptosis was assessed by measuring lipid peroxidation, Ptgs2 mRNA levels, NADPH content, and cell viability. Consistent with iron overload–induced ferroptosis, we found that FAC treatment significantly increased lipid peroxidation (Fig. 1A), increased Ptgs2 mRNA levels (Fig. 1B), and decreased NADPH content (Fig. 1C). Iron overload–induced ferroptosis was confirmed using the specific ferroptosis inhibitors ferrostatin‐1 (Ferr‐1) and deferoxamine (DFO), which significantly reversed FAC‐induced lipid peroxidation, Ptgs2 mRNA levels, NADPH content (Fig. 1A‐C), and cell death (Fig. 1D). In contrast, inhibitors of other forms of cell death, including Z‐VAD‐FMK (an apoptosis inhibitor) and necrostatin‐1 (a necroptosis inhibitor), failed to rescue iron overload–induced ferroptosis (Fig. 1A‐C). These in vitro data indicate that iron overload induces ferroptosis.
Figure 1.
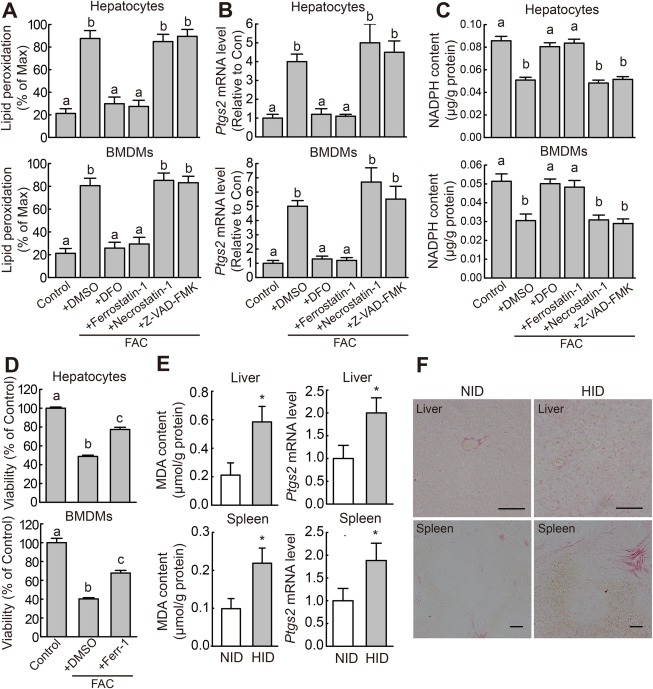
Iron overload induces ferroptosis both in vitro and in vivo. (A) Lipid peroxidation, (B) Ptgs2 mRNA level, and (C) NADPH content were measured in mouse primary hepatocytes and BMDMs treated for 12 hours with or without 100 μM FAC and the indicated specific inhibitors of cell death; lipid peroxidation was measured using C11‐BODIPY staining. (D) Cell viability was measured in mouse primary hepatocytes and BMDMs treated for 48 hours with or without 1 mM FAC and 10 μM Ferr‐1. The data in (A‐D) are representative of three independent experiments. (E) MDA content and Ptgs2 mRNA were measured in the indicated tissues of male wild‐type mice that were fed a normal diet (white bars, n = 6 mice/group) or the HID (gray bars, n = 6 mice/group) for 8 weeks after weaning. (F) Liver sections were obtained from the same animals shown in (E) and stained with sirius red; scale bars represent 100 μm. In (B) and (E), mRNA levels were normalized to β‐actin mRNA and are expressed relative to the mean value of the control cells and normal diet–fed mice, respectively. Significance in (A‐D) was calculated using a one‐way ANOVA with Tukey's post hoc test; groups labeled without a common letter were significantly different (P < 0.05). Significance in (E) and (F) was calculated using the Student t test; *P < 0.05. Abbreviations: DFO, deferoxamine; DMSO, dimethyl sulfoxide; HID, high‐iron diet; NID, normal‐iron diet.
Next, we investigated whether iron overload induces ferroptosis in vivo by measuring ferroptosis in iron‐overloaded mice. To increase their tissue iron content, wild‐type mice were fed the HID after weaning (Supporting Fig. S1A‐D). Based on previous reports,34 tissue MDA content can be used as a measure of lipid peroxidation in tissues. Compared with control mice fed a standard diet, HID‐fed mice had increased levels of lipid peroxidation and Ptgs2 mRNA in the liver and spleen (Fig. 1E) and reduced NADPH content in the liver (Supporting Fig. S1E). We then performed sirius red staining of liver and spleen sections to measure collagen deposits, a marker for fibrosis. HID‐fed mice had increased levels of collagen deposits in the liver and spleen (Fig. 1F), indicating that iron overload causes severe tissue fibrosis. Together, these data indicate that iron overload can induce ferroptosis in vivo.
FERROPTOSIS INDUCTION IN MOUSE MODELS OF HEMOCHROMATOSIS
HH is the most common inherited condition with iron overload–associated tissue damage, including liver damage and fibrosis.30, 33 To investigate whether ferroptosis is associated with HH, we measured ferroptosis in two classic mouse models of HH (Hfe–/– and Hjv–/– mice) and in one HH‐like mouse model (Smad4Alb/Alb mice); these models develop systemic iron overload (Fig. 2A; Supporting Fig. S2A,B) due to impaired hepcidin expression in the liver.24, 25, 26 Under normal dietary iron, the hepatic iron content in these mice was 5‐fold to 10‐fold higher than in wild‐type mice (Fig. 2A). Consistent with increased hepatic iron content, both Hjv–/– and Smad4Alb/Alb mice had significantly higher hepatic MDA and Ptgs2 mRNA levels, as well as lower hepatic NADPH content, compared to both wild‐type and Hfe–/– mice; moreover, no significant difference was found between wild‐type and Hfe–/– mice (Fig. 2B‐D). On the other hand, Hfe–/–, Hjv–/–, and Smad4Alb/Alb mice do not develop splenic iron accumulation (Supporting Fig. S2C). Thus, splenic lipid peroxidation and Ptgs2 mRNA levels were similar between the wild‐type and HH mouse models (Supporting Fig. S2D,E). Next, mice from all three transgenic lines were fed the LID for 3‐5 weeks after weaning; wild‐type mice were fed a standard diet containing normal iron content. Using this protocol, the serum and hepatic iron contents in all three LID‐fed mouse lines were similar to those in wild‐type mice fed a standard diet (Fig. 2A; Supporting Fig. S2A,B). Notably, the LID fully rescued the ferroptosis phenotype in the transgenic mouse models, including hepatic MDA content, Ptgs2 expression, and NADPH content (Fig. 2B‐D).
Figure 2.
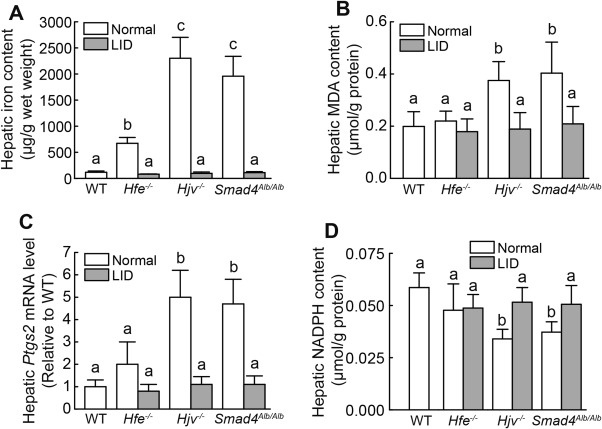
Mouse models of hemochromatosis develop ferroptosis. (A) Iron content, (B) MDA content, (C) Ptgs2 mRNA, and (D) NADPH content were measured in livers of 8‐week‐old male wild‐type, Hfe–/–, Hjv–/–, and Smad4Alb/Alb mice that were fed a normal diet (white bars, n = 6‐10 mice/group) or an LID (gray bars, n = 5 mice/group). mRNA levels in (C) were normalized to β‐actin mRNA and are expressed relative to the mean value of the wild‐type group. Significance was calculated using a one‐way ANOVA with Tukey's post hoc test, and groups labeled without a common letter were significantly different (P < 0.05). Abbreviation: LID, low‐iron diet; WT, wild type.
To confirm that ferroptosis plays a role in hemochromatosis‐associated liver damage, Hjv–/– and Smad4Alb/Alb mice were treated daily with the ferroptosis inhibitor Ferr‐1 for 3 weeks. Compared to untreated Smad4Alb/Alb mice, Ferr‐1‐treated Hjv–/– and Smad4Alb/Alb mice had significantly lower hepatic MDA and Ptgs2 mRNA levels and increased hepatic NADPH content (Fig. 3A‐C). Next, we assessed liver damage by measuring serum ALT levels and hepatic fibrosis. Serum ALT levels in Hjv–/– and Smad4Alb/Alb mice were >2‐fold higher than in wild‐type mice, and Ferr‐1‐treated Hjv–/– and Smad4Alb/Alb mice had significantly lower ALT levels compared to untreated Hjv–/– and Smad4Alb/Alb mice (Fig. 3D). Moreover, Hjv–/– and Smad4Alb/Alb mice had more collagen deposits than wild‐type mice, and Ferr‐1‐treated Hjv–/– and Smad4Alb/Alb mice had significantly fewer collagen deposits compared to untreated knockout mice (Fig. 3E). Interestingly, Ferr‐1 treatment had no effect on hepatic iron content in Hjv–/– or Smad4Alb/Alb mice (Supporting Fig. S2F). Taken together, these findings indicate that iron overload induces ferroptosis in HH mice.
Figure 3.
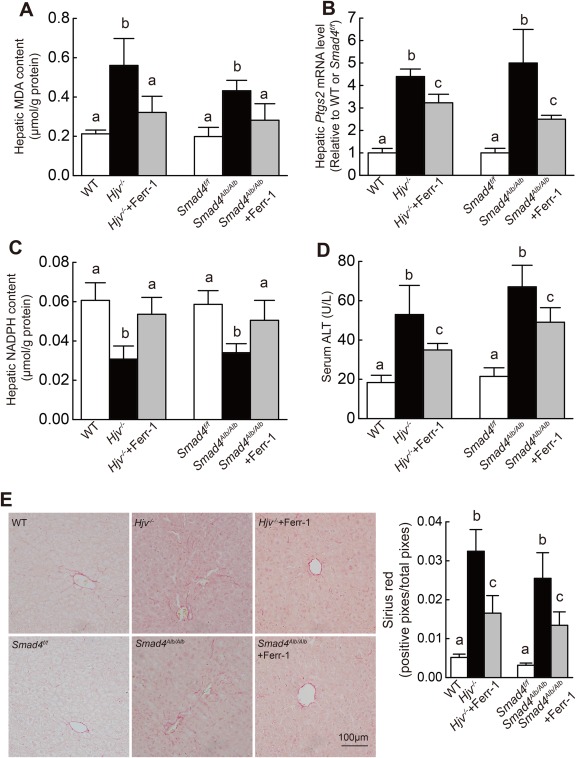
The ferroptosis inhibitor Ferr‐1 attenuates iron overload–induced liver damage in Hjv–/– and Smad4Alb/Alb mice. (A) Hepatic MDA content, (B) hepatic Ptgs2 mRNA levels, (C) hepatic NADPH content, and (D) serum ALT levels were measured in 8‐week‐old wild‐type, Hjv–/–, Smad4flox/flox (Smad4f/f), and/or Smad4Alb/Alb mice treated with or without Ferr‐1; n = 6 mice/group. (E) (left) Liver sections were obtained from the indicated mice and stained with sirius red; (right) summary of staining intensity. mRNA levels in (B) were normalized to β‐actin mRNA and are expressed relative to the mean value of the WT or Smad4flox/flox group. Significance was calculated using a one‐way ANOVA with Tukey's post hoc test, and groups labeled without a common letter were significantly different (P < 0.05). Abbreviation: WT, wild type.
IRON UP‐REGULATES Slc7a11 EXPRESSION THROUGH THE ROS–Nrf2–ARE AXIS
FPN1 is the only known iron exporter, and genetic deletion of Fpn1 in mice increases cellular iron burden.27, 28 To screen for candidate genes involved in iron overload–induced ferroptosis, we performed gene expression profiling using microarray analysis with Fpn1flox/flox and Fpn1LysM/LysM BMDMs treated with FAC or not. We then filtered the resulting data set for genes that were up‐regulated in both Fpn1flox/flox and Fpn1LysM/LysM BMDMs by >2‐fold following FAC treatment. This analysis identified the Slc7a11 gene, which encodes a subunit of the cystine/glutamate antiporter system xc –. Specifically, Slc7a11 expression was up‐regulated >2‐fold in FAC‐treated Fpn1LysM/LysM BMDMs compared to FAC‐treated Fpn1flox/flox BMDMs (Supporting Table S2).
Next, to test whether iron plays a role in regulating Slc7a11 expression, primary hepatocytes and BMDMs were obtained from wild‐type mice and treated with FAC, after which Slc7a11 mRNA and Slc7a11 protein levels were measured. In both primary hepatocytes and BMDMs, FAC treatment significantly increased Slc7a11 expression (Fig. 4A, B) in a time‐dependent and dose‐dependent manner. Moreover, HID‐fed wild‐type mice developed severe iron overload (Supporting Fig. S1) and had increased Slc7a11 expression in the liver and spleen (Fig. 4C). Finally, all three HH mouse lines (Hfe–/–, Hjv–/–, and Smad4Alb/Alb) had increased hepatic Slc7a11 mRNA levels compared to wild‐type mice (Fig. 4C). Taken together, these data indicate that a change in iron levels regulates Slc7a11 expression.
Figure 4.
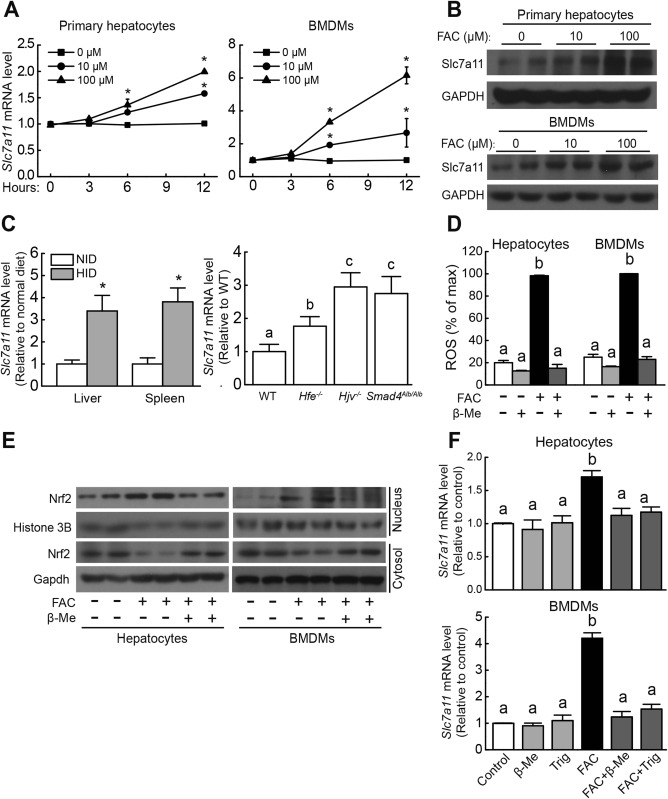
Iron up‐regulates Slc7a11 expression. (A) Slc7a11 mRNA was measured in mouse primary hepatocytes (left) and BMDMs (right) treated with either 10 μM or 100 μM FAC for the indicated times. (B) Slc7a11 protein was measured in mouse primary hepatocytes and BMDMs treated with 0, 10, or 100 μM FAC for 12 hours. (C) (left) Slc7a11 mRNA was measured in the liver and spleen of male wild‐type mice fed either a normal diet (white bars, n = 6 mice/group) or the HID (gray bars, n = 6 mice/group) for 8 weeks after weaning; (right) Slc7a11 mRNA was measured in the liver of 8‐week‐old male wild‐type, Hfe–/–, Hjv–/–, and Smad4Alb/Alb mice fed a normal diet (n = 6 mice/group). (D) Relative ROS levels were measured using H2DCFDA staining in mouse primary hepatocytes and BMDMs treated with either phosphate‐buffered saline or 100 μM β‐mercaptoethanol and/or 100 μM FAC for 12 hours. (E) Cytosolic and nuclear Nrf2 protein was measured in mouse primary hepatocytes and BMDMs treated as in (D). (F) Slc7a11 mRNA was measured in mouse primary hepatocytes (upper panel) and BMDMs (lower panel) treated as indicated for 12 hours. (A,B,D‐F) are representative of three independent experiments. mRNA levels in (A,C,F) were normalized to β‐actin mRNA and are expressed relative to the respective control group or time point. Significance in (A,C[right],D,F) was calculated using a one‐way ANOVA with Tukey's post hoc test. In (A), *P < 0.05 versus 0 μM at each time point; in (C[right],D,F), groups labeled without a common letter were significantly different (P < 0.05). Significance in (C, left) was calculated using the Student t test; *P < 0.05 versus NID. Abbreviations: GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; β‐Me, β‐mercaptoethanol; HID, high‐iron diet; NID, normal‐iron diet; Trig, trigonelline; WT, wild type.
To investigate the mechanism by which iron regulates Slc7a11 expression, we measured Nrf2, a transcription factor that plays a key role in antioxidation processes.35 Under basal conditions, cytosolic thiol antioxidants maintain the stability of inactive KEAP1/Nrf2 heterodimers. When cytosolic thiol is depleted by ROS, the heterodimer dissociates and Nrf2 translocates to the nucleus, where it drives the transcription of genes that contain an ARE, including SLC7A11.36 We found that treating mouse primary hepatocytes and BMDMs with FAC increased the levels of both ROS and nuclear Nrf2, whereas cytosolic Nrf2 levels were decreased (Fig. 4D‐E). β‐Mercaptoethanol, a robust ROS scavenger, inhibited FAC‐induced ROS production, Nrf2 nuclear accumulation, and Slc7a11 up‐regulation (Fig. 4D‐F). Moreover, the Nrf2 inhibitor trigonelline also blocked FAC‐induced Slc7a11 up‐regulation (Fig. 4F). These data suggest that iron regulates Slc7a11 expression through the ROS–Nrf2–ARE axis.
Slc7a11 DEFICIENCY IS NOT SUFFICIENT TO INDUCE FERROPTOSIS IN MICE
Several in vitro studies have suggested that SLC7A11 plays a role in regulating ferroptosis.5, 20, 21 We therefore investigated whether deleting Slc7a11 expression in mice causes ferroptosis in vivo. Slc7a11–/– mice were similar to wild‐type mice with respect to body size, viability, and fertility (data not shown). We next measured ferroptosis (by measuring lipid peroxidation and Ptgs2 mRNA levels) in primary hepatocytes, BMDMs, and tissues obtained from wild‐type and Slc7a11–/– mice. We found that lipid peroxidation (Supporting Fig. S3A), Ptgs2 expression, (Supporting Fig. S3B,D), and MDA content (Supporting Fig. S3C) did not differ significantly between wild‐type and Slc7a11–/– mice, suggesting that deleting Slc7a11 is not sufficient to induce ferroptosis in vivo.
Slc7a11 is an important importer of cysteine for use in GSH synthesis. Slc7a11–/– mice had decreased serum GSH levels compared to wild‐type mice (Supporting Fig. S4A), consistent with previous results.29 Next, we measured the expression of genes that express antioxidant proteins, including catalase, Gpx4, and superoxide dismutase 1. Gpx4 was reported to scavenge lipid peroxides and inhibit ferroptosis through GSH, whereas superoxide dismutase 1 and catalase function independently of GSH. We found higher mRNA levels of these three genes in the liver and spleen of Slc7a11–/– mice compared to wild‐type mice (Supporting Fig. S4B), suggesting that up‐regulation of these antioxidant‐encoding genes might compensate for GSH deficiency, thereby maintaining redox balance in Slc7a11–/– mice. On the other hand, tissue iron content was similar between wild‐type and Slc7a11–/– mice (Supporting Fig. S4C).
Slc7a11 DELETION INCREASES SUSCEPTIBILITY TO IRON OVERLOAD–INDUCED FERROPTOSIS
Based on our microarray results (Supporting Table S2), Slc7a11 expression increases during iron overload. We therefore studied the role of Slc7a11 in iron overload–induced ferroptosis in primary hepatocytes treated with FAC or not. Compared to untreated hepatocytes, both wild‐type and Slc7a11–/– FAC‐treated hepatocytes had smaller, ruptured mitochondria (Fig. 5A); these cellular morphological features are characteristic of ferroptosis.5, 21 In addition, FAC treatment reduced cell viability in a dose‐dependent and time‐dependent manner in hepatocytes and BMDMs obtained from both wild‐type and Slc7a11–/– mice, with higher potency in Slc7a11–/– cells compared to wild‐type cells (Fig. 5B,C). FAC treatment also increased lipid peroxidation, increased Ptgs2 expression, and decreased NADPH content in hepatocytes and BMDMs, again with higher potency in Slc7a11–/– cells (Fig. 5D‐F). Finally, treating cells with the ferroptosis inhibitor Ferr‐1 prevented the FAC‐induced up‐regulation of Ptgs2 expression and down‐regulation of NADPH content (Fig. 5E,F). These results indicate that Slc7a11–/– hepatocytes and BMDMs are more susceptible than wild‐type cells to iron overload–induced ferroptosis.
Figure 5.
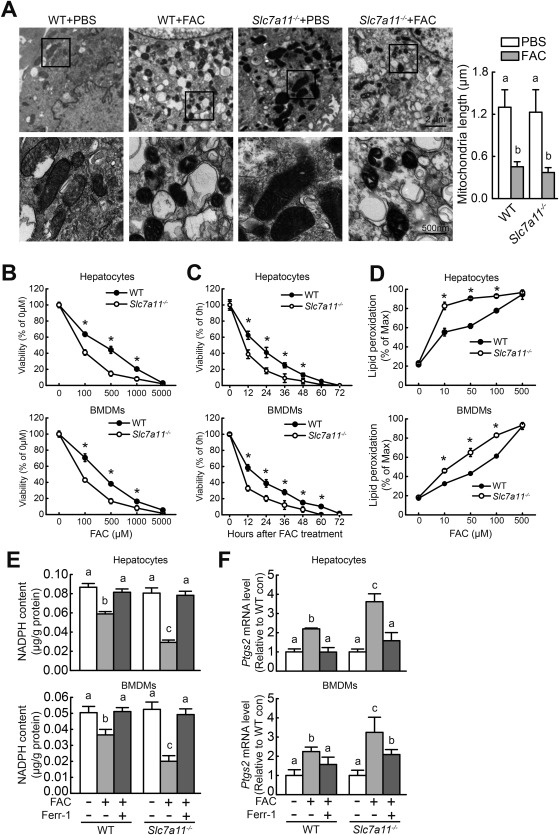
Slc7a11 knockout exacerbates iron overload–induced ferroptosis in vitro. (A) (left) Wild‐type and Slc7a11–/– primary hepatocytes were treated with phosphate‐buffered saline or 10 mM FAC for 12 hours and then examined using transmission electron microscopy; (right) mitochondrial length (along the long axis) was measured and summarized. (B) Cell viability was measured in wild‐type and Slc7a11–/– primary hepatocytes and BMDMs after treatment with the indicated concentration of FAC for 48 hours. (C) Cell viability was measured in wild‐type and Slc7a11–/– primary hepatocytes and BMDMs after treatment with 1 mM FAC for the indicated times. (D) Lipid peroxidation was measured using C11‐BODIPY staining in wild‐type and Slc7a11–/– primary hepatocytes and BMDMs treated with the indicated concentration of FAC for 12 hours. (E) NADPH content and (F) Ptgs2 mRNA were measured in wild‐type and Slc7a11–/– primary hepatocytes and BMDMs treated for 12 hours with or without 100 μM FAC in the presence or absence of 10 μM Ferr‐1. mRNA levels were normalized to β‐actin mRNA and are expressed relative to the respective mean control‐treated wild‐type value. Data are representative of three independent experiments. Significance was calculated using a one‐way ANOVA with Tukey's post hoc test; in (B‐D), * P < 0.05 versus Slc7a11–/–; and in (A,E,F), groups labeled without a common letter were significantly different (P < 0.05). Abbreviations: con, control; PBS, phosphate‐buffered saline; WT, wild type.
Next, we examined whether the loss of Slc7a11 expression increases iron overload–induced ferroptosis in vivo by examining the effect of feeding wild‐type and Slc7a11–/– mice an iron‐rich diet. Feeding wild‐type and Slc7a11–/– mice the HID for 8 weeks after weaning led to significantly higher tissue iron content compared with their respective control (i.e., normal diet–fed) groups (Supporting Fig. S5). Moreover, compared to their respective control groups the HID‐fed mice had significantly higher MDA content and Ptgs2 expression in the liver and spleen, as well as decreased hepatic NADPH content. Notably, all of these HID‐induced ferroptosis parameters were more severe in the Slc7a11–/– mice compared to wild‐type mice (Fig. 6A‐C). In addition, and consistent with the in vitro results, Ferr‐1 treatment significantly reduced HID‐induced ferroptosis in both wild‐type and Slc7a11–/– mice (Fig. 6A‐C).
Figure 6.
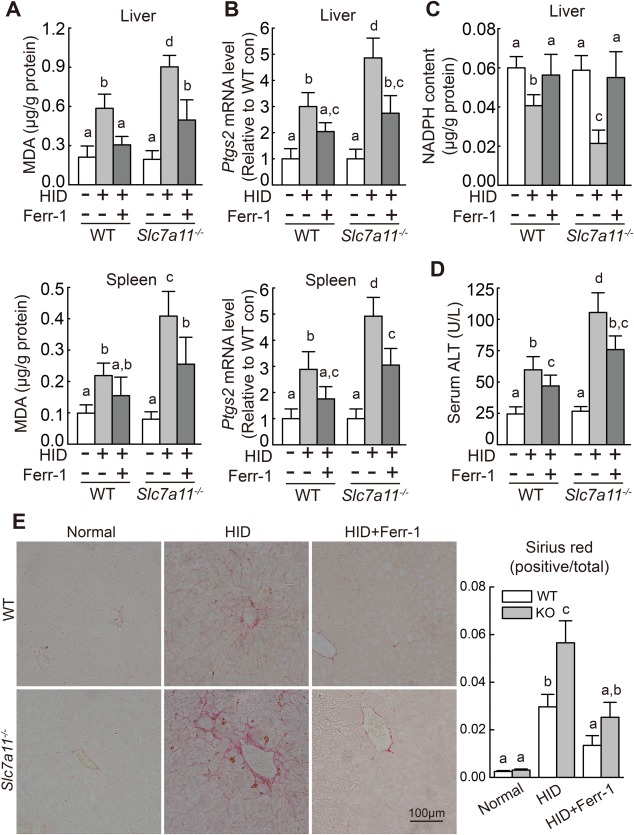
Iron‐overloaded Slc7a11–/– mice have increased levels of tissue ferroptosis. (A) Hepatic and splenic MDA content, (B) hepatic and splenic Ptgs2 mRNA, (C) hepatic NADPH content, and (D) serum ALT activity were measured in male wild‐type and Slc7a11–/– mice that were fed a normal‐iron diet or the HID with or without Ferr‐1 treatment (n = 6 mice/group). (E) (left) Liver sections were obtained from the indicated mice and stained with sirius red; (right) summary of staining intensity. mRNA levels in (B) were normalized to β‐actin mRNA and are expressed relative to the respective mean wild‐type control value. Significance was calculated using a one‐way ANOVA with Tukey's post hoc test, and groups labeled without a common letter were significantly different (P < 0.05). Abbreviations: con, control; KO, knockout; WT, wild type.
Under basal conditions, we found no difference between wild‐type and Slc7a11–/– mice with respect to serum ALT or hepatic collagen deposits (Fig. 6D,E), indicating that Slc7a11–/– mice do not develop liver damage or fibrosis. When fed the HID, however, both wild‐type and Slc7a11–/– mice had increased serum ALT levels and hepatic collagen deposits, and these effects were more severe in the Slc7a11–/– mice (Fig. 6D,E). Importantly, Ferr‐1 treatment reduced the HID‐induced liver injury and fibrosis in both wild‐type and Slc7a11–/– mice (Fig. 6D,E). Thus, consistent with our in vitro results, these data indicate that deleting Slc7a11 expression increases susceptibility to iron overload–induced ferroptosis.
IRON OVERLOAD–INDUCED FERROPTOSIS IS INDEPENDENT OF ER STRESS, THE MAPK PATHWAY, AND AUTOPHAGY
Next, we investigated the mechanisms underlying iron overload–induced ferroptosis. In contrast with erastin‐induced ferroptosis in Ras‐mutated tumor cells,20 all the HH mouse models (Fig. 7A) and Slc7a11–/– mice (Fig. 7B) had wild‐type levels of hepatic spliced XBP1 (sXBP1) mRNA, suggesting that ER stress is not likely involved in iron overload–induced ferroptosis. Because the MAPK pathway has been linked to erastin‐induced ferroptosis,37, 38 we also tested whether this pathway is involved in iron‐induced ferroptosis. Compared to wild‐type mice fed a normal diet, HH mice on a normal diet and HID‐fed wild‐type mice had decreased levels of hepatic P‐JNK protein, and iron overload did not affect the levels of hepatic P‐JNK in Slc7a11–/– mice (Fig. 7C,D). Hepatic P‐ERK levels were similar among Hfe–/– mice, Hjv–/– mice, and wild‐type mice, whereas P‐ERK levels were decreased in Smad4Alb/Alb mice and increased in HID‐fed Slc7a11–/– mice; in contrast, levels of hepatic P‐p38 were similar among all genotypes (Fig. 7C,D). Lastly, FAC‐induced cell death was not affected by treating primary hepatocytes with SP600125 (an inhibitor of JNK phosphorylation), SB202190 (an inhibitor of p38 activation), or PD98059 (an inhibitor of the upstream ERK activators MAPK kinases 1 and 2) (Fig. 7E). Taken together, these results suggest that the MAPK pathway does not likely play a key role in iron‐induced ferroptosis.
Figure 7.
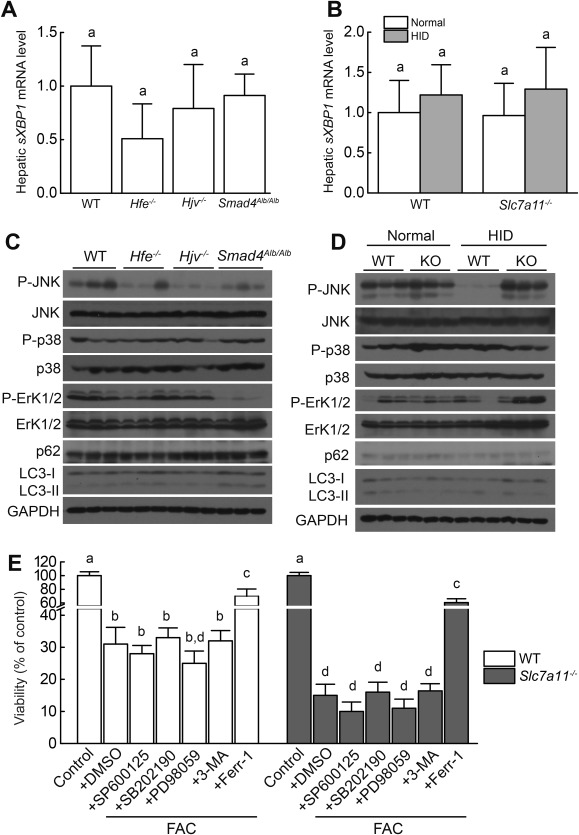
ER stress, MAPK signaling, and autophagy in HH mice and Slc7a11 knockout mice treated with iron. (A,B) Hepatic spliced XBP1 (sXBP1) mRNA was measured in 8‐week‐old male wild‐type, Hfe–/–, Hjv–/–, and Smad4Alb/Alb mice fed a normal diet (n = 6 mice/group) and male wild‐type and Slc7a11–/– mice fed a normal diet or the HID (n = 6 mice/group). (C,D) Hepatic P‐JNK, P‐Erk1/2, P‐p38, LC3‐I, LC3‐II, and p62 were measured in 8‐week‐old male wild‐type, Hfe–/–, Hjv–/–, and Smad4Alb/Alb mice fed a normal diet and male wild‐type and Slc7a11–/– mice fed a normal diet or the HID. (E) Wild‐type and Slc7a11–/– primary hepatocytes were treated with 1 mM FAC in the presence or absence of 10 μM SP600125, 10 μM SB202190, 10 μM PD98059, 2 mM 3‐methylademine, or 10 μM Ferr‐1 for 72 hours, after which cell viability was measured. Data in (C) and (D) are representative of three independent experiments. Significance was calculated using a one‐way ANOVA with Tukey's post hoc test, and groups labeled without a common letter were significantly different (P < 0.05). Abbreviations: DMSO, dimethyl sulfoxide; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; KO, knockout; 3‐MA, 3‐methylademine; WT, wild type.
Finally, autophagy can be activated in erastin‐induced ferroptosis.34, 39 However, LC3‐II and p62, two markers used to measure autophagy,40 were not increased in our HH mice or in HID‐fed Slc7a11–/– mice. Moreover, the autophagy inhibitor 3‐methylademine did not rescue FAC‐induced cell death in primary hepatocytes (Fig. 7C‐E). These results suggest that iron overload–induced ferroptosis may be independent of autophagy.
Discussion
Iron has been linked to several forms of programmed cell death, including apoptosis and autophagy.32, 33 Recently, iron has also been implicated in ferroptosis, a novel form of cell death that plays a role in several pathological conditions, including ischemia/reperfusion.11, 41 Given that iron homeostasis is tightly regulated in order to maintain health,1 it is not surprising that perturbations in iron balance cause a variety of diseases, including HH, an inherited iron‐overload condition with severe complications. The increased production of ROS due to excess iron has been suggested as a major pathogenic mechanism underlying HH‐associated complications, which include hepatic cirrhosis and fibrosis. However, this mechanism—while promising—cannot fully explain the clinical observation that patients with type 2 HH (i.e., patients with a mutation in the HJV gene) present with symptoms earlier, develop more severe tissue damage, and have worse outcome than patients with type 1 HH (i.e., patients with HFE‐HH).1, 4
To investigate the putative role of ferroptosis in HH, we characterized ferroptosis in three transgenic mouse models of HH. We found that two of these models—Hjv –/– and Smad4Alb/Alb mice, both of which develop high iron overload—develop hepatic ferroptosis. In contrast, Hfe–/– mice, which develop only moderate iron overload, do not develop hepatic ferroptosis. Importantly, feeding HH mice an LID can rescue ferroptosis. These observations suggest that high iron levels serve as a driving factor in the induction of ferroptosis. In addition, these results suggest that ferroptosis may underlie the higher disease severity associated with type 2 HH compared to type 1 HH.
We also examined possible regulators of iron overload–induced ferroptosis. Our analysis of gene expression data revealed that iron overload increases the expression of Slc7a11 through the ROS–Nrf2–ARE axis. SLC7A11 is a major component of the glutamate/cystine antiporter system xc –, which regulates the downstream synthesis of GSH, a key antioxidant that scavenges lipid peroxides and prevents ferroptosis.5 A growing body of in vitro data indicate that SLC7A11 plays a role in regulating ferroptosis; these data stem from using either pharmacological inhibitors (e.g., erastin, sulfasalazine) or RNA interference directed against SLC7A11.5, 20 However, we found that Slc7a11–/– mice do not develop ferroptosis under basal iron conditions. Given that Slc7a11–/– mice have decreased GSH levels,29 we propose that iron plays a driving role in ferroptosis induction and that the combination of reduced GSH levels and increased ROS levels may facilitate this process (see Supporting Fig. S6). Nevertheless, our data indicate that iron‐induced ferroptosis is a distinct process from erastin‐induced ferroptosis. Further studies are therefore needed in order to investigate the mechanisms that underlie iron‐induced ferroptosis.
Our results suggest that in addition to ROS, other currently unidentified genes and/or pathways may mediate iron‐dependent ferroptosis in the absence of SLC7A11. First, the GPX4‐catalyzed reaction between GSH and lipid peroxides can prevent ferroptosis, and overexpressing GPX4 reduces ferroptosis.8 We found increased expression of Gpx4 in Slc7a11–/– mice, suggesting a possible compensatory mechanism in the absence of Slc7a11. Second, the amino acid antiporter system xc – is one major source of cellular cystine/cysteine and subsequent GSH synthesis. Although GSH levels in Slc7a11–/– mice are lower than in wild‐type mice (Supporting Fig. S4A),29 the fact that measurable GSH levels remain and the presence of normal lipid peroxidation in Slc7a11–/– mice (Supporting Fig. S3A‐D) suggest that other sources of cysteine might compensate—at least partially—for the loss of Slc7a11. These sources might include alanine, serine, and cysteine transporters.42 Lastly, Slc7a11–/– mice have increased expression of the catalase, Gpx4, and superoxide dismutase 1 genes, which encode GSH‐dependent and GSH‐independent lipid peroxidation scavenging proteins, suggesting that redundant proteins may help maintain redox balance in the absence of Slc7a11.
Under physiological conditions, circulating free iron is bound to transferrin, which renders it nonreactive. When fed an HID, both wild‐type and Slc7a11–/– mice develop severe tissue iron overload, and serum transferrin binding approaches saturation (Supporting Fig. S5), suggesting the presence of non‐transferrin‐bound iron in the circulation. Circulating non‐transferrin‐bound iron depletes GSH and oxidizes cysteine to form cysteine,43 which can then be imported into cells through the Slc7a11 transporter. Consistent with this process, we found increased levels of Slc7a11 mRNA in iron‐overloaded wild‐type mice (Fig. 4C), indicating increased cystine uptake and subsequent GSH synthesis. In contrast, deleting Slc7a11 expression significantly lowers this source of cellular cystine/cysteine, thereby limiting subsequent GSH synthesis and increasing the cell's susceptibility to iron overload–induced ferroptosis. Moreover, genetic mutations in antioxidant enzymes (e.g., GPX1P198L and SOD2A16V) can accelerate the clinical course of HH.44 Thus, taken together, these results suggest that SLC7A11 plays a protective role against iron overload–induced ferroptosis.
These findings have several key implications. First, we provide evidence of ferroptosis in mouse models of hemochromatosis, and we show that targeting ferroptosis using the specific inhibitor Ferr‐1 rescues liver damage in these mice. Recent studies reported that ferroptosis is driven by the peroxidation of polyunsaturated fatty acids, particularly oxidized arachidonic acid and adrenic acid phosphatidylethanolamines.7, 45, 46 Consistent with these findings, high levels of lipid peroxidation in cellular membranes result in cell death and organ damage in patients with HH.1, 3, 4, 30 Although Ferr‐1 did not change iron levels in Hjv–/– and Smad4Alb/Alb mice, it significantly decreased hepatic MDA content, suggesting that scavenging lipid peroxidases partially inhibits ferroptosis‐related damage even under iron‐overload conditions.
Currently, therapeutic strategies for treating hemochromatosis are limited to phlebotomy and iron chelators, which can reduce iron levels.1, 3, 4 Our results indicate that inhibiting ferroptosis may be a feasible approach for treating and/or preventing hemochromatosis. On the other hand, inducing ferroptosis may be an effective strategy for killing oncogenic Ras‐mutated tumor cells, which are usually resistant to apoptosis.5 Given that SLC7A11 may function as an oncogene in various tumor cell types,47 it may be possible to induce ferroptosis by pharmacologically inhibiting SLC7A11 (e.g., with erastin, sulfasalazine, or sorafenib).5, 20 Importantly, our results show that deleting Slc7a11 expression in otherwise normal (i.e., noncancerous) cells does not induce ferroptosis, suggesting that targeting oncogenic SLC7A11 might be clinically safe. Thus, ferroptosis appears to serve several important functions in both health and disease.
In summary, we report that iron homeostasis plays an essential role in Slc7a11‐regulated ferroptosis. From a mechanistic perspective, the iron–ROS–Slc7a11 network drives iron overload–induced ferroptosis. Finally, these findings provide compelling evidence that therapeutic strategies can be developed for targeting ferroptosis and/or SLC7A11 in order to treat several forms of cancer as well as iron overload–associated diseases, including HH.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.29117/suppinfo.
Supporting Information
Acknowledgment
We are grateful to Drs. Nancy C. Andrews, Hideyo Sato, and Chu‐Xia Deng for providing the knockout mice. We thank the members of the Wang and Min laboratories for helpful discussions.
Potential conflict of interest: Nothing to report.
Supported by research grants from the National Natural Science Foundation of China (31330036, 31530034, and 31225013, to F.W.; 31570791 and 91542205, to J.M.; 31500960, to P.A.; 31200892, to Z.Z.; and 31401005, to G.L.), the China Postdoctoral Science Foundation (2016M601932, to H.W.), and the Zhejiang Provincial Natural Science Foundation of China (LZ15H160002, to J.M.).
Contributor Information
Junxia Min, Email: junxiamin@zju.edu.cn.
Fudi Wang, Email: fudiwang.lab@gmail.com, Email: fwang@zju.edu.cn.
REFERENCES
- 1. Ganz T. Systemic iron homeostasis. Physiol Rev 2013;93:1721‐1741. [DOI] [PubMed] [Google Scholar]
- 2. Meynard D, Babitt JL, Lin HY. The liver: conductor of systemic iron balance. Blood 2014;123:168‐176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Powell LW, Seckington RC, Deugnier Y. Haemochromatosis. Lancet 2016;388:706‐716. [DOI] [PubMed] [Google Scholar]
- 4. Pietrangelo A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology 2010;139:393‐408. [DOI] [PubMed] [Google Scholar]
- 5. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell 2012;149:1060‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol 2014;10:9‐17. [DOI] [PubMed] [Google Scholar]
- 7. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA 2016;113:E4966‐E4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014;156:317‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shimada K, Hayano M, Pagano NC, Stockwell BR. Cell‐line selectivity improves the predictive power of pharmacogenomic analyses and helps identify NADPH as biomarker for ferroptosis sensitivity. Cell Chem Biol 2016;23:225‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol 2016;26:165‐176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 2014;16:1180‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62–Keap1–NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016;63:173‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sun X, Niu X, Chen R, He W, Chen D, Kang R, et al. Metallothionein‐1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology 2016;64:488‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell 2015;59:298‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kwon MY, Park E, Lee SJ, Chung SW. Heme oxygenase‐1 accelerates erastin‐induced ferroptotic cell death. Oncotarget 2015;6:24393‐24403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem 1999;274:11455‐11458. [DOI] [PubMed] [Google Scholar]
- 17. Deneke SM, Fanburg BL. Regulation of cellular glutathione. Am J Physiol 1989;257:L163‐L173. [DOI] [PubMed] [Google Scholar]
- 18. Telorack M, Meyer M, Ingold I, Conrad M, Bloch W, Werner S. A glutathione–Nrf2–thioredoxin cross‐talk ensures keratinocyte survival and efficient wound repair. PLoS Genet 2016;12:e1005800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gasol E, Jimenez‐Vidal M, Chillaron J, Zorzano A, Palacin M. Membrane topology of system xc – light subunit reveals a re‐entrant loop with substrate‐restricted accessibility. J Biol Chem 2004;279:31228‐31236. [DOI] [PubMed] [Google Scholar]
- 20. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine‐glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014;3:e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53‐mediated activity during tumour suppression. Nature 2015;520:57‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, et al. Acetylation is crucial for p53‐mediated ferroptosis and tumor suppression. Cell Rep 2016;17:366‐373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 2005;1:191‐200. [DOI] [PubMed] [Google Scholar]
- 24. Huang FW, Pinkus JL, Pinkus GS, Fleming MD, Andrews NC. A mouse model of juvenile hemochromatosis. J Clin Invest 2005;115:2187‐2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nicolas G, Viatte L, Lou DQ, Bennoun M, Beaumont C, Kahn A, et al. Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nat Genet 2003;34:97‐101. [DOI] [PubMed] [Google Scholar]
- 26. Wang RH, Li C, Xu X, Zheng Y, Xiao C, Zerfas P, et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab 2005;2:399‐409. [DOI] [PubMed] [Google Scholar]
- 27. Zhang Z, Zhang F, Guo X, An P, Tao Y, Wang F. Ferroportin1 in hepatocytes and macrophages is required for the efficient mobilization of body iron stores in mice. Hepatology 2012;56:961‐971. [DOI] [PubMed] [Google Scholar]
- 28. Zhang Z, Zhang F, An P, Guo X, Shen Y, Tao Y, et al. Ferroportin1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood 2011;118:1912‐1922. [DOI] [PubMed] [Google Scholar]
- 29. Sato H, Shiiya A, Kimata M, Maebara K, Tamba M, Sakakura Y, et al. Redox imbalance in cystine/glutamate transporter‐deficient mice. J Biol Chem 2005;280:37423‐37429. [DOI] [PubMed] [Google Scholar]
- 30. Delima RD, Chua AC, Tirnitz‐Parker JE, Gan EK, Croft KD, Graham RM, et al. Disruption of hemochromatosis protein and transferrin receptor 2 causes iron‐induced liver injury in mice. Hepatology 2012;56:585‐593. [DOI] [PubMed] [Google Scholar]
- 31. Nabeyama A, Kurita A, Asano K, Miyake Y, Yasuda T, Miura I, et al. xCT deficiency accelerates chemically induced tumorigenesis. Proc Natl Acad Sci USA 2010;107:6436‐6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang GS, Eriksson LC, Xia L, Olsson J, Stal P. Dietary iron overload inhibits carbon tetrachloride–induced promotion in chemical hepatocarcinogenesis: effects on cell proliferation, apoptosis, and antioxidation. J Hepatol 1999;30:689‐698. [DOI] [PubMed] [Google Scholar]
- 33. Lunova M, Goehring C, Kuscuoglu D, Mueller K, Chen Y, Walther P, et al. Hepcidin knockout mice fed with iron‐rich diet develop chronic liver injury and liver fibrosis due to lysosomal iron overload. J Hepatol 2014;61:633‐641. [DOI] [PubMed] [Google Scholar]
- 34. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016;12:1425‐1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ye P, Mimura J, Okada T, Sato H, Liu T, Maruyama A, et al. Nrf2‐ and ATF4‐dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol Cell Biol 2014;34:3421‐3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Antelmann H, Helmann JD. Thiol‐based redox switches and gene regulation. Antioxid Redox Signal 2011;14:1049‐1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS–RAF–MEK‐dependent oxidative cell death involving voltage‐dependent anion channels. Nature 2007;447:864‐868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, et al. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol 2015;2:e1054549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res 2016;26:1021‐1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007;3:542‐545. [DOI] [PubMed] [Google Scholar]
- 41. Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci USA 2014;111:16836‐16841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scopelliti AJ, Heinzelmann G, Kuyucak S, Ryan RM, Vandenberg RJ. Na+ interactions with the neutral amino acid transporter ASCT1. J Biol Chem 2014;289:17468‐17479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Song H, Her AS, Raso F, Zhen Z, Huo Y, Liu P. Cysteine oxidation reactions catalyzed by a mononuclear non‐heme iron enzyme (OvoA) in ovothiol biosynthesis. Org Lett 2014;16:2122‐2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nahon P, Sutton A, Pessayre D, Rufat P, Charnaux N, Trinchet JC, et al. Do genetic variations in antioxidant enzymes influence the course of hereditary hemochromatosis? Antioxid Redox Signal 2011;15:31‐38. [DOI] [PubMed] [Google Scholar]
- 45. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 2017;13:81‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 2017;13:91‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Timmerman LA, Holton T, Yuneva M, Louie RJ, Padro M, Daemen A, et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple‐negative breast tumor therapeutic target. Cancer Cell 2013;24:450‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.29117/suppinfo.
Supporting Information