

Abstract
Aim
To evaluate the pharmacodynamics of lixisenatide once daily vs sitagliptin once daily in Japanese patients with type 2 diabetes receiving insulin glargine U100.
Materials and methods
This multicentre, open‐label, phase IV study (NEXTAGE Study; ClinicalTrials.gov number, NCT02200991) randomly assigned 136 patients to either lixisenatide once daily via subcutaneous injection (10 µg initially increased weekly by 5 up to 20 µg) or once‐daily oral sitagliptin 50 mg. The primary endpoint was the change in postprandial glucose (PPG) exposure 4 hours after a standardized breakfast (PPG area under the plasma glucose concentration–time curve [AUC0 :00‐4:00h]) from baseline to day 29.
Results
Lixisenatide reduced PPG exposure to a statistically significantly greater extent than sitagliptin: least squares (LS) mean change from baseline in PPG AUC0 :00‐4:00h was −347.3 h·mg/dL (−19.3 h·mmol/L) in the lixisenatide group and −113.3 h·mg/dL (−6.3 h·mmol/L) in the sitagliptin group (LS mean between‐group difference −234.0 h·mg/dL [−13.0 h·mmol/L], 95% confidence interval −285.02 to −183.00 h·mg/dL [−15.8 to −10.2 h·mmol/L]; P < .0001). Lixisenatide led to significantly greater LS mean reductions in maximum PPG excursion than sitagliptin (−122.4 vs −46.6 mg/dL [−6.8 vs −2.6 h·mmol/L]; P < .0001). Change‐from‐baseline reductions in exposure to C‐peptide, fasting glycoalbumin levels, and the gastric emptying rate were greater in the lixisenatide than in the sitagliptin group. The incidence of treatment‐emergent adverse events was higher with lixisenatide (60.9%) than with sitagliptin (16.4%), with no serious events or severe hypoglycaemia reported.
Conclusion
Lixisenatide reduced PPG significantly more than sitagliptin, when these agents were added to basal insulin glargine U100, and was well tolerated.
Keywords: gastric emptying, glucagon‐like peptide 1 receptor agonist, lixisenatide, postprandial glucose, randomized trial, type 2 diabetes mellitus
1. INTRODUCTION
Current evidence suggests that controlling the key metrics of blood glucose control (glycated haemoglobin [HbA1c], fasting plasma glucose [FPG] and postprandial glucose [PPG]) may be required to optimize outcomes in patients with type 2 diabetes (T2D).1 Loss of postprandial glycaemic control occurs early in patients with T2D subsequent to sequential loss of glycaemic control during the nocturnal fasting period.2 The absolute contribution that PPG makes to hyperglycaemia in T2D is generally constant at different HbA1c levels, but becomes proportionately greater than the contribution of FPG as HbA1c decreases.2, 3, 4 Hence, targeting PPG control rather than relying on managing FPG alone may increase the opportunity for patients to meet their recommended HbA1c targets.1, 3, 4, 5 Because targeting PPG control may mitigate the risk of diabetes‐related complications,1 the American Association of Clinical Endocrinologists/American College of Endocrinology and the International Diabetes Federation recommend a target level for 2‐hour PPG of ≤140 mg/dL (≤7.8 mmol/L),1, 6 and the American Diabetes Association recommend a higher target level for 2‐hour PPG of ≤180 mg/dL (≤10.0 mmol/L).7
The introduction of insulin glargine represented an advance in basal insulin therapy, because its slow release after administration from subcutaneous tissue to blood results in a relatively constant 24‐hour concentration–time profile, with no pronounced peak.8 This protracted time–action profile of insulin glargine not only allows once‐daily dosing,8 but also confers similar glycaemic control to that of NPH insulin and a lower incidence of hypoglycaemia9, 10; however, therapy with insulin glargine alone does not improve PPG excursions to the same extent as it improves FPG levels.9 More stringent control of PPG is also required to maintain good glycaemic control and delay complications.
Adjunctive incretin‐related therapy with either a glucagon‐like peptide‐1 (GLP‐1) receptor agonist or a dipeptidyl peptidase‐4 (DPP‐4) inhibitor is a pharmacologically rational approach to take for patients with T2D with suboptimum glycaemic control, despite basal insulin therapy. Endogenous incretin hormones mediate food‐stimulated, glucose‐dependent insulin secretion that accounts for up to 60% of the insulin secretory response after ingestion of increasing oral glucose loads.11 GLP‐1 receptor agonists and DPP‐4 inhibitors are, therefore, attractive candidates for improving PPG control as they supplement the attenuated incretin effect often observed in patients with T2D, without significantly increasing the risk of hypoglycaemia.11, 12, 13, 14 Conversely, in vitro data suggest that basal insulin therapy supplementing diurnal endogenous insulin production will allow greater β‐cell recovery time to optimize the postprandial incretin‐induced endogenous insulin response.15, 16, 17 Results of a proof‐of‐concept study showed that further improvement in PPG control is possible when the GLP‐1 receptor agonist exenatide or DPP‐4 inhibitor sitagliptin was added to combination therapy with insulin glargine and metformin.18
Whereas mimetics or analogues of GLP‐1 with resistance to DPP‐4 (the GLP‐1 receptor agonists) provide supraphysiological stimulation of the GLP‐1 receptor, DPP‐4 inhibitors prolong the activity of endogenous GLP‐1 (and glucose‐dependent insulinotropic polypeptide).19 This difference in mode of action may underpin the better glycaemic effects of GLP‐1 receptor agonists compared with DPP‐4 inhibitors,20, 21 as well as rationalize the beneficial non‐glycaemic effects of GLP‐1 receptor agonists, including suppressed gastric emptying rate, suppressed appetite, and weight loss, which are not seen routinely with DPP‐4 inhibitor treatments.22, 23
Lixisenatide is a short‐acting GLP‐1 receptor agonist confirmed to improve glycaemic control when taken once daily in patients with T2D as monotherapy24 and in those insufficiently controlled on a range of antidiabetic background therapies, including metformin, a sulphonylurea, and/or insulin glargine, in combination with diet and exercise.25, 26, 27, 28, 29, 30 Lixisenatide was associated with a pronounced improvement in postprandial hyperglycaemia compared with placebo in these studies,25, 26, 27, 29 including studies in Asian patients exclusively.28, 30 Further, lixisenatide had a greater postprandial effect on blood glucose levels than the longer‐acting GLP‐1 receptor agonist liraglutide in patients with T2D insufficiently controlled on metformin, with or without insulin glargine.31, 32
Currently, there are no direct comparisons between the suppressive effects on PPG of short‐acting GLP‐1 receptor agonists and DPP‐4 inhibitors, in combination with basal insulin. In the present study we compared the PPG reduction and blood glucose profile of Japanese patients with T2D who received either lixisenatide or the DPP‐4 inhibitor sitagliptin adjunctive to background insulin glargine U100 (Gla‐100).
2. MATERIALS AND METHODS
2.1. Study design
This multicentre, randomized, open‐label, parallel‐group, phase IV study was conducted in 15 medical facilities across Japan between August 2014 and November 2015. The study comprised a 2‐week screening period, a 4‐week treatment period and a 3‐day follow‐up period. The study was approved by the institutional review board at each participating site, and was conducted according to the provisions of the Declaration of Helsinki and the Good Clinical Practice Guidelines of the International Conference on Harmonization. All patients provided written informed consent before participation. The NEXTAGE Study was registered with ClinicalTrials.gov (NCT02200991).
2.2. Study population
Adults aged between 20 and 75 years with T2D for at least 5 years, an HbA1c of 7.0% to 10.0%, and a fasting blood glucose level ≤180 mg/dL (≤10 mmol/L) at screening were included. Before screening, all patients had been treated with a stable dose of Gla‐100 for at least 3 months with or without a stable dose of a sulphonylurea.
Key exclusion criteria were: use of any antidiabetic agents (including lixisenatide) other than Gla‐100 or a sulphonylurea within 6 weeks before the screening visit; active liver disease (alanine aminotransferase >3 times the upper limit of the normal laboratory range); positive test for hepatitis B virus antigen and/or hepatitis C virus antibody; clinically relevant history of gastrointestinal disease associated with persistent nausea and vomiting, including gastroparesis, unstable and uncontrolled gastroesophageal reflux disease within 6 months before the screening visit; known history of drug or alcohol abuse within 6 months before the screening visit; initiation of anti‐obesity agents within 3 months before the screening visit; use of systemic glucocorticoids (excluding topical application or inhaled forms) for a total duration of at least 1 week within 3 months before the screening visit; and moderate or severe renal impairment as defined by an estimated glomerular filtration rate of <50 mL/min/1.73 m2 and/or patients on dialysis. Pregnant or lactating women, and women unable to practise an effective contraceptive method, were also excluded.
2.3. Interventions
Patients with T2D were randomized in a 1:1 ratio to receive either lixisenatide or sitagliptin adjunctive to Gla‐100 using an interactive web response system and according to a predefined randomization list. Randomization was stratified by sulphonylurea use at screening. Similar to other studies of lixisenatide in Asian patients,28, 30 lixisenatide was titrated in a stepwise manner to 20 µg once daily. Patients initially received a once‐daily subcutaneous injection of 10 µg, which increased weekly by 5 up to 20 µg once daily. If nausea or vomiting was observed, the lixisenatide dose was maintained without any increase. Sitagliptin was administered once daily as a 50‐mg oral tablet. Both agents were administered daily for 28 days, half an hour before breakfast.
Both groups continued with their established dosage regimen of Gla‐100, which was administered subcutaneously once daily before breakfast or at bedtime. If the HbA1c level at screening was <7.5%, the Gla‐100 dose was reduced by 20% at randomization; however, if the FPG level was not controlled after reducing the Gla‐100 dose, it was increased to the level used at screening. The same dose and timing of administration of Gla‐100 was maintained throughout the study period; however, the dose could be adjusted depending on occurrence or increased risk of hypoglycaemia. The dose of concomitant sulphonylurea therapy was reduced by ≥25% (or discontinued when the minimum dose was used) if HbA1c was <8% at randomization, providing that adequate control of FPG was achieved.
The content and frequency of diet and exercise remained unchanged from the time of provision of consent to the end of study drug administration. At baseline (day 1) and visit 5 (day 29), all participants received a standardized breakfast (68.6 g carbohydrates, 17.6 g protein, 17.6 g fat, 500 kcal in total, consumed within 10 minutes), 30 minutes after study drug administration.
2.4. Endpoints and assessments
The primary objective of the present study was to compare the PPG reduction and blood glucose profile in patients with T2D who received either lixisenatide or sitagliptin in combination with Gla‐100. The primary endpoint was the change in PPG exposure after a standardized breakfast from baseline to day 29, where PPG exposure was defined as the 4‐hour period after the start of the standardized breakfast test meal area under the plasma glucose concentration–time curve (PPG AUC0:00‐4:00h). PPG AUC0:00‐4:00h was calculated, using the linear trapezoidal rule and corrected by the pre‐meal glucose concentration.
The secondary endpoints were changes from baseline to day 29 in maximum PPG excursion, plasma exposure to C‐peptide (AUC0:00‐4:00h) and glucagon (AUC0:00‐4:00h), fasting plasma levels of 1.5‐anhydro‐D‐glucitol (1.5‐AG) and glycoalbumin, and gastric emptying rate. On days 1 and 29 (after the last dose of study medication), blood sampling for measurement of pharmacodynamic variables was performed immediately before the standardized breakfast and at 0.25, 0.5, 1, 1.5, 2, 3, 4 hours after eating. The gastric emptying rate was calculated using the 13C acetic acid breath gas tests at specific study sites on these same study days. Breath gas in a standardized tolerance test was collected just before the standardized breakfast as well as at 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75, 2, 2.25, 2.5, 2.75, 3, 3.25, 3.5, 3.75, and 4 hours after eating. Outputs from the test data were the time to reach peak excretion rate of 13CO2 (tmax) and half of total cumulative excretion of 13CO2 (t1/2b).
Safety was evaluated via physical examination, ECGs, vital signs, clinical laboratory tests, and adverse event (AE) reporting conducted at screening, throughout the treatment period, and at the conclusion of the study. Treatment‐emergent adverse events (TEAEs) were defined as AEs that developed or worsened or became serious during the period from the first administration of study drug up to 3 days after the last administration of study drug. All AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA, Version 18.0) terms at the time of database lock. Specific safety information on hypoglycaemia was recorded, including severe hypoglycaemia (defined as an event during which someone else's help was required to administer glucose or glucagon or perform other emergency medical treatments), and documented symptomatic hypoglycaemia (defined as an event associated with typical hypoglycaemic symptoms with an accompanying plasma glucose 70 mg/dL [≤3.9 mmol/L]).
2.5. Analysis populations
Safety analyses were performed on the intention‐to‐treat (ITT) population, defined as all randomized patients who received ≥1 dose of study drug. Efficacy analyses were performed on the modified ITT (mITT) population, defined as the subset of patients of the ITT population who had both a baseline measurement and ≥1 post‐baseline measurement of any efficacy variables.
A pharmacodynamic population was also assessed (defined as all patients who signed the informed consent for the gastric emptying rate test, who were randomized and exposed to ≥1 dose of study drug, and who performed the gastric emptying rate test on both days 1 and 29); however, those patients whose gastric emptying measurements (tmax and t1/2b) on days 1 or 29 could not be estimated by the appropriate statistical model because of non‐convergence were excluded from the analysis of these variables.
2.6. Statistical analysis
The primary endpoint was change in PPG AUC0:00‐4:00h after a standardized breakfast from baseline to day 29. A target sample size of 148 patients (74 patients per group) was set to ensure statistical power of 95%, assuming that the between‐group difference of the change in PPG AUC0:00‐4:00h in the standardized meal tolerance test from baseline to day 29 was 150 h·mg/dL (8.3 h·mmol/L), the common standard deviation was 250 h·mg/dL (13.9 h·mmol/L) and the 2‐sided significance level was 5%.
All efficacy variables were analysed using an analysis of covariance (ANCOVA) model with treatment groups and randomization strata of screening sulphonylurea use as fixed effects, and the baseline value of the corresponding variable as a covariate. The least squares (LS) mean changes in each variable from baseline to day 29 for each treatment group were provided in the framework of this model, as well as the difference between treatment groups and the 95% confidence interval (CI) for the LS mean. The statistical test for the primary efficacy variable was set at a (2‐sided) 5% significance level. Summaries of safety data (descriptive statistics and frequency tables) were presented by treatment group.
The 2 gastric emptying rate variables (tmax and t1/2b) were analysed using a rank ANCOVA model with treatment groups and randomization strata of screening sulphonylurea use as fixed effects, and the ranked baseline value as a covariate. These variables, estimated by the appropriate statistical model, were used in this analysis. A sample size of 26 patients (13 patients per group) ensured 90% power to detect a 40‐minute difference in the absolute change in t1/2 of 13C‐acetic acid concentration in breath gas during the gastric emptying rate test from baseline to day 29 between lixisenatide and sitagliptin groups. This power analysis assumed a common standard deviation of 30 minutes at a 2‐sided significance level of 5%. Assuming the negative impact of dropouts, a target sample size for the gastric emptying rate test was determined to be 20 patients per group.
All efficacy, pharmacodynamic and safety data analyses were performed using descriptive statistics compiled using SAS version 9.2 or higher (SAS Institute Inc., Cary, North Carolina).
3. RESULTS
3.1. Patient disposition, demographics and clinical characteristics
All 136 patients randomized to the lixisenatide (n = 69) and sitagliptin (n = 67) groups received treatment, and were included in the ITT population (Figure 1). All but 1 patient had baseline and ≥1 post‐baseline efficacy measurements; hence, the mITT comprised 135 patients (lixisenatide, n = 69; sitagliptin, n = 66). Most patients in both the lixisenatide (67 of 69 patients) and sitagliptin (66 of 67 patients) groups completed the 28‐day treatment period. Two patients from the lixisenatide group withdrew because of AEs, while 1 patient from the sitagliptin group discontinued on the patient's request (inability to complete study visits).
Figure 1.
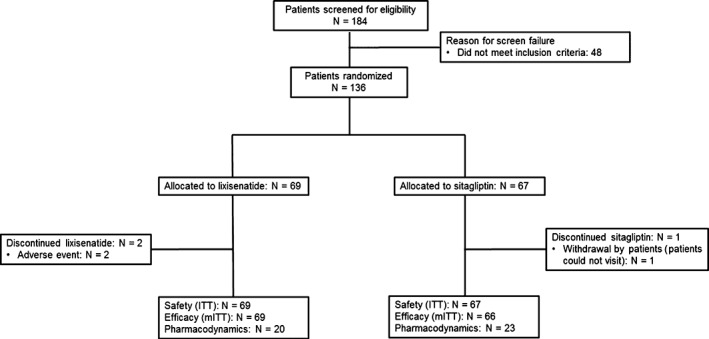
Patient disposition. AE, adverse event; ITT, intent to treat; mITT, modified ITT
Baseline patient characteristics were similar for both treatment groups in the ITT population with respect to gender, age, body mass index and key blood glucose metrics (Table 1). Patients in the lixisenatide group had slightly longer duration of diabetes (11.39 vs 9.82 years), and longer duration of treatment with Gla‐100 (0.85 vs 0.44 years) and sulphonylurea (6.48 vs 4.71 years).
Table 1.
Demographics and baseline characteristics (ITT population)
| Variable | Lixisenatide | Sitagliptin |
|---|---|---|
| (N = 69) | (N = 67) | |
| Male, n (%) | 41 (59.4) | 44 (65.7) |
| Mean ± s.d. age, years | 58.5 ± 10.1 | 58.3 ± 9.6 |
| Median (range) duration of diabetes mellitus, years | 11.39 (5.6, 35.2) | 9.82 (5.2, 41.1) |
| Mean ± s.d. body weight, kg | 65.15 ± 13.64 | 70.48 ± 16.38 |
| Mean ± s.d. BMI, kg/m2 | 24.99 ± 4.08 | 26.08 ± 5.10 |
| Mean ± s.d. HbA1c, % | 8.27 ± 0.75 | 8.32 ± 0.80 |
| Median (range) duration of treatment with insulin glargine, years | 0.85 (0.3, 13.1) | 0.44 (0.3, 9.8) |
| Mean ± s.d. daily insulin glargine dose at screening, U | 14.3 ± 8.4 | 17.0 ± 10.7 |
| SU use at screening, n (%) | 33 (47.8) | 32 (47.8) |
| Median (range) duration of treatment with SU, years | 6.48 (0.3, 22.1) | 4.71 (0.3, 27.4) |
| Mean ± s.d. FPG, mg/dL | 148.1 ± 31.8 | 151.7 ± 35.3 |
| Mean ± s.d. 2‐h PPG, mg/dL | 308.2 ± 63.4 | 299.1 ± 58.5 |
| Mean ± s.d. C‐peptide, ng/mL | 1.18 (0.74) | 1.12 (0.62)a |
Abbreviation: 2‐h PPG, 2‐hour postprandial glucose; BMI, body mass index; FPG, fasting plasma glucose; HbA1c, glycated haemoglobin; s.d., standard deviation; SU, sulphonylurea.
To convert C‐peptide ng/mL to nmol/L, multiply by 0.331.
N = 66.
3.2. Efficacy
Treatment with lixisenatide resulted in a statistically significant decrease in PPG exposure from baseline to day 29 relative to treatment with sitagliptin (Table 2). The LS mean change from baseline in PPG AUC0:00‐4:00h was −347.3 h·mg/dL (−19.3 h·mmol/L) in the lixisenatide group and −113.3 h·mg/dL (−6.3 h·mmol/L) in the sitagliptin group, yielding an LS mean difference between the treatment groups of −234.0 h·mg/dL with a 95% confidence interval (CI) −285.02 to −183.00 h·mg/dL (−13.0 h·mmol/L [95% CI −15.8 to −10.2 h·mmol/L]; P < .0001).
Table 2.
Changes in pharmacodynamic characteristics from baseline to day 29 (mITT population)
| Variable | Measurement | Lixisenatide | Sitagliptin | LS mean treatment difference (95% CI); P value |
|---|---|---|---|---|
| (N = 69) | (N = 66) | |||
| Primary endpoint | ||||
| PPG AUC0:00‐4:00h (h·mg/dL) | Baseline | 423.0 ± 179.3 | 398.5 ± 144.5 | – |
| Day 29 (visit 5) | 69.6 ± 186.1 | 291.5 ± 147.2 | – | |
| LS mean change from baseline | −347.3 ± 18.14 | −113.3 ± 18.28 | −234.0 (−285.02, −183.00); P < .0001 | |
| Secondary endpoints | ||||
| Maximum PPG excursion, mg/dL | Baseline | 325.9 ± 56.2 | 316.8 ± 54.5 | – |
| Day 29 (visit 5) | 200.7 ± 60.0 | 272.8 ± 53.7 | – | |
| LS mean change from baseline | −122.4 ± 6.40 | −46.6 ± 6.45 | −75.8 (−93.80, −57.81); P < .0001 | |
| C‐peptide AUC0:00‐4:00h, h ng/mL | Baseline | 7.9 ± 3.3 | 8.5 ± 3.6 | – |
| Day 29 (visit 5) | 3.2 ± 4.5 | 9.3 ± 4.2 | – | |
| LS mean change from baseline | −4.8 ± 0.47 | 0.9 ± 0.48 | −5.8 (−7.10, −4.44); P < .0001 | |
| Glucagon AUC0:00‐4:00h, h pg/mL | Baseline | 58.8 ± 62.8 | 55.4 ± 55.7 | – |
| Day 29 (visit 5) | 17.6 ± 54.2 | 29.8 ± 58.2 | – | |
| LS mean change from baseline | −40.0 ± 6.60 | −26.8 ± 6.64 | −13.2 (−31.69, 5.35); P = .1620 | |
| Fasting 1.5‐AG, µg/mL | Baseline | 4.05 ± 3.76 | 4.08 ± 3.49 | – |
| Day 29 (visit 5) | 5.21 ± 4.45 | 5.76 ± 4.80 | – | |
| LS mean change from baseline | 1.16 ± 0.194 | 1.68 ± 0.196 | −0.52 (−1.067, 0.025); P = .0612 | |
| Fasting glycoalbumin, % | Baseline | 22.54 ± 3.61 | 22.13 ± 3.47 | – |
| Day 29 (visit 5) | 20.46 ± 3.28 | 20.87 ± 3.19 | – | |
| LS mean change from baseline | −2.03 ± 0.210 | −1.31 ± 0.211 | −0.73 (−1.318, −0.139); P = .0158 |
Abbreviations: 1.5‐AG, 1.5‐anhydro‐D‐glucitol; AUC, area under the curve; CI, confidence interval; LS, least squares; PPG, postprandial glucose. Errors are standard deviation for values at baseline and day 29, and standard error for LS mean change from baseline values. To convert glucose mg/dL to mmol/L, multiply by 0.0555; to convert C‐peptide ng/mL to nmol/L, multiply by 0.331; glucagon pg/mL is equivalent to ng/L. ANCOVA model with treatment group (lixisenatide, sitagliptin) and randomization strata of screening sulphonylurea use (yes, no) as fixed effects and the baseline value of the corresponding variable as a covariate. The comparison between groups was achieved through appropriate contrast. If values at more than or equal to 2 time points of 1, 1.5 and 2 hours after the start of standardized breakfast were missing, AUC0 :00‐4:00h was set as missing.
Lixisenatide also led to significantly greater LS mean reductions in maximum PPG excursion than sitagliptin (−122.4 vs −46.6 mg/dL [−6.8 vs −2.6 h·mmol/L], respectively; P < .0001). Indeed, the post‐meal glycaemic excursion that was evident in the sitagliptin group was not observed in the lixisenatide group (Figure 2).
Figure 2.
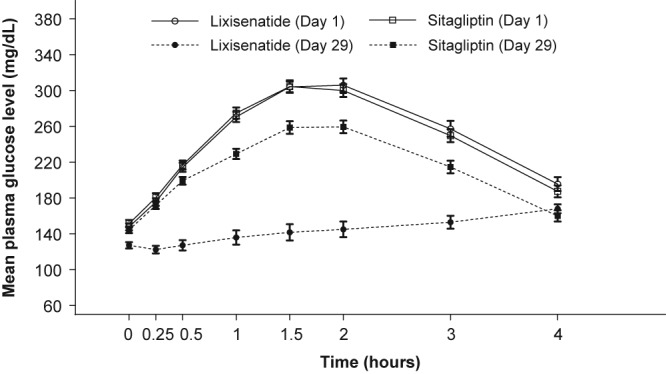
Mean ± standard error PPG change from pre‐meal values at baseline and day 29. PPG, postprandial plasma glucose
Treatment with lixisenatide was also associated with a reduction in C‐peptide exposure and fasting glycoalbumin levels relative to treatment with sitagliptin, but no such statistically significant differences were detected between the 2 groups regarding changes in plasma levels of glucagon and fasting 1.5‐AG (Table 2). In the pharmacodynamic population, gastric emptying was statistically significantly slower in the lixisenatide group than the sitagliptin group, as evidenced by baseline to day 29 mean changes in the gastric emptying rate tmax (baseline: 1.283 vs 1.290 hours; day 29: 3.937 vs 1.292 hours; change‐from‐baseline mean: 2.654 vs 0.002 hours, for lixisenatide and sitagliptin, respectively; P = .0003) and t1/2b (baseline: 2.101 vs 2.161 hours; day 29: 10.247 vs 2.332 hours; change‐from‐baseline mean: 8.146 vs 0.171 hours, for lixisenatide and sitagliptin, respectively; P = .0032).
In the ITT population, there was little change in mean body weight from baseline in the lixisenatide and sitagliptin groups (baseline: 65.15 vs 70.48 kg; day 29: 65.11 vs 70.93 kg; mean change‐from‐baseline mean: −0.41 vs +0.39 kg, for lixisenatide and sitagliptin, respectively).
3.3. Safety and tolerability
In the lixisenatide group TEAEs were reported more frequently (42 of 69 patients, 60.9%) than in the sitagliptin group (11 of 67 patients, 16.4%; Table 3), although all TEAEs were mild or moderate in severity. Gastrointestinal disorders (particularly nausea) and hypoglycaemia occurred more often in the lixisenatide group than in the sitagliptin group (Table 3). TEAEs leading to permanent treatment discontinuation were reported in 2 patients (2.9%) in the lixisenatide group and none in the sitagliptin group. One patient in the lixisenatide group discontinued because of dysgeusia, nausea and oral hypoaesthesia, and the other discontinued because of vomiting. In addition, 1 patient (1.4%) from the lixisenatide group had mild ECG QT prolongation. No patients experienced severe hypoglycaemia, and the hypoglycaemic episodes that occurred in the lixisenatide group occurred during the daytime (Table 3).
Table 3.
Safety and tolerability (ITT population)
| TEAE, n (%) | Lixisenatide | Sitagliptin |
|---|---|---|
| (N = 69) | (N = 67) | |
| Any TEAE | 42 (60.9) | 11 (16.4) |
| Any TEAE possibly related to IMP | 41 (59.4) | 3 (4.5) |
| Any TEAE possibly related to NIMP | 11 (15.9) | 2 (3.0) |
| Any TEAE leading to death | 0 | 0 |
| Any serious TEAE | 0 | 0 |
| Any AE of special interesta | 1 (1.4) | 0 |
| TEAE leading to permanent treatment discontinuation | 2 (2.9) | 0 |
| Types of TEAEs (MedDRA preferred term)b | ||
| Nausea | 22 (31.9) | 0 |
| Hypoglycaemia | 11 (15.9) | 1 (1.5) |
| Severe hypoglycaemiac | 0 | 0 |
| Symptomatic hypoglycaemiad | 7 (10.1) | 1 (1.5) |
| Asymptomatic hypoglycaemiae | 4 (5.8) | 0 |
| Nocturnal hypoglycaemiaf | 0 | 0 |
| Daytime hypoglycaemiag | 11 (15.9) | 1 (1.5) |
| Vomiting | 4 (5.8) | 0 |
| Abdominal discomfort | 3 (4.3) | 0 |
| Constipation | 3 (4.3) | 0 |
| Diarrhoea | 3 (4.3) | 0 |
| Decreased appetite | 3 (4.3) | 0 |
| Eructation | 2 (2.9) | 0 |
| Pruritus | 2 (2.9) | 0 |
| Nasopharyngitis | 2 (2.9) | 3 (4.5) |
| Dizziness | 2 (2.9) | 0 |
Abbreviations: AE, adverse event; IMP, investigational medicinal product; MedDRA, Medical Dictionary for Regulatory Activities; NIMP, non‐investigational medicinal product; TEAE, treatment‐emergent adverse event.
Mild electrocardiogram QT prolongation.
With an incidence ≥2% in the lixisenatide group or the sitagliptin group.
An event during which someone else's help was required to administer glucose or glucagon or perform other emergency medical treatments. Severe hypoglycaemia presents with acute neurological impairment resulting in seizure, unconsciousness or coma directly resulting from the hypoglycaemic event.
An event which was associated with typical hypoglycaemic symptoms with an accompanying plasma glucose 70 mg/dL (≤3.9 mmol/L).
An event which was not associated with typical hypoglycaemic symptoms with an accompanying plasma glucose 70 mg/dL (≤3.9 mmol/L).
Two definitions were used to determine the incidence of nocturnal hypoglycaemia based on time of day and sleep status. Nocturnal hypoglycaemia was defined as an event that: (1) occurred from 12:00 am until 5:59 am (whether the event occurred when the subject was awake, or if the event woke the subject, did not need to be taken into account); and (2) woke patients spanning from the time of sleep in the night until the time of wake‐up in the morning (before administering insulin).
A hypoglycaemic event which occurred from 6:00 am until 23:59 pm.
4. DISCUSSION
In the present study involving Japanese patients with T2D treated with stable doses of Gla‐100, with or without a sulphonylurea, 28 days of treatment with lixisenatide reduced PPG and maximal PPG excursion relative to sitagliptin. Consequently, a post‐meal glycaemic excursion was avoided in the lixisenatide group, which was not the case in the sitagliptin group. This pronounced lowering effect of lixisenatide on PPG is consistent with observations from previous studies in Asian and white patients.24, 25, 26, 27, 28, 29, 30, 33 Further, the PPG‐lowering effect produced by lixisenatide coincided with change‐from‐baseline reductions in exposure to C‐peptide, fasting glycoalbumin levels, and the gastric emptying rate, all of which were statistically significantly greater than that observed for sitagliptin.
The principal effects of GLP‐1 receptor agonists are to induce pancreatic insulin secretion, slow gastric emptying, and suppress postprandial glucagon secretion.15, 34, 35, 36, 37 Since increases in tmax and t1/2b of the gastric emptying rate were observed in the lixisenatide group vs the sitagliptin group, it is likely that slowing of gastric emptying was the driver for PPG reduction in this study, rather than insulinotropic effects. Indeed, greater exposure to postprandial C‐peptide (and hence insulin) was observed in the sitagliptin group than in the lixisenatide group, which is consistent with the inverse association of C‐peptide levels with glycaemic variability and with post‐meal glucose rise in T2D (both of which were higher in the sitagliptin group).38 In previous studies, there was a direct relationship between PPG AUC after breakfast and gastric emptying with lixisenatide 20 µg once daily,32, 39 which was not observed with placebo.39
Similar marked improvements in postprandial glycaemic control were observed with 24 weeks of lixisenatide in 2 randomized, placebo‐controlled studies of Asian patients with T2D inadequately controlled on metformin or basal insulin, with or without a sulphonylurea.28, 30 Our data thus lend support to the theory that incretin‐based therapies (and GLP‐1 receptor agonists in particular) appear well‐suited for use in Asian and Japanese patients with T2D,28, 30 as these populations are predisposed to insulin deficiency rather than insulin resistance, which may manifest via a profound underlying GLP‐1 insufficiency.40 The findings of the present study are also consistent with studies in Western patients with T2D inadequately controlled on metformin or on optimized Gla‐100, as once‐daily pre‐breakfast lixisenatide was associated with significantly greater reductions in PPG, postprandial C‐peptide, and postprandial glucagon relative to pre‐breakfast liraglutide.31, 32 Gastric emptying was also substantially slower in the lixisenatide than the liraglutide study group.31, 32
Statistically significant change‐from‐baseline reductions in exposure to fasting glycoalbumin levels with lixisenatide vs sitagliptin were not accompanied by similar changes in 1.5‐AG. Glycoalbumin reflects the proportion of glycated albumin to total serum albumin and is informative of blood glucose control over the preceding 3 weeks, whereas 1.5‐AG is a marker of glycaemia‐induced glycosuria, and thus is informative of high glycaemic variability over the hour‐to‐day time frame. It is possible that the duration of action of lixisenatide administered before breakfast was not long enough to address PPG rise after lunch and dinner, and this phenomenon was captured by analysis of 1.5‐AG levels only.
Both lixisenatide and sitagliptin were well tolerated in the present study. Gastrointestinal disorders and hypoglycaemia occurred with greater frequency in the lixisenatide than the sitagliptin group. No new lixisenatide safety signals were identified.
The main limitations of the present study were the relatively small sample size, lack of double blinding associated with the open‐label design, and brevity. While sitagliptin 50 mg/d is the standard initial dosage in Japan, which is increased to 100 mg/d in patients poorly controlled on the lower dosage, the present study was not conducted over a sufficiently long time frame to make this decision. It is likely that the PPG outcome data in the sitagliptin arm would have improved had the 100 mg/d dose level been used, because sitagliptin over the dose range of 25 to 100 mg/d is known to reduce 2‐hour PPG in a dose‐dependent manner relative to placebo.41 A future study with a larger scale and longer treatment period is required to determine how freedom from postprandial hyperglycaemia associated with lixisenatide affects HbA1c concentration.
In conclusion, at the doses tested, combined therapy of lixisenatide with Gla‐100 showed greater efficacy, as indicated by a significant reduction in PPG rise, than combined therapy of sitagliptin with Gla‐100. These data support the use of lixisenatide to address PPG excursions when correction of PPG is insufficient with long‐acting insulin.
ACKNOWLEDGEMENTS
Medical writing assistance was provided by Malcolm Darkes, PhD, of ProScribe K.K.– Envision Pharma Group, and was funded by Sanofi K.K. ProScribe's services complied with international guidelines for GPP3. The authors would like to thank all study participants.
Conflict of interest
Y. Y. is a consultant to Sanofi K.K. and Ono, serves on the speakers’ bureau of Sanofi K.K., MSD and Ono, and receives research funding from Sanofi K.K., MSD and Ono. M. S., Y. N., M. T., D. W. and Y. S. are employees of Sanofi K.K, and M. S., Y. N., M. T. and Y. S. own stock in Sanofi K.K. Y. U. is a consultant to and serves on the advisory panel of Sanofi K.K., receives research funding from Sanofi K.K. and Okinawa prefecture, and his institution receives grants and endowments from Daiichi Sankyo, Takeda, Sumitomo Dainippon Pharma, Tsumura, Eisai, and AstraZeneca.
This study was sponsored by Sanofi K.K., manufacturer and licensee of lixisenatide and insulin glargine. Sanofi K.K. was responsible for the design and coordination of this trial, monitoring clinical sites, collecting and managing data, and performing all statistical analyses.
Author contributions
All authors were involved in developing the manuscript, providing critical review of the content and final approval of the version to be published. M. S., Y. N. and M. T. contributed to the design of the study protocol and managed the study, and participated in writing, reviewing and editing the manuscript. D. W. undertook statistical analyses. Y. Y., Y. S. and Y. U. made significant suggestions to the analysis and interpretation of the data.
Yamada Y, Senda M, Naito Y, Tamura M, Watanabe D, Shuto Y and Urita Y. Reduction of postprandial glucose by lixisenatide vs sitagliptin treatment in Japanese patients with type 2 diabetes on background insulin glargine: A randomized phase IV study (NEXTAGE Study). Diabetes Obes Metab. 2017;19:1252–1259. https://doi.org/10.1111/dom.12945
Funding Information This study was sponsored by Sanofi K.K., manufacturer and licensee of lixisenatide and insulin glargine. Medical writing assistance was provided by Malcolm Darkes PhD, of ProScribe K.K.– Envision Pharma Group, and was funded by Sanofi K.K.
REFERENCES
- 1. Ceriello A, Colagiuri S. International Diabetes Federation guideline for management of postmeal glucose: a review of recommendations. Diabet Med. 2008;25:1151‐1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Monnier L, Colette C, Dunseath GJ, Owens DR. The loss of postprandial glycemic control precedes stepwise deterioration of fasting with worsening diabetes. Diabetes Care. 2007;30:263‐269. [DOI] [PubMed] [Google Scholar]
- 3. Monnier L, Lapinski H, Colette C. Contributions of fasting and postprandial plasma glucose increments to the overall diurnal hyperglycemia of type 2 diabetic patients: variations with increasing levels of HbA(1c). Diabetes Care. 2003;26:881‐885. [DOI] [PubMed] [Google Scholar]
- 4. Peter R, Dunseath G, Luzio SD, Chudleigh R, Choudhury SR, Owens DR. Relative and absolute contributions of postprandial and fasting plasma glucose to daytime hyperglycaemia and HbA(1c) in subjects with type 2 diabetes. Diabet Med. 2009;26:974‐980. [DOI] [PubMed] [Google Scholar]
- 5. Woerle HJ, Neumann C, Zschau S, et al. Impact of fasting and postprandial glycemia on overall glycemic control in type 2 diabetes importance of postprandial glycemia to achieve target HbA1c levels. Diabetes Res Clin Pract. 2007;77:280‐285. [DOI] [PubMed] [Google Scholar]
- 6. Rodbard HW, Blonde L, Braithwaite SS, et al. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the management of diabetes mellitus. Endocr Pract. 2007;13(suppl 1):1‐68. [DOI] [PubMed] [Google Scholar]
- 7. Basevi V, Di Mario S, Morciano C, Nonino F, Magrini N. Comment on: American Diabetes Association. Standards of medical care in diabetes 2011. Diabetes Care. 2011;34(suppl. 1):S11–S61. Diabetes Care. 2011;34:e53; author reply e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. LANTUS® (insulin glargine injection) solution for subcutaneous injection. Highlights of prescribing information. 2015 Sanofi‐Aventis U.S. LLC. https://www.lantus.com. Accessed October 6, 2016.
- 9. Riddle MC, Rosenstock J, Gerich J. The treat‐to‐target trial: randomized addition of glargine or human NPH insulin to oral therapy of type 2 diabetic patients. Diabetes Care. 2003;26:3080‐3086. [DOI] [PubMed] [Google Scholar]
- 10. Bazzano LA, Lee LJ, Shi L, Reynolds K, Jackson JA, Fonseca V. Safety and efficacy of glargine compared with NPH insulin for the treatment of Type 2 diabetes: a meta‐analysis of randomized controlled trials. Diabet Med. 2008;25:924‐932. [DOI] [PubMed] [Google Scholar]
- 11. Nauck MA, Homberger E, Siegel EG, et al. Incretin effects of increasing glucose loads in man calculated from venous insulin and C‐peptide responses. J Clin Endocrinol Metab. 1986;63:492‐498. [DOI] [PubMed] [Google Scholar]
- 12. Toft‐Nielsen MB, Damholt MB, Madsbad S, et al. Determinants of the impaired secretion of glucagon‐like peptide‐1 in type 2 diabetic patients. J Clin Endocrinol Metab. 2001;86:3717‐3723. [DOI] [PubMed] [Google Scholar]
- 13. Vaag AA, Holst JJ, Volund A, Beck‐Nielsen HB. Gut incretin hormones in identical twins discordant for non‐insulin‐dependent diabetes mellitus (NIDDM)–evidence for decreased glucagon‐like peptide 1 secretion during oral glucose ingestion in NIDDM twins. Eur J Endocrinol. 1996;135:425‐432. [DOI] [PubMed] [Google Scholar]
- 14. Vilsboll T, Krarup T, Deacon CF, Madsbad S, Holst JJ. Reduced postprandial concentrations of intact biologically active glucagon‐like peptide 1 in type 2 diabetic patients. Diabetes. 2001;50:609‐613. [DOI] [PubMed] [Google Scholar]
- 15. Drucker DJ, Nauck MA. The incretin system: glucagon‐like peptide‐1 receptor agonists and dipeptidyl peptidase‐4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696‐1705. [DOI] [PubMed] [Google Scholar]
- 16. Bosi E. Time for testing incretin therapies in early type 1 diabetes? J Clin Endocrinol Metab. 2010;95:2607‐2609. [DOI] [PubMed] [Google Scholar]
- 17. Tews D, Werner U, Eckel J. Enhanced protection against cytokine‐ and fatty acid‐induced apoptosis in pancreatic beta cells by combined treatment with glucagon‐like peptide‐1 receptor agonists and insulin analogues. Horm Metab Res. 2008;40:172‐180. [DOI] [PubMed] [Google Scholar]
- 18. Arnolds S, Dellweg S, Clair J, et al. Further improvement in postprandial glucose control with addition of exenatide or sitagliptin to combination therapy with insulin glargine and metformin: a proof‐of‐concept study. Diabetes Care. 2010;33:1509‐1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brunton S. GLP‐1 receptor agonists vs. DPP‐4 inhibitors for type 2 diabetes: is one approach more successful or preferable than the other? Int J Clin Pract. 2014;68:557‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vora J. Combining incretin‐based therapies with insulin: realizing the potential in type 2 diabetes. Diabetes Care. 2013;36(suppl 2):S226‐S232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garber AJ. Long‐acting glucagon‐like peptide 1 receptor agonists: a review of their efficacy and tolerability. Diabetes Care. 2011;34(suppl 2):S279‐S284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lovshin JA, Drucker DJ. Incretin‐based therapies for type 2 diabetes mellitus. Nat Rev Endocrinol. 2009;5:262‐269. [DOI] [PubMed] [Google Scholar]
- 23. DeFronzo RA, Okerson T, Viswanathan P, Guan X, Holcombe JH, MacConell L. Effects of exenatide versus sitagliptin on postprandial glucose, insulin and glucagon secretion, gastric emptying, and caloric intake: a randomized, cross‐over study. Curr Med Res Opin. 2008;24:2943‐2952. [DOI] [PubMed] [Google Scholar]
- 24. Fonseca VA, Alvarado‐Ruiz R, Raccah D, et al. Efficacy and safety of the once‐daily GLP‐1 receptor agonist lixisenatide in monotherapy: a randomized, double‐blind, placebo‐controlled trial in patients with type 2 diabetes (GetGoal‐Mono). Diabetes Care. 2012;35:1225‐1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Riddle MC, Forst T, Aronson R, et al. Adding once‐daily lixisenatide for type 2 diabetes inadequately controlled with newly initiated and continuously titrated basal insulin glargine: a 24‐week, randomized, placebo‐controlled study (GetGoal‐Duo 1). Diabetes Care. 2013;36:2497‐2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Riddle MC, Aronson R, Home P, et al. Adding once‐daily lixisenatide for type 2 diabetes inadequately controlled by established basal insulin: a 24‐week, randomized, placebo‐controlled comparison (GetGoal‐L). Diabetes Care. 2013;36:2489‐2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ahren B, Leguizamo Dimas A, Miossec P, Saubadu S, Aronson R. Efficacy and safety of lixisenatide once‐daily morning or evening injections in type 2 diabetes inadequately controlled on metformin (GetGoal‐M). Diabetes Care. 2013;36:2543‐2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Seino Y, Min KW, Niemoeller E, Takami A. Randomized, double‐blind, placebo‐controlled trial of the once‐daily GLP‐1 receptor agonist lixisenatide in Asian patients with type 2 diabetes insufficiently controlled on basal insulin with or without a sulfonylurea (GetGoal‐L‐Asia). Diabetes Obes Metab. 2012;14:910‐917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rosenstock J, Hanefeld M, Shamanna P, et al. Beneficial effects of once‐daily lixisenatide on overall and postprandial glycemic levels without significant excess of hypoglycemia in type 2 diabetes inadequately controlled on a sulfonylurea with or without metformin (GetGoal‐S). J Diabetes Complications. 2014;28:386‐392. [DOI] [PubMed] [Google Scholar]
- 30. Yu Pan C, Han P, Liu X, et al. Lixisenatide treatment improves glycaemic control in Asian patients with type 2 diabetes mellitus inadequately controlled on metformin with or without sulfonylurea: a randomized, double‐blind, placebo‐controlled, 24‐week trial (GetGoal‐M‐Asia). Diabetes Metab Res Rev. 2014;30:726‐735. [DOI] [PubMed] [Google Scholar]
- 31. Kapitza C, Forst T, Coester HV, Poitiers F, Ruus P, Hincelin‐Méry A. Pharmacodynamic characteristics of lixisenatide once daily versus liraglutide once daily in patients with type 2 diabetes insufficiently controlled on metformin. Diabetes Obes Metab. 2013;15:642‐649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meier JJ, Rosenstock J, Hincelin‐Mery A, et al. Contrasting effects of lixisenatide and liraglutide on postprandial glycemic control, gastric emptying, and safety parameters in patients with type 2 diabetes on optimized insulin glargine with or without metformin: a randomized, open‐label trial. Diabetes Care. 2015;38:1263‐1273. [DOI] [PubMed] [Google Scholar]
- 33. Ratner RE, Rosenstock J, Boka G. Dose‐dependent effects of the once‐daily GLP‐1 receptor agonist lixisenatide in patients with type 2 diabetes inadequately controlled with metformin: a randomized, double‐blind, placebo‐controlled trial. Diabet Med. 2010;27:1024‐1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Drucker DJ, Buse JB, Taylor K, et al. Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: a randomised, open‐label, non‐inferiority study. Lancet. 2008;372:1240‐1250. [DOI] [PubMed] [Google Scholar]
- 35. Cersosimo E, Gastaldelli A, Cervera A, et al. Effect of exenatide on splanchnic and peripheral glucose metabolism in type 2 diabetic subjects. J Clin Endocrinol Metab. 2011;96:1763‐1770. [DOI] [PubMed] [Google Scholar]
- 36. Cervera A, Wajcberg E, Sriwijitkamol A, et al. Mechanism of action of exenatide to reduce postprandial hyperglycemia in type 2 diabetes. Am J Physiol Endocrinol Metab. 2008;294:E846‐E852. [DOI] [PubMed] [Google Scholar]
- 37. Linnebjerg H, Park S, Kothare PA, et al. Effect of exenatide on gastric emptying and relationship to postprandial glycemia in type 2 diabetes. Regul Pept. 2008;151:123‐129. [DOI] [PubMed] [Google Scholar]
- 38. Jones AG, Hattersley AT. The clinical utility of C‐peptide measurement in the care of patients with diabetes. Diabet Med. 2013;30:803‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lorenz M, Pfeiffer C, Steinstrasser A, et al. Effects of lixisenatide once daily on gastric emptying in type 2 diabetes–relationship to postprandial glycemia. Regul Pept. 2013;185:1‐8. [DOI] [PubMed] [Google Scholar]
- 40. Seino Y, Fukushima M, Yabe D. GIP and GLP‐1, the two incretin hormones: similarities and differences. J Diabetes Invest. 2010;1:8‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Iwamoto Y, Taniguchi T, Nonaka K, et al. Dose‐ranging efficacy of sitagliptin, a dipeptidyl peptidase‐4 inhibitor, in Japanese patients with type 2 diabetes mellitus. Endocr J. 2010;57:383‐394. [DOI] [PubMed] [Google Scholar]