

Abstract
Aromatic amino acid ammonia‐lyases and aromatic amino acid 2,3‐aminomutases contain the post‐translationally formed prosthetic 3,5‐dihydro‐4‐methylidene‐5H‐imidazol‐5‐one (MIO) group. MIO enzymes catalyze the stereoselective synthesis of α‐ or β‐amino acid enantiomers, making these chemical processes environmentally friendly and affordable. Characterization of novel inhibitors enables structural understanding of enzyme mechanism and recognizes promising herbicide candidates as well. The present study found that both enantiomers of the aminophosphonic acid analogue of the natural substrate phenylalanine and a novel derivative bearing a methylidene at the β‐position inhibited phenylalanine ammonia‐lyases (PAL), representing MIO enzymes. X‐ray methods unambiguously determined the absolute configuration of all tested enantiomers during their synthesis. Enzyme kinetic measurements revealed the enantiomer of the methylidene‐substituted substrate analogue as being a mirror image relation to the natural l‐phenylalanine as the strongest inhibitor. Isothermal titration calorimetry (ITC) confirmed the binding constants and provided a detailed analysis of the thermodynamic driving forces of ligand binding. Molecular docking suggested that binding of the (R)‐ and (S)‐enantiomers is possible by a mirror image packing.

Keywords: amino acids, aminophosphonic acids, bioinformatics, calorimetry, enzyme inhibition, MIO enzymes
Introduction
Phenylalanine ammonia‐lyases catalyze the non‐oxidative ammonia elimination from l‐phenylalanine to yield (E)‐cinnamic acid [(E)‐CA].1 They are most commonly found in fungi and plants, where cinnamic acid is the starting material for the biosynthesis of an array of natural products ranging from flavonoids to lignins.2 Industrial applications make use of the reverse reaction for the synthesis of unnatural amino acids. In 1983, Genex Corporation patented a method for the enzymatic production of l‐phenylalanine, one of the components of the artificial sweetener dipeptide aspartame.3 Currently PALs are more widely used for the synthesis of unnatural amino acids.4 DSM Pharma Chemicals developed a method based on PAL‐catalyzed hydroamination of 2′‐chlorocinnamic acid for the production of (S)‐2‐indolinecarboxylic acid on the ton scale.5
Related and structurally similar enzymes are l‐tyrosine (TAL)6 and l‐histidine ammonia‐lyases (HAL)7 and l‐tyrosine (TAM)8 and l‐phenylalanine 2,3‐amino‐mutases (PAM).9 These enzymes all contain in their catalytic centers the 3,5‐dihydro‐4‐methylidene‐5H‐imidazol‐5‐one (MIO) electrophilic prosthetic group. This uncommon cofactor is formed in an autocatalytic way in a posttranslational process from three adjacent amino acids of the respective protein being usually alanine‐serine‐glycine by cyclization and dehydration.10 In spite of the relatively low similarity between their sequences (<30%), the tertiary and quaternary folds of all MIO‐containing proteins are highly similar. Mutagenesis studies showed that two tyrosines (TyrA and TyrB in Scheme 1) are essential for catalysis in HAL,11 PAL,12 and TAL.13 These structural similarities suggest that ammonia elimination proceeds by similar mechanisms in all MIO enzymes.
Scheme 1.
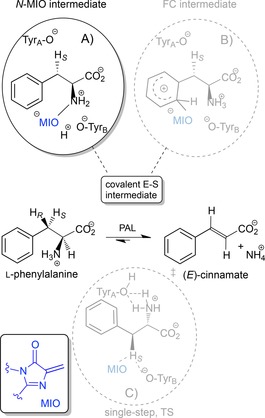
Three proposed mechanisms for the PAL‐catalyzed conversion of l‐phenylalanine to (E)‐CA. The reaction proceeds via covalent intermediates in A) and B) or through a single‐step transition state in C).
Three mechanisms have been proposed for the enzymatic elimination of ammonia involving MIO (Scheme 1).
The N‐MIO intermediate mechanism suggests that the amino group of the substrate forms a covalent bond with the electrophilic MIO residue of the enzyme, that generates a better leaving group (Scheme 1A, N‐MIO intermediate pathway). Most likely, ammonia elimination from the covalent intermediate proceeds by a stepwise process via a carbanion intermediate (E1cB mechanism).14
As the abstraction of the non‐acidic β‐proton was considered to be difficult by an amino acid base, a Friedel–Crafts (FC)‐type mechanism was proposed (Scheme 1B). The β‐hydrogens of phenylalanine could be acidified by the positive charge in the forming σ‐complex. Thus TyrA can deprotonate the β‐position with concomitant elimination of the protonated amino group as NH3. At last, the release of MIO restores the aromaticity.15
Recently, a single‐step mechanism was proposed for TAL. It assumes a single transition state (TS) for the deamination without the formation of a covalent bond between the substrate and the MIO group (Scheme 1C).16
Although some questions remain open about the mechanism of action of MIO‐enzymes, kinetic isotope effect studies,17 crystallographic data18 and computational studies19 support the N‐MIO mechanism.
PALs show high enantioselectivity in the catalysis of their natural substrate l‐phenylalanine and its analogues, but are also capable of binding the d‐enantiomers. It was found that PAL could catalyze ammonia elimination from l‐phenylalanine more than 4500 times faster than from d‐phenylalanine.1 Moreover, d‐phenylalanine was found to be a competitive inhibitor,1 suggesting that the d‐enantiomer can also be bound to PAL. A recent study, involving three PALs, suggested that PALs are able to catalyze the formation of the d‐enantiomers of substrates with an electron‐deficient aromatic moiety by a slower and MIO‐independent pathway.20
A wide range of aromatic compounds inhibit PALs. The d‐phenylalanine is a general inhibitor of PALs, as a mirror image packing is possible. This was suggested based on similarities in K i value of d‐Phe and K m value of l‐Phe.21 Further kinetic studies investigating protection and inhibition provided more evidence on binding of the opposite enantiomers and led to a model suggesting mirror image packing of the substrate enantiomers.22 Interestingly, inhibition by SH group‐containing compounds seemed to be species specific.23 A wide variety of substrate analogues was studied as inhibitors of PAL in vitro and in vivo for potential herbicide use.24 The largest inhibitory effects were found with 2‐aminoindan‐2‐phosphonic acid (AIP) (K i=25 nM) and (S)‐2‐aminooxy‐3‐phenylpropanoic acid (AOPP) (K i=0.38 nM).25
Besides kinetic studies, numerous methods are routinely used to determine the binding affinity of small molecules to proteins. Uniquely among the techniques to measure protein–small molecule interactions, isothermal titration calorimetry (ITC) stands out, as it directly provides quantitative thermodynamic parameters (ΔH is directly measured, K d is calculated from the titration curve, then ΔG and ΔS are estimated).26 This method is becoming widely appreciated in measuring the affinity of potential drug candidates to their protein targets.27
In spite of the usefulness of ITC to assess binding of small molecules to enzymes, to the best of our knowledge, no direct calorimetric measurements were performed for any MIO enzymes.
Here we present detailed enzyme kinetic and equilibrium binding studies with PAL from Petroselinum crispum (PcPAL) using three racemic aminophosphonic acids [(±)‐4, (±)‐5, (±)‐6, in the dashed box] and enantiopure forms of two further ones [(R)‐2, (S)‐2 and (R)‐3, (S)‐3, in the solid box] (Figure 1).
Figure 1.
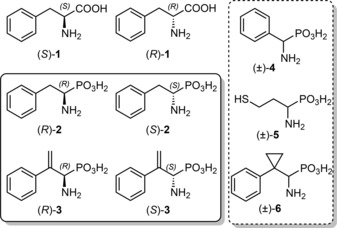
Structures of the investigated compounds. Note that the configuration of (R)‐2 and (R)‐3 corresponds to that of l‐phenylalanine [(S)‐1] due to the higher CIP rank of the phosphonic acid moiety compared to the carboxylate group.
Results and Discussion
Aminophosphonic acids (R)‐2, (S)‐2 and (±)‐4 are known inhibitors of PAL,24 and were included in this study for comparative reasons. In compounds 3 and 6 the β‐carbon bears instead of hydrogens moieties which can accept attack from nucleophiles from the active site (either the exocyclic methylidene of MIO or a catalytic tyrosine). Phosphahomocysteine (5) has in place of the phenyl ring an SH group which may interact with the methylidene group of MIO and thus inhibit PAL.
On top of the detailed study with PcPAL, the inhibitory behavior of the enantiopure inhibitors [(R)‐2, (S)‐2 and (R)‐3, (S)‐3] was tested with other PALs of eukaryotic and prokaryotic origin [Rhodosporidium toruloides (RtPAL) and Anabaena variabilis (AvPAL), respectively]. Evaluation of the inhibitory properties to these three different PALs offers general insight regarding the binding stereochemistry and its perturbation by the addition of the methylidene group.
To complete the kinetic studies, we also provide a full thermodynamic analysis for these inhibitory molecules to the wild type and a specifically designed Y110F mutant of PcPAL. Modulation of the binding compared to the wild‐type protein, especially with the methylidene‐substituted analogues, argues for a specific interaction between the methylidene moiety and Tyr110 of the mobile, catalytically relevant loop (called the Tyr‐loop). As ITC has never been applied for MIO enzymes, the present study contributes to the understanding of the thermodynamics of substrate binding to PAL with significant novelty.
Finally, a detailed docking study on the molecular background of the binding of the various compounds to AvPAL corroborates these conclusions.
Synthesis of α‐Aminophosphonic Acids 2–6
As we aimed for racemic α‐aminophosphonic acids and in some cases also their pure enantiomers with ee ≥99% (Supporting Information, Figures S8–S19), we opted first for the preparation of the racemates. The enantiomers were obtained by HPLC separation on a chiral stationary phase. First, syntheses of aminophosphonic acids (±)‐2, (±)‐4, (±)‐5 and (±)‐6 and the key intermediate (±)‐7 for the preparation of the phosphaphenylalanine analogue (±)‐3 were performed [see the Supporting Information]. For the sake of clarity, only the final steps of the synthesis of (±)‐, (R)‐ and (S)‐3 are given in Scheme 2. Since the use of refluxing 6 M HCl caused extensive decomposition of the labile α‐amino‐2‐propenylphosphonate, full deprotection of the racemate and both enantiomers of 8 had to be performed with TMSBr/allyl‐TMS28 at 55 °C for 18 h. Crystallization of the α‐aminophosphonic acids from water/i‐PrOH gave colorless crystals of (±)‐3, (S)‐(+)‐3 and (R)‐(−)‐3 in 61%, 74% and 68% yields, respectively.
Scheme 2.

Preparation of α‐aminophosphonic acids (R)‐(+)‐3 and (S)‐(−)‐3 with absolute configuration assignment by the X‐ray method. Reaction conditions: a) Me2NH, H2O2; b) TMSBr, allylTMS; c) H2O; d) HCl, dioxane; e) 4‐BrC6H4NCO, pyridine.
The mixture of Boc‐protected aminophosphonates 7 was converted to racemic 2‐propenylphosphonate (±)‐8 in 92% yield using H2O2 (in combination with Me2NH having a beneficial effect on yield29). The enantiomers of 8 were separated by preparative HPLC on a chiral stationary phase (Chiralpak IC column; t R=15.05 and 30.97 min, both enantiomerically pure). Figure 2A shows the (S) absolute configuration of the less polar (+)‐8 as determined by single crystal X‐ray structure analysis.
Figure 2.

Absolute configuration of (A) (S)‐8 and (B) (R)‐9 determined by X‐ray crystallography. Ellipsoid plot (left) and stick (right) representations are shown.
Surprisingly, the (S)‐(+)‐enantiomer of 3 proved to be a more potent inhibitor of this enzyme than the (R)‐(−)‐enantiomer. This finding was contrary to our initial expectation, as we assumed at first that (S)‐(+)‐3, corresponding to d‐Phe [(R)‐1], would be a less potent inhibitor than (R)‐(−)‐3, corresponding to the natural substrate l‐Phe [(S)‐1]. To eliminate any ambiguity, a urea derivative of the more polar enantiomer (−)‐8 was prepared. Figure 2B shows the result of the single crystal X‐ray structure analysis revealing that substituted urea (−)‐9 had the (R)‐configuration. This data corroborated the previous absolute configuration assignment for (R)‐(−)‐3.
Steady State Inhibition Studies with PcPAL
Initial reaction rates for deamination of l‐Phe [(S)‐1] catalyzed by PcPAL were determined by UV spectrometry detecting the formation of the (E)‐CA product at 290 nm.
Initial reaction rates (v 0) as a function of the substrate concentration determined the Michaelis constant (K m) and the turnover number (k cat) of the enzyme. Kinetic measurements in the presence of inhibitors determined the inhibition constants (K i). The mechanism of inhibition could be determined by comparing the fit of experimental data to different inhibition models. Competitive, uncompetitive and non‐competitive inhibition models are most commonly found, hence we tested these models in our experiments. We note that the structure of the tested compounds suggests similar binding as the substrate, potentially resulting in competitive inhibition.
As expected, compound (R)‐2 – the phosphonic acid analogue of the l‐Phe [(S)‐1] – was one order of magnitude stronger as an inhibitor than the opposite enantiomer (S)‐2 (Table 1). In close agreement with data from the literature,24 the competitive inhibition model with K i=0.66 μM fitted best to the experimental data for (R)‐2. The K i=4.28 μM value for (S)‐2 was also in agreement with the previous results on inhibition of PAL from maize.24 The enantiomers of aminophosphonic acids 2 and 3 were purified to ≥99.6% ee values (Supporting Information, Figures S17–S19), hence inhibition by contaminating opposite enantiomers could be ruled out.
Table 1.
Apparent inhibition constants and binding equilibrium constants of the aminophosphonic acids 2–6.
| Inhibitor | Type of inhibition[a] | K i [μM] | K i_lit [μM] | K d,ITC [b] [μM] |
|---|---|---|---|---|
| (R)‐2 | SB‐Comp. | 0.66±0.11 | 1.5[24a],[c] | 2.7 |
| (S)‐2 | Comp. | 4.28±0.11 | 11.6[24a],[c] | 5.2 |
| (R)‐3 | Comp. | 0.64±0.02 | 1.1 | |
| (S)‐3 | SB‐Comp. | 0.04±0.01 | 0.04 | |
| (±)‐4 | Comp. | 41.5±5.6 | 6.524c | n.d. |
| (±)‐5 | No inh. | >1000 | n.d. | |
| (±)‐6 | No inh. | >1000 | n.d. |
[a] Best fitting inhibition model to the experimental data. Comp.: competitive; SB‐Comp: competitive with slow binding of the inhibitor; No inh.: no measurable inhibition.
[b] Details of the ITC measurements are described in the next section. n.d.: not determined.
[c] Inhibition constants measured using PAL from maize (sequence identity with PcPAL: 77%).
Interestingly, introduction of a single methylidene group at the C‐2 position of phosphonic acid analogue 2 resulting in a novel inhibitor 3 reversed the enantiopreference of inhibition of PcPAL. In fact, (S)‐3 was one order of magnitude stronger as an inhibitor than (R)‐3. The inhibition constant of (S)‐3 (K i=0.04 μM) with PcPAL was only slightly higher than that of AIP – the most potent aminophosphonic PAL inhibitor (K i=0.025 μM).25 The fact that (S)‐3 was two orders of magnitude stronger as an inhibitor than (S)‐2, suggested that the methylidene group had a significant effect on binding of the (S)‐enantiomers. In contrast, the inhibition constants of (R)‐3 and (R)‐2 are similar.
The phenylglycine analogue (±)‐4 was a weak competitive inhibitor of PcPAL. The competitive inhibition model with K i=41.5 μM, described best the measured initial reaction rates (v 0). The almost one order of magnitude difference compared to previous data24c is considerable, nonetheless (±)‐4 was the weakest one amongst the effective PAL inhibitors. The aromatic binding pocket of PcPAL suited to accommodate a benzyl moiety seems to be too large for the smaller phenyl ring of (±)‐4, hence the hydrophobic interactions with the aromatic binding region of the active site pocket are less pronounced.
In contrast to the preliminary expectations, (±)‐5 and (±)‐6 showed no measurable inhibition with PcPAL up to 1 mM concentration. This is surprising as Pseudomonas putida HAL (sequence identity to PcPAL: 21%) was irreversibly inhibited by l‐homocysteine at 9 mM concentration after incubation for 120 min in the presence of oxygen.30 Initial velocities of the (E)‐CA formation with PcPAL were identical when the reaction was started with the addition of PcPAL to the test in the presence or in the absence of (±)‐5 or (±)‐6 or the enzyme was preincubated with (±)‐5 for 60 min prior to activity measurement. In the case of (±)‐6, the sterically demanding cyclopropyl substituent at the β‐carbon atom presumably hindered the binding to PcPAL. This hypothesis was later supported by molecular docking.
Slow Binding Inhibition of PcPAL by the Phosphonic Acid Inhibitors (R)‐2 and (S)‐3
Addition of (S)‐3 to the PcPAL‐catalyzed reaction resulted in non‐linear progress curves, as shown on Figure 2A, corresponding to a change in the enzymatic activity in a time‐dependent manner. The non‐linear rate curves observed on the steady‐state time scale due to transient kinetics suggested a slow binding mechanism for the inhibitors.31
This behavior may originate from different mechanisms at the molecular level: (i) if there is a transition state‐mimicking intermediate, (ii) if the inhibitor exists in multiple forms, and only one of which may interact with the enzyme, (iii) if only a rare form of the enzyme binds the inhibitor or (iv) if after an initial fast binding, slow structural rearrangements form a second inhibitory complex.32 In kinetic analysis of slow binding inhibitors, the integrated rate equation for the product concentration (P) was derived by Frieden [Eq. (1)].33

This integrated rate equation determines the velocities at time zero (v
z), the steady‐state velocities (v
s) and the frequency constant of the exponential phase (λ). Steady state velocities determine the apparent inhibitory constants. The two simplest slow binging inhibition mechanisms – shown as Eq. (2) and Eq. (3) – can be distinguished by the velocities at time zero as a function of the inhibitor concentration.

According to the first mechanism represented by Eq. (2), binding of the inhibitor may be a slow process, hence v z is independent of the inhibitor concentration. According to another slow binding mechanism represented by Eq. (3), an EI complex forms rapidly, which subsequently undergoes changes to form the strongly bound (EI)* complex. Due to the first fast step in the second mechanism, v z depends on the inhibitor concentration.
Figure 2B shows the first 30 s of the progress curves of the product formation from l‐Phe catalyzed by PcPAL in the absence and in the presence of (S)‐3. It was clearly visible that even the early slope of the curves depended on the inhibition concentrations, and the fitted parameters confirmed this observation. This favored the two‐step slow binding mechanism described by Eq. (3), where after an initial fast binding process a slow change took place.
Several possibilities could account for this behavior. One possibility may be that deprotonation of the enzyme or the ligand after the fast binding enforces the interactions further. A second possibility may be that in the slow change step a nucleophilic part of the ligand forms a Michael adduct with the methylidene group of the MIO. Finally, TyrA of the mobile loop of the enzyme may also attack the methylidene group of the ligand. It is noteworthy that the frequently used AIP also showed slow binding characteristics with PcPAL, however by the one‐step slow binding mechanism [Eq. (2)].25 The enantiopure (R)‐2 also showed slow binding behavior, but to a smaller extent than (S)‐3. Interestingly, (R)‐3 was not a slow binding inhibitor. Differences in the binding kinetics indicated that although the inhibition constants were almost similar [K i=0.64 μM for (R)‐3 and K i=0.66 μM for (S)‐2], the binding mechanism was perturbed by the addition of the methylidene group at the C‐2 position.
Decreases in the product formation rate after the slow binding step might happen due to an irreversible covalent binding. However, the progress curves determined after preincubation of PcPAL with (S)‐3 suggested reversible binding. The blue progress curve with empty circles shown in Figure 3A was obtained in a reaction started with addition of l‐Phe to the assay after preincubation of PcPAL with (S)‐3 for 5 min. Increases of activity to a steady state level indicated reversible binding of (S)‐3. Additionally, variations of the preincubation time between 1–60 min did not alter the progress curves indicating that the fast binding step was completed within 1 min.
Figure 3.

Production curves of (E)‐cinnamic acid [(E)‐CA] formation from l‐Phe [(S)‐1] catalyzed by PcPAL in the presence of various amounts of enantiopure (S)‐3. Panel A depicts progress curves at various inhibitor concentrations for 5 min (for clear visibility only every 20th data point is plotted). The curve in blue with empty dots shows the production of (E)‐CA in an assay containing PcPAL preincubated with (S)‐3 (2 μM). Panel B shows the initial part of the progress curves at various inhibitor concentrations (for 0.3 min, for better visibility only production curves at 10 μM and 50 μM inhibitor concentrations are plotted).
Inhibition of AvPAL and RtPAL by 2 and 3
In order to ascertain that the effect of the methylidene group on the binding was not PcPAL specific, but that the enantiomers of 2 and 3 are general inhibitors of PALs, two other PALs were tested. In general, MIO enzymes from prokaryotic and eukaryotic sources differ structurally. Although MIO enzymes of both origins have tetrameric structures, eukaryotic MIO enzymes contain an additional domain at their C terminal end, rendering each monomeric chain longer by approximately 200 amino acids. Thus, in addition to a further PAL of eukaryotic origin (RtPAL, 34% sequence identity to PcPAL), another PAL of prokaryotic origin (AvPAL, 27% sequence identity to PcPAL) was also tested with the enantiomers of aminophosphonic acid 3.
Experiments demonstrated that enantiomers of both 2 and 3 were general inhibitors of PALs. The enantiopure (R)‐2 and (S)‐2, furthermore (R)‐3 and (S)‐3 inhibited AvPAL and RtPAL in a similar degree as PcPAL (Supporting Information, Tables S10 and S11). Previous results showed that, although AIP has a non‐substituted aromatic ring, it could efficiently inhibit TAL as well.18a By analogy, it is likely that (S)‐3 is a general, strong MIO enzyme inhibitor.
Thermodynamics of Ligand Binding to PcPAL
Although ITC proved to be a powerful tool to characterize binding behavior of small molecules to enzymes, to the best of our knowledge, no direct calorimetric measurements were performed for any MIO enzymes. During an ITC measurement, a concentrated protein solution is titrated with small portions of the ligand solution in a way that heat change is measurable for each injection producing a curved thermogram34 (representative thermograms for each measured system are included in the Supporting Information, Figures S20–S26). At the beginning of the titration, practically all molecules of the ligand in the first injection aliquots bind to the protein, thus ΔH of the binding could be determined. With the addition of further portions of ligand, saturation is reached, the released heat decreases. The rate of decrease provides the association constant (K a) of the binding from which the binding and dissociation constants and ΔG of the binding can be calculated. The C parameter determining the curvature (C=[E]×K d −1) is suggested to be between 10–1000 for an efficient determination of the thermodynamic parameters. In the present titration experiments of the non‐reactive d‐Phe [(S)‐1], and both enantiomers of the aminophosphonates 2 and 3 with wt‐PcPAL sufficient heat was generated in all cases, however, in some cases weak binding of the ligands limited the accurate determination of the binding enthalpy (Figure 4). Due to precipitation, the protein concentration was limited to 10 mg mL−1 (0.125 μM of monomers). Thus, accurate determination of all thermodynamic parameters was limited to compounds with specific dissociation constants of less than 12.5 μM (corresponding to C=10). Although binding entropies could not be determined reliably for C<10, ITC can still be used for determining dissociation constants and free energies of binding.35
Figure 4.

Thermodynamic binding energies of d‐Phe [(S)‐1], and each enantiomer of the aminophosphonates 2 and 3 with wt‐PcPAL measured by ITC. Free energies for binding (ΔG) were calculated from the measured K a, using the equation ΔG=−RT×lnK a. The enthalpy contributions were obtained by fitting to the titration curves and entropies were calculated by ΔG=ΔH−TΔS.
Comparison of the inhibition constants of both enantiomers of the aminophosphonates 2 and 3 with the binding affinity measured by ITC revealed that K values determined by the two methods were in agreement (Table 1). Reasons of differences could be the different protein concentrations for kinetics and for ITC, or the slight difference in the pH (8.8 for kinetics and 8.0 for ITC).
Binding of either enantiomer of aminophosphonic acids 2 and 3 was enthalpy driven (Figure 4, Supporting Information, Tables S12–S18). Enthalpy contributions of the binding process derive from hydrophobic interactions, hydrophilic interactions and salt bridges between the ligand and the protein. The non‐significant binding enthalpy difference between (R)‐2 and (R)‐3 (−0.4 kcal mol−1) revealed that the methylidene group had negligible effect on the binding affinity of the aminophosphonates with (R) configurations. On the other hand, the presence of the methylidene group at the C‐2 position increased the binding enthalpy of the (S)‐enantiomer of 3 by 3.1 kcal mol−1 compared to that of (S)‐2. This effect is in the range of the formation of an additional hydrogen bond between the protein and the ligand.
Solvent displacement contributes favorably to the entropy of binding. On the other hand, if an initially less ordered region – like a loop – adopts a more rigid conformation, the entropy of the system decreases. Thus, in such an instance the overall binding free energy becomes less negative. The lowest entropy contributions were determined for the slow binding inhibitors (R)‐2 and (S)‐3 (−1.2 kcal mol−1 and −0.6 kcal mol−1, respectively). A somewhat larger entropy contribution to the binding was observed for (S)‐2 and (R)‐3 (−1.7 kcal mol−1 and −2.1 kcal mol−1, respectively). As all of these compounds occupy the same binding pocket, the solvent entropy contributions should be equivalent for their binding. The smaller entropy contribution with the slow binding inhibitors (R)‐2 and (S)‐3 is therefore likely due to a larger reduction of the entropy of the system by more constraining the flexibility of protein loop(s).
A dissociation constant of 12.6 μM was determined for d‐phenylalanine [(R)‐1]. This value was in the same order of magnitude as K m of l‐phenylalanine [(S)‐1]. This argued that the same sets of molecular interactions were involved in the binding of l‐ and d‐phenylalanine to PcPAL. This was in full agreement with the early model suggesting mirror image packing of the substrate enantiomers within PAL,22 and directly proved that d‐phenylalanine [(R)‐1] could bind to the active site in a non‐reactive conformation.
Perturbation of Ligand Binding to PcPAL by the Y110F Mutation
The active site of MIO enzymes is covered by a sequence of loops, including the catalytically essential TyrA residue within the innermost one of them. The possible role of TyrA in the elimination reaction is shown in Figure 1. It was shown that mutation of TyrA (at position 110 in PcPAL) to phenylalanine resulted in an inactive protein.12 By assuming that the Y110F mutation has no effect on the overall protein structure, the effect of the mutation on the binding energy can be determined directly using ITC. As the Y110F mutation led to a catalytically inactive protein,12 ITC measurements with this mutant could provide additional details on ligand binding thermodynamics to PcPAL.
The Y110F mutation in PcPAL weakened the binding for all investigated molecules (Table 2 and Supporting Information, Tables S12–S18). Comparison of the binding characteristics of several compounds to wt‐PcPAL and Y110F‐PcPAL revealed (Table 2) a small contribution of Tyr110 to the binding (smaller than required for a weak hydrogen bond, <0.8 kcal mol−1). The effect of Y110F on the binding of (E)‐CA served as a control. The planar orientation of the β‐hydrogen on (E)‐CA, and its position in the crystal structure18b suggest that there is no direct interaction between Tyr110 and the (E)‐CA product. Thus, differences in the binding properties of (E)‐CA to the two PcPAL variants quantified the maximum perturbation caused by the Y110F mutation in the binding of substrates with no direct interaction with Y110. Hence, effects with ΔΔG <0.6 kcal mol−1 could be due to a small perturbation of the structure and/or natural uncertainty of the experiments. Thus, we can only state that the Y110F mutation influences significantly the binding of (R)‐2, (R)‐3 and (S)‐3.
Table 2.
Equilibrium binding constants and binding free energy perturbation measured by ITC for the enantiomers of substrate (1), for aminophosphonic acid inhibitors 2 and 3, and for (E)‐cinnamic acid with wild type PcPAL and its Y110F mutant.
| Ligand | PcPAL_wt K d [μM] | PcPAL_Y110F K d [μM] | ΔΔG [kcal mol−1][a] |
|---|---|---|---|
| (R)‐1 | 12.6 | 29.1 | −0.5 |
| (S)‐1 | n.a. | 23.0 | n.a. |
| (R)‐2 | 2.7 | 8.9 | −0.7 |
| (S)‐2 | 5.2 | 6.2 | −0.1 |
| (R)‐3 | 1.1 | 35.3 | −2.1 |
| (S)‐3 | 0.04 | 0.17 | −0.9 |
| (E)‐CA | 6.5 | 16.6 | −0.6 |
[a] Differences in the binding free energies show the contribution of the hydroxy group of Tyr110 to the binding energies. ΔΔG=ΔG wt−ΔG Y110F.
A shift in the equilibrium binding constants demonstrated that Tyr110 is directly involved in binding of the enantiomeric forms of 3. The perturbations in the binding free energies [ΔΔG=2.1 kcal mol−1 for (R)‐3 and 0.9 kcal mol−1 for (S)‐3] were in the range of a weak hydrogen bond. It is also possible that the hydroxy group of Tyr110 forms a Michael adduct with the methylidene group of 3, whose formation might be slow, accounting for the slow binding behavior of (S)‐3.
The similarity of the equilibrium binding constants of the enantiomers of phenylalanine, (R)‐1 and (S)‐1 (K d =29.1 μM for d‐Phe and 23.0 μM for l‐Phe, respectively) to the Y110F‐PcPAL mutant provided direct evidence for their mirror image packing within PcPAL.
Tyr110 – an essential residue in catalysis – lies in a mobile loop region that plays an important role in modulation of substrate binding and product release.19 Unfortunately, this loop was found in an inactive “loop‐out” conformation within the only currently available crystal structure of PcPAL36 containing a dithiothreitol covalently bound to MIO. Presumably, a strong interaction of a bound ligand with Tyr110 can lead to stabilization of the loop and thus enhance crystallization in a catalytically relevant conformation. Our attempts to crystallize PcPAL in an apo form were unsuccessful, likely due to the high mobility of these loops. However, the presence of the strongest binding inhibitor (S)‐3 enhanced crystallization of PcPAL, and produced well diffracting crystals. Analysis of the diffraction data is underway; these results will be published in the near future.
Docking of Compounds 1–6 to AvPAL
Molecular docking can contribute to the understanding of binding of various inhibitors within the PAL active site and can provide data on possible distances between the MIO prosthetic group and various parts of the ligands. The ITC results with the Y110F mutant indicated that Tyr110 (Tyr78 in AvPAL) could play an important role not only in the catalysis but also in binding of the enantiomeric forms of 3. As the only crystal structure of PcPAL (PDB code: 1W27)36 contained the critical Tyr‐loop in an inactive conformation, it was essential to find a proper structural model with a catalytically capable “loop‐in” conformation of this essential loop. Fortunately, a crystal structure determined for unliganded AvPAL (PDB code: 3CZO)37 had the Tyr‐loop in a catalytically relevant conformation. As the aminophosphonic acid enantiomers 2 and 3 inhibited AvPAL similarly to PcPAL (see Section 9 in the Supporting Information), the active site of AvPAL (with the MIO from chain A) was selected as model site for docking studies.
Glide algorithm docked all compounds represented on Figure 1 to the structure of AvPAL.38 Docking score of Glide (GScore) is an empirical scoring function used for the ranking of the ligand binding conformations (poses). It was developed to approximate the ligand binding free energy and binding affinity prediction. Its numerous terms include force field contributions and other terms accounting for interactions known to influence ligand binding. Values below −7 usually agree with acceptable binding.
GScores could differentiate between binding and non‐binding compounds. Non‐inhibiting compounds 5 and 6 docked frequently to the protein surface outside of the active site. Even if they docked within the active site, their GScores were poor (the best GScores inside the active site were −6.2 for 5, and −4.2 for 6). In contrast, inhibiting compounds 2, 3 and 4 docked mostly within the active site (Figure 5), and their GScores predicted strong binding (lowest GScores were −8.5 for 2, −9.8 for 3 and −8.6 for 4). d‐Phe and l‐Phe [(R)‐1 and (S)‐1, respectively] and the product [(E)‐CA] docked exclusively inside the active site, with good docking scores (GScores were −11.2 for (S)‐1, −5.9 for (R)‐1, and −8.3 for (E)‐CA). The absence of the salt bridge between the carboxyl group of (S)‐1 and Arg317 of the protein resulted in the poor docking score. GScores were −9.1 for (S)‐1, −10.5 for (R)‐1, when docked to structure where Tyr78 (Tyr110 equivalent in AvPAL) was modelled in the deprotonated form (ligand 1 was docked in the zwitterionic form). The carboxyl group of both enantiomers of phenylalanine (1) formed a salt bridge with Arg317, resulting in significantly better docking scores, and indicating the importance of protonation pattern during docking.
Figure 5.

Ligand binding within PALs. Docking results (within apo AvPAL, in grey) are compared to ligands bound to crystal structures (in green). Panel A shows (E)‐CA within a complexed structure of AvPAL (PDB code 3NZ4,9 in green) overlaid with (E)‐CA (in magenta) docked into apo AvPAL (PDB code 3CZO, in grey). Panel B depicts the poses of (R)‐2 (cyan) and (S)‐2 (yellow) with the best docking scores within AvPAL (in grey). Panel C shows the poses of (R)‐3 (cyan) and (S)‐3 (yellow) with the best docking scores AvPAL (in grey). Panel D shows the binding of AIP within a homologous protein structure (PDB code 2O7E,18a in green).
Docking poses found for the aminophosphonic acids 2 and 3 revealed poses with the phosphonic acid function of the ligands forming three hydrogen bonds within the carboxylate binding part of the active site within AvPAL. Residues Q311(B), R317(B) and N347(A) located at the carboxylate binding region are conserved in all known PALs, and also in many other MIO enzymes as well.19 Panels B, C and D in Figure 5 show hydrogen bonding of the phosphonic acid functional group of various inhibitors. Apparently, the larger flexibility of the enantiomers of aminophosphonates 2 and 3 allows their mode of binding (Figure 5B and C, respectively) to be similar to that of cinnamic acid [(E)‐CA, Figure 5A] with hydrogen bonding distance to the conserved carboxylate binding residues Q311, R317 and N347. In contrast, the constrained structure of AIP enforced a different mode of binding with less interactions to the carboxylate binding residues within a TAL structure (Figure 5D).
Docking found no significant differences for the different enantiomers of 2 and 3, and could not account for the direct effect of the methylidene group on the binding free energies. This suggests that these differences are due to differences in the protonation state patterns and/or induced fitting effects of the protein structure.
Conclusions
Synthesis of and enzyme kinetic and calorimetric studies with racemic and enantiopure aminophosphonic acids 2–6 as specific inhibitors of phenylalanine ammonia‐lyase and further MIO enzymes provided significant insights into the details of ligand binding to PAL. Here we propose that for MIO enzymes having synthetic importance even on an industrial scale, compound (S)‐3 may act as general inhibitor with comparable inhibition to the less substrate‐like AIP which is the strongest aminophosphonic acid‐type inhibitor for MIO enzymes so far. This compound binds by an interesting slow binding mechanism, that may be due to Michael adduct formation with Tyr110 and/or to the sequential to stabilization of the Tyr loop.
Our study indicated for the first time that isothermal titration calorimetry (ITC) could provide valuable information for ligand binding to PAL and further MIO enzymes as well. With the aid of suitable point mutations – such as the Y110F mutation in PcPAL – ITC was capable of estimating the contribution of an individual residue and thus provided significant contribution to the understanding of ligand binding. By ITC with the catalytically inactive Y110F‐PcPAL, we could directly prove that the non‐reactive d‐phenylalanine (R)‐1 binds to PcPAL almost as strong as the natural substrate l‐phenylalanine (S)‐1.
Although compound (S)‐3 is a weaker inhibitor than l‐AOPP, a non‐aminophosphonic acid‐type transition state‐mimicking compound being the strongest inhibitor of PALs, but it is also less sensitive and less reactive. Therefore, (S)‐3 may have high potential in ligand binding and structural studies of MIO enzymes. The usefulness of (S)‐3 in structural studies was indicated by the formation of well diffracting crystals of PcPAL in the presence of the strongest binding inhibitor, while trials to crystallize PcPAL in its apo form were unsuccessful. Moreover, similarly to AIP, both enantiomers of 3 could be further investigated as promising potential herbicide candidates.
Experimental Section
General
All chemicals used for the biochemical measurements were purchased from Sigma–Aldrich and Merck. Details of the syntheses of aminophosphonic acids (±)‐2, (±)‐4, (±)‐5 and (±)‐6 and the key intermediate (±)‐7 for the preparation of the phosphaphenylalanine analogue (±)‐3 and characterization of the compounds are included as Supporting Information. CCDC 1537722 http://www.ccdc.cam.ac.uk/cgi‐bin/catreq.cgi, CCDC 1537723 http://www.ccdc.cam.ac.uk/cgi‐bin/catreq.cgi, CCDC 1537725 http://www.ccdc.cam.ac.uk/cgi‐bin/catreq.cgi, and CCDC 1537829 http://www.ccdc.cam.ac.uk/cgi‐bin/catreq.cgi contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Protein Production and Purification
PcPAL (Uniprot code: P24481) was produced by heterologous expression in E. coli‐Rosetta, and was purified with Ni‐affinity chromatography, as described by Dima et al.39 The Y110F mutant of PcPAL was created by site‐directed mutagenesis following the protocol described by Naismith and Liu40 using the forward primer: CTGACTCCTTCGGAGTAACTACTGGATTCGGAGCCACT and reverse primer: GTTACTCCGAAGGAGTCAGTTCCCTTGTTCATGGATCC. Mutagenesis was verified by sequencing using T7‐for CAGAACATTACTCCCTGCTTG and T7‐rev TTGACGTATCCAGAAACAAGG primers. Both primers and sequencing were performed at the Sequencing Service of Genomed (Debrecen, Hungary). The length of the insert required the use of four primers to cover the whole transcribed DNA region. Protein production and purification followed the same protocol as used for PcPAL.39
AvPAL (Uniprot code: Q3M5Z3; with two surface cysteines mutated to serine, C503S, C565S) and RtPAL (Uniprot sequence code: P11544) were produced and purified using a similar procedure as described for PcPAL.
Kinetic Assays
Initial reaction velocities of the PAL‐catalyzed reaction of l‐phenylalanine [(S)‐1] were determined in 100 mM TRIS, pH 8.8 at 30 °C, using 0.01 mg mL−1 PcPAL by measuring the (E)‐cinnamic acid formation at 290 nm in a UV/Vis spectrophotometer (Specord 200; Analytic Jena AG, Jena, Germany). The extinction coefficient for (E)‐cinnamic acid was determined by 7‐point calibration using (E)‐CA solutions in 100 mM TRIS (pH 8.8 at 30 °C). To determine the Michaelis constant and the turnover number, initial velocities were measured at nine substrate concentrations between 75 μM and 10 mM. Inhibition constants for the aminophosphonic acids 2 and 3 were determined by measuring the initial product formation rates at the same substrate concentration in the presence of the inhibitor. When the regression coefficient of the linear fit (R2) was lower than 0.999, in the slow binding experiments with (S)‐3 and (R)‐2, the combination of a linear and an exponential equation was fit to describe the production curves. Steady state velocities were used for determining the inhibition constant values. All measurements were performed twice.
Non‐inhibited kinetic parameters were obtained by fitting the Michaelis–Menten equation to all data points using the nls (non‐linear least squares) fit in program R41 (with keeping all settings at their default values). The mechanism of inhibition was determined by fitting the competitive, non‐competitive and uncompetitive inhibition models to the measured data. The lowest value of the residuals indicated the best fitting model, and thus the mechanism of the inhibition. Visual comparison of the fitted curves to the experimental data confirmed in all cases the choice of mechanism.
Isothermal Titration Calorimetry
ITC measurements were performed in a microcalorimeter (MicroCal 200; GE Healthcare, Chicago, USA). Protein solutions (≈100 μM monomer units of PcPAL) were titrated with 5 times more concentrated ligand solutions using 20–25 injections. Initial delay, and time between two injections was set to 180 s. Binding was measured at 30 °C in TRIS (50 mM, pH 8.0, containing 1 mM of tris(2‐carboxyethyl)phosphine, TCEP).
Titration curves were analyzed using the software and methods described by Brautigam et al.42 Thermograms were integrated using NITPIC.43 Binding thermodynamic data were determined by fitting the A+B heteroassociation model in SEDPHAT.44 Confidence intervals for ΔH, K d and the stoichiometry of the binding were calculated at p values corresponding to one σ level. The confidence interval of K d determined the one for ΔG, while the minimal and maximal differences between the ΔH and ΔG confidence intervals yielded the one for −TΔS. Data were plotted using GUSSI.45
Molecular Docking
Docking was carried out in the crystal structure of AvPAL (PDB code: 3CZO), having a catalytically active closed conformation with intact Tyr‐loop. Binding of l‐ and d‐phenylalanine [(S)‐1 and (R)‐1, respectively], the aminophosphonic acid inhibitors [(S)‐2, (R)‐2, (S)‐3, and (R)‐3] as well as (E)‐cinnamic acid to the active site was modelled by molecular docking.
Prior to docking, hydrogens were added to the structure of AvPAL, and their positions were optimized using the Protein Preparation Wizard in Maestro.46 Protonation states of the amino acid residues at pH 8.8 were set on the basis of pK a estimation by ProPka.47
Docking was carried out using a rigid protein model with a flexible ligand, by extra precision docking in Glide.38 The center of mass of the ligand was restricted to a 20×20×20 Å box centered on the midpoint between the MIO residue and Tyr78 of the tyrosine loop. The entire ligand was restricted to a larger 40×40×40 Å box centered at the same midpoint. The van der Waals (vdW) radii of the hydrophobic residues (partial charge less than 0.2) were scaled down to 80%. This reduced vdW radii of the hydrophobic residues approximated a small degree of enzyme flexibility. The vdW radii of the ligands were not scaled. For other docking settings the default values were kept. Ligand poses that encountered steric clashes, defined as a sum of Coulomb and vdW interactions energies >10 kcal mol−1, were discarded. Duplicate poses, defined by an RMS deviation less than 1.0 Å or by a maximum atomic displacement less than 1.5 Å from existing poses, were also discarded.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support for project NEMSyB, ID P37 273, Cod MySMIS 103413 [funded by the Romanian Ministry for European Funds, through the National Authority for Scientific Research and Innovation (ANCSI) and co‐funded by the European Regional Development Fund, Competitiveness Operational Program 2014–2020 (POC), Priority axis 1, Action 1.1] is gratefully acknowledged. LCB thanks for the financial support from the Swiss National Science Foundation (PROMYS grant). LP, BGV and CP thank the support from COST Action CM1303 (SysBiocat). ZB thanks the Minnesota Supercomputing Institute for granting access to their resources for the computational studies, during the Fulbright Visiting Student Researcher period. FH thanks Susanne Felsinger for recording NMR spectra, Elena Macoratti for performing HPLC separations and Johannes Theiner for combustion analyses. We would like to acknowledge Klaudia Kovács and Lilla Vida for their contributions to the preliminary inhibition studies, and Gergely Nagy for his advice on the evaluation of ITC thermograms.
Z. Bata, R. Qian, A. Roller, J. Horak, L. C. Bencze, C. Paizs, F. Hammerschmidt, B. G. Vértessy, L. Poppe, Adv. Synth. Catal. 2017, 359, 2109.
Contributor Information
Friedrich Hammerschmidt, Email: friedrich.hammerschmidt@univie.ac.at.
Beáta G. Vértessy, Email: vertessy@mail.bme.hu
László Poppe, Email: poppe@mail.bme.hu.
References
- 1. Hodgins D. S., J. Biol. Chem. 1971, 246, 2977–2985. [PubMed] [Google Scholar]
- 2. Petersen M., Hans J., Matern U., in: Annual Plant Reviews: Biochemistry of Plant Secondary Metabolism, 2 nd edn., Vol. 40, (Ed.: M. Wink), Wiley-Blackwell, Oxford, 2010, pp 182–257. [Google Scholar]
- 3. Corporation Genex, U.S. Patent 4,584,269 A, 1986.
- 4. Poppe L., Paizs C., Kovács K., Irimie F. D., Vértessy B. G., Meth. Mol. Biol. 2012, 794, 3–19. [DOI] [PubMed] [Google Scholar]
- 5. de Lange B., Hyett D. J., Maas P. J. D., Mink D., van Assema F. B. J., Sereinig N., de Vries A. H. M., de Vries J. G., ChemCatChem 2011, 3, 289–292. [Google Scholar]
- 6. Kyndt J. A., Meyer T. E., Cusanovich M. A., Van Beeumen J. J., FEBS Lett. 2002, 512, 240–244. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Givot L., Smith T. A., Abeles R. H., J. Biol. Chem. 1969, 244, 6341–6353; [PubMed] [Google Scholar]
- 7b. Wickner R. B., J. Biol. Chem. 1969, 244, 6550–6552. [PubMed] [Google Scholar]
- 8.
- 8a. Christenson S. D., Liu W., Toney M. D., Shen B., J. Am. Chem. Soc. 2003, 125, 6062–6063; [DOI] [PubMed] [Google Scholar]
- 8b. Christianson C. V., Montavon T. J., Van Lanen S. G., Shen B., Bruner S. D., Biochemistry 2007, 46, 7205–7214. [DOI] [PubMed] [Google Scholar]
- 9. Feng L., Wanninayake U., Strom S., Geiger J., Walker K. D., Biochemistry 2011, 50, 2919–2930. [DOI] [PubMed] [Google Scholar]
- 10. Schwede T. F., Rétey J., Schulz G. E., Biochemistry 1999, 38, 5355–5361. [DOI] [PubMed] [Google Scholar]
- 11. Röther D., Poppe L., Viergutz S., Langer B., Rétey J., Eur. J. Biochem. 2001, 268, 6011–6019. [DOI] [PubMed] [Google Scholar]
- 12. Röther D., Poppe L., Morlock G., Viergutz S., Rétey J., Eur. J. Biochem. 2002, 269, 3065–3075. [DOI] [PubMed] [Google Scholar]
- 13. Schroeder A. C., Kumaran S., Hicks L. M., Cahoon R. E., Halls C., Yu O., Jez J. M., Phytochemistry 2008, 69, 1496–1506. [DOI] [PubMed] [Google Scholar]
- 14. Hermes J. D., Weiss P. M., Cleland W. W., Biochemistry 1985, 24, 2959–2967. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Schuster B., Rétey J., Proc. Natl. Acad. Sci. USA 1995, 92, 8433–8437; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Poppe L., Rétey J., Angew. Chem. 2005, 117, 3734–3754; [Google Scholar]; Angew. Chem. Int. Ed. 2005, 44, 3668–3688. [DOI] [PubMed] [Google Scholar]
- 16. Pinto G. P., Ribeiro A. J. M., Ramos M. J., Fernandes P. A., Toscano M., Russo N., Arch. Biochem. Biophys. 2015, 582, 107–115. [DOI] [PubMed] [Google Scholar]
- 17. Ratnayake N. D., Wanninayake U., Geiger J. H., Walker K. D., J. Am. Chem. Soc. 2011, 133, 8531–8533. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Louie G. V., Bowman M. E., Moffitt M. C., Baiga T. J., Moore B. S., Noel J. P., Chem. Biol. 2006, 12, 1327–1338; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Wybenga G. G., Szymanski W., Wu B., Feringa B. L., Janssen D. B., Dijkstra B. W., Biochemistry 2014, 53, 3187–3198. [DOI] [PubMed] [Google Scholar]
- 19. Pilbák S., Farkas Ö., Poppe L., Chem. Eur. J. 2012, 18, 7793–7802. [DOI] [PubMed] [Google Scholar]
- 20. Lovelock S. L., Lloyd R. C., Turner N. J., Angew. Chem. 2014, 126, 4740–4744; [Google Scholar]; Angew. Chem. Int. Ed. 2014, 53, 4652–4656. [DOI] [PubMed] [Google Scholar]
- 21. Havir E. A., Hanson K. R., Biochemistry 1968, 7, 1904–1914. [DOI] [PubMed] [Google Scholar]
- 22. Hanson K. R., Arch. Biochem. Biophys. 1981, 211, 575–588. [DOI] [PubMed] [Google Scholar]
- 23. MacDonald M. J., D′Cunha G. B., Biochem. Cell Biol. 2007, 85, 273–282. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Laber B., Kiltz H.-H., Amrhein N., Z. Naturforsch. 1986, 41C, 33–39; [Google Scholar]
- 24b. Zoń J., Amrhein N., Liebigs Ann. Chem. 1992, 625–628; [Google Scholar]
- 24c. Zoń J., Amrhein N., Gancarz R., Phytochemistry 2002, 59, 9–21. [DOI] [PubMed] [Google Scholar]
- 25. Appert C., Zoń J., Amrhein N., Phytochemistry 2003, 62, 415–422. [DOI] [PubMed] [Google Scholar]
- 26. Lewis E. A., Muphy K. P., in: Protein-Ligand Interactions, 1 st edn., (Ed.: G. U. Nienhaus), Humana Press Inc., Totowa, New Jersey, 2005. pp 1–15. [Google Scholar]
- 27. Ladbury J. E., Klebe G., Freire E., Nat. Rev. Drug Discov. 2010, 9, 23–27. [DOI] [PubMed] [Google Scholar]
- 28. Reich H. J., Wollowitz S., Trend J. E., Chow F., Wendelborn D. F., J. Org. Chem. 1978, 43, 1698–1705. [Google Scholar]
- 29. Hammerschmidt F., Liebigs Ann. Chem. 1991, 469–475. [Google Scholar]
- 30. Klee C. B., Biochemistry 1974, 13, 4501–4507. [DOI] [PubMed] [Google Scholar]
- 31. Morrison J. F., Trends Biochem. Sci. 1982, 7, 102–105. [Google Scholar]
- 32. Baici A., Proc. 5th Beilstein ESCEC Symposium 2011, 5, 55–73. [Google Scholar]
- 33. Frieden C., J. Biol. Chem. 1970, 245, 5788–5799. [PubMed] [Google Scholar]
- 34. Freyer M. W., Lewis E. A., Meth. Cell. Biol. 2008, 84, 79–113. [DOI] [PubMed] [Google Scholar]
- 35. Ternbull W. B., Daranas A. H., J. Am. Chem. Soc. 2003, 125, 14859–14866. [DOI] [PubMed] [Google Scholar]
- 36. Ritter H., Schulz G. E., Plant Cell 2004, 16, 3426–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang L., Gamez A., Archer H., Abola E. E., Sarkissian C. N., Fitzpartick P., Wendt D., Zhang Y., Vellard M., Bliesath J., Bell S. M., Lemontt J. F., Scriver C. R., Stevens R. C., J. Mol. Biol. 2008, 380, 623–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.
- 38a. Friesner R. A., Murphy R. B., Repasky M. P., Frye L. L., Greenwood J. R., Halgren T. A., Sanschagrin P. C., Mainz D. T., J. Med. Chem. 2006, 49, 6177–6196; [DOI] [PubMed] [Google Scholar]
- 38b. Schrödinger Release 2015–3: Glide, Schrödinger, LLC, New York, NY, 2015.
- 39. Dima N. A., Filip A., Bencze L. C., Oláh M., Sátorhelyi P., Vértessy B. G., Poppe L., Paizs C., Stud. Univ. Babes-Bolyai Ser. Chem. 2016, 61, 21–34. [Google Scholar]
- 40. Liu H., Naismith J. H., BMC Biotechnol. 2008, 8, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. R Core Team, R: A language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria, 2016: https://www.R-project.org/.
- 42. Brautigam C. A., Zhao H., Vargas C., Keller S., Schuck P., Nat. Protoc. 2016, 11, 882–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.
- 43a. Keller S., Vargas C., Zhao H., Piszczek G., Brautigam C. A., Schuck P., Anal. Chem. 2012, 11, 5066–5073; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43b. Scheuermann T. H., Brautigam C. A., Methods 2015, 76, 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Houtman J. C. D., Brown P. H., Bowden B., Yamagushi H., Appella E., Samelson L. E., Schuck P., Prot. Sci. 2007, 16, 30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brautigam C. A., Meth. Enzymol. 2015, 562, 109–133. [DOI] [PubMed] [Google Scholar]
- 46. Schrödinger Release 2015–3: Maestro, Schrödinger, LLC, New York, NY, 2016.
- 47. Olsson M. H. M., Sondergaard C. R., Rostkowski M., Jensen J. H., J. Chem. Theory Comput. 2011, 7, 525–537. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary