

Abstract
Brentuximab vedotin, a CD30‐directed antibody‐drug conjugate (ADC), is approved for treating certain patients with CD30‐expressing hematologic malignancies. Its primary mechanism of action is the targeted delivery of a microtubule‐disrupting agent, monomethyl auristatin E (MMAE), to CD30‐expressing cells. A population pharmacokinetic (PopPK) analysis was conducted to characterize the PK of ADC and unconjugated MMAE in patients with CD30‐expressing hematologic malignancies by compartmental analysis and to evaluate the effects of covariates on PK of the ADC. A nonlinear mixed‐effects modeling approach was used to evaluate data from 314 patients in 5 clinical studies. ADC PK was described by a linear, 3‐compartment model with first‐order elimination. MMAE PK was described by a semimechanistic, linear, 2‐compartment model with first‐order elimination. The estimated typical values for a 75‐kg male patient were 1.56 L/d and 4.29 L for ADC systemic clearance (CL) and volume of central compartment (V1), respectively, with weight effect exponents of 0.698 and 0.503, respectively. Typical V1 in 75‐kg females was 87% of that in males, with no impact on systemic ADC exposure. Typical values of MMAE clearance (CLM) and volume of central compartment (V4) were 55.7 L/d and 79.8 L, respectively, with weight effect exponents fixed to 0.75 and 1.0, respectively. This is the first PopPK model of brentuximab vedotin to semimechanistically link the PK of ADC and that of the unconjugated small molecule MMAE. Both ADC and MMAE PK data were adequately described by the final integrated model, which supports weight‐based dosing of brentuximab vedotin in adult patients with CD30‐expressing hematologic malignancies.
Keywords: population pharmacokinetics, clinical pharmacology, biologics, clinical trials, oncology, brentuximab vedotin, antibody‐drug conjugate, CD30
Brentuximab vedotin is a CD30‐directed antibody‐drug conjugate (ADC) that is currently indicated for the treatment of patients with relapsed systemic anaplastic large‐cell lymphoma (sALCL) after failure of at least 1 prior multiagent chemotherapy regimen and for the treatment of patients with classical Hodgkin lymphoma (HL) after failure of autologous hematopoietic stem cell transplantation (auto‐HSCT), after failure of at least 2 prior multiagent chemotherapy regimens in patients who are not auto‐HSCT candidates, and for treatment of patients with classical HL at high risk of relapse or progression as post–auto‐HSCT consolidation. In clinical trials, including 2 pivotal phase 2 studies,1, 2 brentuximab vedotin showed substantial efficacy and an acceptable safety profile when administered at 1.8 mg/kg every 3 weeks. Additional ongoing studies continue to investigate the utility of brentuximab vedotin in other clinical settings.
The ADC brentuximab vedotin consists of a CD30‐directed antibody conjugated by a protease‐cleavable linker to a microtubule‐disrupting agent, monomethyl auristatin E (MMAE). The primary mechanism of action of brentuximab vedotin is the targeted delivery of MMAE to CD30‐expressing cells.3 After the ADC binds to CD30‐expressing cells, the ADC‐CD30 complex is internalized, and MMAE is released via proteolytic cleavage. MMAE then binds to tubulin, disrupting the microtubule network within the cell and inducing cell‐cycle arrest and apoptotic cell death.4, 5 Additional mechanisms of tumor cell killing that may contribute to the clinical activity of brentuximab vedotin include antibody‐dependent cellular phagocytosis, immunogenic cell death, and bystander effects on nearby cells in the tumor microenvironment.6, 7, 8, 9, 10
Noncompartmental analyses of the pharmacokinetics (PK) of the brentuximab vedotin ADC and unconjugated MMAE in phase 1 and phase 2 studies11, 12, 13 indicate that exposure of both analytes is approximately dose proportional in the therapeutic dose range.
After intravenous administration of ADC, peak concentrations generally occur at the end of infusion, and a multiexponential decline in serum concentrations is observed. The estimated half‐life of ADC is 4 to 6 days, and steady state is achieved by approximately 21 days. When administered every 3 weeks, minimal to no accumulation of ADC occurs.14, 15, 16 The mean steady‐state volume of distribution is approximately 6 to 10 L. Similar to other antibody‐based therapeutics, elimination of ADC is thought to occur through proteolytic degradation into amino acids and recycling into other proteins.17
For MMAE, peak concentrations are observed within approximately 2 to 3 days postdose, and steady state also is achieved by approximately 21 days, consistent with the estimated half‐life of 3 to 4 days for MMAE when administered as a component of the ADC. MMAE exposures decrease with continued administration, with approximately 50% to 80% of the exposure of the first dose being observed at subsequent doses.14, 15, 16 MMAE is widely distributed in tissues including bone marrow, spleen, lung, and liver.17 In vivo data suggest that a small fraction of MMAE is metabolized, and in vitro data indicate that the metabolism occurs primarily via oxidation by CYP3A4/5.14, 15, 16 Elimination of the unconjugated small molecule MMAE occurs primarily through the liver and kidneys. The primary excretion route of MMAE is via feces (median 72% of recovered MMAE over a 1‐week period), with the remainder recovered in urine.18
The objectives of the work reported here were to characterize the PK of ADC and unconjugated MMAE in patients with CD30‐expressing hematologic malignancies by population‐based compartmental analysis in an integrated model and to evaluate the effects of covariates on the PK of the ADC.
Methods
Study Approvals and Informed Consent
Protocols for the 5 brentuximab vedotin (ADCETRIS®) clinical trials that contributed to the population PK (PopPK) analysis were approved by the institutional review board for each study site, and all patients provided written informed consent before study‐specific procedures began, in accordance with the Declaration of Helsinki. The study sites and institutional review boards for each trial are provided in Supplementary Appendix S1.
Study Designs and Patient Populations
Data from 5 phase 1 and 2 clinical studies in patients with relapsed/refractory CD30‐expressing hematologic malignancies contributed to the PopPK analyses (Table 1). All 5 studies (2 phase 1 dose‐ranging studies, 2 pivotal phase 2 studies, and 1 clinical pharmacology study) were open‐label trials conducted at multiple study centers. Studies 1 through 4 used a single‐arm design. For study 5, a clinical pharmacology study with multiple arms, ADC and MMAE concentration data that could have been affected by potential drug–drug interactions were excluded from the PopPK analysis. Study 5 contributed 4 patients with renal impairment and no patient with hepatic impairment to the PopPK data set.
Table 1.
Overview of Clinical Trials Included in Population PK Analysis
| Study | Description | Dose Regimen | Patient Population | No. of Patientsa | PK Samples | ClinicalTrials.gov No. |
|---|---|---|---|---|---|---|
| 111 | Phase 1 dose‐ranging study | 0.1 to 3.6 mg/kg intravenously every 3 weeks (2‐hour infusion) | Relapsed/refractory CD30‐expressing hematologic malignancies | 48b | Rich | NCT00430846 |
| 212 | Phase 1 dose‐ranging study | 0.4 to 1.4 mg/kg intravenously on days 1, 8, and 15 of every 28‐day cycle (2‐hour or 30‐minute infusion)c | Relapsed/refractory CD30‐expressing hematologic malignancies | 46b | Rich | NCT00649584 |
| 32 | Phase 2 pivotal study in HL | 1.8 mg/kg intravenously every 3 weeks (30‐minute infusion) | Relapsed/refractory HL; previous autologous stem cell transplant | 102 | Sparse | NCT00848926 |
| 41 | Phase 2 pivotal study in sALCL | 1.8 mg/kg intravenously every 3 weeks (30‐minute infusion) | Relapsed/refractory sALCL; previous frontline chemotherapy | 58 | Sparse | NCT00866047 |
| 518, 25 | Phase 1 clinical pharmacology study | 1.2 or 1.8 mg/kg intravenously every 3 weeks (30‐minute infusion) | Relapsed/refractory CD30‐expressing hematologic malignancies | 60d | Rich | NCT01026415 |
HL, Hodgkin lymphoma; sALCL, systemic anaplastic large cell lymphoma.
Number of patients contributing to population PK analyses.
Includes 5 patients who were reenrolled with new patient numbers and retreated in study 1 (n = 3) or in study 2 (n = 2).
Protocol amended to reduce infusion time.
Includes 56 patients from the drug–drug interaction portion of the study and 4 patients with renal impairment.
Brentuximab vedotin was administered as an intravenous infusion over a 30‐minute or 2‐hour period according to each protocol. Administration was weight based, but the dose was capped for patients with body weight > 100 kg. For example, for patients who received 1.8 mg/kg, the maximum dose was 180 mg for individuals with body weight 100 kg or higher. Infusions were given every 3 weeks in studies 1, 3, 4, and 5 and weekly in the first 3 weeks of every 4‐week cycle in study 2.
Sample Collection and Bioanalytical Methods
PK sampling times for each study are described in detail in Supplementary Table S1. The ADC was measured in serum, and MMAE was measured in plasma using validated assay methods that were described previously.18 Briefly, concentrations of the ADC were measured with an enzyme‐linked immunosorbent assay in a sandwich format; the lower limit of quantitation was 12.5 ng/mL. Concentrations of MMAE in plasma were measured by liquid chromatography and tandem mass spectrometry following solid‐phase extraction; the lower limit of quantitation was 25 pg/mL.
Antitherapeutic antibody (ATA) assessments for studies 3 and 4 used a validated electrochemiluminescence assay as previously described18 to test serum samples collected prior to infusion every 3 weeks. The assay had a sensitivity of 4 ng/mL anti–brentuximab vedotin monoclonal antibody and drug tolerance of 3125 ng/mL brentuximab vedotin. Screen‐positive samples were further evaluated for titer and specificity.
Population PK Analysis
The PopPK analysis was performed using a nonlinear mixed‐effects modeling approach: NONMEM VII software (version 7.3.0; ICON Development Solutions, Ellicott City, Maryland) with the first‐order conditional estimation method with interaction. The open‐source statistical software R (version 3.1.2; R Foundation for Statistical Computing, Vienna, Austria; available at: http://www.R-project.org) was used for exploratory analysis, postprocessing of NONMEM output, and generating model diagnostic plots (eg, goodness‐of‐fit [GOF] and visual predictive check [VPC] plots). Modeling was done in molar units (concentration in units of pmol/mL, amount in μmol, and rate of infusion in μmol/h). ADC and MMAE have a molecular weight of 153 kDa and 718 Da, respectively. It is presumed that MMAE does not alter the ADC PK, so to reduce the computing time, a sequential approach was undertaken. A flow chart illustrating the PopPK model development is provided in Supplementary Figure S1. Briefly, data were divided into model development (studies 1 and 2) and validation (studies 3, 4, and 5) data sets. Following initial model development, external validation, and refinement, simulations were performed to evaluate the effect of covariates on the PK of the ADC.
ADC Model Development
The initial ADC model was a 2‐compartment model with zero‐order input and first‐order elimination. More complex models (eg, a 3‐compartment model with linear clearance [CL]; 2‐compartment models with time‐dependent CL or parallel linear and nonlinear CL) were then considered and tested to best describe the PK of ADC.
Following establishment of the ADC base model, the covariate model was built using the full covariate model approach,19 whereby all covariate‐parameter relationships of interest were entered in the model simultaneously, and parameters were estimated. Highly correlated covariates were not simultaneously included in the full model but were subsequently investigated separately with 1 of the competing correlated covariates. Generally, interpretation and further refinement of the covariate model was based on point estimates, confidence intervals, objective function values (OFVs), and diagnostic plots of the covariate effects. The statistical significance of each covariate was assessed using the χ2 test. A covariate was excluded from the model if less than a 10.8‐point increase (based on the critical value of the χ2 distribution at α level of 0.001 for 1 degree of freedom) in the OFV (P < .001) occurred after removing the covariate. Covariate effects not supported by the data (effects close to null value and/or with high relative standard error of the estimate [%RSE]) were also excluded.
The following covariates were evaluated: baseline age, alanine aminotransferase, albumin, aspartate aminotransferase, bilirubin, body weight (BW), creatinine clearance (CRCL), disease type, and tumor size; race and sex; and manufacturing process. Continuous covariates such as BW were centered to their medians, and relationships with PK parameters, P, were defined as equation (1):
| (1) |
where Pi is the typical value of the parameter for an individual i with a body weight of BWi, PTV is the typical value of the parameter for an individual with a median BW (BWmed), and θBW is an the exponent determining the shape of the relationship between BW and parameter P. Categorical covariates such as sex were tested in the model according to equation (2):
| (2) |
where PTV is the typical value of parameter P for males (for females, INDi = 1 and for males, INDi = 0), and θsex is the ratio of parameter P in females to males.
MMAE Model Development
The ADC covariate PK model was then used to develop the MMAE PK model. MMAE data were fitted alone using individual post hoc ADC PK parameter estimates to predict the ADC concentrations. Multiple MMAE models were investigated, including 1‐ or 2‐compartment models with linear or nonlinear elimination and linear or nonlinear formation rate with or without delay. Final model selection was based on OFVs, precise and meaningful estimates of parameters, GOF plots, and VPC plots. The GOF plots included observed versus model‐predicted concentrations as well as conditionally weighted residuals with η‐ɛ interaction (CWRESI) versus model‐predicted values and time postdose. The VPC plots were used to internally evaluate the predictive performance of the final model. Three hundred data sets were simulated from parameter estimates of the final model, and the median and 5th and 95th percentiles of simulated data were compared with observed data. Similar model selection and/or interval validation approaches were also applied to ADC model development.
Model External Validation
To further validate the model developed with the model development data set, external validation was carried out using 2 methods: predicting the observed PK data in the validation data set (method 1) and comparing the results of Monte Carlo simulations with the observed PK data in the validation data set (method 2).
Predicted ADC and MMAE concentrations (method 1) for patients in the validation data set were obtained by setting POSTHOC and MAXEVAL = 0 options in the NONMEM $ESTIMATION command without model fitting. Bias in model prediction was assessed by calculating the prediction error (PE%) as equation (3):
| (3) |
where PRED is the population prediction and OBS represents the observed PK data in the validation data set.
The Monte Carlo simulations (method 2) generated a total of 200 data sets for the 220 patients in the validation data set. Simulations combined estimated PK parameters from the model development data set with the patients’ characteristics, dosing, and sampling information from the validation data set. The median and 5th and 95th percentiles of simulated data were plotted alongside the observed data. This simulation was performed in NONMEM VII.
Final Model Refinement and Simulation
Final model parameter estimates were refined using both the model development (studies 1 and 2) and validation (studies 3, 4, and 5) data sets, and the final model was evaluated internally using GOF and VPC plots. The influence of statistically significant covariates on ADC exposure (ie, AUC) was assessed by simulating ADC exposure in a typical patient for each of the statistically significant covariates using the final model.
Pharmacostatistical Model
Distribution of between‐subject variability was assumed to be log‐normal and was described by an exponential error model as equation (4):
| (4) |
where Pi is the parameter value for individual i, PTV is the typical value of the parameter, and ηi is individual‐specific between‐subject variability for individual i and is assumed to be normally distributed (η∼N(0, ω2)).
A partial variance‐covariance matrix was used where correlation between CL and V1 was estimated. The residual error model was initially described using both combined additive and proportional error terms for both ADC and MMAE.
Results
Analysis Data Sets
For the initial analysis, a total of 94 patients from studies 1 and 2 contributed 3677 ADC and 3796 MMAE concentrations. The validation data set included 220 patients from studies 3, 4, and 5, which contributed 3404 ADC and 3656 MMAE concentrations. All 314 patients from the 5 studies contributed 7081 ADC and 7452 MMAE concentrations to the final analysis. All patients had CD30‐expressing hematologic malignancies. Baseline characteristics are summarized by study and for the population as a whole in Table 2.
Table 2.
Patient Characteristics
| Study 1 (n = 48) | Study 2 (n = 46) | Study 3 (n = 102) | Study 4 (n = 58) | Study 5 (n = 60) | All Patients (n = 314)a | |
|---|---|---|---|---|---|---|
| Age (years), median (range) | 36 (20–87) | 31.5 (12–82) | 31 (15–77) | 52 (14–76) | 36.5 (16–71) | 35 (12–87) |
| Female, n (%) | 19 (40) | 14 (30) | 54 (53) | 25 (43) | 24 (40) | 136 (43) |
| White, n (%) | 42 (88) | 38 (83) | 89 (87) | 48 (83) | 50 (83) | 267 (85) |
| Weight (kg), median (range) | 81.1 (45.9–154) | 79.6 (41.4–118) | 70.4 (44.6–168) | 69.8 (42.5–126) | 79.2 (41.5–165) | 74.8 (41.4–168) |
| Body surface area (m2), median (range) | 1.99 (1.43–2.69) | 1.96 (1.33–2.38) | 1.82 (1.43–2.86) | 1.81 (1.42–2.54) | 1.96 (1.35–2.64) | 1.86 (1.33–2.86) |
| Disease | ||||||
| HL, n (%) | 44 (92) | 40 (87) | 102 (100) | 0 (0) | 55 (92) | 241 (77) |
| sALCL, n (%) | 3 (6) | 5 (11) | 0 (0) | 56 (97) | 3 (5) | 67 (21) |
| Other, n (%) | 1 (2) | 1 (2) | 0 (0) | 2 (3) | 2 (3) | 6 (2) |
| Renal function | ||||||
| Creatinine clearance (mL/min), median (range)b | 115 (44–150) | 129 (22–150) | 124 (59–150) | 112 (28–150) | 126 (22–150) | 122 (22–150) |
| Hepatic function | ||||||
| Albumin (g/dL), median (range) | 3.7 (1.6–4.7) | 3.9 (2.4–4.8) | 3.6 (2.0–4.8) | 3.5 (1.8–4.6) | 3.8 (2.6–4.7) | 3.7 (1.6–4.8) |
| Aspartate aminotransferase (U/L), median (range) | 22 (10–55) | 24 (12–54) | 19 (9–109) | 22 (11–113) | 22 (9–73) | 21 (9–113) |
| Alanine aminotransferase (U/L), median (range) | 21.5 (5–98) | 29 (10–65) | 17 (4–80) | 20 (5–232) | 21 (6–115) | 21 (4–232) |
| Bilirubin (mg/dL), median (range) | 0.4 (0.2–1.0) | 0.4 (0.1–1.1) | 0.4 (0.2–1.3) | 0.4 (0.2–7.2) | 0.4 (0.1–1.0) | 0.4 (0.1–7.2) |
| Baseline tumor size (cm2), median (range) | 22.6 (2.83–180) | 16.7 (1.60–101) | 23.7 (1.50–276) | 14.0 (1.95–105) | NA | 20.3 (1.50–276)c |
| No. of concentrations | ||||||
| ADC | 1598 | 2079 | 2232 | 848 | 324 | 7081 |
| MMAE | 1698 | 2098 | 2241 | 840 | 575 | 7452 |
ADC, antibody‐drug conjugate; HL, Hodgkin lymphoma; MMAE, monomethyl auristatin E; sALCL, systemic anaplastic large‐cell lymphoma.
Includes 5 patients who were reenrolled with new patient numbers and retreated in study 1 (n = 3) or in study 2 (n = 2).
Capped at 150 mL/min.
n = 254; baseline tumor measurements were not collected in study 5.
ADC Final Model
Linear 2‐ and 3‐compartment models with first‐order elimination were tested: the 3‐compartment model described ADC concentration–time data better, with a significant reduction in OFV by 320 points compared with the 2‐compartment model. A 2‐compartment model with parallel linear and Michaelis‐Menten elimination, to account for any nonlinearity between the doses, was also evaluated but failed to converge. Drug load was shown to affect ADC clearance,20 so an alternative 2‐compartment model with drug load–dependent clearance was also evaluated. Although this model showed improvement over the 2‐compartment model, it provided no improvement over the 3‐compartment model with first‐order elimination in terms of OFV and diagnostic plots. Therefore, the linear 3‐compartment model with first‐order elimination was selected as the final ADC base model (Figure 1).
Figure 1.
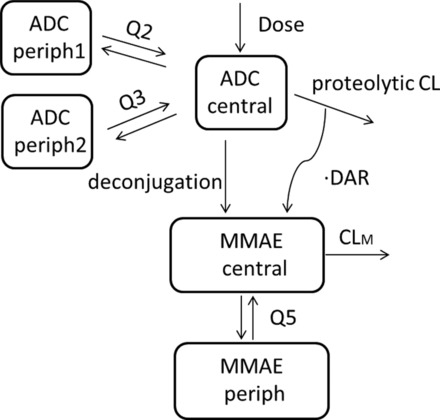
ADC and MMAE PK model schema after an intravenous infusion of brentuximab vedotin. ADC, antibody‐drug conjugate; DAR, drug‐antibody ratio; CL, clearance of ADC by proteolytic degradation; Q2 and Q3, ADC intercompartmental clearance; CLM, MMAE apparent clearance; MMAE, monomethyl auristatin E; Q5, MMAE apparent intercompartmental clearance.
The 3‐compartment model was parameterized in terms of clearance (CL), volume of the central compartment (V1), volume of peripheral compartment 1 (V2), volume of peripheral compartment 2 (V3), distributional clearance between V1 and V2 (Q2), and distributional clearance between V1 and V3 (Q3). Body weight and body surface area were highly correlated; of these 2 covariates, BW was chosen to be explored in the model, as it was the simpler measure to obtain. The final covariate model included the influence of BW on ADC clearance (CL, Q2, Q3) and volume (V1, V2, V3) and the influence of sex on central volume (V1). All other covariates (eg, age, race, and manufacturing process) were found not be statistically significant and were therefore excluded from the final model.
GOF plots of population and individual model‐predicted versus observed ADC concentrations showed satisfactory agreement (Supplementary Figure S2a); conditionally weighted residuals were evenly distributed on both sides of the identity line (CWRESI = 0). Predictive performance of the final model was also assessed using VPC plots. The 90% prediction interval overlapped the observed data (Figure 2a), supporting the conclusion that the final model adequately described the observed data.
Figure 2.
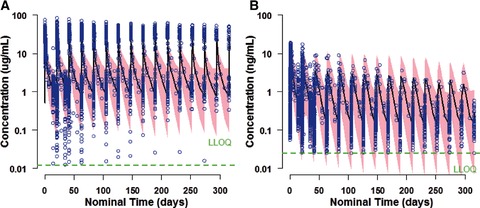
Visual predictive check plots of ADC (a) and MMAE (b) for brentuximab vedotin administered intravenously at 1.8 mg/kg every 3 weeks (studies 1, 3, and 4). Pink area, 90%CI of model prediction; blue dots, observed data; green dotted line, lower limit of assay quantification. ADC, antibody‐drug conjugate; MMAE, monomethyl auristatin E.
The final ADC PopPK model parameter estimates based on all 5 studies are summarized in Table 3. These PK parameters were precisely estimated with a relative standard error (%RSE) < 10% for all fixed‐effect parameters and < 31% for all random‐effect parameters. The typical values of ADC CL and V1 for a 75‐kg male patient were 1.56 L/d and 4.29 L, respectively, with BW effect exponents of 0.698 and 0.503, respectively. The V1 in females was 87% of that in males. Inclusion of BW in the model resulted in decreases of 208 points for OFV, 2.6% CV for between‐subject variability (BSV) for CL, and 8% CV for the BSV of V1. Adding sex to the model further reduced the OFV by 32 points and reduced the BSV of V1 by 1.5% CV, whereas a relatively high BSV remained in the final ADC covariate model for CL, Q3, and V3 (estimates of 46.9% CV, 45.2% CV, and 106% CV, respectively). The model condition number was less than 1000. The η‐shrinkage was low, ranging from 6% (CL) to 27% (V1) for all parameters except Q3, which had a shrinkage value of 45%, indicating potential difficulty for the model to precisely estimate this parameter. However, overall, the final ADC PK model was deemed appropriate for use in the sequential MMAE modeling.
Table 3.
Parameter Estimates of Final ADC Model
| Parametera | Units | Estimate (%RSE) | BSV %CV (%RSE) |
|---|---|---|---|
| CL | L/d | 1.56 (2.9) | 46.9 (20) |
| V1 | L | 4.29 (1.9) | 13.5 (24) |
| Q2 | L/d | 2.83 (5.9) | 15 fixed |
| V2 | L | 3.83 (6.3) | 25 fixed |
| Q3 | L/d | 0.708 (8.2) | 45.2 (31) |
| V3 | L | 9.52 (8.8) | 106 (19) |
| ΘBW,CL,Q2,Q3 b | — | 0.698 (8.8) | — |
| ΘBW,V1,V2,V3 c | — | 0.503 (8.0) | — |
| ΘSEX,V1 | — | 0.873 (2.5) | — |
| Corr (CL, V1) | — | 0.229 (53) | — |
| σ1 (additive) | μg/mL | 0.0125 fixed | — |
| σ2 (proportional) | %CV | 32.9 (9.8) | — |
σ1 and σ2, variance of the additive and proportional components of the residual error, respectively; ADC, antibody‐drug conjugate; BSV, between‐subject variability; BW, body weight; CL, clearance; %CV, percent coefficient of variation; Q2 and Q3, intercompartmental clearance; %RSE, percent relative standard error of the estimate; V1, central compartment volume; V2 and V3, peripheral compartment volumes.
Reference population = 75‐kg male patient.
Dependence on BW as CL·(BW/75)0.698, Q2·(BW/75)0.698, Q3·(BW/75)0.698.
Dependence on BW as V1·(BW/75)0.503, V2·(BW/75)0.503, V3·(BW/75)0.503.
MMAE Final Model
The final MMAE model consisted of a semimechanistic linear 2‐compartment model with first‐order elimination (Figure 1). MMAE was assumed to form from both ADC proteolytic degradation and deconjugation processes. Deconjugation was considered a multistep time‐dependent process to eventually form unconjugated MMAE. However, a model with an additional compartment representing thiol‐vc‐MMAE, the intermediate product of deconjugation, could not be supported by current data. The average drug load (or drug‐antibody ratio [DAR]) was not measured but was assumed to decrease exponentially21 after each dose as equation (5):
| (5) |
where DARt is the average DAR at time postdose, DAR0 is the average DAR at time of dosing and fixed to 4 for brentuximab vedotin at the beginning of each dosing cycle. α is fixed to 0.25 because the assay only quantifies ADC with at least 1 drug so that DARt would be no less than 1. β is a macro rate constant of DAR decline, resulting from both deconjugation and differential ADC clearance.
Differential equations of the final model are provided in Supplementary Material M1. BW was included in the model as a covariate to MMAE clearance and volume of distribution, and the weight effect exponent was fixed to 0.75 and 1.0 for clearance and volume of distribution, respectively. By adding BW as a covariate, the median PE% was improved by approximately 30%. The final MMAE PopPK model parameter estimates based on all 5 studies are summarized in Table 4. These PK parameters were estimated with a %RSE < 30% for all fixed‐ and random‐effect parameters. Typical values of MMAE apparent CL (CLM) and apparent volume of central compartment (V4) were 55.7 L/d and 79.8 L, respectively.
Table 4.
Parameter Estimates of Final MMAE Model
| Parametera | Units | Estimate (%RSE) | BSV %CV (%RSE) |
|---|---|---|---|
| CLM | L/d | 55.7 (5.2) | 60.7 (9.7) |
| V4 | L | 79.8 (11) | 78.2 (11) |
| DAR0 | — | 4 fixed | — |
| α | — | 0.25 fixed | — |
| β | 1/d | 0.0785 (12) | 98.1 (24) |
| Q5 | L/d | 65.0 (30) | 0 fixed |
| V5 | L | 28.1 (14) | 0 fixed |
| Fm by cycle | Fraction | −0.261 (14) | 130 (17) |
| ΘBW,CLM,Q5 b | — | 0.75 fixed | — |
| ΘBW,V4,V5 c | — | 1.0 fixed | — |
| Corr (CLM, V4) | — | 0.634 (11) | — |
| σ1 (additive) | ng/mL | 0.0119 (24) | — |
| σ2 (proportional) | %CV | 36.8 (5.3) | — |
α and β, intercept and slope of DAR decline function; σ1 and σ2, variance of the additive and proportional components of the residual error, respectively; ADC, antibody‐drug conjugate; BSV, between‐subject variability; BW, body weight; CLM, MMAE apparent clearance; %CV, percent coefficient of variation; DAR0, baseline drug‐antibody ratio; Fm by cycle, exponent to describe the decline in the conversion of ADC to MMAE by ADC proteolytic degradation by cycles; MMAE, monomethyl auristatin E; Q5, MMAE apparent intercompartmental clearance; %RSE, percent relative standard error of the estimate; V4, MMAE apparent central compartment volume; V5, MMAE apparent peripheral compartment volume.
Reference population = 75‐kg male patient.
Dependence on BW as CLM·(BW/75)0.75, Q5·(BW/75)0.75.
Dependence on BW as V4·(BW/75), V5·(BW/75).
The first‐order rate constant of DAR decline, β, was estimated to be 0.0785 d−1, which suggested that approximately 13% of unconjugated MMAE is formed by the deconjugation pathway (derivation provided in Supplementary Material M1). A relatively high BSV was observed for CLM, V4, and β, with estimates of 60.7% CV, 78.2% CV and 98.1% CV, respectively. MMAE exposures have been shown to decrease after multiple doses, with approximately 50% to 80% of the exposure of the first dose observed at subsequent doses.14, 15, 16 The decline in MMAE exposure was assumed to be the consequence of tumor reduction from brentuximab vedotin treatment. The Fmc, the fraction of ADC converted to MMAE by cycle, is modeled as equation (6):
| (6) |
where Fm is estimated to be −0.261 with a large BSV of 130% CV.
Overall, the final model fit the data well. The model condition number was less than 1000. Diagnostic plots showed unbiased prediction (Supplementary Figure S2b). As shown in the VPC plot (Figure 2b), the 90% prediction interval was consistent with the observed data.
Model External Validation
Two methods were applied to the model validation data set to externally validate the model: prediction of observed PK data (method 1) and comparison of Monte Carlo simulations with the observed data (method 2). The results from both methods demonstrated that the final PopPK model adequately predicted the observed ADC and MMAE concentration–time data of the model validation data set. For the model prediction, the median PE% for ADC and MMAE was ‐15% and 17%, respectively. As shown in Supplementary Figure S3, most of the model validation data for both ADC and MMAE fell within the 90% confidence interval of the model‐predicted values derived from the PK parameters estimated from the model development data set.
Influence of Covariates on ADC Exposure
Among the covariates evaluated, only BW and sex had statistically significant effects on ADC PK parameters. However, based on simulations, it was found that only BW had an effect on ADC exposures. The median weight of patients in the analysis was 75 kg (range, 41 to 168 kg), and 45 of the 314 patients weighed >100 kg. ADC concentration–time profiles were simulated for typical patients weighing 41, 75, and 168 kg who received 1.8 mg/kg intravenous brentuximab vedotin every 3 weeks with dose capped at 180 mg. Relative to a patient weighing 75 kg (1.0), the ratio of predicted AUC0–21 days was 0.81 and 0.78 for patients weighing 41 and 168 kg, respectively. The ratio below 1.0 for the heaviest patient is the result of capping the dose at 180 mg. However, importantly, model‐predicted exposure distributions for 1.8 mg/kg intravenous every‐3‐week dosing (with the dose capped at 180 mg) indicate similar ADC exposures across weight quartiles for all 314 patients in the 5 studies (Figure 3a). Figure 3b illustrates the model‐predicted typical patient ADC concentration–time profiles for male and female patients receiving brentuximab vedotin 1.8 mg/kg intravenously every 3 weeks. As expected, ADC exposures are the same for male and female patients, whereas Cmax is slightly higher in female patients because sex only affected ADC V1.
Figure 3.
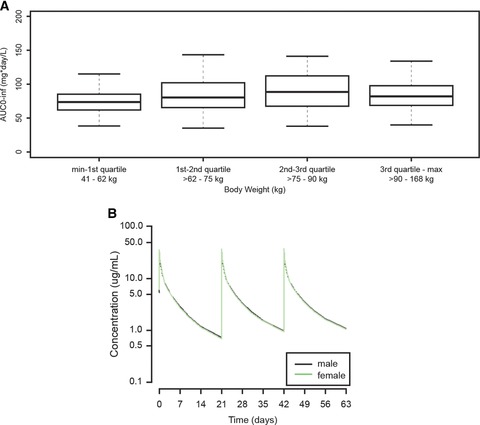
(a) Model‐predicted ADC exposure (AUC0–inf) for patients (n = 314) receiving brentuximab vedotin administered intravenously at 1.8 mg/kg every 3 weeks with the dose capped at 180 mg. The box‐and‐whisker plots indicate minimum and maximum values (lower and upper whiskers), median (heavy band), and first and third quartiles (bottom and top of box). (b) Model‐predicted typical patient ADC concentration–time profiles for male (black line) and female (green line) patients receiving brentuximab vedotin administered intravenously at 1.8 mg/kg every 3 weeks. ADC, antibody‐drug conjugate.
Immunogenicity
A total of 149 patients in the pooled phase 2 population (studies 3 and 4) tested negative for ATA at baseline and had postbaseline ATA data available. Ninety‐six of these 149 patients (64%) remained ATA negative throughout treatment; 42 patients (28%) became transiently positive (1 or 2 positive postbaseline time points) after exposure to brentuximab vedotin, and 11 patients (7%) became persistently positive for ATA (3 or more positive postbaseline time points). In addition, 8 patients were ATA positive at baseline; following exposure to brentuximab vedotin 2 of these patients were negative, 5 were transiently positive, and 1 was persistently positive for ATA. ATA was directed against the antibody component of brentuximab vedotin in all positive samples.
The development of persistently positive ATA appeared to impact brentuximab vedotin PK in a titer‐dependent manner with large variability. Two of the 11 patients who developed persistently positive ATA, corresponding to 1.3% of those who were ATA negative at baseline, showed substantial decreases in ADC Cmax. ATA did not appear to meaningfully affect the efficacy or the tolerability of brentuximab vedotin (data not shown). The overall incidence and severity of adverse events did not appear to be greater in the small number of patients with persistently positive ATA, compared with transiently positive or ATA‐negative patients. However, a higher incidence of infusion‐related reactions was observed among the patients who developed persistently positive ATA (3 of 11, 27%) relative to patients who were transiently (5 of 42, 12%) or never (7 of 96, 7%) positive. The same 2 patients with substantial decreases in ADC Cmax discontinued treatment because of adverse events consistent with infusion‐related reactions.
Discussion
This is the first report of a PopPK analysis of brentuximab vedotin in patients with CD30‐expressing hematologic malignancies and the first PopPK model of brentuximab vedotin to semimechanistically link the PK of ADC and that of the unconjugated small molecule MMAE. This analysis paves the way for future PK analyses of brentuximab vedotin and other vc‐MMAE‐based ADCs in different diseases and patient populations and in combination treatment settings.
It is challenging to characterize brentuximab vedotin PK because of the complex disposition of both ADC and the unconjugated MMAE, as well as the multiple processes of MMAE formation from ADC. In addition, although an average of 4 MMAE molecules are attached to each antibody molecule, brentuximab vedotin is a heterogeneous entity. The drug load of individual species ranges from 0 to 8,22 but the predominant species has a drug load of 4. The number of small molecules conjugated to an ADC may impact ADC elimination, leading to a shorter serum half‐life and lower exposure for the higher drug load species.17 ADC may also lose conjugated small molecules through deconjugation at a low rate while in circulation. Both processes contribute to a decrease in average drug load over time, which was taken into account for MMAE formation in the model. Although there have been similar efforts to describe ADC PK using mechanistic approaches,23, 24 our work represents the first attempt to link ADC and the released small‐molecule drug semimechanistically in an integrated population PK model.
The final model described in this work made certain assumptions to accommodate the complexity of the ADC disposition and the data available for model building. Various alternative models were tested, and a sensitivity analysis (ie, testing different initial estimates of model parameters) was performed during model development. A parsimonious model, which comprised a 3‐compartment model with linear elimination for ADC and a 2‐compartment model with linear elimination for MMAE, was achieved. MMAE was assumed to form from both ADC proteolytic degradation and deconjugation processes, and the average drug load was assumed to decrease exponentially. The estimated rate constant (β) for average DAR decline in the current model cannot be directly compared with the in vivo observations of Sanderson et al21 because they calculated DAR as a function of total antibody, whereas ours was calculated relative to ADC. The final model adequately described the observed concentration–time profiles of ADC and MMAE in patients administered brentuximab vedotin intravenously. As demonstrated by the predicted versus observed values and by the PE% values for the results, the a priori prediction of the concentrations of ADC and MMAE by the model was reasonably accurate.
The potential effects of intrinsic and extrinsic factors on ADC PK were evaluated using the PopPK approach: body weight and sex were the only statistically significant covariates. Simulations supported the recommended weight‐based dosing of brentuximab vedotin in adult patients with CD30‐expressing hematologic malignancies (typically 1.8 mg/kg, with a maximum of 180 mg for individuals with BW > 100 kg);14, 15, 16 sex did not result in different ADC exposure. This PopPK analysis found that alanine aminotransferase, albumin, aspartate aminotransferase, bilirubin, and CRCL did not significantly affect ADC exposure. Study 525 showed a trend of decreased ADC exposure and increased MMAE exposure in patients with hepatic impairment (defined as Child‐Pugh classes A to C) and patients with severe renal impairment (CRCL < 30 mL/min); however, that study contributed just 4 patients with renal impairment and none with hepatic impairment to the PopPK data set. The majority of patients in the PopPK data set had normal hepatic and renal function, so it is possible that the small proportion of patients with organ impairment did not provide adequate power to detect the association.
The effects of covariates on MMAE could not be formally assessed because MMAE is a metabolite when administered as part of the ADC during a brentuximab vedotin infusion, rather than administered directly as the small molecule. However, when BW was added empirically as a covariate to the MMAE model with effect exponents fixed to values that are physiologically meaningful for small molecules,26 the median PE% improved by approximately 30%.
Conclusions
This is the first PopPK model of brentuximab vedotin to semimechanistically link the PK of ADC and that of the unconjugated small molecule MMAE. Both ADC and MMAE PK data were adequately described by the final integrated PopPK model, which supports weight‐based dosing of brentuximab vedotin in adult patients with CD30‐expressing hematologic malignancies.
Supporting information
Supporting Information
Acknowledgments
Medical writing assistance was provided by Roberta Connelly and sponsored by Seattle Genetics, Inc. Presented in part as a poster at the American Society for Clinical Pharmacology and Therapeutics (ASCPT) Annual Meeting, March 8–12, 2016, San Diego, California.
Declaration of Conflicting Interests
H.L., N.N.H., G.J., and B.Z. are employed by and have equity ownership in Seattle Genetics, Inc. T.H.H. was employed by Seattle Genetics, Inc. at the time the work was performed and has equity ownership in Seattle Genetics, Inc.
Funding
Direct funding for this research was issued by Seattle Genetics, Inc. through the joint financial support of Seattle Genetics, Inc. and Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceuticals Limited.
References
- 1. Pro B, Advani R, Brice P, et al. Brentuximab vedotin (SGN‐35) in patients with relapsed or refractory systemic anaplastic large cell lymphoma: results of a phase II study. J Clin Oncol. 2012;30(18):2190–2196. [DOI] [PubMed] [Google Scholar]
- 2. Younes A, Gopal AK, Smith SE, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin's lymphoma. J Clin Oncol. 2012;30(18):2183–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sutherland MS, Sanderson RJ, Gordon KA, et al. Lysosomal trafficking and cysteine protease metabolism confer target‐specific cytotoxicity by peptide‐linked anti‐CD30‐auristatin conjugates. J Biol Chem. 2006;281(15):10540–10547. [DOI] [PubMed] [Google Scholar]
- 4. Francisco JA, Cerveny CG, Meyer DL, et al. cAC10‐vcMMAE, an anti‐CD30‐monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood. 2003;102(4):1458–1465. [DOI] [PubMed] [Google Scholar]
- 5. Okeley NM, Miyamoto JB, Zhang X, et al. Intracellular activation of SGN‐35, a potent anti‐CD30 antibody‐drug conjugate. Clin Cancer Res. 2010;16(3):888–897. [DOI] [PubMed] [Google Scholar]
- 6. Oflazoglu E, Stone IJ, Gordon KA, et al. Macrophages contribute to the antitumor activity of the anti‐CD30 antibody SGN‐30. Blood. 2007;110(13):4370–4372. [DOI] [PubMed] [Google Scholar]
- 7. Li F, Zhang X, Emmerton K, et al. Relationship between in vivo antitumor activity of ADC and payload release in preclinical models [abstract 3694]. Cancer Res. 2014;74:3694. [Google Scholar]
- 8. Muller P, Martin K, Theurich S, et al. Microtubule‐depolymerizing agents used in antibody‐drug conjugates induce antitumor immunity by stimulation of dendritic cells. Cancer Immunol Res. 2014;2(8):741–755. [DOI] [PubMed] [Google Scholar]
- 9. Gardai SJ, Epp A, Law C‐L. Brentuximab vedotin‐mediated immunogenic cell death [abstract 2469]. Cancer Res. 2015;75:2469. [Google Scholar]
- 10. Kim YH, Tavallaee M, Sundram U, et al. Phase II investigator‐initiated study of brentuximab vedotin in Mycosis fungoides and Sezary syndrome with variable CD30 expression level: a multi‐institution collaborative project. J Clin Oncol. 2015;33(32):3750–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Younes A, Bartlett NL, Leonard JP, et al. Brentuximab vedotin (SGN‐35) for relapsed CD30‐positive lymphomas. N Engl J Med. 2010;363(19):1812–1821. [DOI] [PubMed] [Google Scholar]
- 12. Fanale MA, Forero‐Torres A, Rosenblatt JD, et al. A phase I weekly dosing study of brentuximab vedotin in patients with relapsed/refractory CD30‐positive hematologic malignancies. Clin Cancer Res. 2012;18(1):248–255. [DOI] [PubMed] [Google Scholar]
- 13. Han TH, Kennedy D, Hayes S, Lynch CM. The pharmacokinetics of brentuximab vedotin (SGN‐35), an antibody‐drug conjugate (ADC) [abstract PII‐1]. Clin Pharmacol Ther. 2012;91(suppl 1):PII‐1. [Google Scholar]
- 14. ADCETRIS [EU summary of product characteristics]. Taastrup, Denmark: Takeda Pharma A/S; November 2015.
- 15. ADCETRIS [Canadian product monograph]. Bothell, WA: Seattle Genetics, Inc.; 2016.
- 16. ADCETRIS [US package insert]. Bothell, WA: Seattle Genetics, Inc.; 2016.
- 17. Han TH, Zhao B. Absorption, distribution, metabolism, and excretion considerations for the development of antibody‐drug conjugates. Drug Metab Dispos. 2014;42:1914–1920. [DOI] [PubMed] [Google Scholar]
- 18. Han TH, Gopal AK, Ramchandren R, et al. CYP3A‐mediated drug‐drug interaction potential and excretion of brentuximab vedotin, an antibody‐drug conjugate, in patients with CD30‐positive hematologic malignancies. J Clin Pharmacol. 2013;53(8):866–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gastonguay MR. Full covariate models as an alternative to methods relying on statistical significance for inferences about covariate effects: a review of methodology and 42 case studies [abstract 2229]. Population Approach Group Europe (PAGE 20);2011. www.page-meeting.org/?abstract=2229. Accessed March 3, 2017.
- 20. Hamblett KJ, Senter PD, Chace DF, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10(20):7063–7070. [DOI] [PubMed] [Google Scholar]
- 21. Sanderson RJ, Hering MA, James SF, et al. In vivo drug‐linker stability of an anti‐CD30 dipeptide‐linked auristatin immunoconjugate. Clin Cancer Res. 2005;11(2 Pt 1):843–852. [PubMed] [Google Scholar]
- 22. Senter PD, Sievers EL. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat Biotechnol. 2012;30(7):631–637. [DOI] [PubMed] [Google Scholar]
- 23. Shah DK, Haddish‐Berhane N, Betts A. Bench to bedside translation of antibody drug conjugates using a multiscale mechanistic PK/PD model: a case study with brentuximab‐vedotin. J Pharmacokinet Pharmacodyn. 2012;39(6):643–659. [DOI] [PubMed] [Google Scholar]
- 24. Lu D, Jin JY, Girish S, et al. Semi‐mechanistic multiple‐analyte pharmacokinetic model for an antibody‐drug‐conjugate in Cynomolgus monkeys. Pharm Res. 2015;32(6):1907–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao B, Chen R, O'Connor OA, et al. Brentuximab vedotin, an antibody‐drug conjugate, in patients with CD30‐positive haematologic malignancies and hepatic or renal impairment. Br J Clin Pharmacol. 2016;82(3):696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Anderson BJ, Holford NH. Mechanism‐based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol. 2008;48:303–332. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information