

Abstract
Haptophyta encompasses more than 300 species of mostly marine pico‐ and nanoplanktonic flagellates. Our aims were to investigate the Oslofjorden haptophyte diversity and vertical distribution by metabarcoding, and to improve the approach to study haptophyte community composition, richness and proportional abundance by comparing two rRNA markers and scanning electron microscopy (SEM). Samples were collected in August 2013 at the Outer Oslofjorden, Norway. Total RNA/cDNA was amplified by haptophyte‐specific primers targeting the V4 region of the 18S, and the D1‐D2 region of the 28S rRNA. Taxonomy was assigned using curated haptophyte reference databases and phylogenetic analyses. Both marker genes showed Chrysochromulinaceae and Prymnesiaceae to be the families with highest number of Operational Taxonomic Units (OTUs), as well as proportional abundance. The 18S rRNA data set also contained OTUs assigned to eight supported and defined clades consisting of environmental sequences only, possibly representing novel lineages from family to class. We also recorded new species for the area. Comparing coccolithophores by SEM with metabarcoding shows a good correspondence with the 18S rRNA gene proportional abundances. Our results contribute to link morphological and molecular data and 28S to 18S rRNA gene sequences of haptophytes without cultured representatives, and to improve metabarcoding methodology.
Keywords: Abundance, coccolithophores, high‐throughput sequencing, Oslofjorden, phylogeny, richness
THE protist division Haptophyta encompasses more than 300 morphospecies of mostly pico‐ and nanoplanktonic flagellates (Edvardsen et al. 2016; Jordan et al. 2004; Thomsen et al. 1994). The group inhabits all seas and some also thrive in freshwater, exhibiting a high degree of morphological, physiological and functional diversity (Jordan and Chamberlain 1997). Haptophytes share common structural features, notably the production of unmineralized organic scales and possession of two flagella and a haptonema, although the latter was lost in a few members (e.g. Isochrysidales). Haptophytes are major primary producers in open oceans (Andersen et al. 1996; Liu et al. 2009) and the calcifying coccolithophores play a key role in the biogeochemical carbon cycle (Holligan et al. 1993; Iglesias‐Rodríguez et al. 2002; Robertson et al. 1994). In addition, blooms of noncalcifying haptophytes can have a strong impact on coastal ecosystems through toxin production (Granéli et al. 2012; Moestrup 1994).
Being a large and diverse group, haptophytes commonly exhibit species‐ and even strain‐specific physiological traits, which ultimately define their ecological and biogeochemical performance (Edvardsen and Paasche 1998; Langer et al. 2006; Ridgwell et al. 2009). Therefore, identifying haptophytes to a low taxonomic level is of great importance in ecological surveys. Morphological identification to the species level is particularly difficult in noncalcifying groups, which lack hard and mineralized body parts, and can only be accurately identified by the time‐consuming examination of organic scales using electron microscopy (EM) (Edvardsen et al. 2011; Eikrem 1996; Eikrem and Edvardsen 1999). On the other hand, scanning electron microscopy (SEM)‐based methods allow for precise analysis of taxonomic composition and species abundance of coccolithophore communities (Bollmann et al. 2002; Cros and Fortuño 2002; Young et al. 2003).
With the advance of molecular techniques, notably environmental sequencing of clone‐libraries and metabarcoding using high‐throughput sequencing (HTS), it has become possible to overcome some of the limitations of microscopical analysis in investigating haptophyte communities (Edvardsen et al. 2016). Most importantly, molecular methods allow for detection of rare or fragile species that commonly remain unnoticed in ecological surveys. A number of studies specifically investigated haptophyte communities using molecular methods (Bittner et al. 2013; Egge et al. 2015a,b; Liu et al. 2009), each of them further improving the methodology and providing new insights about the diversity and distribution of the group. The major shift in understanding the diversity of haptophytes was provided by the early clone‐library studies (Moon‐van der Staay et al. 2000, 2001; Edvardsen et al. 2016 for comprehensive list of studies), as well as HTS‐based works of Bittner et al. (2013) and Egge et al. (2015a,b). These studies identified an abundance of new haptophyte sequences (OTUs) and new haptophyte lineages, which could not be assigned to a cultured and genetically characterized taxon. This indicated that the haptophyte diversity in the modern oceans was largely underestimated in previous microscopic investigations, and that there are many morphospecies still to be described.
However, despite the rigorous processing of data during analysis of 454‐reads, a significant portion of ribotypes may still represent chimeric sequences or amplification artefacts (Haas et al. 2011; Huse et al. 2010; Speksnijder et al. 2001). The lack of studies combining qualitative and quantitative morphological analysis of haptophyte communities with molecular approaches complicates the estimation of the actual species diversity and abundance. Conducting such investigations on haptophyte communities is difficult due to the mentioned methodological limitations in identifying noncalcifying species using a morphological approach. Therefore, coccolithophores are arguably the most appropriate group of haptophytes for such comparative investigation. A study by Young et al. (2014) aimed at comparing the molecular (environmental sequencing of clone‐libraries) approach based on the 28S rRNA gene with the morphological (LM and SEM) analysis of coccolithophore communities. A good match between the morphological and molecular results was observed in terms of taxonomy, but the study found no strong correlation of the relative OTU and major morphotype abundances. Weak or no correlation between relative OTU abundance and biomass or cell number was also found by Egge et al. (2013) using 454 pyrosequencing in a haptophyte mock community experiment. Since HTS generates much more reads from environmental samples than sequences from clone‐libraries, there is a need to compare it with the traditional SEM based approach to assess if this is an appropriate method for studying the diversity and proportional abundance of coccolithophores.
Previous metabarcoding studies of haptophytes (Bittner et al. 2013) and protists (Massana et al. 2015) comparing DNA vs. RNA as templates did not find any significant differences in community structure. However, Egge et al. (2013) found that RNA captured more of the diversity than with DNA where larger cells were favored. Using RNA may significantly reduce the bias due to variability in rDNA copy numbers among taxonomic groups (Not et al. 2009). Massana et al. (2015) found that Haptophyta were underrepresented in DNA compared to RNA surveys (average read ratio DNA/RNA was 7.4). In addition, compared to DNA, RNA is thought to more accurately picture which protists are metabolically active at the time of collection (Stoeck et al. 2007).
Further, comparison of different studies is complicated due to use of different marker genes. Of both 18S and 28S rRNA genes there are more than 600 different reference sequences available in gene databases from cultured material and environmental clone‐libraries (Edvardsen et al. 2016). The number of described haptophyte species for which there are reference sequences available are higher for the 18S than 28S rRNA gene (96 vs. 76) (Edvardsen et al. 2016) which makes 18S a more useful marker for identifying species in an environmental sample. However, in haptophytes the 18S rRNA gene may differ in only a few base pairs between closely related species, and short variable regions used for HTS metabarcoding, such as the V4, may be identical (Bittner et al. 2013; Edvardsen et al. 2016). The 28S rRNA gene has more variable regions than 18S, and regions of the 28S such as the D1‐D2 have therefore been suggested to be more appropriate barcodes for distinguishing recently diverged species (Bittner et al. 2013; Liu et al. 2009), although some intraspecies variation may occur, which will overestimate species richness (Liu et al. 2009). As these two markers offer complementary views of environmental haptophyte communities, it is important to compare richness and taxonomic composition of 18S and 28S metabarcoding data sets obtained from the same samples. To our knowledge, no study has so far done such a rigorous comparison for haptophytes.
The aim of our study was to investigate the Oslofjorden haptophyte community diversity and vertical distribution by using HTS with two RNA markers supplemented with scanning electron microscopy. We also aim at testing and improving the approach to study haptophyte diversity and proportional abundance. We addressed the following questions (i) Do we find novel taxa or species that have not previously been recorded in the Oslofjorden? (ii) Is there a difference in the community composition and proportional abundance by depth and size fraction? (iii) Do we obtain the same results with the 18 and 28S rRNA marker genes? (iv) Can we place 28S OTUs without a cultured representative in 18S rRNA gene‐defined clades? (v) How does the qualitative and quantitative composition of the coccolithophore community compare between HTS and scanning electron microscopy?
Materials and Methods
Sampling
Water samples from subsurface (1 m) and deep‐chlorophyll maximum (DCM, 8 m) for high‐throughput sequencing were collected at the OF2 station (59.19 N, 10.69 E) in the outer Oslofjorden, Skagerrak on August 21, 2013. Twenty litres of seawater from each sampling depth were collected with Niskin bottles and prefiltered through a 45‐μm nylon mesh. Subsequently, an in‐line filtration through 3‐μm and 0.8‐μm pore size polycarbonate (PC) filters (142‐mm Millipore, Darmstads, Germany) was performed with a Millipore Tripod unit and a peristaltic pump for maximum 40 min. This procedure yielded two size fractions: nanoplankton (3–45 μm) and picoplankton (0.8–3 μm). Filters were fast frozen in liquid nitrogen and stored at −80 °C until RNA extraction. Water column profiling was carried out using a conductivity‐temperature‐depth device (CTD, Falmouth Scientific Inc., Cataumet, MA) and TD‐700 fluorometer sensor (Turner Designs, Sunnyvale, CA) attached to a rosette. Nutrients measurements were performed on samples collected at eight different depths (1, 2, 4, 8, 12, 16, 20, and 40 m) as described in Egge et al. (2015b).
RNA extraction, PCR, and 454‐pyrosequencing
RNA extraction, PCR, and 454 pyrosequencing of filtered samples were conducted following a modified protocol by Egge et al. (2015a). Briefly, RNA was isolated from 1 m and 8 m filter samples using RNA NucleoSpin II (Macherey‐Nagel, Düren, Germany) and converted to cDNA by reverse transcription with the High‐Fidelity 1st Strand cDNA Synthesis Kit (Agilent, Santa Clara, CA). For PCR amplification of the ribosomal 18S V4 RNA gene region, we used the forward 528Flong and reverse PRYM01+7 primers described in Egge et al. (2013), whereas for amplification of the 28S D1–D2 region we used the LSU 1 primer pair from Bittner et al. (2013). Amplification protocol for both markers was the same as in Egge 2013, with the exception of the annealing temperature (55 and 53 °C for 18S and 28S respectively). The amplicon library was processed as described by Roche (Basel, Switzerland) and sequenced with the 454 GS‐FLX Titanium system at the Norwegian Sequencing Centre (NSC) at the Department of Biosciences, University of Oslo (www.sequencing.uio.no). Two technical replicates were analysed for each depth, size fraction and marker gene (total of 16 samples).
Processing and analyses of raw pyrosequencing data
Sequences were denoised using AmpliconNoise v.1.6.0 in QIIME (Quince et al. 2009). Putative chimeras were identified and removed using Perseus in AmpliconNoise. Further bioinformatic processing was done in Mothur v. 1.36.1 (Schloss et al. 2009), unless otherwise stated. Sequences shorter than 365 bp and with homopolymers > 8 bp were removed using the “trim.seqs”‐command. Additional chimera check was done using the “chimera.uchime”‐command, with default settings. Sequence clustering was done using the “cluster”‐command with the average neighbour algorithm. To be able to compare our 18S rRNA results to previous studies (i.e. Egge et al. 2015a,b) we clustered our 18S OTUs at 99% similarity. Bittner et al. (2013) showed that the more variable 28S rRNA needed clustering at a lower similarity level to balance between species detection and spurious diversity and found 97% to be reasonable, which we used here too. To remove nonhaptophyte reads, the “classify.seqs”‐command was used to perform a first OTU taxonomical assignation to phylum against the full Protist Ribosomal Data Base (PR2, Guillou et al. 2013) for the 18S OTUs, and against the SILVA LSU reference database (v. 123) for the 28S rRNA OTUs. The haptophyta OTUs were extracted with the “get.lineage”‐command. Sequence similarity and a first taxonomic assignation of haptophyte reads was done by blast against the curated Haptophyta 18S rRNA gene database by Edvardsen et al. (2016) (Haptophyta‐PiP) and a curated Haptophyta 28S rRNA gene database from this study, based on Bittner et al. (2013), consisting of 184 unique sequences (see description in Data S1). Both databases with alignments are available at figshare (https://figshare.com/account/home#/projects/11914). Single‐tons and double‐tons (OTUs with only one or two reads across all samples) were removed before further analysis. To compare our 18S OTUs to the OTUs obtained by HTS of samples from the same station taken in two earlier years (Egge et al. 2015a,b), we ran MegaBLAST (Morgulis et al. 2008) with the OTUs of the previous study as query sequences, and our OTUs as subject sequences. MegaBLAST was run on the University of Oslo Lifeportal (www.lifeportal.uio.no). Detection of an OTU from the previous study is defined as ≥ 99% sequence identity. To be able to compare between samples, they were rarefied (subsampled) to the smallest sample size.
Phylogenetic analyses
Phylogenies were performed following EUKREF RAxML‐EPA (Evolutionary Placement Algorithm) pipeline (del Campo, pers. commun.) for a more reliable taxonomic assignation of reads than by BLAST. Curated haptophyte 18S and 28S rRNA gene reference alignments of the reference databases described above were created with MAFFT G‐INS‐i (http://mafft.cbrc.jp/alignment/server/). Reads were aligned against the reference alignments using “align_seqs.py”. Gaps and hypervariable positions were removed using “filter_alignment.py” in QIIME. The alignment was checked manually in Geneious v.7.1.9, and positions that did not align well were edited. All known members of Prymnesiophyceae have a six A homopolymer (position 751–756 in reference sequence AJ004866 Prymnesium polylepis 18S rRNA gene) (Egge et al. 2015a), but in our 18S data set this homopolymer varied between 5 and 6 bp. Too short or long homopolymers is a common error with 454 pyrosequencing (e.g. Gilles et al. 2011). To avoid inflated OTU richness, we truncated this A homopolymer to 5 bp in all the sequences. Maximum‐likelihood analyses (RAxML v.8.0.26; Stamatakis 2006) was performed on the two reference alignments with substitution model GTR + CAT with 100 bootstraps run on the UiO Abel computer cluster. Finally, the program raxmlHPC‐PTHREADS‐SSE3 was run to place our 18S and 28S rRNA gene OTUs on our RAxML reference trees, where the alignments and the tree‐files in newick format were the input files.
SEM analysis
Water samples were collected at 8 depths (1, 2, 4, 8, 12, 16, 20, and 40 m) using 5‐l Niskin water samplers. A known volume from each sample, ranging between 250 (2, 4, 12, 16, 20, and 40 m samples) and 300 ml (1 m sample), was filtered under weak vacuum onto the polycarbonate filter (0.8‐μm Cyclopore, 25‐mm diameter, Whatman, Kent, UK) that was placed on cellulose nitrate membranes filter (0.8‐μm Whatman) to ensure an even distribution of material. After the filtration, filters were dried in oven at 50 °C. Before the analysis under a Zeiss Supra35‐VP scanning electron microscope, a piece of filter was mounted on an aluminium stub and sputter‐coated with gold. Quantitative analysis of the coccolithophore community was conducted following Bollmann et al. (2002). The same number of fields of view (600 for 1 m and DCM samples, 300 for other samples) was analysed on each filter. Using 600 fields of view covered 6.89 mm2 of the filter area, corresponding to 4.90 ml of analysed seawater at 1 m depth and 4.15 ml at DCM. Using 300 fields of view covered 3.45 mm2, analysing 2.07–2.48 ml of seawater. Number of cells counted for each analysed sample ranged between 1,045 and 1,122 using 600 fields of view and 76–570 using 300 fields of view. Taxonomy of coccolithophores was determined to the lowest possible level using the standard taxonomic literature (Cros and Fortuño 2002; Jordan et al. 2004; Young et al. 2003).
Statistical analyses
For each 454‐pyrosequencing sample, Shannon–Wiener's species diversity index (H′ = −Σp i(ln p i)) and Pielou's evenness index (J′ = H′/ln S) were calculated, where p i is the proportion of individuals of species i and S is the total number of species. Student's one sample t‐test was then conducted to determine the significance between pseudo‐replicates diversities for the two different marker data sets (Pielou 1966). OTU proportional abundances were normalized by performing a square root transformation, for each of the 16 samples. The Mean‐difference (Bland–Altman) method was used on the normalized data to test replicate reproducibility. Differences between replicates were plotted and standard deviation of the differences were computed and added as dotted lines.
Results and Discussion
Our aims were to investigate the Oslofjorden haptophyte community diversity and vertical distribution and to improve the approach to study haptophyte diversity by metabarcoding. We amplified 18S and 28S rRNA/cDNA genes with haptophyte‐specific primers to identify the summer community in the Oslofjorden at two different depths. Most of the haptophyte species that have been morphologically described are found in the size fraction between 2 and 40 μm. In order to cover the entire range, we collected samples between 0.8 and 45 μm divided into two size fractions here called picoplankton (0.8–3 μm) and nanoplankton (3–45 μm). We used maximum‐likelihood phylogenetic placement (RAxML‐EPA) of OTUs and curated 18S and 28S rRNA gene sequence reference databases to determine their taxonomical assignation. The Oslofjorden haptophyte community has previously been described by high‐throughput sequencing (Egge et al. 2015a,b). However, these studies used only 18S rRNA as a marker and only included one depth. Electron microscopy on coccolithophores was also performed on samples taken at eight different depths to assess the semi‐quantitative capacity of metabarcoding data.
Hydrography
Figure 1 shows environmental variables at the OF2 sampling site at the day of sampling that may influence haptophyte species composition. At the surface (0–1 m) the salinity was 23.7 PSU and increased by depth down to 90 m where it stabilized at 34.6 PSU. The temperature was 18.5 °C at the surface and gradually decreased down to 80 m stabilizing to 6.8 °C. The density plot indicates a shallow upper mixed‐layer in 0–2 m with a pycnocline most pronounced at 2–8 m. The fluorescence (an estimate for relative phytoplankton biomass) increased with depth, reaching maximum values at 8–10 m, (deep‐chlorophyll maximum, DCM). Nutrient concentrations by depths are shown in Fig. 2. Concentration of dissolved inorganic nitrogen ([NO3 −] + [NO2 −]) was near the detection limit (0–0.29 μM) in 1–20 m. Also phosphate (PO4 3−) concentrations were low (0.26–0.29 μM) in the euphotic zone above 20 m. The silicate concentration peaked at 3 m (up to 3.34 μM), probably originated from a fresh water inflow from land.
Figure 1.
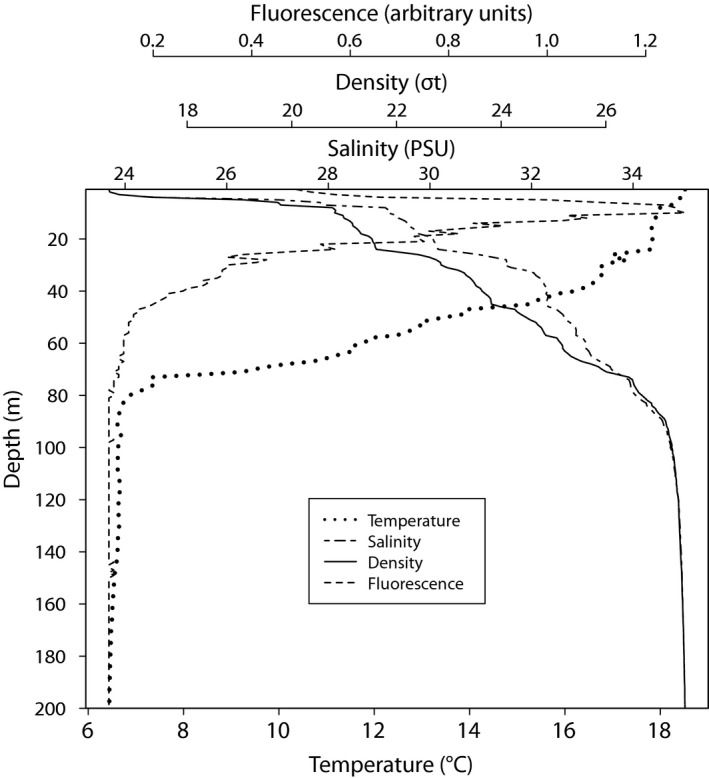
Depth profiles of temperature, salinity, density and fluorescence at outer Oslofjorden, OF2 station on 21 August 2013.
Figure 2.
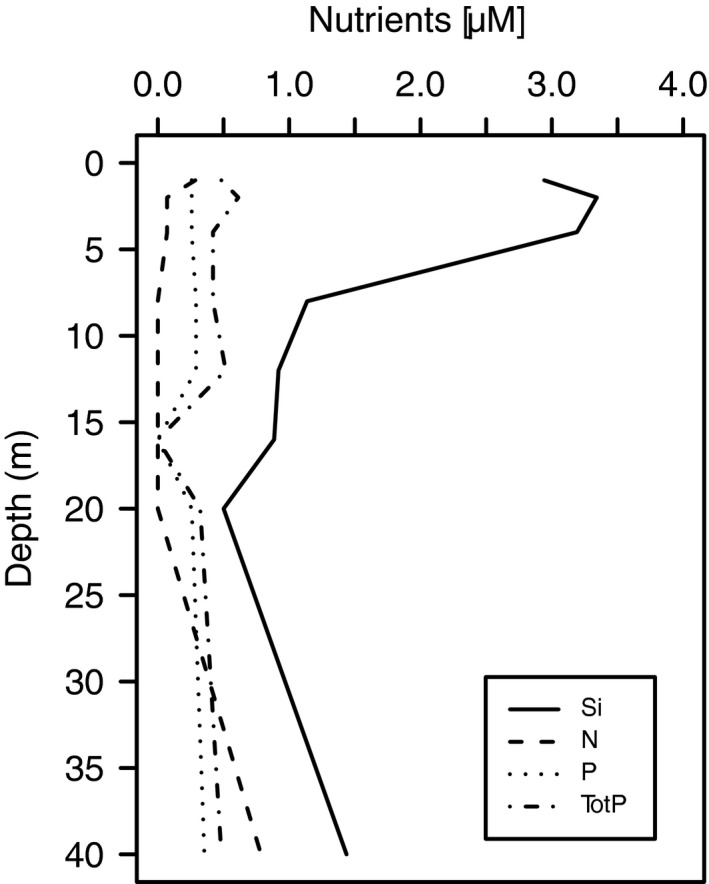
Concentrations (μM) of dissolved inorganic silicate (Si), nitrate + nitrite (N) and phosphate (P), and total phosphorus (dissolved and particulate, inorganic and organic, Tot‐P) at outer Oslofjorden OF2 station on the sampling date (21 August 2013).
Haptophyta richness
Denoising of initial reads and removal of putative chimaeras using AmpliconNoise generated a total of 120,282 amplicon reads of the V4 18S rRNA gene and 38,795 of the D1‐D2 region of 28S rRNA, with similar numbers of reads among the replicates within each marker (Table S1). Removal of short (< 365 bp) reads and reads with homopolymers > 8 bp from the pooled within‐marker data sets yielded 112,958 of reads of 18S rRNA and 30,981 reads of 28S rRNA. Of those, 112,399 18S rRNA reads were assigned to haptophytes (95.5%) compared to 30,892 of the 28S rRNA reads (99.7%). Subsequent removal of chimeric reads discarded 7,454 (6.6%) of the 18S rRNA and 288 (0.9%) of the 28S rRNA haptophyte reads. Finally, after clustering at 99% and 97% similarity and removal of singletons and double‐singletons, the 18S rRNA data set contained 215 OTUs (104,345 reads) while the 28S rRNA data set contained 432 OTUs (29,751 reads), respectively.
The haptophyte richness in the Skagerrak area is relatively high. To date, a total of 85 haptophyte species based on microscopy and 156 OTUs obtained by HTS, estimating species, have been recorded from the Skagerrak area (Egge et al. 2015a and references therein). Haptophyta currently comprises 312 morphologically described species (Edvardsen et al. 2016). We recovered considerably higher haptophyte diversity (215 OTUs) in the Oslofjorden than has previously been observed with microscopy in this area. We also recovered a higher 18S OTU richness on one single day than the only previous study applying HTS on monthly samples from 2 yrs (Egge et al. 2015a). In our study, both 1 m and deep‐chlorophyll maximum were sampled, whereas in Egge et al. (2015a) only 1 m depth was analysed. In total 104 of the OTUs recovered from the DCM in this study were not detected (i.e. < 99% similar to any OTU) in Egge et al. (2015a), which contributed to a higher richness in this study (Table S2a).
The higher number of 18S OTUs in this study compared to Egge et al. (2015a) may also in part be due to differences in the bioinformatic filtering procedure. The RNA extraction, cDNA synthesis and PCR amplification protocols were identical; however, Egge et al. (2015a) included a very stringent manual OTU filtering step (manual editing of homopolymers and chimera check to GenBank‐sequences by BLAST). This stringent filtering is not feasible with a higher number of OTUs, and also not fully reproducible, which is why we did not include it in this study. PCR and HTS techniques such as 454 are well known to introduce sequencing errors and chimeras, and it is near impossible to remove all such errors (e.g. Huse et al. 2010). On the other hand, too stringent filtering may theoretically remove genuine phylotypes.
Taxonomic composition and proportional abundance
Phylogenetic trees inferred from 18S and 28S reference sequences, with our OTUs added, are shown in Fig. 3. The taxonomic assignation is based on these trees. The number of OTUs within each clade are marked to the right of the collapsed and supported clades with bootstrap values > 50%. Some OTUs did not fall within a supported clade and are shown individually in the tree (number of reads per OTU are marked to the right).
Figure 3.
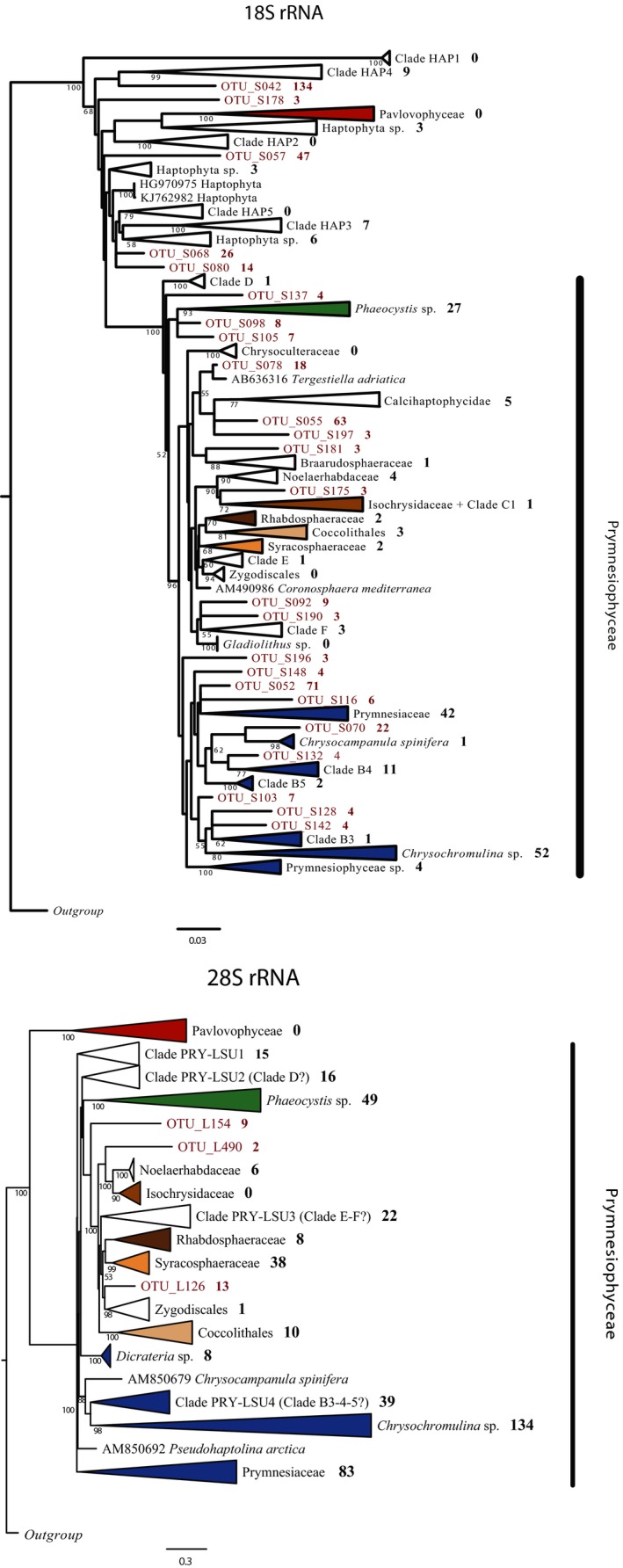
Maximum‐likelihood (RAxML) haptophyte phylogeny for (a) 18S rRNA and (b) 28S rRNA. Support values are inferred using GTRCAT model with 100 bootstraps. Bold numbers indicate the number of OTUs presented in each clade. Numbers next to OTU names indicate the number of reads in each OTU.
18S rRNA
At 99% sequence similarity, we detected OTUs representing 18 supported clades at taxonomic levels from class to family. Within the class Prymnesiophyceae, the family Chrysochromulinaceae hosted the highest number of OTUs (52, 24.2%), followed by Prymnesiaceae (42) and Phaeocystaceae (27) (Fig. 3a and Table S3). Noelaerhabdaceae and Syracosphaeraceae, were represented by only four and two OTUs, respectively. A total of 33 OTUs were placed outside the class Prymnesiophyceae, seven in Clade HAP3, nine in HAP4 and 12 were matching haptophyte environmental sequences without a clade name. The five remaining OTUs did not cluster with any reference sequence.
The proportional read abundance within each major clade is shown in Table S3, and of the 30 most abundant OTUs in Table 1. Chrysochromulinaceae was the family with the highest proportion of reads (33%) followed by Noelaerhabdaceae (18%), Prymnesiaceae (15%) and Syracosphaeraceae (12%). Phaeocystaceae represented 9% of the reads. A total of 3.1% of the reads belonged to OTUs that could not be assigned further than Prymnesiophyceae. OTUs that matched clades HAP3, HAP4, and sequences without phylogenetic placement to any supported clade accounted for 0.9% of the total reads.
Table 1.
List of the 30 most abundant haptophyte V4 18S rRNA OTUs detected
| OTU ID | Total reads (N) | Total reads (%) | Total reads after subsampling (N) | Total reads after subsampling (%) | Depth | Size fraction | Group | Lowest taxonomic level possible to determine |
|---|---|---|---|---|---|---|---|---|
| OTU_S001 | 19,473 | 18.66 | 13,715 | 18.74 | Both | Both | Chrysochromulinaceae | Chrysochromulina |
| OTU_S002 | 18,849 | 18.06 | 11,413 | 15.59 | Both | Both | Noelaerhabdaceae | Emiliania huxleyi |
| OTU_S003 | 10,500 | 10.06 | 6,669 | 9.11 | Both | Both | Syracosphaeraceae | Syracosphaeraceae |
| OTU_S004 | 5,186 | 4.97 | 3,893 | 5.32 | Both | Both | Prymnesiaceae | Prymnesium |
| OTU_S005 | 4,234 | 4.06 | 3,364 | 4.60 | Both | Both | Prymnesiaceae | Haptolina |
| OTU_S006 | 4,231 | 4.05 | 3,252 | 4.44 | Both | Both | Phaeocystaceae | Phaeocystis |
| OTU_S007 | 3,349 | 3.21 | 2,847 | 3.89 | Both | Both | Chrysochromulinaceae | Chrysochromulina |
| OTU_S008 | 3,126 | 3.00 | 2,293 | 3.13 | Both | Both | Chrysochromulinaceae | Chrysochromulina |
| OTU_S009 | 2,784 | 2.67 | 1,976 | 2.70 | Both | Both | Chrysochromulinaceae | Chrysochromulina |
| OTU_S010 | 2,757 | 2.64 | 2,255 | 3.08 | Both | Both | Chrysochromulinaceae | Chrysochromulina |
| OTU_S011 | 2,650 | 2.54 | 1,963 | 2.68 | Both | Both | Prymnesiaceae | Prymnesiaceae |
| OTU_S012 | 2,592 | 2.48 | 1,928 | 2.63 | Both | Both | Prymnesiophyceae | Prymnesiophyceae |
| OTU_S013 | 2,334 | 2.24 | 1,614 | 2.21 | Both | Both | Phaeocystaceae | Phaeocystis |
| OTU_S014 | 2,148 | 2.06 | 1,258 | 1.72 | Only DCM | Only nano | Syracosphaeraceae | Syracosphaeraceae |
| OTU_S015 | 2,132 | 2.04 | 1,690 | 2.31 | Both | Both | Prymnesiales Clade B3–B4–B5 | Prymnesiales Clade B4 |
| OTU_S016 | 1,679 | 1.61 | 1,235 | 1.69 | Both | Both | Prymnesiaceae | Prymnesium polylepis |
| OTU_S017 | 1,378 | 1.32 | 810 | 1.11 | Both | Both | Calyptrosphaeraceae | Calyptrosphaera sphaeroidea |
| OTU_S018 | 1,284 | 1.23 | 927 | 1.27 | Both | Both | Phaeocystaceae | Phaeocystis |
| OTU_S019 | 1,037 | 0.99 | 775 | 1.06 | Both | Both | Prymnesiales Clade B3–B4–B5 | Prymnesiales Clade B4 |
| OTU_S020 | 871 | 0.83 | 632 | 0.86 | Both | Both | Chrysochromulinaceae | Chrysochromulina |
| OTU_S021 | 866 | 0.83 | 640 | 0.87 | Both | Both | Phaeocystaceae | Phaeocystis cordata |
| OTU_S022 | 797 | 0.76 | 520 | 0.71 | Both | Both | Rhabdosphaeraceae | Algirosphaera robusta |
| OTU_S023 | 778 | 0.75 | 559 | 0.76 | Both | Both | Prymnesiales Clade B3–B4–B5 | Prymnesiales Clade B4 |
| OTU_S024 | 765 | 0.73 | 557 | 0.76 | Both | Both | Prymnesiales Clade B3–B4–B5 | Prymnesiales Clade B4 |
| OTU_S025 | 722 | 0.69 | 579 | 0.79 | Both | Both | Prymnesiales Clade B3–B4–B5 | Prymnesiales Clade B4 |
| OTU_S026 | 633 | 0.61 | 577 | 0.79 | Only 1 m | Both | Calcihaptophycidae | Calcihaptophycidae |
| OTU_S027 | 479 | 0.46 | 276 | 0.38 | Only DCM | Both | Prymnesiaceae | Prymnesium |
| OTU_S028 | 449 | 0.43 | 298 | 0.41 | Both | Both | Prymnesiophyceae | Prymnesiophyceae |
| OTU_S029 | 443 | 0.42 | 287 | 0.39 | Both | Both | Chrysochromulinaceae | Chrysochromulina |
| OTU_S030 | 364 | 0.35 | 276 | 0.38 | Both | Both | Phaeocystaceae | Phaeocystis globosa |
The taxonomic assignment was based on phylogenetic placement.
The 10 most abundant OTUs were present in all samples (Fig. S1a). The first three 18S OTUs were nested within Chrysochromulina sp. (OTU_S001), Emiliania huxleyi (OTU_S002), and Syracosphaeraceae sp. (OTU_S003). They constituted 47% of the final reads (Tables 1, S2a). The 126 rarest OTUs (< 10 reads) represented 0.5% of the reads (Fig. S2).
28S rRNA
After clustering at 97% similarity we obtained OTUs within 12 supported clades. Chrysochromulinaceae was the most diverse family with 134 OTUs, followed by Prymnesiaceae (91), Phaeocystaceae (49), and Syracosphaeraceae (38) (Fig. 3b and Table S3). Ten or less OTUs were detected in each of the five remaining clades with cultured representatives. A major part of the OTUs (22%) formed clades without affiliation to any reference sequences from cultures (here called PRY‐LSU1, PRY‐LSU2, PRY‐LSU3, and PRY‐LSU4), and only three were not placed in any clade. These may possibly represent the clades without a cultured and sequenced representative as defined by the 18S rRNA gene (see Fig. 3b).
Highest proportion of reads (34%) was found in Chrysochromulinaceae, while Syracosphaeraceae represented the second most abundant family (17%) (Table S3). A similar proportion (17%) was also found for Prymnesiaceae followed by Phaeocystaceae (11.5%). A high proportion (15%) could not be assigned further than Prymnesiophyceae.
We found three 28S OTUs with more than 1,500 reads each, constituting 18% of the total. These clustered within the reference sequences of Syracosphaera pulchra (OTU_L001 and OTU_L002) and Phaeocystis sp. (OTU_L003) (Tables 2, S2b and Fig. S2). The 12 next most abundant OTUs contained between ~ 500 and 1,000 sequences. Thirty percentage of the 280 rarest OTUs (< 10 reads) belonged to the genus Chrysochromulina (Table S2b).
Table 2.
List of the 30 most abundant haptophyte D1–D2 28S rRNA OTUs detected
| OTU ID | Total reads (N) | Total reads (%) | Total reads after subsampling (N) | Total reads after subsampling (%) | Depth | Size fraction | Group | Lowest taxonomic level possible to determine |
|---|---|---|---|---|---|---|---|---|
| OTU_L001 | 1,980 | 6.66 | 740 | 5.87 | Both | Both | Syracosphaeraceae | Syracosphaera pulchra |
| OTU_L002 | 1,830 | 6.15 | 685 | 5.43 | Both | Both | Syracosphaeraceae | Syracosphaera pulchra |
| OTU_L003 | 1,555 | 5.23 | 679 | 5.38 | Both | Both | Phaeocystaceae | Phaeocystis |
| OTU_L004 | 957 | 3.22 | 494 | 3.92 | Both | Both | Chrysochromulinaceae | Chrysochromulina acantha |
| OTU_L005 | 862 | 2.90 | 365 | 2.89 | Both | Both | Prymnesiaceae | Haptolina ericina/hirta/fragaria |
| OTU_L006 | 782 | 2.63 | 339 | 2.69 | Both | Both | Prymnesiaceae | Haptolina |
| OTU_L007 | 710 | 2.39 | 318 | 2.52 | Both | Both | Chrysochromulinaceae | Chrysochromulina |
| OTU_L008 | 676 | 2.27 | 299 | 2.37 | Both | Both | Chrysochromulinaceae | Chrysochromulina throndsenii/C. campanulifera |
| OTU_L009 | 659 | 2.22 | 319 | 2.53 | Both | Both | Prymnesiophyceae | Clade PRY‐LSU3 (Clade‐E–F?) |
| OTU_L010 | 648 | 2.18 | 269 | 2.13 | Both | Both | Phaeocystaceae | Phaeocystis |
| OTU_L011 | 645 | 2.17 | 224 | 1.78 | Both | Both | Chrysochromulinaceae | Chrysochromulina camella |
| OTU_L012 | 597 | 2.01 | 232 | 1.84 | Both | Both | Prymnesiophyceae | Clade PRY‐LSU2 (Clade‐D?) |
| OTU_L013 | 576 | 1.94 | 257 | 2.04 | Both | Both | Prymnesiaceae | Prymnesium polylepis |
| OTU_L014 | 560 | 1.88 | 257 | 2.04 | Both | Both | Chrysochromulinaceae | Chrysochromulina |
| OTU_L015 | 538 | 1.81 | 234 | 1.85 | Both | Both | Prymnesiaceae | Prymnesium kappa |
| OTU_L016 | 516 | 1.73 | 234 | 1.85 | Both | Both | Chrysochromulinaceae | Chrysochromulina camella |
| OTU_L017 | 491 | 1.65 | 198 | 1.57 | Both | Both | Prymnesiaceae | Prymnesium |
| OTU_L018 | 468 | 1.57 | 160 | 1.27 | Both | Both | Syracosphaeraceae | Syracosphaera pulchra |
| OTU_L019 | 467 | 1.57 | 188 | 1.49 | Only DCM | Both | Phaeocystaceae | Phaeocystis |
| OTU_L020 | 426 | 1.43 | 180 | 1.43 | Both | Both | Prymnesiophyceae | Clade PRY‐LSU2 (Clade‐D?) |
| OTU_L021 | 412 | 1.38 | 186 | 1.47 | Both | Both | Prymnesiaceae | Haptolina |
| OTU_L022 | 406 | 1.36 | 174 | 1.38 | Both | Both | Prymnesiaceae | Dicrateria rotunda |
| OTU_L023 | 367 | 1.23 | 137 | 1.09 | Both | Both | Syracosphaeraceae | Syracosphaera pulchra |
| OTU_L024 | 355 | 1.19 | 178 | 1.41 | Both | Both | Coccolithales | Coccolithaceae |
| OTU_L025 | 352 | 1.18 | 155 | 1.23 | Both | Both | Chrysochromulinaceae | Chrysochromulina simplex |
| OTU_L026 | 351 | 1.18 | 130 | 1.03 | Both | Both | Chrysochromulinaceae | Chrysochromulina throndsenii |
| OTU_L027 | 339 | 1.14 | 140 | 1.11 | Both | Both | Rhabdosphaeraceae | Algirosphaera robusta |
| OTU_L028 | 327 | 1.10 | 157 | 1.24 | Both | Both | Chrysochromulinaceae | Chrysochromulina |
| OTU_L029 | 305 | 1.03 | 117 | 0.93 | Both | Both | Chrysochromulinaceae | Chrysochromulina simplex |
| OTU_L030 | 291 | 0.98 | 133 | 1.05 | Both | Both | Chrysochromulinaceae | Chrysochromulina simplex |
The taxonomic assignment was based on phylogenetic placement.
In samples from the Oslofjorden collected in August–September 2009 and 2010, Egge et al. (2015b) found that Prymnesiaceae and Chrysochromulinaceae were the most OTU‐rich groups, followed by Phaeocystaceae and Calcihaptophycidae (de Vargas et al. 2007). The novel lineages HAP3 and HAP4 were also represented with 3–4 OTUs in the 2009 and 2010 late summer samples. Chrysochromulinaceae, Prymnesiaceae, E. huxleyi, Syracosphaerace, and Phaeocystaceae were proportionally the most abundant groups in these samples. Thus, both in terms of taxonomic distribution of 18S rRNA OTUs and proportional abundance of the different groups, our results are consistent with Egge et al. (2015b).
Previous microscopy surveys have also reported Prymnesiaceae and Chrysochromulinaceae to be very species rich in the Skagerrak and Kattegat. Jensen (1998) recorded c. 30 morphological species of Chrysochromulina (sensu lato), and scales of 20 undescribed forms, which morphologically resembled this group. Members of Chrysochromulinaceae and Prymnesiaceae have been reported as the most abundant noncalcifying haptophyte groups in June–September (Dahl and Johannessen 1998; Kuylenstierna and Karlson 1994; Lekve et al. 2006).
Novel taxa or records for the area
We performed taxonomical assignation of OTUs by blast against the Haptophyta‐PiP database. Detection of a cultured species or an environmental sequence is here defined as ≥ 99% or ≥ 97% sequence identity to one of the 18S or 28S rRNA OTUs, respectively. Of the 215 18S OTUs, only 20 had ≥ 99% match to a cultured species, whereas 47 had ≥ 99% match to an environmental sequence present in the Haptophyta‐PiP database (Table S4). Thirty‐six (16%) did not nest within any specific haptophyte clade in the phylogenetic tree (Fig. 3). Of the 432 28S OTUs, 47 had ≥ 97% match to a cultured species, (Table S4a, b). Comparing our 18S OTUs to Egge et al. (2015a,b) we found that 68 of our OTUs were ≥ 99% identical to any OTU recovered in Egge et al. 2015a (Tables S2a, S4a). The majority of these (62 OTUs) were present with ≥ 10 reads. Out of these 68 OTUs, 26 were ≥ 99% identical to OTUs from Egge et al. 2015a and at the same time < 99% identical to any sequence in the Haptophyta‐PiP database. This suggests that these 26 OTUs have only been detected in the Oslofjorden. For instance, OTU_S072, whose closest match in the Haptophyta‐PiP database was Tergestiella adriatica (95.8%), was 99.7% identical to an OTU nesting within a clade of environmental sequences classified as Calcihaptophycidae in Egge et al. (2015a) (cf. OTU113, fig. 4 in Egge et al. 2015a). Thus, it may represent a coccolithophore species that has not yet been sequenced. Of the most OTU‐rich groups, the group with the highest proportion of OTUs matching environmental sequences was Phaeocystaceae and Chrysochromulinaceae (Table S4a). Within both of these groups, several morphological forms have been recognized that are not yet in culture and genetically characterized (Jensen 1998; Medlin and Zingone 2007). Our result, that several OTUs had best match with DNA sequences or reads from environmental samples, supports previous studies showing that there is a large diversity of haptophytes that remains to be cultured and DNA sequence determined (e.g. Bittner et al. 2013; Liu et al. 2009; de Vargas et al. 2015).
Sample comparisons
After subsampling to the lowest number of reads (9,148 and 1,577 for 18S and 28S rRNA genes), four and 42 OTUs were removed, respectively. In the Venn diagram (Fig. 4) unique and shared OTUs of the 18S and 28S samples are presented separately. Only ~ 13% of the OTUs were present in all samples (28 for 18S and 53 for 28S). A considerable number of OTUs were found in only one sample (60% for 18S and 52% for 28S rRNA gene).
Figure 4.
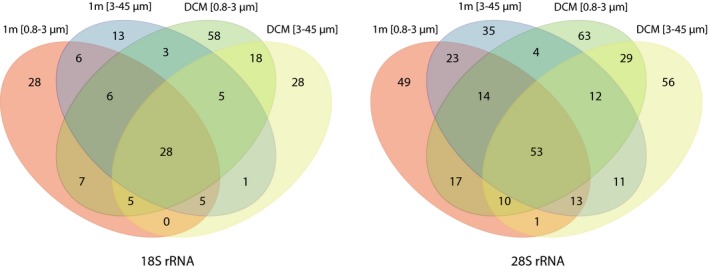
Four‐way Venn diagram illustrating the number of unique and shared haptophyte 18S and 28S OTUs for the four set of samples studied at OF2 station.
Reproducibility of PCR and 454 sequencing
High‐throughput sequencing studies have often been criticized for lack of replication, as experimental errors can arise during sample and library preparation or sequencing and filtering (Robasky et al. 2014). In our study, we tested technical replicates (TR), dividing each filter in two and performing extraction, PCR, and pyrosequencing separately. The TR were highly similar for all pairs of samples (R 2 > 0.9, p < 0.001) presenting similar proportional abundances (Fig. 5b, S1). The Bland–Altman plot (Fig. S3) performed for normalized OTU abundances clearly showed that larger disagreements between TR were found among the rare OTUs. With increasing abundances, the plots get consistently linear indicating high similarity. The TR had similar taxonomic composition and proportional abundance for the different major taxonomic groups (Fig. 5a, b).
Figure 5.
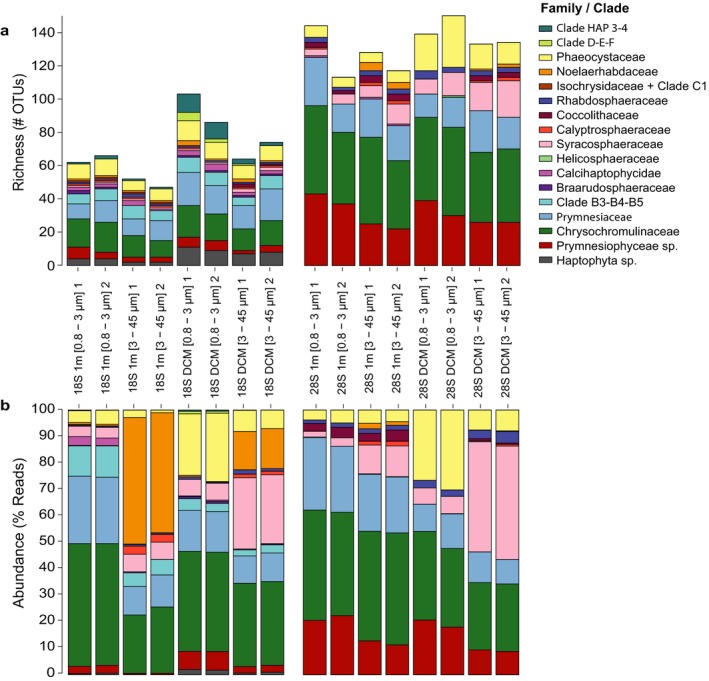
Haptophyte taxonomic distribution for all 18S rRNA (left) and 28S rRNA (right) samples and replicates 1 and 2. (a) OTU richness. (b) Proportional read abundances.
We suggest that TR do not bring considerable extra information to studies focusing on the most abundant groups. However, many of the rare OTUs appeared in only one TR (Fig. S1), indicating the importance of TR in recovering low abundant OTUs. This agrees with findings by Massana et al. (2015).
Comparing markers
There are no previous studies comparing 18S and 28S rRNA gene as markers in metabarcoding studies of haptophytes. Here, we wanted to examine if the 28S marker gives the same resolution or higher than 18S. Compared to 18S rRNA, we found a higher haptophyte diversity using 28S haptophyte‐specific primers (Fig. 6). However, it is not understood if this diversity represents intra‐ or interspecific variation. The number of OTUs detected was higher in the 28S than in the 18S samples (Table S1). In the 18S data set, members of Chrysochromulinaceae dominated in all samples followed by Prymnesiaceae, both in number of OTUs and read proportion. The next most abundant families in the 18S data set were Noelaerhabdaceae, Syracosphaeraceae, and Phaeocystaceae, whereas in the 28S data set members of Noelaerhabdaceae were almost missing (0.5%, Fig. 5b). This is likely due to mismatches between the 28S sequence of members of Isochrysidales and the LSU1 forward primer, one of which occurs at the 3′ end of the primer, which may prevent elongation. OTUs belonging to the families Isochrysidaceae and Braarudosphaeraceae were only detected in the 18S samples. In contrast, Helicosphaeraceae OTUs were only observed in the 28S data set. Respectively 4% and 15.5% of 18S and 28S OTUs could not be assigned to any known haptophyte family and was therefore assigned to either Prymnesiophyceae sp. or Haptophyta sp. The 18S marker provided a more accurate and extensive assessment of species identity than the 28S marker, due to the lower number of 28S reference sequences. In addition, for the 18S reference data set there are a number of defined clades (from class‐ to genus‐level) without cultured representatives. We asked whether we could place 28S OTUs without a cultured representative in 18S‐defined clades. We compared our 18S and 28S phylogenetic trees (Fig. 3) and identified three 28S clades (PRY‐LSU2, ‐LSU3, and –LSU4) that may represent 18S defined clades without culture representatives (Clade‐D, Clade‐E + F, Clade B3–B5, respectively). Although differences in both number of OTUs and sequences abundances are found between the markers, both rRNA data sets revealed that the majority of haptophyte species can be assigned to a defined clade with either the 18S or the 28S rRNA gene.
Figure 6.
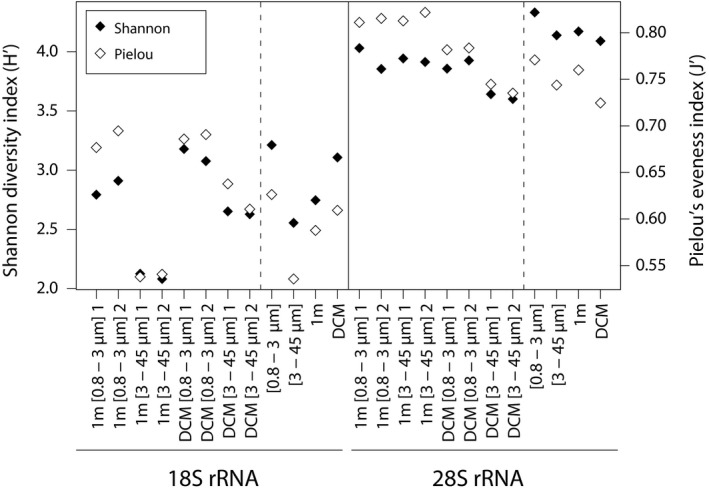
Shannon diversity index and Pielou's evenness index for all 18S and 28S samples studied at the OF2 station. The continued line splits the two different markers’ data sets, whereas the dotted lines split the single samples from the pooled ones in size and depth.
Comparing depths and size fractions
Haptophyta communities may have different species composition according to depth: in the Mediterranean Sea (Bittner et al. 2013), in the South Pacific (Shi et al. 2009) and in the Red Sea (Man‐Aharonovich et al. 2010). We aimed to examine if these differences also occur on the Norwegian coast. The highest number of OTUs in our data sets was found at the DCM in picoplankton (0.8–3 μm) samples for both markers, and pooled pico‐ and nanoplankton 1 m samples contained fewer OTUs than at DCM, corresponding with previous findings (Bittner et al. 2013).
Sixty‐three 18S OTUs were present at both depths (Table S2a), whereas 46 were only found at 1 m and 102 only in the DCM. OTUs that clustered with Isochrysis sp. and Tergestiella adriatica were only found at 1 m (Table S2a). Contrarily, those belonging to Clades Prymnesiales B3, D, E, F, and HAP4, and Coccolithus sp. were only present in the DCM sample within 18S rRNA marker (Table S2a). The highest number of OTUs was found in DCM samples for both markers. The proportion of OTUs of each taxonomic group was similar among all samples (Fig. 5a). There is, however, a clear difference in proportional abundances by depth (Fig. 5b). As described in previous studies (Malinverno et al. 2003), differences in communities by depth can be explained by temperature, phosphorus and light availability. On our sampling date, temperature and phosphate at 1 m and DCM were equal and thus light availability and differences in salinity (23.7 PSU for 1 m and 29.1 PSU for DCM) could explain the differences found in community structure. Comparing size fractions, we observed that the picoplankton had higher number of OTUs than the nanoplankton, corresponding with previous findings by Bittner et al. (2013). OTUs nested within Clades B3, E, and F were exclusive in the pico‐fraction and OTUs from Chrysochromulina rotalis, Tergestiella adriatica, and Coccolithus sp. were unique in the nano‐fraction.
The 28S data set contained 171 OTUs present at both depths (Table S2b). Of the remaining OTUs, 88 were only found at 1 m and 131 at the DCM. OTUs best matching Chrysotila stipitata and Umbilicosphaera sp. were uniquely found at 1 m, and Coccolithus braarudii, Coronosphaera mediterranea, whereas Helicosphaera sp. were unique for the DCM samples. As for the 18S data, the 28S contained larger number of OTUs in the picoplankton size fraction compared to the nanoplankton. The unique OTU for the picoplankton‐fraction was clustered with Helicosphaera sp. OTUs assigned to C. stipitata, C. braarudii, C. mediterranea, E. huxleyi, and Umbilicosphaera sp. were only found in the nanoplankton samples. Coccolithophores were found in both size fractions, although almost all known species are larger than 3 μm. An explanation can be that during filtration cells break and ribosomes pass through the filter pores. However, some haploid stages of coccolithophores (i.e. E. huxleyi) and some members of Syracosphaeraceae are known to be < 3 μm in size.
Diversity and evenness
Tables with proportional abundances per sample and OTU (not shown, see Fig. S1) were used to calculate the Shannon's diversity and Pilou's evenness indexes. Shannon index (18S t‐test: t = 0.29, df = 3, p‐value = 0.79, and 28S t‐test: t = 0.88, df = 3, p‐value = 0.44) and Pilou's index (18S t‐test: t = 0.05, df = 3, p‐value = 0.97, and 28S t‐test: t = −0.37, df = 3, p‐value = 0.74) were similar among the technical replicates (Fig. 6). These results suggest that the metabarcoding methodology used in this study is adequate for obtaining robust beta‐diversity and taxonomic descriptions. Similar findings were obtained by Massana et al. (2015). When looking at single samples in the 28S data sets, the nano‐DCM samples appear less diverse than the rest. However, for 18S the 1 m nanoplankton seems to harbour the lowest diversity. When pooling the samples from the same depth, we see the same pattern, that the pico‐fraction had both higher OTU richness and evenness than the nano‐fraction, resulting in higher diversity in the picoplankton. However, regarding depth, the 18S DCM sample was more diverse than the 1 m one, whereas the opposite was found for the 28S samples. In the 18S data set, we observed few Noelaerhabdaceae OTUs (E. huxleyi as the most abundant) representing a high proportion of the total reads (Fig. 5). The Noelaerhabdaceae OTUs occurred in very low read proportions in the 28S data, due to a mismatch in the primers (discussed above). This difference is probably the reason why we observe an opposite diversity pattern by depth for the two markers.
Comparison of SEM and metabarcoding for analyses of coccolithophore communities
Species diversity estimation
Taxonomic analysis of coccolithophore community at OF2 using SEM detected 26 distinct coccolithophore morphotypes (from eight depths). When corrected for life‐cycle phases and combination coccospheres (intermediate life‐cycle forms), the complete taxonomic list numbered 22 coccolithophore species, two of which could not be precisely identified (Fig. 7 and Table 3). Our observations expand the checklist of species detected in the Scandinavian waters by 6, and the number of morphotypes by 12 (Egge et al. 2015a; Eikrem 1999). The total number of species detected at the two depths used for the method comparison (subsurface (1 m) and DCM (8 m)) was 14, with an increase in species number observed from 1 m (8 species) to DCM (14 species). The metabarcoding at these two depths generated a total of 29 coccolithophore OTUs based on the 18S rRNA, and 89 OTUs based on 28S rRNA. The increase in coccolithophore OTU diversity by depth was also detected with metabarcoding. The number of 18S rRNA OTUs increased from 13 to 23 and the number of 28S rRNA OTUs from 51 to 69 from 1 m to DCM. In the SEM analysis, Syracosphaeraceae was the most species‐rich family at the two depths with eight species, followed by various holococcolithophore taxa (2 species) and Rhabdosphaeraceae (2 species). On the other hand, the 18S rRNA OTUs were mostly assigned to Noelaerhabdaceae (4), Rhabdosphaeraceae (2), and Syracosphaeraceae (3). The 28S rRNA OTUs showed highest richness of Syracosphaeraceae (38), Rhabdosphaeraceae (8) and Calyptrosphaeraceae (6) (Table 4).
Figure 7.
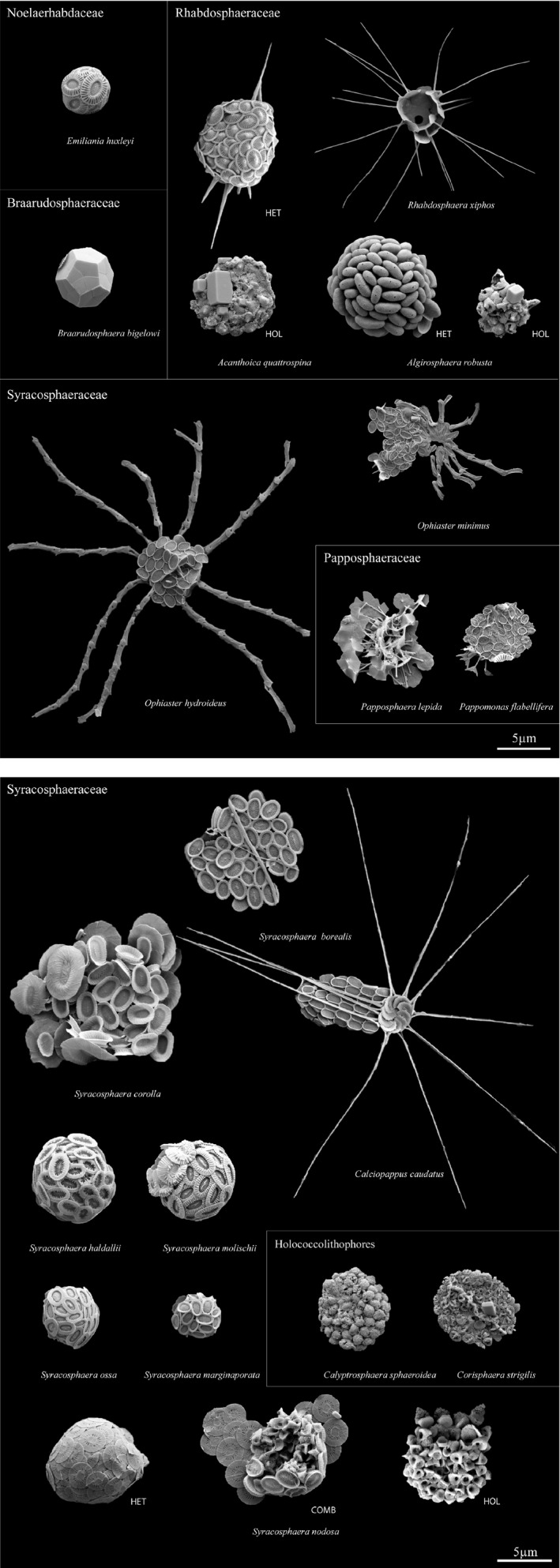
Scanning electron micrographs of coccolithophore morphotypes detected in this study. Abbreviations used for species detected in more than one life‐cycle phase: HET, heterococcolith phase; COMB, combination coccosphere with both heterococcoliths and holococcoliths representing a transition phase; HOL, holococcolith phase.
Table 3.
Species list of coccolithophores detected by SEM and their abundances (cells/l) at the sampled depths
| Species | Author | 1 m | 2 m | 4 m | 8 m | 12 m | 16 m | 20 m | 40 m |
|---|---|---|---|---|---|---|---|---|---|
| Noelaerhabdaceae | |||||||||
| Emiliania huxleyi TYPE A | Young & Westbroek, 1991 | 214,077 | 237,131 | 253,518 | 170,860 | 140,227 | 99,769 | 80,972 | 16,387 |
| Rhabdosphaeraceae | |||||||||
| Acanthoica quattrospina | Lohmann, 1903 | 602 | 2,410 | 964 | 2,410 | 2,009 | 1,928 | 964 | 482 |
| Acanthoica quattrospina HOLa | Cros et al. 2000 | 241 | 482 | ||||||
| Algirosphaera robusta | (Lohmann 1902) Norris, 1984 | 402 | |||||||
| Algirosphaera robusta HOLa | (Schiller 1913) Deflandre, 1952 | 482 | |||||||
| Rhabdosphaera xiphos a | (Deflandre & Fert 1954) Norris, 1984 | 241 | 1,205 | 964 | 482 | 1,928 | |||
| Syracosphaeraceae | |||||||||
| Calciopappus caudatus | Gaarder & Ramsfjell, 1954 | 482 | |||||||
| Ophiaster hydroideus | (Lohmann 1903) Lohmann, 1913 | 6,266 | 2,009 | 9,157 | 5,784 | 5,302 | |||
| Ophiaster minimus a | Manton & Oates, 1983 | 1,446 | 402 | 964 | 482 | 964 | |||
| Syracosphaera anthos | (Lohmann 1912) Janin, 1987 | ||||||||
| Syracosphaera borealis | Okada & McIntyre, 1977 | 1,607 | 482 | 964 | 241 | 804 | 964 | ||
| Syracosphaera corolla a | Lecal, 1966 | 482 | 402 | 482 | 1,446 | ||||
| Syracosphaera halldalii a | Gaarder in Gaarder & Hasle 1971 ex Jordan, 1994 | 803 | 1,446 | 1,928 | 5,784 | 4,822 | 9,639 | 10,121 | 1,928 |
| Syracosphaera marginaporata | Knappertsbusch, 1993 | 7,230 | 7,712 | 13,977 | 46,992 | 23,706 | 15,423 | 18,797 | 7,712 |
| Syracosphaera molischii | Young, 2014 | 201 | 7,953 | 4,420 | 4,338 | 2,892 | 482 | ||
| Syracosphaera nodosa | Kamptner, 1941 | 1,446 | 1,607 | 964 | 1,928 | ||||
| Syracosphaera nodosa COMBa | This study | 402 | 482 | 482 | |||||
| Syracosphaera nodosa HOLa | This study | 1,687 | 1,607 | 4,820 | 3,374 | ||||
| Syracosphaera ossa a | (Lecal 1966) Loeblich & Tappan, 1968 | 602 | 964 | 1,928 | 1,687 | 804 | 482 | ||
| Papposphaeraceae | |||||||||
| Papposphaera lepida | Tangen, 1972 | 964 | |||||||
| Pappomonas flabellifera | Manton & Oates, 1975 | 482 | |||||||
| Calyptrosphaeraceae (holococcoliths) | |||||||||
| Calyptrosphaera sphaeroidea | Schiller 1913 | 964 | 2,410 | 4,822 | 1,446 | ||||
| Corisphaera strigilis a | Gaarder, 1962 | 482 | 723 | 804 | |||||
| Undetermined taxa | |||||||||
| Undetermined HOLa | 482 | ||||||||
| Undetermined HETa | 964 | ||||||||
| Braarudosphaeraceae | |||||||||
| Braarudosphaera bigelowii | (Gran & Braarud 1935) Deflandre, 1947 | 201 | 1,446 | 8,840 | 5,784 | 3,856 | |||
Taxa previously not reported from the Oslofjorden area.
Columns in grey mark the stations used for the method comparison.
Table 4.
Number of OTUs and morphospecies within coccolithophore families obtained using the two molecular markers and the SEM
| 18S rRNA (OTUs) | 28S rRNA (OTUs) | SEM (morphospecies) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 m | DCM | Total | 1 m | DCM | Total | 1 m | DCM | Total | Total (OF2) | |
| Noelaerhabdaceae | 1 | 4 | 4 | 6 | 2 | 6 | 1 | 1 | 1 | 1 |
| Calcidiscaceae | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Coccolithaceae | 0 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Helicosphaeraceae | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| Pontosphaeraceae | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Rhabdosphaeraceae | 2 | 1 | 2 | 5 | 8 | 8 | 1 | 2 | 2 | 3 |
| Syracosphaeraceae | 1 | 2 | 2 | 18 | 36 | 38 | 5 | 8 | 8 | 11 |
| Papposphaeraceae | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 |
| Calyptrosphaeraceae | 1 | 1 | 1 | 4 | 5 | 6 | 0 | 2 | 2 | 2 |
| Pleurochrysidaceae | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Braarudosphaeraceae | 2 | 1 | 2 | 0 | 0 | 0 | 1 | 1 | 1 | 1 |
| Othera | 6 | 13 | 17 | 15 | 17 | 25 | 0 | 0 | 0 | 2 |
| Sum: | 13 | 23 | 29 | 51 | 69 | 87 | 8 | 14 | 14 | 22 |
The number of species per family obtained using SEM data includes species from 8 analysed depths. Values in bold represent the total number of coccolithophore OTUs detected by HTS at 1 m depth and at DCM as well as the total number of morphospecies detected by SEM at the 8 analysed depths.
Category “Other” includes OTUs placed within the Watznaueriaceae and Isochrysidaceae families as well as OTUs placed within clades Calcihaptophycidae, Coccolithales, Zygodiscales, Clade‐E, Clade‐F and OTUs with an unclear tree placement. Taxa marked as “Other” in the SEM data represent two unidentified morphotypes with an unclear taxonomy.
The number of 18S rRNA OTUs obtained was similar to the number of observed species, but failed to account for the high level of richness within the Syracosphaeraceae and Rhabdosphaeraceae families. Also, the number of coccolithophore OTUs in our study corresponded to the number of OTUs assigned to the subclass Calcihaptophycidae (29) in the study by (Egge et al. 2015a) at the same location. On the other hand, the 28S rRNA marker seemed to overestimate the coccolithophore species richness, generating higher number of coccolithophore OTUs compared to the number of species identified in the SEM analysis. Similar results showing an overestimation of richness using the 28S rRNA gene as marker were obtained in the study of Young et al. (2014) using environmental sequencing of clone‐libraries. This could be due to faster evolutionary rates in the D1‐D2 region of the 28S than the V4 region of the 18S rRNA gene, giving a higher resolution for species‐level identification, but also overestimating diversity (Liu et al. 2009). More 28S rRNA reference sequences are needed to obtain a better link between OTU and species based on this gene.
Due to the low number of reference sequences obtained from cultures of Syracosphaerales and cf. Syracospaherales incertae sedis (Edvardsen et al. 2016), only few direct matches between SEM and metabarcoding data were obtained at the species level. The most abundant species in our samples, E. huxleyi, was represented by the type A morphotype in SEM counts. The most abundant OTU assigned to E. huxleyi (OTU_S002) by phylogeny was 99.7% similar to sequences of cultures of this species (e.g. EU106795). One base differed, as the OTU_S002 had 5. As instead of 6 in a homopolymere region. Using 28S rRNA generated six OTUs matching the cultured E. huxleyi reference. A good correspondence between SEM and metabarcoding was observed in Braarudosphaeara bigelowii, for which the OTU_S033 clustered with a clade including reference sequences from morphologically verified picked cells of B. bigelowii.
The Syracosphaeraceae family is represented with only two cultured species in the Haptophyta‐PiP database (S. pulchra and C. mediterranea), neither of which were observed in the SEM analysis. Only two 18S rRNA OTUs clustered within Syracosphaeraceae, and none was placed close to the two species (Fig. 3a). However, 38 28S rRNA OTUs were assigned to this family, exhibiting different degrees of similarity with the two cultured references. The OTU_L098 was placed close to C. mediterranea 28S culture reference indicating that C. mediterranea was present in our samples. The OTUs clustering with the S. pulchra reference sequence likely represent different Syracosphaera species or members of Syracosphaeraceae. Similarly, a number of OTUs were placed within Rhabdosphaeraceae, represented by one cultured species, Algirosphaera robusta in both 18S and 28S rRNA gene reference databases. Both markers provided OTUs highly similar to this species’ reference sequence, confirming the finding by SEM.
The Haptophyta‐PiP database contained only two reference sequences for holococcolithophore taxa (both markers for Calyptrosphaera sphaeroidea and 28S rRNA for Helladosphaera sp.) obtained from cultured material. We detected OTUs assigned to C. sphaeroidea with both markers (OTU_S017, 18S and OTU_L053, L233, L350, 28S) and also detected this species by SEM. However, no OTUs clustered with Helladosphaera sp. Finally, a number of OTUs detected in our study matched the sequences of coccolithophore species that were not observed in the SEM survey. Those included C. braarudi (OTU_L137), Coccolithus pelagicus/braarudi (OTU_S075, 18S, which is identical for the two species), Helicosphaera carteri/wallichii (OTU_L377, 28S) and Tergestiella adriatica (OTU_S078, 18S). In addition, molecular analysis confirmed the presence of the members of Calcidiscaceae (OTU_L224) and Pleurochrysidaceae (OTU_L222), none of which were observed in the SEM analysis. Finally, a number of OTUs (pooled in category “Other” in Table 4 were placed within the Calcihaptophycidae clade but could not be placed to a clade with cultured representatives. Most of these were placed in defined clades consisting of environmental sequences only, such as Clades E and F for 18S and Clade PRY‐ LSU3.
Overall, the taxonomic assignment using metabarcoding is strongly constrained for some taxa, such as coccolithophores other than Coccolithales and Isochrysidales, by the relatively low number of available reference sequences obtained from taxonomically verified material, as was shown in Young et al. (2014). This lack of reference sequences from morphologically characterized cells (from cultures or picked cells) resulted in overall good assignment of OTUs to the family level, but did not allow for detailed species‐level identification for most of the coccolithophore OTUs in this study. The issue was especially pronounced in highly diverse families such as Syracosphaeraceae and Rhabdosphaeraceae, where 28S rRNA marker yielded high numbers of OTUs, but only two reference sequences were available for species‐level taxonomic assignment. The power of metabarcoding combined with a curated reference sequence database was illustrated by our detection of a very rare species Tergestiella adriatica, that was considered to be extinct after K/Pg boundary (66 million years ago) until it was recently described in modern plankton (Hagino et al. 2015).
Abundance estimation
The quantitative analysis of coccolithophore community using SEM revealed a peak in abundance at 4 m (2.8 × 105 cells/l) decreasing gradually towards the 40 m depth (Fig. S4 and Table 3). The community was dominated by E. huxleyi, accounting for over 90% of the coccolithophore cell abundance in the top 4 m layer and decreasing in abundance and relative contribution (44–70%) in the deeper layers. Syracosphaeraceae showed an increase in contribution (up to 28%) in layers below 8 m depth, while other families, such as Rhabdosphaeraceae, Braarudosphaeraceae, and holococcolith taxa were present in lower numbers all along the vertical profile. It is important to note that the coccolithophore abundance was slightly lower at 1 m compared to the DCM.
The quantitative analysis using metabarcoding showed large variation in the proportional abundance of reads between the markers (Fig. 8). The 18S rRNA showed overall similar trends to SEM in describing proportional abundance of the coccolithophore families. The OTUs belonging to Noelaerhabdaceae accounted for 72% at the 1 m depth and decreased to 28% at the DCM. Syracosphaeraceae were the second most abundant group, increasing in proportional abundance from 16% at the 1 m depth to 61% at the DCM. On the other hand, the 28S rRNA highly underestimated the proportional abundance of Noelaerhabdaceae, likely due to mismatches with the LSU1 primer pair, and showed high relative contribution (34%) of taxonomically unidentified 28S rRNA OTUs. However, the trends in the proportional abundance of Syracosphaeraceae and Rhabdosphaeraceae 28S rRNA OTUs followed the same pattern as the SEM counts. Unlike the SEM counts, both 18S and 28S rRNA metabarcoding showed a significant contribution of Calyptrosphaeraceae, notably at 1 m depth.
Figure 8.
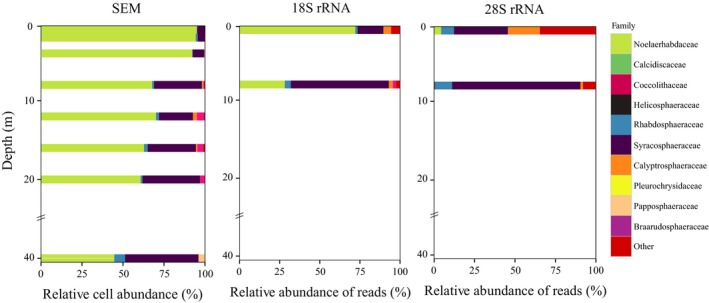
Proportional abundance of coccolithophore families inferred from SEM counts at eight depths and number of reads obtained using 18S and 28S rRNA markers at 1 and 8 m depths.
Using HTS for quantitative analysis of haptophyte communities is a challenging task, as the amount of RNA extracted from each species varies with size, growth rate and is likely group‐specific (Egge et al. 2013). Scanning electron microscopy has been a standard method for qualitative and quantitative analysis of coccolithophore communities for many years (Bollmann et al. 2002). This study was the first one using SEM quantitative analysis to test the usage of HTS for coccolithophore proportional abundance estimation. Environmental sequencing of clone‐libraries has previously showed weak correspondence between SEM counts and proportional abundance of 28S rRNA gene OTUs (Young et al. 2014), largely owing to poor representation of Noelaerhabdaceae sequences. We observed the same marked underrepresentation compared to SEM counts. In this study, the low representation of Noelaerhabdaceae is likely related to primer mismatch, and the marker could still be suitable for proportional abundance estimation if primers without any mismatches are used. The main advantages of the SEM technique; a high degree of taxonomic precision, well‐established, morphology‐based taxonomy of the group (Cros and Fortuño 2002; Young et al. 2003) and availability of absolute abundance data, were all confirmed in this study.
Methodological considerations
In this study we used RNA as template because we were interested in living cells of haptophytes that seem to be more represented in the total RNA than DNA (Massana et al. 2015). However, RNA requires reverse transcription into cDNA which may introduce additional chimeras (Egge et al. 2013) that need to be identified and removed. In monitoring surveys of total protist community DNA could therefore be the preferred template.
Here, we contribute with a curated 28S rRNA gene reference database based on cultures with updated taxonomy verified by phylogeny. With this gene some haptophyte taxa could not be taxonomically assigned to a major clade consisting of sequences from cultures or only environmental samples (e.g. Clades HAP‐3), due to a constrained reference database. We observed, however, a considerably higher richness using the 28S rRNA than 18S. Both markers showed, however, the same percentage of OTUs and reads for the most diverse and abundant families. We also tested the need for technical replicates in metabarcoding and found that it do not add considerable information in studies focusing on the most abundant groups, but is important in recovering low abundant OTUs.
Some of the possible drawbacks of the SEM method were also observed. Most importantly, the method is time consuming and requires taxonomic expertise, meaning that it is not suitable for routine monitoring of coccolithophore communities or surveys with a high number of samples. And the smaller sample volume that is practical to examine result in that some species are overlooked.
We suggest that 28S can be a useful complement to 18S in haptophyte metabarcoding studies. While the 28S rRNA gene seems to better distinguish between closely related coccolithophore species, the 18S rRNA gene has more reference sequences of defined clades and better assign to major clades. The observed mismatch to Isochrysidales with the LSU1 primer pair could account for the difference in relative abundance estimates between markers. Therefore, future studies should use modified 28S primers without this mismatch. To improve the resolution and specificity of metabarcoding protist communities we recommend that more reference sequences of both the 28S and 18S rRNA genes are produced from cultures and isolated single cells. We also suggest the construction of a curated concatenated 18S and 28S rRNA reference database, that also will confirm the link between 18S and 28S rRNA clades without cultured representatives.
Conclusions
A tenth of the OTUs with both markers matched a cultured species. More than half of the 18S OTUs had not previously been recorded in the area, showing that the majority of the OTUs had a best match with an environmental sequence and were recorded for the first time for the Outer Oslofjorden and Skagerrak. Six coccolithophore species were recorded for the first time in the area by SEM. The species composition differed significantly at the two depths and the diversity revealed by 18S rRNA was significantly higher at DCM than at subsurface. This shows that there is a need to sample more than one depth to reveal the full diversity. Further, the picoplankton size fraction contained more OTUs than the nanoplankton, even if only a few of the described haptophyte species are 3 μm or smaller (Edvardsen et al. 2016). We conclude from this that there is a large haptophyte diversity that remains to be described both morphologically and genetically, especially in the picoplankton, even in a relatively well‐studied area for phytoplankton diversity, as the Skagerrak.
Supporting information
Figure S1. (a) Heat map showing proportional abundances of all 18S rRNA gene OTUs for the different samples and replicates. Proportional read abundance was scaled by colour (white indicates that no reads were recorded). (b) Heat map showing proportional abundances of all 28S rRNA gene OTUs for the different samples and replicates. Proportional read abundance was scaled by colour (white indicates that no reads were recorded).
Figure S2. Rank‐abundance curves for 18S (a) and 28S rRNA gene (b).
Figure S3. Mean‐difference (Bland–Altman) plot showing level of agreement between technical replicates for OTU proportional abundances in the 18S and 28S rRNA gene data sets.
Figure S4. Vertical distribution of coccolithophore families observed at the OF2 station.
Table S1. Total number of reads at the beginning and end of the bioinformatics analysis and changes in the number of unique sequences (OTUs, operational taxonomic units) along the analysis process.
Table S2. (a) Haptophyte V4 18S rRNA OTUs recorded in the Skagerrak in August 2013. Red OTUs were removed after subsampling. Taxonomic assignations are based on phylogenetic placement. (b) Haptophyte D1‐D2 28S rRNA OTUs recorded in the Skagerrak in August 2013. Red OTUs were removed after subsampling. Taxonomic assignations are based on phylogenetic placement.
Table S3. Total and proportional read abundances and OTUs within each major clade for 18S and 28S rRNA genes.
Table S4. (a) Overview over matching of 18S OTUs to other databases. Total: Total number of OTUs in each group. ≥ 99% any sequence: Number of OTUs that have ≥ 99% BLAST match with either any sequence in the Haptophyta‐PiP database, or an OTU from Oslofjorden from Egge et al. 2015a. ≥ 99% Hapto‐PiP_ENV: Number of OTUs that have ≥ 99% BLAST match with an “environmental sequence” in the Haptophyta‐PiP database. ≥ 99% Hapto‐PiP_CULT: Number of OTUs that have ≥ 99% BLAST match with a sequence from a cultured species in the Haptophyta‐PiP database. ≥ 99% OF OTUs: Number of OTUs that have ≥ 99% BLAST match with an OTU previously obtained by HTS of samples from Oslofjorden (these may represent either cultured species, environmental sequences obtained by Sanger sequencing, or novel sequences from the Egge et al. 2015a study). ≥ 99% OF OTU & < 99% with any Hapto‐PiP sequence: Number of OTUs that have ≥ 99% match to an OTU from Egge et al. (2015a,b), but at the same time is < 99% similar to any sequence present in Hapto‐PiP. The numbers from ≥ 99% Hapto‐PiP_ENV and ≥ 99% Hapto‐PiP_CULT may not add up, because environmental sequences in the Haptophyta‐PiP database may also come from species that exist in culture. (b) Overview over matching of 28S OTUs to the 28S haptophyta reference database, consisting of sequences from cultured strains.
Data S1. Description of how the haptophyte 28S reference database was created.
Acknowledgments
This research was supported by the Research Council of Norway (FRIMEDBIO project 197823 to Jorijntje Henderiks, HAPTODIV project 190307 to BE and EE, and MICROPOLAR project 225956 to BE and EE), and the University of Oslo strategic funding (to SGS and BE). We thank captain Sindre Holm and crew on board R/V Trygve Braarud for assistance during field work, Ramiro Logares for advice on bioinformatics protocols and Tom Andersen for statistical suggestions. AmpliconNoise was run within QIIME on the Biocluster at the Institut de Ciències del Mar (CSIC) in Barcelona, and Mothur on the Abel cluster at the University of Oslo.
Literature Cited
- Andersen, R. A. , Bidigare, R. R. , Keller, M. D. & Latasa, M. 1996. A comparison of HPLC pigment signatures and electron microscopic observations for oligotrophic waters of the North Atlantic and Pacific Oceans. Deep Sea Res. II Top. Stud. Oceanogr., 43:517–537 [Google Scholar]
- Bittner, L. , Gobet, A. , Audic, S. , Romac, S. , Egge, E. S. , Santini, S. , Ogata, H. , Probert, I. , Edvardsen, B. & de Vargas, C. 2013. Diversity patterns of uncultured Haptophytes unravelled by pyrosequencing in Naples Bay. Mol. Ecol., 22:87–101. [DOI] [PubMed] [Google Scholar]
- Bollmann, J. , Cortés, M. Y. , Haidar, A. T. , Brabec, B. , Close, A. , Hofmann, R. , Palma, S. , Tupas, L. & Thierstein, H. R. 2002. Techniques for quantitative analyses of calcareous marine phytoplankton. Mar. Micropaleontol., 44:163–185. [Google Scholar]
- Cros, L. & Fortuño, J. M. 2002. Atlas of Northwestern Mediterranean coccolithophores. Sci. Mar., 66:7–182. [Google Scholar]
- Dahl, E. & Johannessen, T. 1998. Temporal and spatial variability of phytoplankton and chlorophyll a: lessons from the south coast of Norway and the Skagerrak. ICES J. Mar. Sci., 55:680–687. [Google Scholar]
- Edvardsen, B. , Egge, E. S. & Vaulot, D. 2016. Diversity and distribution of haptophytes revealed by environmental sequencing and metabarcoding – a review. Perspect. Phycol., 3:77–91. [Google Scholar]
- Edvardsen, B. , Eikrem, W. , Throndsen, J. , Sáez, A. G. , Probert, I. & Medlin, L. K. 2011. Ribosomal DNA phylogenies and a morphological revision provide the basis for a revised taxonomy of the Prymnesiales (Haptophyta). Eur. J. Phycol., 46:202–228. [Google Scholar]
- Edvardsen, B. & Paasche, E. 1998. Bloom dynamics and physiology of Prymnesium and Chrysochromulina . NATO ASI Ser. G Ecol. Sci., 41:193–208. [Google Scholar]
- Egge, E. , Bittner, L. , Andersen, T. , Audic, S. , de Vargas, C. & Edvardsen, B. 2013. 454 pyrosequencing to describe microbial eukaryotic community composition, diversity and relative abundance: a test for marine haptophytes. PLoS One, 8:e74371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egge, E. S. , Eikrem, W. & Edvardsen, B. 2015a. Deep‐branching novel lineages and high diversity of haptophytes in the Skagerrak (Norway) uncovered by 454 pyrosequencing. J. Eukaryot. Microbiol., 62:121–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egge, E. S. , Johannessen, T. V. , Andersen, T. , Eikrem, W. , Bittner, L. , Larsen, A. , Sandaa, R.‐A. & Edvardsen, B. 2015b. Seasonal diversity and dynamics of haptophytes in the Skagerrak, Norway, explored by high‐throughput sequencing. Mol. Ecol., 24:3026–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikrem, W. 1996. Chrysochromulina throndsenii sp. nov. (Prymnesiophyceae). Description of a new haptophyte flagellate from Norwegian waters. Phycologia, 35:377–380. [Google Scholar]
- Eikrem, W. 1999. The class Prymnesiophyceae (Haptophyta) in Scandinavian waters. Dissertation. University of Oslo, Oslo, Norway: 214 p. [Google Scholar]
- Eikrem, W. & Edvardsen, B. 1999. Chrysochromulina fragaria sp. nov. (Prymnesiophyceae), a new haptophyte flagellate from Norwegian waters. Phycologia, 38:149–155. [Google Scholar]
- Gilles, A. , Meglécz, E. , Pech, N. , Ferreira, S. , Malausa, T. & Martin, J.‐F. 2011. Accuracy and quality assessment of 454 GS‐FLX Titanium pyrosequencing. BMC Genom., 12:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granéli, E. , Edvardsen, B. , Roelke, D. L. & Hagström, J. A. 2012. The ecophysiology and bloom dynamics of Prymnesium spp. Harmful Algae, 14:260–270. [Google Scholar]
- Guillou, L. , Bachar, D. , Audic, S. , Bass, D. , Berney, C. , Bittner, L. , Boutte, C. , Burgaud, G. , de Vargas, C. , Decelle, J. , del Campo, J. , Dolan, J. R. , Dunthorn, M. , Edvardsen, B. , Holzmann, M. , Kooistra, W. H. C. F. , Lara, E. , Le Bescot, N. , Logares, R. , Mahé, F. , Massana, R. , Montresor, M. , Morard, R. , Not, F. , Pawlowski, J. , Probert, I. , Sauvadet, A.‐L. , Siano, R. , Stoeck, T. , Vaulot, D. , Zimmermann, P. & Christen, R. 2013. The Protist Ribosomal Reference database (PR2): a catalog of unicellular eukaryote small sub‐unit rRNA sequences with curated taxonomy. Nucleic Acids Res., 41:D597–D604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, B. J. , Gevers, D. , Earl, A. M. , Feldgarden, M. , Ward, D. V. , Giannoukos, G. , Ciulla, D. , Tabbaa, D. , Highlander, S. K. , Sodergren, E. , Methé, B. , DeSantis, T. Z. , The Human Microbiome Consortium , Petrosino, J. F. , Knight, R. & Birren, B. W. 2011. Chimeric 16S rRNA sequence formation and detection in Sanger and 454‐pyrosequenced PCR amplicons. Genome Res., 21:494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagino, K. , Young, J. R. , Bown, P. R. , Godrijan, J. , Kulhanek, D. K. , Kogame, K. & Horiguchi, T. 2015. Re‐discovery of a “living fossil” coccolithophore from the coastal waters of Japan and Croatia. Mar. Micropaleontol., 116:28–37. [Google Scholar]
- Holligan, P. M. , Fernández, E. , Aiken, J. , Balch, W. M. , Boyd, P. , Burkill, P. H. , Finch, M. , Groom, S. B. , Malin, G. , Muller, K. , Purdie, D. A. , Robinson, C. , Trees, C. C. , Turner, S. M. & van der Wal, P. 1993. A biogeochemical study of the coccolithophore, Emiliania huxleyi, in the North Atlantic. Global Biogeochem. Cycles, 7:879–900. [Google Scholar]
- Huse, S. M. , Welch, D. M. , Morrison, H. G. & Sogin, M. L. 2010. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol., 12:1889–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias‐Rodríguez, M. D. , Brown, C. W. , Doney, S. C. , Kleypas, J. , Kolber, D. , Kolber, Z. , Hayes, P. K. & Falkowski, P. G. 2002. Representing key phytoplankton functional groups in ocean carbon cycle models: Coccolithophorids. Global Biogeochem. Cycles, 16:1100. [Google Scholar]
- Jensen, M. Ø. 1998. The genus Chrysochromulina (Prymnesiophyceae) in Scandinavian coastal waters – diversity, abundance and ecology. Dissertation. University of Copenhagen, Copenhagen, Denmark: 200 p. [Google Scholar]
- Jordan, R. W. & Chamberlain, A. H. L. 1997. Biodiversity among haptophyte algae. Biodivers. Conserv., 6:131–152. [Google Scholar]
- Jordan, R. W. , Cros, L. & Young, J. R. 2004. A revised classification scheme for living haptophytes. Micropaleontology, 50:55–79. [Google Scholar]
- Kuylenstierna, M. & Karlson, B. 1994. Seasonality and composition of Pico‐ and Nanoplanktonic cyanobacteria and protists in the Skagerrak. Bot. Mar., 37:17–33. [Google Scholar]
- Langer, G. , Geisen, M. , Baumann, K.‐H. , Kläs, J. , Riebesell, U. , Thoms, S. & Young, J. R. 2006. Species‐specific responses of calcifying algae to changing seawater carbonate chemistry. Geochem. Geophys. Geosyst., 7:Q09006. [Google Scholar]
- Lekve, K. , Bagøien, E. , Dahl, E. , Edvardsen, B. , Skogen, M. D. & Stenseth, N. C. 2006. Environmental forcing as a main determinant of bloom dynamics of the Chrysochromulina algae. Proc. Biol. Sci., 273:3047–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H. , Probert, I. , Uitz, J. , Claustre, H. , Aris‐Brosou, S. , Frada, M. , Not, F. & de Vargas, C. 2009. Extreme diversity in noncalcifying haptophytes explains a major pigment paradox in open oceans. Proc. Natl Acad. Sci., 106:12803–12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinverno, E. , Ziveri, P. & Corselli, C. 2003. Coccolithophorid distribution in the Ionian Sea and its relationship to eastern Mediterranean circulation during late fall to early winter 1997. J. Geophys. Res., 108:8115. [Google Scholar]
- Man‐Aharonovich, D. , Philosof, A. , Kirkup, B. C. , Le Gall, F. , Yogev, T. , Berman‐Frank, I. , Polz, M. F. , Vaulot, D. & Béjà, O. 2010. Diversity of active marine picoeukaryotes in the Eastern Mediterranean Sea unveiled using photosystem‐II psbA transcripts. ISME J., 4:1044–1052. [DOI] [PubMed] [Google Scholar]
- Massana, R. , Gobet, A. , Audic, S. , Bass, D. , Bittner, L. , Boutte, C. , Chambouvet, A. , Christen, R. , Claverie, J.‐M. , Decelle, J. , Dolan, J. R. , Dunthorn, M. , Edvardsen, B. , Forn, I. , Forster, D. , Guillou, L. , Jaillon, O. , Kooistra, W. H. , Logares, R. , Mahe, F. , Not, F. , Ogata, H. , Pawlowski, J. , Pernice, M. C. , Probert, I. , Romac, S. , Richards, T. , Santini, S. , Shalchian‐Tabrizi, K. , Siano, R. , Simon, N. , Stoeck, T. , Vaulot, D. , Zingone, A. & de Vargas, C. 2015. Marine protist diversity in European coastal waters and sediments as revealed by high‐throughput sequencing. Environ. Microbiol., 17:4035–4049. [DOI] [PubMed] [Google Scholar]
- Medlin, L. & Zingone, A. 2007. A taxonomic review of the genus Phaeocystis . Biogeochemistry, 83:3–18. [Google Scholar]
- Moestrup, Ø. 1994. Economic aspects: ‘blooms’, nuisance species, and toxins In: Green J. E. & Leadbeater B. S. C. (eds), The Haptophyte Algae. Clarendon Press, Oxford: 51:265–285. [Google Scholar]
- Moon‐van der Staay, S. Y. , de Wachter, R. & Vaulot, D. 2001. Oceanic 18S rDNA sequences from picoplankton reveal unsuspected eukaryotic diversity. Nature, 409:607–610. [DOI] [PubMed] [Google Scholar]
- Moon‐van der Staay, S. Y. , van der Staay, G. W. M. , Guillou, L. , Vaulot, D. , Claustre, H. H. & Medlin, L. K. 2000. Abundance and diversity of prymnesiophytes in the picoplankton community from the equatorial Pacific Ocean inferred from 18S rDNA sequences. Limnol. Oceanogr., 45:98–109. [Google Scholar]
- Morgulis, A. , Coulouris, G. , Raytselis, Y. , Madden, T. L. , Agarwala, R. & Schäffer, A. A. 2008. Database indexing for production MegaBLAST searches. Bioinformatics, 24:1757–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Not, F. , del Campo, J. , Balagué, V. , de Vargas, C. & Massana, R. 2009. New insights into the diversity of marine picoeukaryotes. PLoS One, 4(9):e7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielou, E. C. 1966. The measurement of diversity in different types of biological collections. J. Theor. Biol., 13:131–144. [Google Scholar]
- Quince, C. , Lanzén, A. , Curtis, T. P. , Davenport, R. J. , Hall, N. , Head, I. M. , Read, L. F. & Sloan, W. T. 2009. Accurate determination of microbial diversity from 454 pyrosequencing data. Nat. Methods, 6:639–641. [DOI] [PubMed] [Google Scholar]
- Ridgwell, A. , Schmidt, D. N. , Turley, C. , Brownlee, C. , Maldonado, M. T. , Tortell, P. & Young, J. R. 2009. From laboratory manipulations to Earth system models: scaling calcification impacts of ocean acidification. Biogeosciences, 6:2611–2623. [Google Scholar]
- Robasky, K. , Lewis, N. E. & Church, G. M. 2014. The role of replicates for error mitigation in next‐generation sequencing. Nat. Rev. Genet., 15:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson, J. E. , Robinson, C. , Turner, D. R. , Holligan, P. , Watson, A. J. , Boyd, P. , Fernandez, E. & Finch, M. 1994. The impact of a coccolithophore bloom on oceanic carbon uptake in the northeast Atlantic during summer 1991. Deep Sea Res. I: Oceanogr. Res. Pap., 41:297–314. [Google Scholar]
- Schloss, P. D. , Westcott, S. L. , Ryabin, T. , Hall, J. R. , Hartmann, M. , Hollister, E. B. , Lesniewski, R. A. , Oakley, B. B. , Parks, D. H. , Robinson, C. J. , Sahl, J. W. , Stres, B. , Thallinger, G. G. , Van Horn, D. J. & Weber, C. F. 2009. Introducing mothur: open‐source, platform‐independent, community‐supported software for describing and comparing microbial communities. Appl. Environ. Microbiol., 75:7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, X. L. , Marie, D. , Jardillier, L. , Scanlan, D. J. & Vaulot, D. 2009. Groups without cultured representatives dominate eukaryotic picophytoplankton in the oligotrophic South East Pacific Ocean. PLoS One, 4:e7657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speksnijder, A. G. C. L. , Kowalchuk, G. A. , De Jong, S. , Kline, E. , Stephen, J. R. & Laanbroek, H. J. 2001. Microvariation artifacts introduced by PCR and cloning of closely related 16S rRNA gene sequences. Appl. Environ. Microbiol., 67:469–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis, A. 2006. RAxML‐VI‐HPC: maximum likelihood‐based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics, 22:2688–2690. [DOI] [PubMed] [Google Scholar]
- Stoeck, T. , Zuendorf, A. , Breiner, H. & Behnke, A. W. 2007. A molecular approach to identify active microbes in environmental eukaryote clone libraries. Microb. Ecol. 53:328–339. [DOI] [PubMed] [Google Scholar]
- Thomsen, H. A. , Buck, K. R. & Chavez, F. P. 1994. Haptophytes as components of marine phytoplankton In: Green J. E. & Leadbeater B. S. C. (eds), The Haptophyte Algae. Clarendon Press, Oxford: 51:187–208. [Google Scholar]
- de Vargas, C. , Aubry, M. P. , Probert, I. & Young, J. 2007. Origin and evolution of coccolithophores: from coastal hunters to oceanic farmers In: Falkowski P. G. & Knoll A. H. (eds.), Evolution of Primary Producers in the Sea. Elsevier, Boston, MA: p. 251–285. [Google Scholar]
- de Vargas, C. , Audic, S. , Henry, N. , Decelle, J. , Mahé, F. , Logares, R. , Lara, E. , Berney, C. , Le Bescot, N. , Probert, I. , Carmichael, M. , Poulain, J. , Romac, S. , Colin, S. , Aury, J.‐M. , Bittner, L. , Chaffron, S. , Dunthorn, M. , Engelen, S. , Flegontova, O. , Guidi, L. , Horák, A. , Jaillon, O. , Lima‐Mendez, G. , Lukeš, J. , Malviya, S. , Morard, R. , Mulot, M. , Scalco, E. , Siano, R. , Vincent, F. , Zingone, A. , Dimier, C. , Picheral, M. , Searson, S. , Kandels‐Lewis, S. , Acinas, S. G. , Bork, P. , Bowler, C. , Gorsky, G. , Grimsley, N. , Hingamp, P. , Iudicone, D. , Not, F. , Ogata, H. , Pesant, S. , Raes, J. , Sieracki, M. E. , Speich, S. , Stemmann, L. , Sunagawa, S. , Weissenbach, J. , Wincker, P. & Karsenti, E. 2015. Eukaryotic plankton diversity in the sunlit ocean. Science, 348:1261605‐1‐11. [DOI] [PubMed] [Google Scholar]
- Young, J. R. , Geisen, M. , Cros, L. , Kleijne, A. , Sprengel, C. , Probert, I. & Østergaard, J. 2003. A guide to extant coccolithophore taxonomy. J. Nannoplankton Res. Spec., 1:1–124. [Google Scholar]
- Young, J. R. , Liu, H. , Probert, I. , Aris‐Brosou, S. & de Vargas, C. 2014. Morphospecies versus phylospecies concepts for evaluating phytoplankton diversity: the case of the Coccolithophores. Cryptogamie Algol., 35:353–377. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (a) Heat map showing proportional abundances of all 18S rRNA gene OTUs for the different samples and replicates. Proportional read abundance was scaled by colour (white indicates that no reads were recorded). (b) Heat map showing proportional abundances of all 28S rRNA gene OTUs for the different samples and replicates. Proportional read abundance was scaled by colour (white indicates that no reads were recorded).
Figure S2. Rank‐abundance curves for 18S (a) and 28S rRNA gene (b).
Figure S3. Mean‐difference (Bland–Altman) plot showing level of agreement between technical replicates for OTU proportional abundances in the 18S and 28S rRNA gene data sets.
Figure S4. Vertical distribution of coccolithophore families observed at the OF2 station.
Table S1. Total number of reads at the beginning and end of the bioinformatics analysis and changes in the number of unique sequences (OTUs, operational taxonomic units) along the analysis process.
Table S2. (a) Haptophyte V4 18S rRNA OTUs recorded in the Skagerrak in August 2013. Red OTUs were removed after subsampling. Taxonomic assignations are based on phylogenetic placement. (b) Haptophyte D1‐D2 28S rRNA OTUs recorded in the Skagerrak in August 2013. Red OTUs were removed after subsampling. Taxonomic assignations are based on phylogenetic placement.
Table S3. Total and proportional read abundances and OTUs within each major clade for 18S and 28S rRNA genes.
Table S4. (a) Overview over matching of 18S OTUs to other databases. Total: Total number of OTUs in each group. ≥ 99% any sequence: Number of OTUs that have ≥ 99% BLAST match with either any sequence in the Haptophyta‐PiP database, or an OTU from Oslofjorden from Egge et al. 2015a. ≥ 99% Hapto‐PiP_ENV: Number of OTUs that have ≥ 99% BLAST match with an “environmental sequence” in the Haptophyta‐PiP database. ≥ 99% Hapto‐PiP_CULT: Number of OTUs that have ≥ 99% BLAST match with a sequence from a cultured species in the Haptophyta‐PiP database. ≥ 99% OF OTUs: Number of OTUs that have ≥ 99% BLAST match with an OTU previously obtained by HTS of samples from Oslofjorden (these may represent either cultured species, environmental sequences obtained by Sanger sequencing, or novel sequences from the Egge et al. 2015a study). ≥ 99% OF OTU & < 99% with any Hapto‐PiP sequence: Number of OTUs that have ≥ 99% match to an OTU from Egge et al. (2015a,b), but at the same time is < 99% similar to any sequence present in Hapto‐PiP. The numbers from ≥ 99% Hapto‐PiP_ENV and ≥ 99% Hapto‐PiP_CULT may not add up, because environmental sequences in the Haptophyta‐PiP database may also come from species that exist in culture. (b) Overview over matching of 28S OTUs to the 28S haptophyta reference database, consisting of sequences from cultured strains.
Data S1. Description of how the haptophyte 28S reference database was created.