

Abstract
Visible‐light photoredox catalysis has been utilized in a new multicomponent reaction forming β‐functionalized δ‐diketones under mild conditions in an operationally convenient manner. Single‐electron reduction of in situ generated carboxylic acid derivatives forms acyl radicals that react further via 1,2‐acylalkylation of olefins in an intermolecular, three‐components cascade reaction, giving valuable synthetic entities from readily available starting materials. A diverse set of substrates has been used, demonstrating robust methodology with broad substrate scope.
Keywords: acyl radicals, carboxylic acids, cascade transformation, multicomponent reactions, photoredox catalysis
In recent years, synthetic chemistry has evolved at such a pace that molecular complexity can be rapidly achieved with high selectivity. One of the most efficient methods to construct molecular complexity is by utilizing multicomponent reactions (MCRs) and cascade‐type transformations.1 MCRs are convergent reactions, in which three or more starting materials are reacted together to form a single product with high atom efficiency.1a Opposed to the classical sequential synthetic approach to construct complex molecules, MCRs enable the assembly of complex molecules in a single operation. The product is formed via a cascade of elementary chemical reactions through a series of reaction equilibrium, ideally ending in an irreversible step to give the desired product. MCRs have become a very popular tool in many areas of applied chemistry due to its labor efficient way to access a vast array of structural space.1e,1f
Recently, visible‐light‐mediated photoredox catalysis has emerged as a competitive method for functional‐group interconversions generating reactive radical intermediates under mild and environmentally benign conditions.2 Contributions to this field from our group include the formation of acyl radicals via single‐electron reduction of aromatic carboxylic acid derivatives (Figure 1 b).3 This approach combines the benefits of using readily available and inexpensive starting materials with a synthetic method that extends beyond the existing routes to access acyl radicals.4 Mixed anhydride intermediates, formed in situ from simple carboxylic acids by reaction with dimethyl dicarbonate (DMDC) have proven to be efficient oxidative quenchers of the excited state of the photocatalyst fac‐Ir(ppy)3 (tris[2‐phenylpyridinato‐C2,N]iridium(III)) to generate the desired acyl radical species, along with CO2 and methanolate as the only byproducts.3b Capitalizing on this mild way to access acyl radical intermediates, we envisioned that MCRs could be engaged in a radical context under redox‐neutral conditions.
Figure 1.
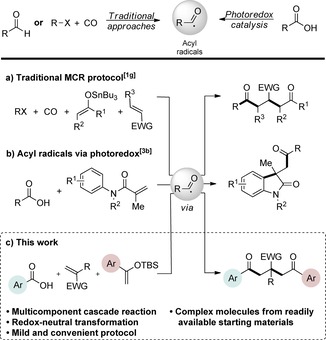
a) Tin enolate‐mediated carbonylative coupling reaction. b) Acylarylation of N‐aryl acrylamides by means of visible‐light photoredox catalysis. c) Photoredox‐catalyzed intermolecular three component synthesis of β‐functionalized δ‐diketones.
Electron‐rich acyl radicals have a pronounced reactivity preference towards electron‐deficient alkenes, a property that can be exploited in a multicomponent setting by achieving selectivity towards electron‐poor coupling partners in the presence of electron‐rich olefins. Strategically, such a combination of coupling partners would provide access to the synthetically challenging 1,2‐dicarbofunctionalization of alkenes (Figure 1 c).
The majority of published photoredox‐mediated MCRs rely on radical polar crossover transformations, in which free radical‐based bond formations are combined with nucleophilic trapping of an intermediate carbocation, typically by a solvent or co‐solvent.5 Photoredox‐mediated MCRs, in which two or more C−C bonds are formed in a pure radical fashion, remain relatively unexplored.6 Moreover, MCRs based on classical free‐radical chemistry are typically characterized by the need of external oxidants, UV irradiation, high CO pressure, high temperature or tin‐, borane, or lead‐based reagents, as well as stannylated and silylated phosphanes as precursors.7 Herein, we report the first redox‐neutral approach for the mild visible‐light‐mediated multicomponent 1,2‐acylalkylation of alkenes giving convenient access to β‐functionalized δ‐diketones (Figure 1 c).
Preliminary studies of the three‐component reaction were investigated by using benzoic acid 1 a, methyl acrylate 2 a, and tert‐butyldimethyl((1‐phenylvinyl)oxy)silane 3 a as the acyl radical precursor, electron‐deficient and electron‐rich coupling partners, respectively. In addition to the specified substrates, fac‐Ir(ppy)3, DMDC, and 2,6‐lutidine were added to the protocol, and the reaction was conducted under visible‐light irradiation (Table S1, the Supporting Information). The choice of the photocatalyst was based on previous studies, which demonstrated that fac‐Ir(ppy)3 is capable of reducing mixed anhydride I to the corresponding acyl radical species III as depicted in Figure 2.3b To our delight, initial experiments validated our approach and provided the corresponding product 4 a in a modest yield (Table S1, the Supporting Information). Subsequent optimizations revealed that employing 3 as the limiting reagent in combination with a catalyst loading of 2–5 mol % provided robust and optimal conditions for the transformation.
Figure 2.
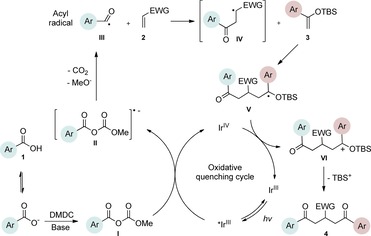
Proposed mechanism.
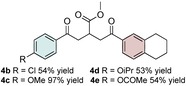
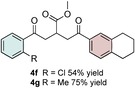
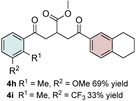
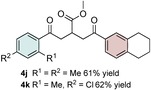
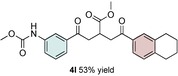
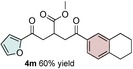
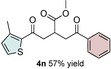
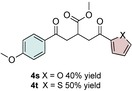
Control experiments performed in the absence of the photocatalyst, DMDC, or light irradiation did not result in the formation of the desired product (entries 14–16 in Table S1, the Supporting Information), observations that validated our mechanistic rational. With optimized reaction conditions in hand, the scope of the reaction in terms of structural diversity of all components was examined. As shown in Table 1, the reaction proceeded in modest to excellent yields (4 a–n, Table 1) for a wide range of mono‐ or disubstituted benzoic acids (1 a–l, Table S2, the Supporting Information), as well as for heteroaromatic substrates, such as 5‐methylthiophene‐2‐carboxylic acid and 2‐furoic acid (1 m–n, Table S2, the Supporting Information). In general, unfunctionalized and electron‐rich carboxylic acids provided higher yields compared to their electron‐deficient counterparts (e.g., 4 a and c). However, the inclusion of strongly electron‐withdrawing groups, such as trifluoromethyl or nitro moieties, at the para‐position of the carboxylic acid gave very low yields (≤10 %). For these substrates, the background reaction (i.e., the conversion of the acid into the corresponding unreactive methyl esters) was evidently faster than the redox chemistry (Table S2 and NMR spectra in the Supporting Information).8 Notably, carboxylic acids bearing free hydroxy or amino groups (1 e and l, Table S2, the Supporting Information) smoothly furnished the corresponding carbonate and carbamate derivatives without affecting the yields in a deleterious way (4 e and l, Table 1).
Table 1.
Scope of the carboxylic acids 1 and electron‐rich olefins 3.[a]
| |
|---|---|
|
|
|
|
|
|
|
|
|
|
[a] Reaction was performed by using 2–5 mol % of fac‐Ir(ppy)3, 2,6‐lutidine (0.5 equiv), DMDC (3 equiv), 1 (2 equiv), 2 a (2 equiv) and 3 (1 equiv) on a 0.2 mmol scale (detailed description is given in the Supporting Information). Isolated yields.
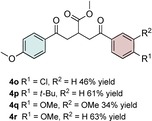
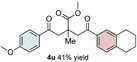
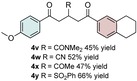
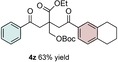
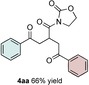
The scope of the reaction was further explored with regards to the olefin substrates 2 and 3 (Table 1 and 2). p‐Anisic acid (1 c) and methyl acrylate (2 a) together with various electron‐rich olefins (3 c–f, Table S3, the Supporting Information) gave the desired products in generally good yields (4 o–t, Table 1). The reaction appears to tolerate both mono‐ and disubstituted aryls, as well as heteroaromatic derivates of the silylenolether 3, although possible limitations include strongly electron‐deficient aryl groups (trifluromethyl (4 ad), Table S3, the Supporting Information) or non‐aromatic silylenolethers (tert‐butyl (4 ae), Table S3, the Supporting Information). In a similar manner, different electron‐poor olefins with a number of electron‐withdrawing groups (2 b–h, Table S4 in the Supporting Information) were evaluated as coupling partners together with benzoic acid (1 a) or p‐anisic acid (1 c) and silylenolethers 3 a or 3 b. Common acrylic derivatives were compatible with the optimized conditions providing the corresponding products in fair yields (4 u–aa, Table 2). Taken together, the protocol gives a new entry to 1,2‐acylalkylated alkenes in the form of β‐functionalized δ‐diketones. The protocol demonstrates a relatively broad scope with respect to the different substrates, with yields ranging from 58 to 99 % for each individual C−C bond‐forming reaction.
Table 2.
Scope of the electron‐poor olefins 2.[a]

| |
|---|---|
|
|
|
|
[a] Reaction was performed by using 2–5 mol % of fac‐Ir(ppy)3, 2,6‐lutidine (0.5 equiv), DMDC (3 equiv), 1 (2 equiv), 2 (2 equiv) and 3 (1 equiv) on a 0.2 mmol scale (detailed description is given in the Supporting Information). Isolated yields.
To demonstrate the robustness and thus synthetic value of this photoredox‐mediated multicomponent reaction, its scalability was evaluated. Satisfyingly, the reaction was successfully scaled‐up to 1 mmol scale by using p‐anisic acid (1 c), methyl acrylate (2 a), and tert‐butyldimethyl((1‐(5,6,7,8‐tetrahydronaphthalen‐2‐yl)vinyl)oxy)silane (3 b), providing 4 c in a very good yield (87 %). Furthermore, to illustrate the diverseness of δ‐diketones in synthetic elaborations, 4 c was converted to a trisubstituted pyridine (5a), and a trisubstituted cyclopentene (5b) derivative in 46 and 48 % yield, respectively, by following literature procedures (Scheme 1).[9] It is worth noting that these compounds were accordingly synthesized in two steps from commercially available and structurally simple starting materials. A plausible catalytic cycle for the presented transformation is proposed in Figure 2. Photoexcitation of fac‐IrIII(ppy)3 (depicted as IrIII) under visible light, generates the strong reductant fac‐*IrIII(ppy)3.
Scheme 1.
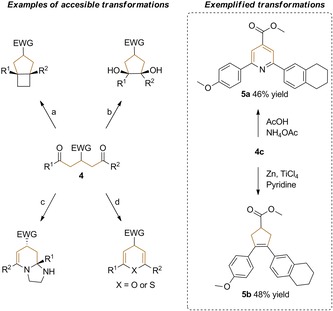
Examples of accessible transformations of 1,5‐diones: a) Mojr et al.10 b) Foster et al.11 c) Wang et al.12 d) Padmavathi et al.13 Transformations of 4 c into cyclized compounds 5 a and b following published literature procedures.9
Single‐electron reduction of mixed anhydride I (generated in situ from carboxylic acid 1 in the presence of DMDC under basic conditions) by fac‐*IrIII(ppy)3 gave fac‐IrIV(ppy)3 and radical anion II that, after fragmentation, gave acyl radical III along with CO2 and methanolate. Subsequently, acyl radical III underwent selective radical addition to the electron‐poor olefin 2 giving radical intermediate IV. Next, IV reacted selectively with the silylenolether 3, forming intermediate V, which in turn was oxidized by fac‐IrIV(ppy)3 to give intermediate VI, while regenerating the ground‐state of the catalyst. Finally, the elimination of TBS+ from VI formed the desired product in an irreversible step.
In conclusion, a new photoredox‐catalyzed multicomponent reaction for the formation of variously β‐functionalized δ‐diketones under mild visible‐light conditions was presented. The use of simple carboxylic acids as acyl radical precursors promoted by visible light allowed the development of this intermolecular cascade reaction, which combines three compounds in a radical process. The overall reaction proceeds in a redox neutral fashion avoiding the need for stoichiometric amounts of external electron donors or oxidants. Furthermore, the possibility of accessing a large variety of structures by the straightforward derivatization of compound 4 c showcases the synthetic utility of the presented method. Exploration of the reactivity of acyl radicals under photoredox catalyzed conditions and exploitation of this chemistry in unprecedented multicomponent reactions are currently under investigation in our laboratory.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors thank the Olle Engkvist Byggmästare foundation, the Wilhelm and Martina Lundgren research foundation, the Ollie and Elof foundation, the Magnus Bergvall foundation, the Adlerbertska foundation and the Swedish Research Council. C.C. is grateful to the European Commission for a Marie Skłodowska‐Curie fellowship (H2020‐MSCA‐IF‐2014 n: 660668). We would also like to thank Dr. Cassandra L. Fleming for proof reading the manuscript.
F. Pettersson, G. Bergonzini, C. Cassani, C.-J. Wallentin, Chem. Eur. J. 2017, 23, 7444.
References
- 1.
- 1a. Dömling A., Chem. Rev. 2006, 106, 17–89; [DOI] [PubMed] [Google Scholar]
- 1b. Ramón D. J., Yus M., Angew. Chem. Int. Ed. 2005, 44, 1602–1634; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 1628–1661; [Google Scholar]
- 1c. Rotstein B. H., Zaretsky S., Rai V., Yudin A. K., Chem. Rev. 2014, 114, 8323–8359; [DOI] [PubMed] [Google Scholar]
- 1d. Ruijter E., Scheffelaar R., Orru R. V., Angew. Chem. Int. Ed. 2011, 50, 6234–6246; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 6358–6371; [Google Scholar]
- 1e. Touré B. B., Hall D. G., Chem. Rev. 2009, 109, 4439–4486; [DOI] [PubMed] [Google Scholar]
- 1f. Multicomponent Reactions in Organic Synthesis ( Eds.: J. Zhu, Q. Wang, M.-X. Wang), Wiley-VCH, Weinheim, 2014; [Google Scholar]
- 1g. Miura K., Tojino M., Fujisawa N., Hosomi A., Ryu I., Angew. Chem. Int. Ed. 2004, 43, 2423–2425; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 2477–2479; [Google Scholar]
- 1h. Volla C. M. R., Atodiresei I., Rueping M., Chem. Rev. 2014, 114, 2390–2431. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Ischay M. A., Yoon T. P., Eur. J. Org. Chem. 2012, 18, 3359–3372; [Google Scholar]
- 2b. Narayanam J. M. R., Stephenson C. R. J., Chem. Soc. Rev. 2011, 40, 102–113; [DOI] [PubMed] [Google Scholar]
- 2c. Prier C. K., Rankic D. A., MacMillan D. W. C., Chem. Rev. 2013, 113, 5322–5363; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d. Romero N. A., Nicewicz D. A., Chem. Rev. 2016, 116, 10075–10166; [DOI] [PubMed] [Google Scholar]
- 2e. Shaw M. H., Twilton J., MacMillan D. W. C., J. of Org. Chem. 2016, 81, 6898–6926; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2f. Teplý F., Collect. Czech. Chem. Commun. 2011, 76, 859–917; [Google Scholar]
- 2g. Fabry D. C., Rueping M., Acc. Chem. Res. 2016, 49, 1969–1979; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2h. Ghosh I., Marzo L., Das A., Shaikh R., König B., Acc. Chem. Res. 2016, 49, 1566–1577. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Bergonzini G., Cassani C., Lorimer-Olsson H., Hörberg J., Wallentin C.-J., Chem. Eur. J. 2016, 22, 3292–3295; [DOI] [PubMed] [Google Scholar]
- 3b. Bergonzini G., Cassani C., Wallentin C.-J., Angew. Chem. Int. Ed. 2015, 54, 14066–14069; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14272–14275. [Google Scholar]
- 4.
- 4a. Bath S., Laso N. M., Lopez-Ruiz H., Quiclet-Sire B., Zard S. Z., Chem. Commun. 2003, 204–205; [DOI] [PubMed] [Google Scholar]
- 4b. Benati L., Calestani G., Leardini R., Minozzi M., Nanni D., Spagnolo P., Strazzari S., Org. Lett. 2003, 5, 1313–1316; [DOI] [PubMed] [Google Scholar]
- 4c. Esposti S., Dondi D., Fagnoni M., Albini A., Angew. Chem. Int. Ed. 2007, 46, 2531–2534; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 2583–2586; [Google Scholar]
- 4d. Guo W., Lu L.-Q., Wang Y., Wang Y.-N., Chen J.-R., Xiao W.-J., Angew. Chem. Int. Ed. 2015, 54, 2265–2269; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 2293–2297; [Google Scholar]
- 4e. Lv L., Lu S., Guo Q., Shen B., Li Z., J. Org. Chem. 2015, 80, 698–704; [DOI] [PubMed] [Google Scholar]
- 4f. Majek M., Jacobi von Wangelin A., Angew. Chem. Int. Ed. 2015, 54, 2270–2274; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 2298–2302; [Google Scholar]
- 4g. Ryu I., Sonoda N., Curran D. P., Chem. Rev. 1996, 96, 177–194. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Dagousset G., Carboni A., Magnier E., Masson G., Org. Lett. 2014, 16, 4340–4343; [DOI] [PubMed] [Google Scholar]
- 5b. Duan Y., Li W., Xu P., Zhang M., Cheng Y., Zhu C., Org. Chem. Front. 2016, 3, 1443–1446; [Google Scholar]
- 5c. Fumagalli G., Boyd S., Greaney M. F., Org. Lett. 2013, 15, 4398–4401; [DOI] [PubMed] [Google Scholar]
- 5d. Tomita R., Yasu Y., Koike T., Akita M., Angew. Chem. Int. Ed. 2014, 53, 7144–7148; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7272–7276. [Google Scholar]
- 6. Li M., Yang J., Ouyang X.-H., Yang Y., Hu M., Song R.-J., Li J.-H., J. Org. Chem. 2016, 81, 7148–7154. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Liautard V., Landais Y. in Multicomponent Reactions in Organic Synthesis (Eds.: J. Zhu, Q. Wang, M. Wang), Wiley-VCH, Weinheim, 2014, pp. 401–435; [Google Scholar]
- 7b. Beeson T. D., Mastracchio A., Hong J.-B., Ashton K., MacMillan D. W. C., Science 2007, 316, 582–585; [PubMed] [Google Scholar]
- 7c. Fukuyama T., Nakashima N., Okada T., Ryu I., J. Am. Chem. Soc. 2013, 135, 1006–1008; [DOI] [PubMed] [Google Scholar]
- 7d. Keck G. E., Kordik C. P., Tetrahedron Lett. 1993, 34, 6875–6876; [Google Scholar]
- 7e. Ollivier C., Renaud P., Chem. Eur. J. 1999, 5, 1468–1473; [Google Scholar]
- 7f. Vaillard S. E., Mück-Lichtenfeld C., Grimme S., Studer A., Angew. Chem. Int. Ed. 2007, 46, 6533–6536; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 6653–6656. [Google Scholar]
- 8. Gooßen L., Döhring A., Adv. Synth. Catal. 2003, 345, 943–947. [Google Scholar]
- 9.
- 9a. Boivin J., Carpentier F., Jrad R., Synthesis 2006, 10, 1664–1672; [Google Scholar]
- 9b. Duan X.-F., Zeng J., Lü J.-W., Zhang Z.-B., J. Org. Chem. 2006, 71, 9873–9876. [DOI] [PubMed] [Google Scholar]
- 10. Mojr V., Svobodova E., Strakova K., Nevesely T., Chudoba J., Dvorakova H., Cibulka R., Chem. Commun. 2015, 51, 12036–12039. [DOI] [PubMed] [Google Scholar]
- 11. Foster S. L., Handa S., Krafft M., Rowling D., Chem. Commun. 2007, 4791–4793. [DOI] [PubMed] [Google Scholar]
- 12. Wang R.-L., Zhu P., Lu Y., Huang F.-P., Hui X.-P., Adv. Synth. Catal. 2013, 355, 87–92. [Google Scholar]
- 13. Padmavathi V., Balaiah A., Jagan Mohan Reddy B., Padmaja A., Heterocycl. Commun. 2003, 6, 599–604. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary