

ABSTRACT
Although deregulation of MEK/extracellular signal-regulated kinase (ERK) activity is a key feature in cancer, high-magnitude MEK/ERK activity can paradoxically induce growth inhibition. Therefore, additional mechanisms may exist to modulate MEK/ERK activity in favor of tumor cell proliferation. We previously reported that mortalin/HSPA9 can facilitate proliferation of certain KRAS and BRAF tumor cells by modulating MEK/ERK activity. In this study, we demonstrated that mortalin can regulate MEK/ERK activity via protein phosphatase 1α (PP1α). We found that PP1α inhibition increases steady-state levels of phosphorylated MEK1/2 in various tumor cells expressing B-RafV600E or K-RasG12C/D. Intriguingly, coimmunoprecipitation and in vitro binding assays revealed that mortalin facilitates PP1α-mediated MEK1/2 dephosphorylation by promoting PP1α-MEK1/2 interaction in an ATP-sensitive manner. The region spanning Val482 to Glu491 in the substrate-binding cavity and the substrate lid of mortalin were necessary for these physical interactions, which is consistent with conventional heat shock protein 70 (HSP70)-client interaction mechanisms. Nevertheless, mortalin depletion did not affect cellular PP1α levels or its regulatory phosphorylation, suggesting a nonconventional role for mortalin in promoting PP1α-MEK1/2 interaction. Of note, PP1α was upregulated in human melanoma and pancreatic cancer biopsy specimens in correlation with mortalin upregulation. PP1α may therefore have a role in tumorigenesis in concert with mortalin, which affects MEK/ERK activity in tumor cells.
KEYWORDS: MEK1/2, ERK1/2, protein phosphatase 1, heat shock protein, mortalin
INTRODUCTION
The Raf/MEK/extracellular signal-regulated kinase (ERK) pathway is a highly specific three-layer kinase cascade that consists of the Ser/Thr kinases, Raf (i.e., A-, B-, and C-Raf), the highly homologous dual-specificity kinases MEK1 and MEK2 (collectively referred to as MEK1/2), and the ubiquitously expressed Ser/Thr kinases ERK1/2 (1). Upon activation, Raf phosphorylates two Ser residues in the activation segment of MEK1/2, which in turn activate ERK1/2 by sequentially phosphorylating Tyr and Thr in their activation loop (2). ERK1/2 then activate/inactivate various targets, including transcription factors, other kinases, phosphatases, cytoskeletal proteins, scaffolds, receptors, and signaling components (1). The Raf/MEK/ERK pathway has pivotal roles in regulating cell survival, cell cycle progression, and differentiation (3), and its deregulation often leads to the onset of cancer, including malignancies of skin, thyroid, pancreas, and colon, wherein mutations in BRAF and its upstream activator, RAS, are prevalent (4).
Diverse physiological outputs of Raf/MEK/ERK signaling are determined by the duration and strength of signals, physical interactions with specific scaffolds, subcellular compartmentalization, cross talk with other signaling pathways, and distinct functions of an isoform in the kinase cascade (1). For example, although known mainly for its role in facilitating cell proliferation (5), high-magnitude activity of the Raf/MEK/ERK pathway can also elicit antiproliferative signaling, i.e., growth arrest or cell death, in different normal cell types and certain tumor cells which do not exhibit aberrant Raf/MEK/ERK activity (6–8). This paradoxical Raf/MEK/ERK signaling has significance in that it may serve as a tumor-suppressive mechanism in the face of aberrant pathway activation (9). For carcinogenesis to occur in this context, antiproliferative signaling of Raf/MEK/ERK should be inactivated, and indeed, a number of inactivation mechanisms have been identified at different downstream effector levels, e.g., mutations in TP53, retinoblastoma (Rb), and cyclin-dependent kinase (CDK) inhibitors (10). Of note, recent studies, including ours, suggest that modulation of the magnitude of Raf/MEK/ERK activity may also be a mechanism by which antiproliferative MEK/ERK signaling is inactivated in tumor cells (11–13). As such, identification of a regulator that modulates the pathway activity in favor of tumor cell proliferation/survival would broaden our understanding of the pathway signaling in cancer.
Mortalin (HSPA9/GRP75/PBP74) is a member of the heat shock protein 70 (HSP70) family (14–16), which is often upregulated and mislocalized in cancer (13, 17–19). Mortalin has been noted for its potential significance in multiple aspects of tumorigenesis, including tumor cell proliferation/survival, stemness, epithelial-mesenchymal transition, and angiogenesis, although the molecular mechanisms underlying these effects require better understanding (17–21). We have reported that mortalin has a role in determining proliferative versus antiproliferative outputs of Raf/MEK/ERK signaling in tumor cells (13, 22, 23). Briefly, mortalin directly interacted with MEK1/2, and its depletion significantly increased MEK1/2 phosphorylation levels in different tumor cells (13, 22). Very intriguingly, mortalin depletion triggered robust growth-inhibitory responses in B-RafV600E and K-RasG13D tumor cells in a MEK/ERK-dependent manner (13). These findings suggested that mortalin is a negative regulator of the Raf/MEK/ERK pathway and has a role in modulating MEK/ERK activity to a level optimal for tumor cell proliferation and survival. Given this novel observation and the potential of mortalin as a target to reactivate latent tumor-suppressive MEK/ERK signaling in tumor cells, we sought in this study to uncover by what mechanism mortalin regulates MEK1/2 phosphorylation.
Protein phosphatase 1 (PP1) is a Ser/Thr protein phosphatase superfamily member that shares similar catalytic core structures and mechanisms with PP2A, PP2B/calcineurin, and PP4 to -7 (24, 25). PP1 is evolutionarily conserved, and mammalian cells express four closely related PP1 isoforms, i.e., PP1α, PP1β, PP1γ1, and PP1γ2 (26). Unlike the case for other siblings in the phosphoprotein phosphatase superfamily, e.g., PP2A (27–31), the role of PP1 isoforms in mediating MEK1/2 dephosphorylation is not well known. In this report, we demonstrate that PP1α has a role in determining steady-state levels of phosphorylated MEK1/2 in certain KRAS- and BRAF-mutated tumor cells and that mortalin regulates this process by facilitating the physical interaction between PP1α and MEK1/2. Our data thus address a novel mechanism by which the magnitude of MEK/ERK activity is regulated in tumor cells.
RESULTS
Inhibitors of PP1 and PP2A upregulate steady-state levels of phosphorylated MEK1/2 in B-RafV600E melanoma and K-RasG12C/D pancreatic cancer cells.
To determine the significance of dephosphorylation in determining the steady-state levels of MEK1/2 activity in RAS- or RAF-mutated tumor cells, we examined the effects of PPIC3, a broad-spectrum protein phosphatase inhibitor cocktail containing cantharidin, calyculin A, and (−)-p-bromolevamisole oxalate, on the levels of MEK1/2 phosphorylated at the two key Ser residues in the activation segment, i.e., Ser218/Ser222 of MEK1 and Ser222/Ser226 of MEK2. This dual Ser phosphorylation is necessary and sufficient for activation of MEK1/2 (32, 33). Calyculin A inhibits protein phosphatase 1 (PP1) and PP2A with similar selectivity (50% inhibitory concentration [IC50] = 0.4 nM for PP1 and 0.25 nM for PP2A), whereas cantharidin inhibits PP2A more selectively than PP1 (IC50 = 0.194 μM for PP2A and 1.1 μM for PP1) (34). However, these inhibitors do not inhibit PP2B (34). (−)-p-Bromolevamisole oxalate is an alkaline phosphatase-specific inhibitor (35).
Western blot analysis revealed that incubation of the whole-cell lysates of SK-MEL 28, a human B-RafV600E melanoma line, with PPIC3 significantly increased the level of phosphorylated MEK1/2 (Fig. 1A, lanes 1 and 2). Subsequently, we evaluated each inhibitor in the PPIC3 inhibitor cocktail. Addition of calyculin A robustly increased the level of phosphorylated MEK1/2 in SK-MEL 28 cell lysates (Fig. 1A, lanes 8 to 10) whereas cantharidin was not effective unless used at a high concentration (Fig. 1A, lanes 5 to 7). (−)-p-Bromolevamisole oxalate, which was used as a negative control, did not show any effects on MEK1/2 phosphorylation. Importantly, the B-Raf-specific inhibitor PLX4032 did not significantly affect these effects of PPIC3 (Fig. 1A, lanes 3 and 4) and calyculin A (Fig. 1B), excluding any potential interference by B-RafV600E activity with the increased phosphorylated MEK1/2 levels under these conditions.
FIG 1.

Effects of protein phosphatase inhibitors on MEK1/2 phosphorylation in B-RafV600E and K-RasG12C/D tumor cells. (A) Total lysates of SK-MEL 28 cells were incubated with different phosphatase inhibitors for 60 min prior to Western blotting. PPIC3 was diluted 1,000-fold for use as instructed by the manufacturer. PLX4032 (PLX) was used at 20 nM either singly or in combination with PPIC3. Cantharidin, calyculin A, and (−)-p-bromolevamisole oxalate (BO) were used at the indicated concentrations. Dimethyl sulfoxide (DMSO) was the vehicle control. Actin was the control for equal protein loading. pMEK1/2 indicates phosphorylations at Ser217/221 of MEK1 and Ser222/226 of MEK2. (B) Total lysates of SK-MEL 28 cells were incubated with 100 μM cantharidin or 50 nM calyculin A for 60 min, with or without addition of 20 nM PLX4032, prior to Western blotting. (C to F) Total lysates of SK-MEL 28 (C), A375 (D), MiaPaCa-2 (E), and PANC-1 (F) cells were incubated with increasing doses of calyculin A for 15 min or cantharidin for 25 min prior to Western blotting. Right panels, densitometry of phosphorylated MEK1/2 signals normalized for actin.
We next determined the effects of calyculin A and cantharidin in cell cultures. Consistent with their in vitro effects described above, calyculin A treatment effectively increased the levels of phosphorylated MEK1/2 in SK-MEL 28 cells, whereas cantharidin treatment was ineffective (Fig. 1C [densitometry is shown at the right]). Calyculin A, but not cantharidin, also effectively increased cellular levels of phosphorylated MEK1/2 in another human B-RafV600E melanoma line, A375 (Fig. 1D), and in two KRAS-mutated human pancreatic cancer lines, MiaPaCa-2 (K-RasG12C) (Fig. 1E) and PANC-1 (K-RasG12D) (Fig. 1F). These data suggest that dephosphorylation is an important mechanism in regulating the steady-state levels of phosphorylated MEK1/2 in RAS- or RAF-mutated tumor cells and that this regulation may require PP1 and PP2A. In this regulation, PP1 may be more important than PP2A, because calyculin A (similarly selective to PP1 and PP2A) was more effective than cantharidin (more selective to PP2A than to PP1). Indeed, lentiviral small hairpin RNA (shRNA)-mediated PP1α knockdown in these BRAF- and KRAS-mutated tumor cells consistently increased phosphorylation of MEK1/2 and ERK1/2 (Thr202/Tyr204 of ERK1 and Thr185/Tyr187 of ERK2), the bona fide readout of MEK1/2 activity, without affecting phosphorylation of B-Raf Ser445, C-Raf Ser338, and A-Raf Ser299, or phosphorylation of C-Raf Ser259 (Fig. 2A to D [densitometry of phospho-MEK1/2 is shown in Fig. 2E]). These phosphorylation signatures serve as surrogate markers for the activation status of Raf proteins (36, 37).
FIG 2.
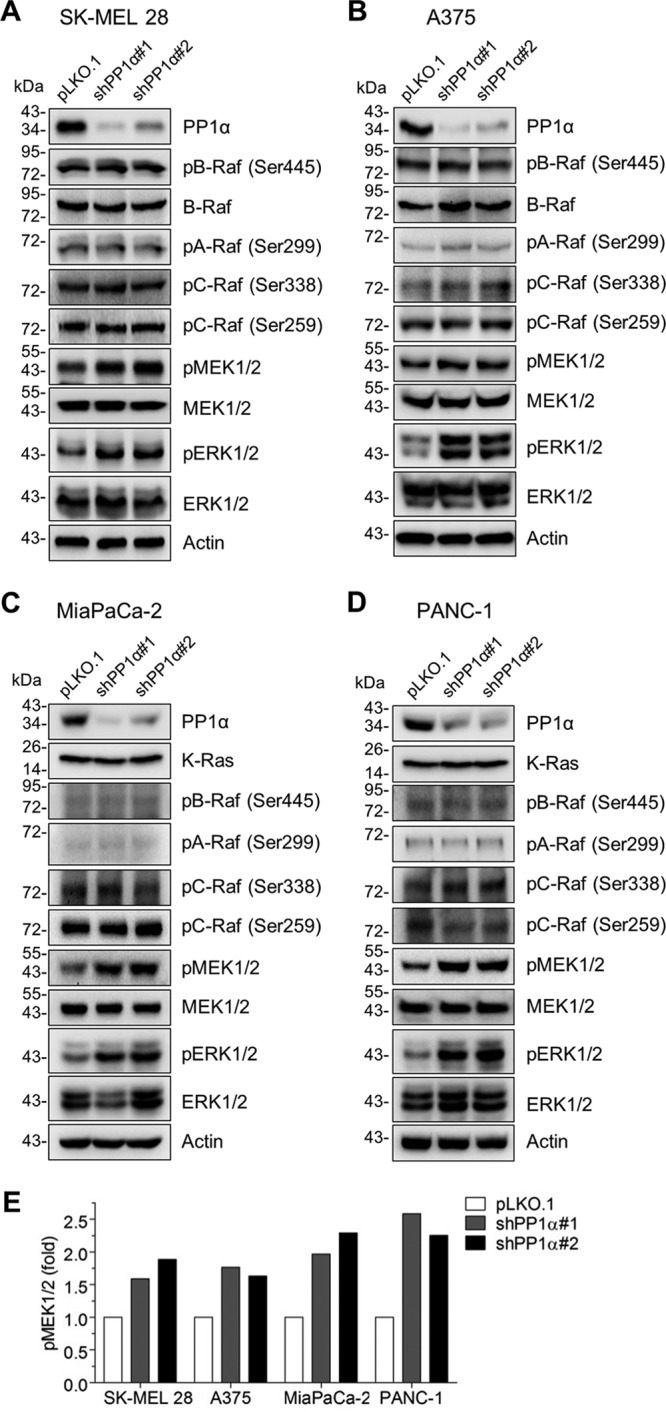
Effects of PP1α depletion on MEK/ERK activation in B-RafV600E and K-RasG12C/D tumor cells. (A to D) Total cell lysates were analyzed by Western blotting. SK-MEL 28 (A), A375 (B), MiaPaCa-2 (C), and PANC-1 (D) cells were infected with two different lentiviral shRNA systems targeting PP1α (shPP1α#1 and shPP1α#2) for 3 days before harvest. pLKO.1 was the control virus. (E) Densitometry of phospho-MEK1/2 signals normalized to total MEK1/2 levels.
Mortalin depletion does not downregulate cellular levels of PP1α and PP2A.
We next examined the effects of mortalin depletion on expression levels of PP1α and PP2A in SK-MEL 28, A375, MiaPaCa-2, and PANC-1 cells. In these cells, mortalin knockdown using different lentiviral shRNA constructs or doxycycline-inducible shRNA constructs consistently increased the levels of phosphorylated MEK1/2, which was accompanied by increased ERK1/2 phosphorylation (Fig. 3). Mortalin knockdown, however, did not significantly affect B-Raf Ser445 phosphorylation and total B-Raf levels in SK-MEL 28 and A375 cells (Fig. 3A and B) or K-Ras protein levels in MiaPaCa-2 and PANC-1 cells (Fig. 3C and D). Mortalin knockdown also did not affect the activation phosphorylation signature of B-Raf, C-Raf, and A-Raf as well as the inhibitory C-Raf Ser259 phosphorylation in these cells (Fig. 3). These data suggest that mortalin may not regulate MEK1/2 phosphorylation at Raf or Ras levels. Under these conditions, neither protein levels of PP1α nor its Thr320 phosphorylation, an inactivating modification (38–40), was significantly affected (Fig. 3). Moreover, catalytic subunits of PP2A were mildly upregulated in SK-MEL 28 and A375 cells (Fig. 3A and B), while not being affected in MiaPaCa-2 and PANC-1 cells (Fig. 3C and D). These data indicate that downregulation of PP1 and PP2A is not required for mortalin depletion to upregulate phosphorylated MEK1/2 levels in RAS/RAF mutant tumor cells.
FIG 3.

Effects of mortalin depletion on MEK/ERK activation and PP1α/PP2A expression in B-RafV600E and K-RasG12C/D tumor cells. Total cell lysates were analyzed by Western blotting. (A) SK-MEL 28 cells were infected with two different lentiviral shRNA systems targeting mortalin (shMort#1 and shMort#2) for the indicated periods before harvest. pLL3.7 was the control virus. (B) A375 cells stably expressing doxycycline-inducible pTRIPZ shRNA targeting mortalin (shMort-mir) were treated with doxycycline for 5 days. (C and D) MiaPaCa-2 (C) and PANC-1 (D) cells were infected with shMort#1 or shMort#2 for 6 days. pPP1 indicates phosphorylation at Thr320 of PP1.
Mortalin facilitates PP1α-mediated MEK1/2 dephosphorylation by increasing the physical interaction between PP1α and MEK1/2.
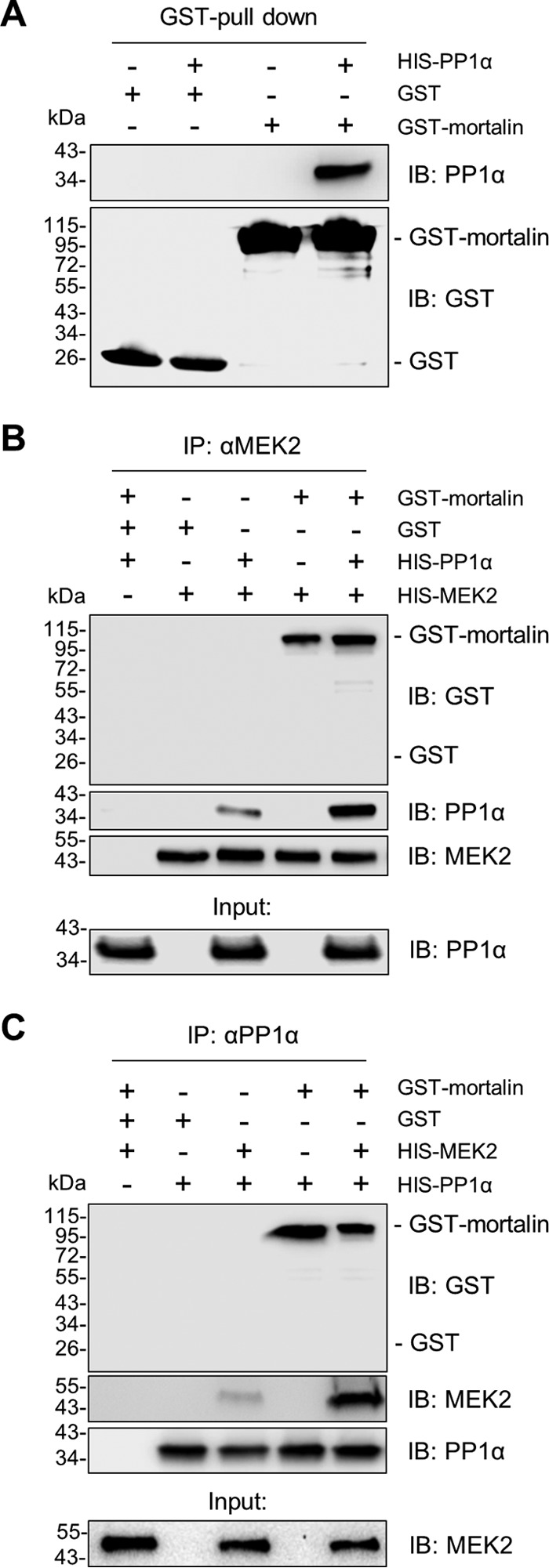
Because mortalin can physically interact with MEK1/2 (13), we asked whether mortalin affects cellular levels of phosphorylated MEK1/2 in RAS/RAF mutant tumor cells by regulating the access of a phosphatase to MEK1/2. Because the data above suggested potential significance of PP1α in RAS/RAF tumor cells and because its role in MEK1/2 dephosphorylation was previously unclear, we focused primarily on PP1α in this study. First, we determined whether PP1α can physically interact with MEK1/2 and mortalin. Indeed, our immunoprecipitation (IP) assays using different antibodies specific to PP1α, MEK1, and MEK2 consistently suggested that PP1α interacts with MEK1/2 in SK-MEL 28 cells and that mortalin is coimmunoprecipitated with these proteins (Fig. 4A to C). Moreover, our co-IP analysis of a panel of B-RafV600E melanoma and thyroid cancer lines suggested that the degree of MEK2-PP1α interaction is correlated with the degree of mortalin-MEK2 interaction, while the degree of these protein interactions is inversely correlated with MEK1/2 phosphorylation levels (Fig. 4D to G). These data led us to hypothesize that mortalin regulates PP1α-mediated MEK1/2 dephosphorylation via direct physical interactions.
FIG 4.

Mortalin physically interacts with PP1α and MEK1/2 in vivo. (A to C) Total lysates of SK-MEL 28 cells were subject to IP using the monoclonal antibody specific to MEK1 (A), MEK2 (B), or PP1α (C). Normal IgG corresponding to each antibody was used as the control. IP fractions were immunoblotted (IB) to determine co-IP of the indicated proteins. (D) Total lysates of different human B-RafV600E tumor lines were subject to IP using the anti-MEK2 antibody. Input and IP fractions were immunoblotted for the indicated proteins. (E) Correlation between PP1α-MEK2 interaction and mortalin-MEK2 interaction. For analysis, densitometry data of PP1α and mortalin signals in the IP fractions in panel D were normalized to the corresponding MEK2 pulldown signals. Fold changes of the normalized data are relative to HT29 (P = 0.0254; r = 0.8155). (F) Inverse correlation between PP1α-MEK2 interaction and phospho-MEK1/2 levels. Data for PP1α-MEK2 interactions are from panel E. Data for phospho-MEK1/2 levels were quantitated in the input in panel D by densitometry and normalized to the corresponding MEK1/2 signals. Fold changes of the normalized phospho-MEK1/2 data are relative to A375 (P = 0.0431; r = −0.7694). (G) Inverse correlation between mortalin-MEK2 interaction and phospho-MEK1/2 levels. Data are from panels E and F (P = 0.079; r = −0.7015).
To test this hypothesis, we conducted in vitro binding assays using recombinant proteins. In these assays, recombinant PP1α directly bound to recombinant mortalin (Fig. 5A) and MEK2 (Fig. 5B and C). Very intriguingly, when recombinant mortalin was added to these reaction mixtures, the physical interaction between MEK2 and PP1α was substantially increased (Fig. 5B and C; see Fig. 8 for mortalin dose-dependent effects). Consistent with these effects, mortalin knockdown substantially decreased PP1α-MEK1/2 interactions in SK-MEL 28 cells, as determined by IP (Fig. 6A). Importantly, this decrease was accompanied by increased levels of phosphorylated MEK1/2 in mortalin-depleted cells (Fig. 6A, lower panel). Moreover, when incubated with recombinant PP1α in an in vitro reaction, MEK2 immunoprecipitates extracted from mortalin-depleted SK-MEL 28 cells exhibited significantly decreased MEK2 phosphorylation (Fig. 6B). These data strongly suggest that mortalin can downregulate cellular levels of phosphorylated MEK1/2 by promoting the access of PP1α to MEK1/2.
FIG 5.
Mortalin physically interacts with PP1α and MEK2 in vitro. (A) In vitro binding assay using 0.5 μM recombinant PP1α and GST-mortalin. GST was used as the control. (B and C) In vitro binding assay using 0.5 μM recombinant MEK2, PP1α, and GST-mortalin to determine whether mortalin affects the interaction between MEK2 and PP1α.
FIG 8.
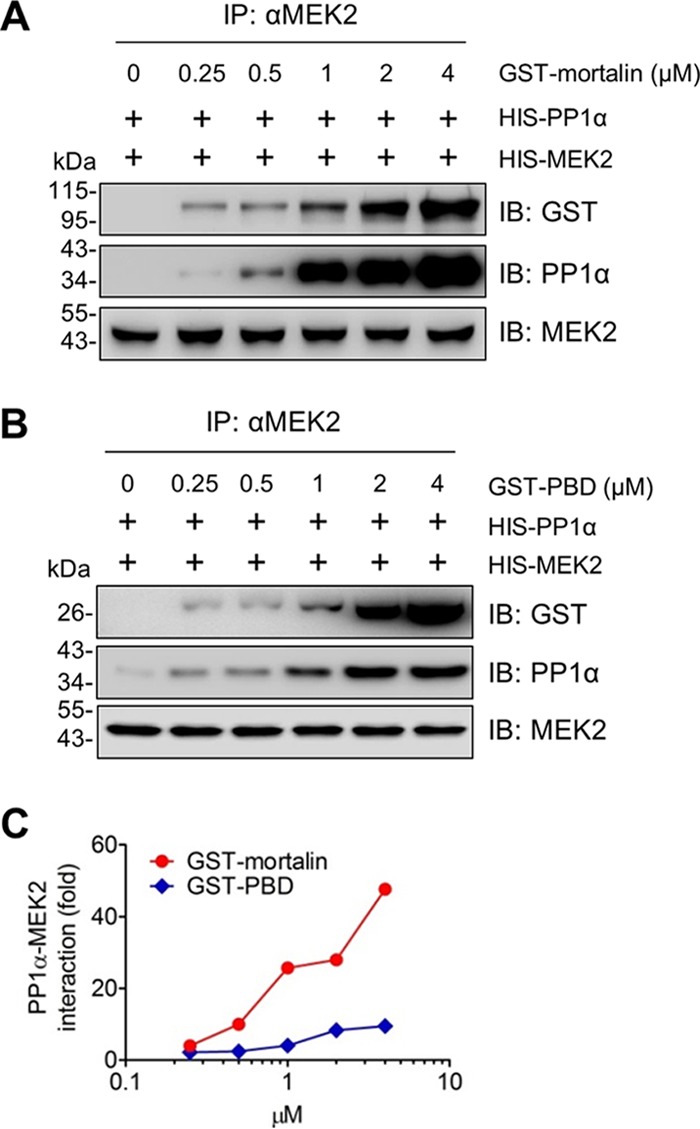
The PBD mutant facilitates PP1α-MEK2 interaction less efficiently than full-length mortalin. (A and B) Dose-dependent effects of mortalin on MEK2-PP1α interaction. Recombinant MEK2 and PP1α (0.5 μM) were incubated for 60 min in the presence of different doses of GST-mortalin (A) or GST-PBD (B) before IP using anti-MEK2 antibody and protein G-agarose. (C) The degree of PP1α-MEK2 interaction affected by GST-mortalin or GST-PBD was determined by densitometry of the PP1α signals normalized by MEK2 signals in panels A and B.
FIG 6.
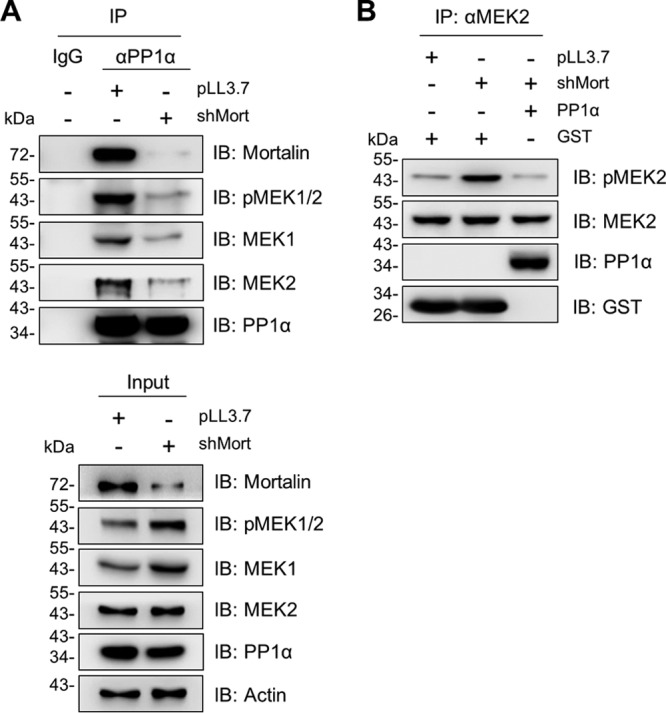
Mortalin depletion decreases PP1α-MEK1/2 interaction in SK-MEL 28 cells. (A) Total lysates of SK-MEL 28 cells infected with shMort for 4 days were subject to IP using PP1α-specific antibody. Co-IP of the indicated proteins was determined by Western blotting. (B) Endogenous MEK2 was pulled down using an anti-MEK2 antibody. After three stringent washes with the IP buffer, MEK2 IP fractions were incubated with 0.5 μM recombinant PP1α for 60 min before Western blotting. GST was used as the control for recombinant PP1α.
Mortalin interacts with PP1α and MEK1/2 via its PBD.
HSP70 chaperones interact with different client proteins through their C-terminal peptide-binding domains (PBDs), wherein an alpha-helical tail serves as the lid for the binding cleft (41). However, they interact with different regulators through the regulatory subdomains in their N-terminal ATPase domain (42). We determined which domain(s) of mortalin is important for its interaction with PP1α and MEK2.
Based upon previous studies (43, 44) and information from the Protein Data Bank database (PDB 4KBO and 3N8E; http://www.wwpdb.org/), we generated different mortalin domain mutants expressing the ATPase domain (AD), the regulatory subdomain 2 (SD2), PBD, and a tail-truncated PBD mutant (PBD-Δtail), as depicted in Fig. 7A. We expressed N-terminally hemagglutinin (HA)-tagged constructs of these mutants in SK-MEL 28 cells and examined their ability to bind to PP1α and MEK2 by IP in comparison with the full-length mortalin (Fig. 7B). This IP using an HA-specific antibody pulled down a similar amount of HA-tagged proteins. Under this condition, full-length mortalin and the PBD mutant, but not AD, SD2, and PBD-Δtail, were coprecipitated with PP1α and MEK2 (Fig. 7B, lanes 1 to 5 and 11). These data indicate that mortalin interacts with PP1α and MEK2 through its PBD, for which its tail is necessary.
FIG 7.

Analysis of mortalin mutants for PP1α or MEK2 interaction. (A) Schematic of the mortalin mutants used in this study. AD, ATPase domain; SD2, subdomain 2; PBD, peptide-binding domain; V482F, Val482Phe; V487A/Y, Val487Ala or Val487Tyr; Δtail, tail deletion. (B) Total lysates of SK-MEL 28 cells infected with lentiviral pHAGE expressing N-terminally HA-tagged mortalin mutants for 2 days were analyzed by immunoblotting for co-IP of indicated proteins. (C) In vitro binding assay using 0.5 μM recombinant GST-PBD mutants and HIS-PP1α. (D) In vitro binding assay using 0.5 μM recombinant GST-PBD mutants and HIS-MEK2. (E) Schematic of the peptide aptamers (APT) designed from the groove region of PBD. (F) SK-MEL 28 cells infected with pHAGE expressing N-terminally HA-tagged PBD mutants were treated with 4 μM synthetic peptide aptamers for 2 days. Equal volumes of dimethylformamide were added to untreated cells. Total cell lysates were analyzed by immunoblotting for co-IP of the indicated proteins.
Val482 in the substrate-binding cavity is a key residue for mortalin interaction with PP1α and MEK1/2.
It was previously demonstrated that a Val-to-Phe exchange in the central hydrophobic pocket of the substrate-binding cavity of DnaK, a bacterial HSP70 homologue, significantly lowers its affinity to clients without causing global structural changes or altering substrate specificity (45). This Val residue is conserved in human HSP70 family members, and exchange of the conserved Val482 to Phe (V482F) impaired mortalin interaction with the tumor suppressor TP53 (46). Indeed, switching Val482 to Phe (V482F) in the PBD mutant substantially abolished its interactions with PP1α and MEK2 (Fig. 7B, lanes 5 and 6). Moreover, consistent with these data, recombinant PBD-V482F proteins exhibited significantly lower affinity to recombinant PP1α and MEK2 than recombinant PBD proteins in our in vitro binding assay (Fig. 7C and D). However, in contrast, switching Cys487 to Tyr (C487Y) or Ala (C487A) did not significantly affect the affinity of PBD to PP1α and MEK2 (Fig. 7B, lanes 8 to 10). These data suggest that Val482 is a key residue for mortalin interaction with PP1α and MEK2.
We further examined the region surrounding Val482 for its importance to mortalin interaction with MEK1/2 and PP1α. For this, we designed multiple peptide aptamers (APTs) based upon the amino acid sequence near Val482 in PBD (Fig. 7E) and determined which aptamer could interfere with PBD-PP1α and PBD-MEK2 interactions in cells. Our IP assays revealed that APT2 (amino acids [aa] 482 to 493) and APT3 (aa 479 to 491) effectively inhibited PBD interactions with MEK2 and PP1α, whereas APT1 (aa 477 to 488) did not (Fig. 7F), suggesting that Gln488-Gly489-Glu490 are also important for mortalin interaction with MEK2 and PP1α. Of note, consistent with the data above, switching Val482 with Phe disabled the interfering capacity of APT3, as demonstrated by APT4 (Fig. 7F). These data suggest that the region spanning amino acids 482 to 490 in the central hydrophobic pocket of PBD comprises a key part of the binding interfaces required for mortalin interaction with PP1α and MEK1/2.
Mortalin interactions with PP1α and MEK1/2 are sensitive to ATP.
The PBD mutant exhibited remarkably higher affinity to PP1α and MEK2 than full-length mortalin, as determined by IP (Fig. 7B, lane 2 versus lane 5). Of note, whereas in vitro PP1α-MEK2 interaction was effectively facilitated by recombinant glutathione S-transferase (GST)-mortalin in a dose-dependent manner (Fig. 8A), the interaction was not as effectively facilitated by recombinant PBD (Fig. 8B and C). Because this suggested a role for the N-terminal domain of mortalin in regulating PP1α-MEK2 interaction and because N-terminal ATPase activity induces conformation changes of HSP70 (42), we determined whether mortalin interactions with PP1α and MEK1/2 are sensitive to adenine nucleotides. Indeed, in our in vitro binding assay, addition of ATP but not of ADP substantially decreased mortalin-PP1α and mortalin-MEK2 interactions (Fig. 9A and B). However, in contrast, PBD-PP1α and PBD-MEK2 interactions were not sensitive to ATP (Fig. 9C and D). These data suggest that the N-terminal ATPase activity has a role in mortalin-mediated regulation of PP1α-MEK2 interaction.
FIG 9.

Effects of adenosine nucleotides on mortalin interactions with PP1α and MEK2. GST-mortalin (A and B) or GST-PBD (C and D) (0.5 μM) was incubated for 60 min with 0.5 μM recombinant PP1α (A and C) or MEK2 (B and D) in the presence of 1 μM ATP or ADP before GST pulldown using glutathione-Sepharose and Western blotting.
PP1α levels are upregulated in human melanoma and pancreatic cancer.
Observing the ability of PP1α to modulate MEK/ERK activity in BRAF melanoma and KRAS pancreatic cancer cell lines, we asked how its expression is altered in biopsy samples from human melanoma and pancreatic cancer patients. By searching a large-scale cancer genomics data set deposited in the cBioPortal database (http://www.cbioportal.org/), we found that PPP1CA (encoding PP1α) gene amplification occurs more significantly than amplification of PPP1CB and PPP1CC (encoding PP1β and PP1γ, respectively) in melanoma and pancreatic cancer (Fig. 10A). Transcriptome sequencing (RNA-seq) analyses of melanoma specimens also revealed significantly upregulated PPP1CA mRNA levels in association with PPP1CA gene amplification (Fig. 10B). Consistent with these data, human cancer microarray data deposited in the Oncomine database (http://www.oncomine.org/) suggested that mRNA levels of PPP1CA but not of PPP1CB and PPP1CC are significantly upregulated during melanoma progression (Fig. 10C). Of note, in this data set, mRNA levels of HSPA9 (encoding mortalin) and PPP1CA were strongly correlated (Fig. 10D). Immunohistochemical analyses of PP1 expression in cancer (Human Protein Atlas, http://www.proteinatlas.org/) also suggested PP1α upregulation in melanoma and pancreatic cancer (Fig. 10E, left panels). Consistent with the genomics data (Fig. 10A to C), PP1γ protein expression was not altered in melanoma and pancreatic cancer specimens (Fig. 10E, right panels), although PP1β protein levels were upregulated (Fig. 10E, middle panels), as previously reported (47). Together, these data suggest a role for PP1α in these tumors, which may be exerted in concert with mortalin.
FIG 10.

Expression of protein phosphatase 1 in human melanoma and pancreatic cancer. (A) Copy number alterations of PPP1C genes encoding PP1α, PP1β, and PP1γ in melanoma and pancreatic ductal adenocarcinoma patient biopsy specimens determined by the copy number analysis algorithms defined by genomic identification of significant targets in cancer (GISTIC) (63) and RAE (64). Deep deletion and shallow deletion are caused mainly by homozygous deletion and heterozygous deletion, respectively. The skin cutaneous melanoma TCGA data set and the pancreatic cancer UTSW data set, originally generated by The Cancer Genome Atlas (TCGA) Research Network (http://cancergenome.nih.gov/) and by Erik Knudsen and coworkers (65), respectively, were obtained from the cBioPortal for Cancer Genomics database (http://www.cBioportal.org). (B) RNA-seq analysis of the melanoma biopsy specimens in panel A. The boxes, whiskers, and asterisks indicate the interquartile range (25 to 75%), the 10 to 90% range, and the minimums, maximums, and outliers, respectively. Fold changes between amplification and diploid were 2.05 (P, <0.0001; t, 6.642) for PPP1CA and 3.41 (P, <0.0001; t, 9.012) for PPP1CB (two-tailed Mann-Whitney test). Fold changes between gain and diploid were 1.222 (P, 0.0043; t, 2.889) for PPP1CA, 1.241 (P, <0.0001; t, 4.016) for PPP1CB, and 1.236 (P, 0.0005; t, 3.542) for PPP1CC (two-tailed Mann-Whitney test). Fold changes between shallow deletion and diploid were 0.732 (P, <0.0001; t, 5.44) for PPP1CA, 0.763 (P, <0.0001; t, 4.111) for PPP1CB, and 0.804 (P, <0.0001; t, 5.353) for PPP1CC (two-tailed Mann-Whitney test). The P value for PPP1CA (amplification, gain, and shallow deletion versus diploid) was <0.05 (with q = 7.344, 3.134, and 4.961, respectively) (one-way ANOVA with Dunnett's multiple-comparison test). (C) Microarray analysis of PPP1C mRNA levels in patient biopsy specimens. Data are from the Talantov melanoma data set from the Oncomine database (www.oncomine.org). Fold changes between benign nevus versus normal skin were 2.517 (P, 0.0052; t, 3.087) for PPP1CA, 0.275 (P, 0.0066; t, 3.002) for PPP1CB, and 0.926 (P, 0.1645; t, 1.436) for PPP1CC (two-tailed t test). Fold changes between cutaneous melanoma versus normal skin were 3.799 (P, <0.0001; t, 13.38) for PPP1CA, 0.19 (P, <0.0001; t, 5.59) for PPP1CB, and 0.832 (P, 0.0006; t, 3.657) for PPP1CC (two-tailed t test). (D) Pearson correlation coefficient analysis of the HSPA9 (encoding mortalin) and PPP1CA mRNA levels in panel C (P < 0.0001; r = 0.6413). (E) Immunohistochemistry of PP1α, PP1β, and PP1γ expression in patient biopsy specimens. Data were obtained from the Human Protein Atlas database (66). PP1α was stained by antibody HPA046833 (www.proteinatlas.org/ENSG00000172531-PPP1CA/cancer), PP1β was stained by HPA065425 (www.proteinatlas.org/ENSG00000213639-PPP1CB/cancer), and PP1γ was stained by HPA013661 (www.proteinatlas.org/ENSG00000186298-PPP1CC/cancer).
DISCUSSION
This study reveals a novel mechanism by which the magnitude of MEK/ERK activity is regulated in tumor cells. Our data demonstrate (i) that mortalin directly interacts with PP1α and MEK1/2 and (ii) that mortalin facilitates PP1α-mediated dephosphorylation of MEK1/2 by promoting their physical interaction. These mechanisms appear to be important in determining cellular steady-state levels of phosphorylated MEK1/2 and, subsequently, MEK/ERK activity. We speculate that mortalin-mediated regulation of PP1α-MEK1/2 interaction may have significance in certain MEK/ERK-dependent cancers in which mortalin and PP1α are upregulated, e.g., melanoma and pancreatic cancer. Because Raf/MEK/ERK activity of different magnitudes induces different cellular responses (6–8), it is conceivable that mortalin regulation of PP1α-MEK1/2 interaction has a role in modulating MEK/ERK activity (Fig. 11) and, subsequently, in determining cell physiological outputs, e.g., growth arrest or proliferation. Indeed, upregulated MEK/ERK activity was necessary for mortalin depletion to induce growth arrest in B-RafV600E and K-RasG13D tumor cells (13).
FIG 11.
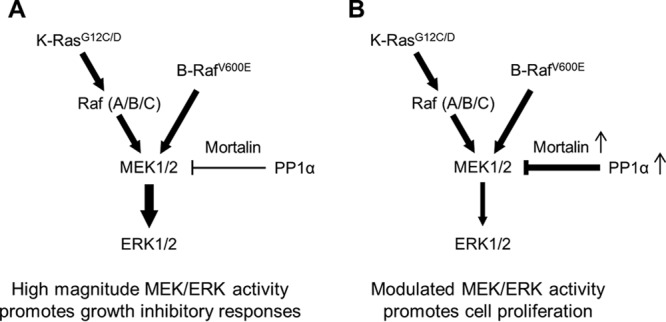
Modulation of MEK/ERK activity by mortalin and PP1α in BRAF- or KRAS-mutated cancer cells. (A) Mutations in BRAF or KRAS can induce high-magnitude MEK/ERK activity. (B) Upregulated mortalin and PP1α may have a role in modulating MEK/ERK activity in certain BRAF/KRAS tumor cells because mortalin can facilitate PP1α-mediated MEK1/2 dephosphorylation. This mechanism may be important for determining the physiological outputs of MEK/ERK activity, i.e., proliferation versus growth arrest.
What is the molecular mechanism by which mortalin facilitates PP1α-mediated MEK1/2 dephosphorylation? Our data suggest that mortalin interacts with PP1α and MEK1/2 via a conventional mechanism for HSP70-client interaction in that its interaction with these proteins is sensitive to the mutations disabling its central hydrophobic pocket or substrate lid, the intercepting aptamers designed from the peptide sequence of its substrate-binding cavity, and the presence of ATP. Given its canonical function as a molecular chaperone, one may suspect that mortalin mediates maturation of PP1α and MEK1/2 independently of any direct enhancement of the dephosphorylation of MEK1/2 by PP1α, consequently contributing to increased rates of the enzyme-substrate interaction and catalysis. However, because mortalin depletion did not affect cellular PP1α levels, mortalin does not appear to regulate PP1α proteostasis. Moreover, we previously demonstrated that although MEK1 levels are mildly upregulated under conditions of mortalin depletion (Fig. 6, bottom panel), this is mainly due to MEK1 transcription upregulated via an ERK1/2-mediated feedback loop, which is activated by mortalin depletion (48). Of note, a recent report has demonstrated that HSP70 can also regulate the stability of folded proteins via a mechanism wherein its lid and substrate-binding cavity act synergistically in a nucleotide-dependent manner (49), suggesting that HSP70 function is not limited to unfolded proteins. Given this, we speculate that serving as a platform for signal transduction may be a primary function of mortalin in regulating PP1α and MEK1/2. In support of this notion, growing evidences suggest that mortalin can regulate different proteins via a chaperone-independent role. For example, mortalin can sequester certain key regulators of cellular processes, including TP53 and mevalonate pyrophosphate decarboxylase, without affecting their cellular levels (50–52). Mortalin can also function as a constituent of noncaveolar membrane raft-associated endocytic vesicles that mediate macromolecule endocytosis (53).
How, then, does mortalin facilitate PP1α-MEK1/2 interaction? Given our data suggesting that mortalin utilizes identical domains and motifs to interact with PP1α and MEK1/2, it is unlikely that mortalin interacts with these proteins simultaneously. We suspect that mortalin interacts with these proteins separately and that the resulting protein complexes, i.e., mortalin-PP1α and mortalin-MEK1/2, may form a signalosome to facilitate PP1α-MEK1/2 interaction in response to a stimulus. In support, recent studies suggest that HSP70 can form a transient homodimer, which is regulated by ATP hydrolysis and affects its interaction with cochaperones and clients, although the orientation of these dimers is currently discrepant (i.e., parallel in bacteria versus antiparallel in eukaryotes) (54, 55). As an alternative model, one may suspect that PP1α displaces MEK1/2 from mortalin, making them accessible to other phosphatases. However, if this model were viable, mortalin would have favorably interacted with PP1α rather than facilitating the PP1-MEK interaction in our in vitro binding assay for Fig. 8. Regardless of these possibilities, given the sensitivity of mortalin-PP1α and mortalin-MEK1/2 interactions to ATP and the requirement of its N-terminal domain for mortalin to promote PP1α-MEK1/2 interaction, the ATPase activity of mortalin may dynamically regulate these processes in response to cellular ATP levels. Indeed, cellular bioenergetics and MEK/ERK signaling are often altered in tumor cells, and it is a tempting question for future studies whether mortalin is involved in a cross talk between energy metabolism and MEK/ERK signaling in tumor cells.
The human genome harbors 518 genes encoding protein kinases, including 428 Ser/Thr kinases and 90 Tyr kinases (56). In contrast, the human genome harbors only 147 genes encoding protein phosphatases, including 40 Ser/Thr phosphatases and 107 Tyr phosphatases (57). Accordingly, substrate specificity of Ser/Thr phosphatases is secured mainly by a wide array of regulators. Indeed, evidence supports that the catalytic activity and substrate specificity of PP1 are also determined by diverse physically interacting regulators (58, 59). It has been reported that PP1 has more than 50 known regulatory proteins, although more regulators are expected to be identified (25). Of note, although it was previously shown that Shoc, a Ras effector, regulates PP1-mediated dephosphorylation of Raf at Ser259, an inhibitory 14-3-3 binding site (60), no other PP1 regulators have been identified in the context of Ras/Raf/MEK/ERK signaling. Our present study strongly suggests that mortalin is a novel regulator of PP1 in the signaling context, where it facilitates PP1α-mediated MEK1/2 dephosphorylation.
MATERIALS AND METHODS
Cell culture and reagents.
The human B-RafV600E melanoma lines SK-MEL 28 (ATCC, Manassas, VA), SK-MEL 1 (ATCC), and RPMI-7951 (ATCC) were cultured in minimal essential medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, 1% sodium pyruvate, 1% nonessential amino acids, 100 U/ml of penicillin, and 100 μg/ml of streptomycin. The human B-RafV600E melanoma line A375 (ATCC), the human B-RafV600E thyroid cancer lines 8505C and B-CPAP (obtained from Barry Nelkin at Johns Hopkins University), the human K-RasG12C pancreatic cancer line MiaPaCa-2 (ATCC), and the human K-RasG12D pancreatic cancer line PANC-1 (ATCC) were cultured in Dulbecco's minimal essential medium (Invitrogen) supplemented with 10% fetal bovine serum, 100 U/ml of penicillin, and 100 μg/ml of streptomycin. All cell lines were authenticated by the ATCC. PLX4032 and cantharidin were purchased from Selleck Chemicals (Houston, TX) and Sigma-Aldrich (St. Louis, MO), respectively. Calyculin A and (−)-p-bromolevamisole oxalate were purchased from Enzo Life Sciences (Farmingdale, NY). Peptide aptamers were custom synthesized by GenScript (Piscataway, NJ).
Plasmids, RNA interference, and recombinant viruses.
Construction of N-terminally HA-tagged mortalin in pHAGE (pHAGE-HA-Mort) was previously described (13). To construct mortalin domain mutants, the N-terminal ATPase domain (AD) (aa 1 to 435), the C-terminal peptide-binding domain (PBD) (aa 435 to 679), the C-terminal peptide-binding domain without the tail (PBD-Δtail) (aa 435 to 552), and the regulatory subdomain 2 (SD2) (aa 221 to 435) were PCR amplified using primers TTATCCGCTAGCATAAGTGCCAGCCGAGCTGCAGCAGCCCG and TTTCTGGGATCCTTACACATCCGTGACATCGCCGGCC, TTATCCGCTAGCGATGTGCTGCTCCTTGATGTCACTCCCC and TTTCTGGGATCCTTACTGTTTTTCCTCCTTTTGATCTTCC, TTATCCGCTAGCGATGTGCTGCTCCTTGATGTCACTCCCC and TTTCTGGGATCCTTATCCACCAGAAGACTGGATTACAATCTG, and TTATCCGCTAGCAATGAGCCCACAGCTGCTGCTCTTGCC and TTTCTGGGATCCTTACACATCCGTGACATCGCCGGCC, respectively. These constructs were used to replace the full-length mortalin in the NheI/BamHI site of pHAGE-HA-Mort. pHAGE-HA-PBD-V482F, -V487A, and -V487Y were generated by mutagenizing pHAGE-HA-PBD using the QuikChange II site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) and the primers GCCGCTGATGGTCAAACGCAATTCGAAATTAAAGTGTGTCAGGG and CCCTGACACACTTTAATTTCGAATTGCGTTTGACCATCAGCGGC, CAAGTGGAAATTAAAGTGGCTCAGGGTGAAAGAGAGAT and ATCTCTCTTTCACCCTGAGCCACTTTAATTTCCACTTG, and CAAGTGGAAATTAAAGTGTATCAGGGTGAAAGAGAGAT and ATCTCTCTTTCACCCTGATACACTTTAATTTCCACTTG, respectively. The lentiviral shRNA expression systems targeting human mortalin, pLL3.7-shMort#1, and pLL3.7-shMort#2, and the pTRIPZ doxycycline-inducible microRNA-adapted shRNA targeting human mortalin (pTRIPZ-shMort-mir) were previously described (13, 22). pLKO-shPP1α constructs and were purchased from Sigma-Aldrich (TRCN0000002452 and TRCN0000002454).
For lentivirus production, 293T cells were cotransfected with pHAGE or pLL3.7 and packaging vectors, as previously described (61, 62). Viral supernatants were collected after 72 h and mixed with Polybrene (Sigma-Aldrich) at 4 to 8 μg/ml before use. The viral titer was determined by scoring cells expressing green fluorescent protein (GFP).
Immunoblot analysis.
Cells harvested at various times were lysed in 62.5 mM Tris (pH 6.8)–2% SDS mixed with protease and phosphatase inhibitor cocktail (Sigma-Aldrich) containing 4-(2-aminoethyl)benzenesulfonyl fluoride, pepstatin A, E-64, bestatin, leupeptin, aprotinin, cantharidin, calyculin A, and (−)-p-bromolevamisole oxalate and briefly sonicated before determining the protein concentration using bicinchoninic acid (BCA) reagent (Pierce, Rockford, IL). Then, 50 μg of protein was resolved by SDS-PAGE and transferred to a polyvinylidene difluoride membrane filter (Bio-Rad, Hercules, CA). Membrane filters were blocked in 0.1 M Tris (pH 7.5)–0.9% NaCl–0.05% Tween 20 with 5% nonfat dry milk and incubated with appropriate antibodies.
Antibodies and their dilutions were as follows: phospho-MEK1/2 (Ser217/221 for MEK1 and Ser222/226 for MEK2, no. 9121), 1:2,500; MEK1/2 (no. 9122), 1:2,500; MEK2 (no. 9125), 1:2,500; ERK1/2 (no. 9102), 1:2,500; phospho-B-Raf (Ser445, no. 2696), 1:2,500; phospho-A-Raf (Ser299, no. 4431), 1:2,500; phospho-C-Raf (Ser338, no. 9427), 1:2,500; phospho-C-Raf (Ser259, no. 9421), 1:2,500; phospho-PP1α (Thr320, no. 2581), 1:2,500; PP2A-A, -B, and -C subunits (no. 2041, 2290, and 2259, respectively), 1:2,500; and glutathione S-transferase (GST) (no. 2625), 1:2,500 (Cell Signaling Technology, Boston, MA); MEK1 (sc-6250), 1:2,500; phospho-ERK1/2 (Thr202/Tyr204, sc-16982), 1:2,500; B-Raf (sc-9002), 1:2,500; mortalin (sc-133137), 1:5,000; PP1α (sc-7482), 1:2,500; hemagglutinin (HA) (sc-7392), 1:2,500; and polyhistidine (HIS) (sc-8036), 1:2,500 (Santa Cruz Biotechnology, Santa Cruz, CA); and β-actin, 1:1,0000 (A2228) (Sigma-Aldrich). SuperSignal West Pico and Femto chemiluminescence kits (Pierce, Rockford, IL) were used for visualization of the signal. For densitometry, immunoblots were scanned and analyzed using Image Lab (Bio-Rad).
IP.
For immunoprecipitation (IP), cells were lysed in 50 mM Tris (pH 7.5)–150 mM NaCl–1% NP-40 and mixed with protease and phosphatase inhibitor cocktail. One milligram of cell lysate was incubated with 2 μg of specific monoclonal antibodies for 2 h. Antibody-protein complexes were then pulled down with Protein G Plus-agarose beads (Santa Cruz Biotechnology), as described previously (13, 62). Normal IgG derived from corresponding species (Santa Cruz Biotechnology) was used as the control. HA-tagged mortalin and its domains were immunoprecipitated using anti-HA-conjugated agarose (Thermo Fisher Scientific, Waltham, MA).
For in vitro binding assays, equal concentrations (0.5 μM) of N-terminally GST-tagged mortalin (Sigma-Aldrich), N-terminally HIS-tagged PP1α (ProSpec, East Brunswick, NJ), and N-terminally HIS-tagged MEK2 (ProSpec) were incubated in 50 mM Tris (pH 7.4)–100 mM KCl–100 mM NaCl–5 mM MgCl2–5% glycerol–10 mM β-mercaptoethanol for 30 min at room temperature. GST (Sigma-Aldrich) was used as the control. Fifty microliters of glutathione-Sepharose 4B (GE Healthcare Life Sciences, Marlborough, MA) was then added to the mixture and left for 1 h at 4°C with gentle agitation. After an extensive wash with the binding buffer, protein-bound beads were collected for Western blotting.
In vitro dephosphorylation assays.
Cells were lysed in 50 mM Tris (pH 7.4)–120 mM NaCl–1 mM EGTA–1% NP-40 and mixed with the protease inhibitor cocktail. For in vitro dephosphorylation assays, 50 μg cell lysate was mixed with 20 mM Tris (pH 7.5)–5 mM MgCl2–1 mM EGTA–0.02% β-mercaptoethanol containing 0.1 μg/μl bovine serum albumin. These mixtures were incubated with different phosphatase inhibitors at the indicated concentrations at room temperature for 60 min. At the end of the incubation, 2× Laemmli buffer (120 mM Tris [pH 6.8]–20% glycerol–4% SDS–0.01% bromophenol blue–10% 2-mercaptoethanol) was added to stop the reaction. For the assay using recombinant HIS-PP1α, MEK2 immunoprecipitated from SK-MEL 28 cell lysates was washed two times with 10 volumes of 50 mM Tris (pH 7.5)–500 mM NaCl–1 mM EDTA, followed by a wash with 10 volumes of 50 mM Tris (pH 7.5)–60 mM NaCl–1 mM EDTA–0.1% NP-40 and a final wash with 10 volumes of phosphate-buffered saline. Immunoprecipitated MEK2 proteins were then incubated with 0.5 μM HIS-PP1α or GST under the conditions described above. MEK1/2 phosphorylation was analyzed by Western blotting.
GST-PBD expression and purification.
A cDNA encoding the PBD of mortalin was cloned into pGEX-6P (GE Healthcare Life Sciences) to generate an N-terminally GST-tagged PBD (GST-PBD). One liter of BL21(DE3) transformed with this pGEX-6P-PBD plasmid was grown in Luria-Bertani medium at 37°C for 16 h. Expression of GST-PBD was induced with 1 mM isopropyl-β-d-thiogalactopyranoside for 16 h at 16°C. Cell pellets were then collected by centrifugation, resuspended in 20 ml of 50 mM Tris (pH 7.5)–150 mM NaCl–0.5% NP-40–1 mM EDTA–1 mM dithiothreitol (DTT) containing protease inhibitor cocktail, and homogenized by sonication. The supernatant containing soluble GST-PBD was incubated with 0.5 ml (vol/vol) glutathione-agarose (Gold Biotechnology, St. Louis, MO) at 4°C for 2 h. After this, the resins were washed three times with 10 volumes of 50 mM Tris (pH 7.5)–500 mM NaCl–1 mM EDTA and two times with 10 volume of 50 mM Tris (pH 7.5)–60 mM NaCl–0.1% NP-40–1 mM EDTA before elution of the recombinant protein using 10 volume of 50 mM Tris (pH 7.0)–40 mM glutathione. Purified GST-PBD was concentrated with a 10-kDa-cutoff Centricon filter unit (EMD Millipore, Billerica, MA).
Statistical analysis.
Statistical significance was determined by the t test, the Mann-Whitney test, one-way analysis of variance (ANOVA), and Pearson correlation coefficient analysis using Prism (GraphPad Software, La Jolla, CA). P values of <0.05 were considered significant.
ACKNOWLEDGMENTS
We thank Sudhakar Veeranki and Mansi Karkhanis for technical support. We also thank Barry Nelkin at Johns Hopkins University for 8505C and B-CPAP.
This work was supported by National Cancer Institute grant R01CA138441 to J.-I.P.
We declare no conflict of interest.
P.-K.W. and S.-K.H. performed experiments. P.-K.W., S.-K.H., and J.-I.P. interpreted the data. P.-K.W. and J.-I.P. conceived and designed the research and prepared the manuscript. J.-I.P. supervised the research and secured funding. All authors reviewed the results and approved the final version of the manuscript.
REFERENCES
- 1.Shaul YD, Seger R. 2007. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta 1773:1213–1226. doi: 10.1016/j.bbamcr.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 2.Canagarajah BJ, Khokhlatchev A, Cobb MH, Goldsmith EJ. 1997. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell 90:859–869. doi: 10.1016/S0092-8674(00)80351-7. [DOI] [PubMed] [Google Scholar]
- 3.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, Evangelisti C, Martelli AM, Franklin RA. 2007. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts PJ, Der CJ. 2007. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 5.Chambard JC, Lefloch R, Pouyssegur J, Lenormand P. 2007. ERK implication in cell cycle regulation. Biochim Biophys Acta 1773:1299–1310. doi: 10.1016/j.bbamcr.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 6.Park JI. 2014. Growth arrest signaling of the Raf/MEK/ERK pathway in cancer. Front Biol 9:95–103. doi: 10.1007/s11515-014-1299-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mebratu Y, Tesfaigzi Y. 2009. How ERK1/2 activation controls cell proliferation and cell death: is subcellular localization the answer? Cell Cycle 8:1168–1175. doi: 10.4161/cc.8.8.8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cagnol S, Chambard JC. 2010. ERK and cell death: mechanisms of ERK-induced cell death–apoptosis, autophagy and senescence. FEBS J 277:2–21. doi: 10.1111/j.1742-4658.2009.07366.x. [DOI] [PubMed] [Google Scholar]
- 9.Mooi WJ, Peeper DS. 2006. Oncogene-induced cell senescence—halting on the road to cancer. N Engl J Med 355:1037–1046. doi: 10.1056/NEJMra062285. [DOI] [PubMed] [Google Scholar]
- 10.McDuff FK, Turner SD. 2011. Jailbreak: oncogene-induced senescence and its evasion. Cell Signal 23:6–13. doi: 10.1016/j.cellsig.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 11.Cheung M, Sharma A, Madhunapantula SV, Robertson GP. 2008. Akt3 and mutant V600E B-Raf cooperate to promote early melanoma development. Cancer Res 68:3429–3439. doi: 10.1158/0008-5472.CAN-07-5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z, Liu Y, Takahashi M, Van Hook K, Kampa-Schittenhelm KM, Sheppard BC, Sears RC, Stork PJ, Lopez CD. 2013. N terminus of ASPP2 binds to Ras and enhances Ras/Raf/MEK/ERK activation to promote oncogene-induced senescence. Proc Natl Acad Sci U S A 110:312–317. doi: 10.1073/pnas.1201514110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu PK, Hong SK, Veeranki S, Karkhanis M, Starenki D, Plaza JA, Park JI. 2013. A mortalin/HSPA9-mediated switch in tumor-suppressive signaling of Raf/MEK/extracellular signal-regulated kinase. Mol Cell Biol 33:4051–4067. doi: 10.1128/MCB.00021-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daugaard M, Rohde M, Jaattela M. 2007. The heat shock protein 70 family: highly homologous proteins with overlapping and distinct functions. FEBS Lett 581:3702–3710. doi: 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- 15.Lee AS. 2014. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer 14:263–276. doi: 10.1038/nrc3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaul SC, Deocaris CC, Wadhwa R. 2007. Three faces of mortalin: a housekeeper, guardian and killer. Exp Gerontol 42:263–274. doi: 10.1016/j.exger.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 17.Wadhwa R, Takano S, Kaur K, Deocaris CC, Pereira-Smith OM, Reddel RR, Kaul SC. 2006. Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int J Cancer 118:2973–2980. doi: 10.1002/ijc.21773. [DOI] [PubMed] [Google Scholar]
- 18.Rozenberg P, Kocsis J, Saar M, Prohaszka Z, Fust G, Fishelson Z. 2013. Elevated levels of mitochondrial mortalin and cytosolic HSP70 in blood as risk factors in patients with colorectal cancer. Int J Cancer 133:514–518. doi: 10.1002/ijc.28029. [DOI] [PubMed] [Google Scholar]
- 19.Chen J, Liu WB, Jia WD, Xu GL, Ma JL, Huang M, Deng YR, Li JS. 2014. Overexpression of Mortalin in hepatocellular carcinoma and its relationship with angiogenesis and epithelial to mesenchymal transition. Int J Oncol 44:247–255. doi: 10.3892/ijo.2013.2161. [DOI] [PubMed] [Google Scholar]
- 20.Na Y, Kaul SC, Ryu J, Lee JS, Ahn HM, Kaul Z, Kalra RS, Li L, Widodo N, Yun CO, Wadhwa R. 2016. Stress chaperone mortalin contributes to epithelial-mesenchymal transition and cancer metastasis. Cancer Res doi: 10.1158/0008-5472.CAN-15-2704. [DOI] [PubMed] [Google Scholar]
- 21.Yun CO, Bhargava P, Na Y, Lee JS, Ryu J, Kaul SC, Wadhwa R. 2017. Relevance of mortalin to cancer cell stemness and cancer therapy. Sci Rep 7:42016. doi: 10.1038/srep42016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Starenki D, Hong SK, Lloyd RV, Park JI. 2015. Mortalin (GRP75/HSPA9) upregulation promotes survival and proliferation of medullary thyroid carcinoma cells. Oncogene 34:4624–4634. doi: 10.1038/onc.2014.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karkhanis M, Park JI. 2015. Sp1 regulates Raf/MEK/ERK-induced p21 transcription in TP53-mutated cancer cells. Cell Signal 27:479–486. doi: 10.1016/j.cellsig.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi Y. 2009. Serine/threonine phosphatases: mechanism through structure. Cell 139:468–484. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 25.Gallego M, Virshup DM. 2005. Protein serine/threonine phosphatases: life, death, and sleeping. Curr Opin Cell Biol 17:197–202. doi: 10.1016/j.ceb.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 26.Gibbons JA, Kozubowski L, Tatchell K, Shenolikar S. 2007. Expression of human protein phosphatase-1 in Saccharomyces cerevisiae highlights the role of phosphatase isoforms in regulating eukaryotic functions. J Biol Chem 282:21838–21847. doi: 10.1074/jbc.M701272200. [DOI] [PubMed] [Google Scholar]
- 27.Heriche JK, Lebrin F, Rabilloud T, Leroy D, Chambaz EM, Goldberg Y. 1997. Regulation of protein phosphatase 2A by direct interaction with casein kinase 2alpha. Science 276:952–955. doi: 10.1126/science.276.5314.952. [DOI] [PubMed] [Google Scholar]
- 28.Gomez N, Cohen P. 1991. Dissection of the protein kinase cascade by which nerve growth factor activates MAP kinases. Nature 353:170–173. doi: 10.1038/353170a0. [DOI] [PubMed] [Google Scholar]
- 29.Anderson NG, Maller JL, Tonks NK, Sturgill TW. 1990. Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase. Nature 343:651–653. doi: 10.1038/343651a0. [DOI] [PubMed] [Google Scholar]
- 30.Sontag E, Fedorov S, Kamibayashi C, Robbins D, Cobb M, Mumby M. 1993. The interaction of SV40 small tumor antigen with protein phosphatase 2A stimulates the map kinase pathway and induces cell proliferation. Cell 75:887–897. doi: 10.1016/0092-8674(93)90533-V. [DOI] [PubMed] [Google Scholar]
- 31.Osborne JK, Zaganjor E, Cobb MH. 2012. Signal control through Raf: in sickness and in health. Cell Res 22:14–22. doi: 10.1038/cr.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng CF, Guan KL. 1994. Activation of MEK family kinases requires phosphorylation of two conserved Ser/Thr residues. EMBO J 13:1123–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Vande Woude GF, Ahn NG. 1994. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 265:966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- 34.Honkanen RE, Golden T. 2002. Regulators of serine/threonine protein phosphatases at the dawn of a clinical era? Curr Med Chem 9:2055–2075. doi: 10.2174/0929867023368836. [DOI] [PubMed] [Google Scholar]
- 35.Onsgard-Meyer M, McCoy AL, Knox FG. 1996. Effect of bromotetramisole on renal phosphate excretion. Proc Soc Exp Biol Med 213:193–195. doi: 10.3181/00379727-213-44050. [DOI] [PubMed] [Google Scholar]
- 36.Mason CS, Springer CJ, Cooper RG, Superti-Furga G, Marshall CJ, Marais R. 1999. Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. EMBO J 18:2137–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chong H, Lee J, Guan KL. 2001. Positive and negative regulation of Raf kinase activity and function by phosphorylation. EMBO J 20:3716–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dohadwala M, da Cruz e Silva EF, Hall FL, Williams RT, Carbonaro-Hall DA, Nairn AC, Greengard P, Berndt N. 1994. Phosphorylation and inactivation of protein phosphatase 1 by cyclin-dependent kinases. Proc Natl Acad Sci U S A 91:6408–6412. doi: 10.1073/pnas.91.14.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu CW, Wang RH, Dohadwala M, Schonthal AH, Villa-Moruzzi E, Berndt N. 1999. Inhibitory phosphorylation of PP1alpha catalytic subunit during the G(1)/S transition. J Biol Chem 274:29470–29475. doi: 10.1074/jbc.274.41.29470. [DOI] [PubMed] [Google Scholar]
- 40.Berndt N. 1999. Protein dephosphorylation and the intracellular control of the cell number. Front Biosci 4:D22–D42. [DOI] [PubMed] [Google Scholar]
- 41.Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, Hendrickson WA. 1996. Structural analysis of substrate binding by the molecular chaperone DnaK. Science 272:1606–1614. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kampinga HH, Craig EA. 2010. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol 11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amick J, Schlanger SE, Wachnowsky C, Moseng MA, Emerson CC, Dare M, Luo WI, Ithychanda SS, Nix JC, Cowan JA, Page RC, Misra S. 2014. Crystal structure of the nucleotide-binding domain of mortalin, the mitochondrial Hsp70 chaperone. Protein Sci 23:833–842. doi: 10.1002/pro.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang P, Leu JI, Murphy ME, George DL, Marmorstein R. 2014. Crystal structure of the stress-inducible human heat shock protein 70 substrate-binding domain in complex with peptide substrate. PLoS One 9:e103518. doi: 10.1371/journal.pone.0103518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laufen T, Mayer MP, Beisel C, Klostermeier D, Mogk A, Reinstein J, Bukau B. 1999. Mechanism of regulation of hsp70 chaperones by DnaJ cochaperones. Proc Natl Acad Sci U S A 96:5452–5457. doi: 10.1073/pnas.96.10.5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iosefson O, Azem A. 2010. Reconstitution of the mitochondrial Hsp70 (mortalin)-p53 interaction using purified proteins—identification of additional interacting regions. FEBS Lett 584:1080–1084. doi: 10.1016/j.febslet.2010.02.019. [DOI] [PubMed] [Google Scholar]
- 47.Sun D, Zhou M, Kowolik CM, Trisal V, Huang Q, Kernstine KH, Lian F, Shen B. 2011. Differential expression patterns of capping protein, protein phosphatase 1, and casein kinase 1 may serve as diagnostic markers for malignant melanoma. Melanoma Res 21:335–343. doi: 10.1097/CMR.0b013e328346b715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hong SK, Wu PK, Karkhanis M, Park JI. 2015. ERK1/2 can feedback-regulate cellular MEK1/2 levels. Cell Signal 27:1939–1948. doi: 10.1016/j.cellsig.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mashaghi A, Bezrukavnikov S, Minde DP, Wentink AS, Kityk R, Zachmann-Brand B, Mayer MP, Kramer G, Bukau B, Tans SJ. 2016. Alternative modes of client binding enable functional plasticity of Hsp70. Nature 539:448–451. doi: 10.1038/nature20137. [DOI] [PubMed] [Google Scholar]
- 50.Wadhwa R, Takano S, Robert M, Yoshida A, Nomura H, Reddel RR, Mitsui Y, Kaul SC. 1998. Inactivation of tumor suppressor p53 by mot-2, a hsp70 family member. J Biol Chem 273:29586–29591. doi: 10.1074/jbc.273.45.29586. [DOI] [PubMed] [Google Scholar]
- 51.Wadhwa R, Yaguchi T, Hasan MK, Taira K, Kaul SC. 2003. Mortalin-MPD (mevalonate pyrophosphate decarboxylase) interactions and their role in control of cellular proliferation. Biochem Biophys Res Commun 302:735–742. doi: 10.1016/S0006-291X(03)00226-2. [DOI] [PubMed] [Google Scholar]
- 52.Lu WJ, Lee NP, Kaul SC, Lan F, Poon RT, Wadhwa R, Luk JM. 2011. Mortalin-p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Differ 18:1046–1056. doi: 10.1038/cdd.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wittrup A, Zhang SH, Svensson KJ, Kucharzewska P, Johansson MC, Morgelin M, Belting M. 2010. Magnetic nanoparticle-based isolation of endocytic vesicles reveals a role of the heat shock protein GRP75 in macromolecular delivery. Proc Natl Acad Sci U S A 107:13342–13347. doi: 10.1073/pnas.1002622107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morgner N, Schmidt C, Beilsten-Edmands V, Ebong IO, Patel NA, Clerico EM, Kirschke E, Daturpalli S, Jackson SE, Agard D, Robinson CV. 2015. Hsp70 forms antiparallel dimers stabilized by post-translational modifications to position clients for transfer to Hsp90. Cell Rep 11:759–769. doi: 10.1016/j.celrep.2015.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sarbeng EB, Liu Q, Tian X, Yang J, Li H, Wong JL, Zhou L, Liu Q. 2015. A functional DnaK dimer is essential for the efficient interaction with Hsp40 heat shock protein. J Biol Chem 290:8849–8862. doi: 10.1074/jbc.M114.596288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. 2002. The protein kinase complement of the human genome. Science 298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 57.Sacco F, Perfetto L, Castagnoli L, Cesareni G. 2012. The human phosphatase interactome: an intricate family portrait. FEBS Lett 586:2732–2739. doi: 10.1016/j.febslet.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peti W, Nairn AC, Page R. 2013. Structural basis for protein phosphatase 1 regulation and specificity. FEBS J 280:596–611. doi: 10.1111/j.1742-4658.2012.08509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heroes E, Lesage B, Gornemann J, Beullens M, Van Meervelt L, Bollen M. 2013. The PP1 binding code: a molecular-lego strategy that governs specificity. FEBS J 280:584–595. doi: 10.1111/j.1742-4658.2012.08547.x. [DOI] [PubMed] [Google Scholar]
- 60.Rodriguez-Viciana P, Oses-Prieto J, Burlingame A, Fried M, McCormick F. 2006. A phosphatase holoenzyme comprised of Shoc2/Sur8 and the catalytic subunit of PP1 functions as an M-Ras effector to modulate Raf activity. Mol Cell 22:217–230. doi: 10.1016/j.molcel.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 61.Hong SK, Yoon S, Moelling C, Arthan D, Park JI. 2009. Noncatalytic function of ERK1/2 can promote Raf/MEK/ERK-mediated growth arrest signaling. J Biol Chem 284:33006–33018. doi: 10.1074/jbc.M109.012591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu PK, Hong SK, Yoon SH, Park JI. 2015. Active ERK2 is sufficient to mediate growth arrest and differentiation signaling. FEBS J 282:1017–1030. doi: 10.1111/febs.13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, Vivanco I, Lee JC, Huang JH, Alexander S, Du J, Kau T, Thomas RK, Shah K, Soto H, Perner S, Prensner J, Debiasi RM, Demichelis F, Hatton C, Rubin MA, Garraway LA, Nelson SF, Liau L, Mischel PS, Cloughesy TF, Meyerson M, Golub TA, Lander ES, Mellinghoff IK, Sellers WR. 2007. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A 104:20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taylor BS, Barretina J, Socci ND, Decarolis P, Ladanyi M, Meyerson M, Singer S, Sander C. 2008. Functional copy-number alterations in cancer. PLoS One 3:e3179. doi: 10.1371/journal.pone.0003179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, Choti MA, Yeo CJ, McCue P, White MA, Knudsen ES. 2015. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 6:6744. doi: 10.1038/ncomms7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F. 2015. Proteomics tissue-based map of the human proteome. Science 347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]