

ABSTRACT
Transforming growth factor β (TGF-β)-induced migration of triple-negative breast cancer (TNBC) cells is dependent on nuclear export of the orphan receptor NR4A1, which plays a role in proteasome-dependent degradation of SMAD7. In this study, we show that TGF-β induces p38α (mitogen-activated protein kinase 14 [MAPK14]), which in turn phosphorylates NR4A1, resulting in nuclear export of the receptor. TGF-β/p38α and NR4A1 also play essential roles in the induction of epithelial-to-mesenchymal transition (EMT) and induction of β-catenin in TNBC cells, and these TGF-β-induced responses and nuclear export of NR4A1 are blocked by NR4A1 antagonists, the p38 inhibitor SB202190, and kinase-dead [p38(KD)] and dominant-negative [p38(DN)] forms of p38α. Inhibition of NR4A1 nuclear export results in nuclear export of TGF-β-induced β-catenin, which then undergoes proteasome-dependent degradation. TGF-β-induced β-catenin also regulates NR4A1 expression through formation of the β-catenin–TCF-3/TCF-4/LEF-1 complex on the NR4A1 promoter. Thus, TGF-β-induced nuclear export of NR4A1 in TNBC cells plays an essential role in cell migration, SMAD7 degradation, EMT, and induction of β-catenin, and all of these pathways are inhibited by bis-indole-derived NR4A1 antagonists that inhibit nuclear export of the receptor and thereby block TGF-β-induced migration and EMT.
KEYWORDS: TGF-β, breast cancer, migration, NR4A1, p38, TGF-beta, p38 kinases
INTRODUCTION
Transforming growth factor β (TGF-β) signaling plays an important and complex role in cancer and exhibits both tumor-promoting and tumor suppressor functions that are dependent on the cancer cell context (1–3). For example, loss of TGF-β in an immortalized but nontumorigenic MCF-10A cell subtype resulted in enhanced growth due to a less differentiated phenotype, and this was accompanied by a decreased population of early progenitor cells (4). In contrast, TGF-β stimulates later-stage cancer cells to migrate and invade, and this is linked to induction of epithelial-to-mesenchymal transition (EMT) and a more aggressive cancer cell phenotype (1, 5, 6). TGF-β signaling is initiated by ligand-dependent TGF-β receptor activation, which results in formation of an activated SMAD2/SMAD3-SMAD4 nuclear transcription factor complex (7). In contrast, SMAD7 functions as an inhibitory SMAD by recruiting factors leading to proteasome-dependent degradation of TGF-β receptor 1 (TGF-βR1) (8, 9).
Arkadia, RNF12, and axin2 also play key roles in activation of TGF-β signaling by inducing polyubiquitination and degradation of inhibitory SMAD7 (9, 10), and a recent study demonstrated that the orphan nuclear receptor NR4A1 (Nur77 or TR3) also has a critical role in SMAD7 degradation (11). It was reported that NR4A1 induces axin2 expression, and NR4A1 directly interacts with axin2 and SMAD7 and is an obligatory factor for activation of SMAD7 degradation. This model was derived from studies on inflammatory-cytokine-induced MDA-MB-231 breast cancer cell invasion and showed that cytokine-induced NR4A1 was necessary for SMAD7 degradation and TGF-β-induced migration/invasion.
Studies in our laboratory have investigated NR4A1-dependent prooncogenic pathways in breast and other cancer cell lines, and we have developed 1,1-bis(3-indolyl)-1-(p-substituted phenyl) methane (C-DIM) NR4A1 ligands that act as NR4A1 antagonists in cancer cell lines and tumors (12–19). In MDA-MB-231 breast cancer cells, we demonstrated that basal migration/invasion are dependent on NR4A1-regulated expression of β1-integrin, which can be inhibited by C-DIM/NR4A1 antagonist (19). TGF-β markedly enhances MDA-MB-231 cell migration/invasion, and as previously reported (11), we observed that this response is NR4A1 dependent and could also be inhibited by C-DIM/NR4A1 antagonists (12). Our studies demonstrated that the key inhibitory effect of the NR4A1 antagonist is to block nuclear export of NR4A1 and thereby prevent interactions with the cytosolic axin2/RNF12/Arkadia/SMAD7 complex.
In this study, we further investigated the critical and essential role of NR4A1 in regulating TGF-β-induced migration/invasion, and we show that TGF-β-induced nuclear export of NR4A1 is dependent on activation of the p38 mitogen-activated protein kinase (MAPK) pathway. We also show that TGF-β induces expression of both NR4A1 and β-catenin, and these responses are also p38 dependent. However, induction of NR4A1 by TGF-β is dependent on β-catenin and its interactions with LEF1/TCF3 and TCF4 bound to the NR4A1 promoter. These studies define a unique role for NR4A1 in TGF-β-induced migration and EMT in estrogen receptor (ER)-negative breast cancer cells and also demonstrate that NR4A1 antagonists block this response, indicating potential clinical applications for treatment of both early and late stages of the disease.
RESULTS
TGF-β-dependent inhibition of migration is dependent on activation of p38-mediated nuclear export of NRA41.
It was initially reported that TGF-β-induced EMT and migration/invasion were dependent on NR4A1, which formed an E3 ligase-axin2/RNF12/Arkadia complex that degraded SMAD7 and thereby activated TGF-βR1 (11). Subsequent studies in our laboratory indicated that a key step in this pathway involved TGF-β-induced nuclear export of NR4A1 (19), and the mechanisms of TGF-β-NR4A1 cross talk were further investigated in ER-negative breast cancer cells. The results shown in Fig. 1A demonstrate that among a series of kinase inhibitors, LY294002 and PD98059 inhibited basal migration of MDA-MB-231 cells, but only the p38 MAPK inhibitor SB202190 significantly inhibited TGF-β-induced migration of MDA-MB-231 cells. We also observed that the p38 inhibitor SB202190 also inhibited TGF-β-induced migration in H5587T and SUM159 triple-negative breast cancer (TNBC) cells (Fig. 1A), whereas LY294002 and PD98059 decreased basal but not TGF-β-induced migration, as observed in MDA-MB-231 cells. TGF-β induces expression and nuclear export of NR4A1 (19), and we now show that this response is significantly inhibited in MDA-MB-231 cells cotreated with the p38 inhibitor SB202190 (Fig. 1B). In contrast, SP600125, LY294002, and PD98059 did not inhibit TGF-β-dependent nuclear export of NR4A1; however, the last two inhibitors decreased induction of NR4A1 by TGF-β (Fig. 1B). These results suggest that TGF-β induces activation of p38, and this was confirmed in MDA-MB-231 cells, where treatment with TGF-β enhanced phosphorylation of p38α (T180/Y182) and also increased phospho-NR4A1 (p-NR4A1) (S355) expression (Fig. 1B). TGF-β also induced NR4A1 expression and nuclear export in H5587T and SUM159 cells (Fig. 1C and D), and this was inhibited by SB202190. SP600125, LY294002, and PD98059 did not inhibit TGF-β-mediated nuclear export of NR4A1 but enhanced expression of the receptor. TGF-β also induced phosphorylation of NR4A1 (S355) and p38 (T180/Y182) in H5587T and SUM159 cells, and SB202190 inhibited this response (Fig. 1C and D), confirming that the TGF-β-induced NR4A1 nuclear export was p38 dependent in the three ER-negative breast cancer cell lines.
FIG 1.

TGF-β-induced migration and nuclear export of NR4A1 are p38 dependent. (A) Cells were treated with DMSO or 5 ng/ml TGF-β for 5 h in the presence or absence of several kinase inhibitors, and migration was determined as outlined in Materials and Methods. The results are means and standard errors (SE) for at least 3 replicate experiments, and significantly (*, P < 0.05) induced (TGF-β versus DMSO) results are indicated. (B to D) MDA-MB-231 (B), H5587T (C), or SUM159 (D) cells were treated with DMSO or 5 ng/ml TGF-β for 5 h in the presence of several kinase inhibitors, and nuclear and cytosolic or whole-cell lysates were analyzed by Western blotting. β-Actin, p84 and GAPDH served as loading controls for whole-cell lysates, nuclear extracts, and cytosolic extracts, respectively, for all studies.
The role of p38 in mediating TGF-β-induced nuclear export of NR4A1 was further investigated using constructs expressing constitutively active [p38(CA)], kinase domain inactive [p38(KD)], and dominant-negative [p38(DN)] p38 constructs in MDA-MB-231 cells, which were used as a model triple-negative breast cancer cell line for subsequent studies. p38(KD) and p38(DN) slightly increased expression of nuclear NR4A1 but did not induce nuclear export of the receptor, whereas both p38(CA) and TGF-β induced expression and nuclear export of NR4A1 protein (Fig. 2A). Figure 2B shows that p38(CA) alone and in combination with TGF-β induced nuclear export of NR4A1, whereas p38(KD) and p38(DN) alone and in combination with TGF-β did not induce nuclear export of the receptor, demonstrating that TGF-β did not rescue/enhance nuclear export of NR4A1 in cells expressing p38(KD) or p38(DN). Previous studies showed that the nuclear export inhibitor leptomycin B (LMB) and NR4A1 antagonists CDIM8 [1,1-bis(3′-indolyl)-1-(p-hydroxyphenyl)methane (DIM-C-pPhOH)] and CDIM14 [1,1-bis(3′-indolyl)-1-(p-carboxymethylphenyl)methane (DIM-C-pPhCO2Me)] block TGF-β-induced nuclear export of NR4A1 (19). In this study, the same treatments plus LMB/TGF-β also inhibited p38(CA)-dependent nuclear export of NR4A1 (Fig. 2C). Transfection of MDA-MB-231 cells with p38(CA) alone or treatment with 5 ng/ml TGF-β alone and in combination induced cell migration after treatment for 5 and 12 h, and the combination of p38 and TGF-β did not induce cell migration greater than that observed for each treatment alone (Fig. 2D). Both p38(KD) and p38(DN) alone did not induce MDA-MB-231 cell migration, and in combination with TGF-β, this inhibitory effect was not rescued, indicating that both kinases inhibited TGF-β-induced migration. The results shown in Fig. 2E demonstrate that TGF-β or p38(CA) (transfected) alone or in combination enhanced MDA-MB-231 cell invasion, and TGF-β cotreatment with agents that inhibit nuclear export of NR4A1 (LMB, CDIM8, CDIM14, and SB202190) (Fig. 1B and 2C) also inhibited of p38/TGF-β-induced invasion (Fig. 2F). We also used immunostaining and confocal microscopy to investigate treatment-related cytosolic and nuclear localization of NR4A1 (Fig. 3). p38CA and TGF-β treatment of MDA-MB-231 cells induced nuclear export of NR4A1, and immunostaining for the receptor showed that nuclear export was significantly abrogated by pretreatment with SB202190, leptomycin B, CDIM8, and CDIM14. Both p38(KD) and p38(DN) alone did not induce NR4A1 nuclear export, and supplementation with TGF-β did not rescue this effect, corroborating the results illustrated in Fig. 2A and B.
FIG 2.

Correlation between nuclear export of NR4A1 and TGF-β-induced cell migration. (A to C) MDA-MB-231 cells were treated with DMSO or 5 ng/ml TGF-β and transfected with p38(CA), p38(KD), or p38(DN) (A) or the same p38 constructs with or without TGF-β (B) or transfected with p38(CA) with or without TGF-β, LMB, and CDIM8/CDIM14 (C), and nuclear and cyctosolic extracts were obtained and analyzed by Western blotting. (D and E) MDA-MB-231 cells were transfected with p38(CA), p38(DN), or p38(KC) alone and in combination with TGF-β (5 and 12 h of treatment) (D) or transfected with p38(CA) with or without TGF-β, LMB, CDIM8/CDIM14, or SB202190 (E), and cell migration was determined as outlined in Materials and Methods. The error bars indicate SE.
FIG 3.
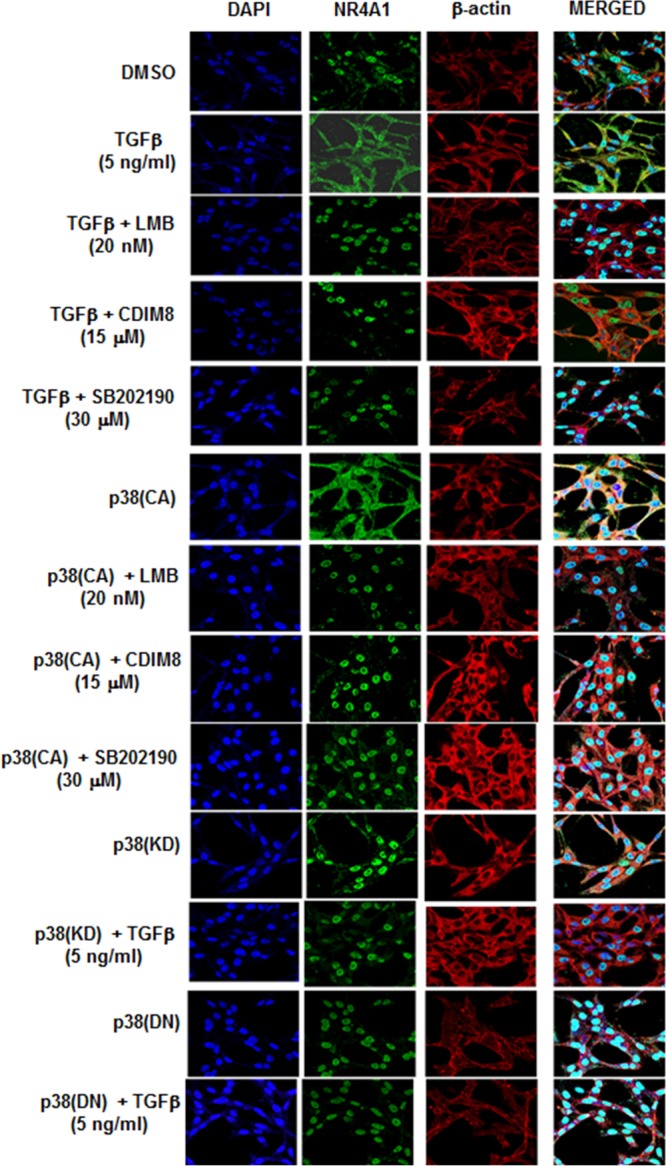
Confocal microscopy analysis. MDA-MB-231 cells were transfected and treated with the various agents, and nuclear export of NR4A1 was determined by immunostaining and confocal microscopy analysis as outlined in Materials and Methods.
NR4A1 interactions with axin2, NRF12, Arkadia, and SMAD7.
It was previously reported that NR4A1 played a role in axin2-RNF12/Arkadia-induced SMAD7 degradation (11), and in this study, we investigated the interactions of NR4A1 with these proteins. MDA-MB-231 cells were treated with 5 ng/ml TGF-β for 4 h, and cell protein lysates were immunoprecipitated with NR4A1 antibodies. Western blots of the immunoprecipitated proteins showed that NR4A1 was associated with axin2, Arkadia, RNF12, and SMAD7 (Fig. 4A). NR4A1 interactions with this cytosolic protein complex were abrogated after treatment with the NR4A1 ligands CDIM8, CDIM14, and LMB alone and in combination with TGF-β (Fig. 4A). These treatments maintained NR4A1 in the nucleus (19), suggesting that NR4A1 interacts with these proteins in the cytosol. We also examined interactions of the C- and N-terminal regions of N4RA1 with the axin2/RNF12/Arkadia/SMAD7 complex by transfecting cells with FLAG-NR4A1(C-F) or FLAG-NR4A1(A-B) and immunoprecipitated protein lysates with FLAG antibodies (Fig. 4B and C). The C-terminal ligand binding domain [FLAG-NR4A1(C-F)] interacted with axin2 and Arkadia and, to a lesser extent, RNF12 (Fig. 4B), whereas TGF-β induced interactions of the N-terminal domain of NR4A1 [FLAG-NR4A1(A-B)] with axin2 and SMAD7 (Fig. 4C and D). These interactions were abrogated by leptomycin B, CDIM8, CDIM14, and SB202190 (Fig. 4D). Moreover, association of NR4A1 with the complex was not observed after treatment with p38KD or p38DN alone or in combination with TGF-β. We also observed in MDA-MB-231 cells transfected with FLAG-NR4A1(full length) that FLAG antibodies coimmunoprecipitated axin2, Arkadia, RNF12, and SMAD7 (Fig. 4E) only in cells transfected with p38(CA) but not p38(KD) or p38(DN) and that these interactions were similar to those observed in TGF-β-treated cells using antibodies against constitutive NR4A1 (Fig. 4A). Transfection of MDA-MB-231 cells with FLAG-NR4A1(LBD) and overexpression of p38(CA) (with or without TGF-β), followed by immunoprecipitation with FLAG antibodies, demonstrated that the C-terminal domain of NR4A1 interacts with axin2, Arkadia, RNF12, and SMAD7 (Fig. 4F), whereas p38(KD) or p38(DN) with or without TGF-β did not induce interactions of NR4A1 with these proteins (Fig. 4F), indicating that both p38 mutants inhibited the TGF-β-induced response. These results demonstrate that NR4A1 export is important for interactions with the ubiquitination complex proteins and that the C-terminal domain of NR4A1 is necessary for interactions with all the members of this complex. The C-DIM/NR4A1 antagonists and the p38 inhibitor block NR4A1 nuclear export and interactions with the ubiquitination complex proteins.
FIG 4.

Interactions of NR4A1 with E3 ligase proteins. (A) MDA-MB-231 cells were treated with DMSO, 5 ng/ml TGF-β, CDIM8, and CDIM14 alone or in combination, and LMB plus TGF-β for 5 h. Whole-cell lysates were immunoprecipitated with NR4A1 antibodies, and the immunoprecipitate was analyzed on a Western blot. IB, immunoblotting. (B to G) MDA-MB-231 cells were transfected with FLAG-NR4A1(C-F) (B), FLAG-NR4A1(A-B) (C), FLAG-NR4A1(A-B) (D), FLAG-NR4A1 (E), FLAG-NR4A1(C-F) (F), and FLAG-NR4A1(C-F) (G), and after various treatments, whole-cell lysates were precipitated with FLAG antibodies. The solubilized precipitates were analyzed by Western blotting as outlined in Materials and Methods.
SMAD7 as a target of TGF-β-induced NR4A1 nuclear export.
The E3 ubiquitin ligase complex and NR4A1 are required for TGF-β-induced activation of TGF-βRI through SMAD7 degradation, and this is confirmed in Fig. 5A, which shows that TGF-β-induced migration of MDA-MB-231 cells is significantly decreased by knockdown of axin2, RNF12, and Arkadia. The specificity of the knockdown (Fig. 5A) also shows that loss of RNF12 knockdown results in decreased expression of both RNF12 and Arkadia. Since NR4A1 is an integral part of the E3 ubiquitin ligase complex and is required for its function, we further investigated the roles of NR4A1 and NR4A1 nuclear export in mediating SMAD7 degradation. SMAD7 is highly expressed in MDA-MB-231 cells (Fig. 5B), and treatment with TGF-β decreased SMAD7. However, treatment with TGF-β plus agents that inhibit NR4A1 export (LMB, CDIM8, and CDIM14) or p38 activity (SB202190) blocked proteasome-dependent downregulation of SMAD7, and the proteasome inhibitor MG132 also blocked the TGF-β-induced response. Moreover, TGF-β and p38(CA) alone or in combination also decreased SMAD7 protein, and this was blocked by MG132, whereas p38(KD) and p38(DN) alone or in combination with TGF-β did not affect SMAD7 expression (Fig. 5C). Thus, treatment with TGF-β did not overcome or rescue the cells from the effects of p38(KD) or p38(DN). These results show that TGF-β- and p38-induced nuclear export of NR4A1 is necessary to activate the E3 ubiquitin ligase complex (Arkadia, NR4A1, RNF12, and axin2) and for proteasome-dependent degradation of SMAD7. This was also confirmed by examination of SMAD7 ubiquitination. Inhibitors that blocked nuclear export of NR4A1 (Fig. 5D) and inactivation of p38 (Fig. 5E and F) all inhibited TGF-β-induced ubiquitination of SMAD7, which appears as a streaking band in the gel, which is typical of blots containing multiple polyubiquitinated proteins. In addition, we also show that TGF-β-induced ubiquitination is partially blocked by axin2 knockdown and completely inhibited by knockdown of Arkadia and RNF12 (Fig. 5G). The results demonstrate the importance of members of the ubiquitin ligase complex in mediating ubiquitination and subsequent degradation of SMAD7 and the inhibition of SMAD7 degradation by NR4A1 antagonists and inhibition of p38.
FIG 5.

Roles of NR4A1 and E3 ligase proteins in SMAD7 degradation. (A) MDA-MB-231 cells were treated with DMSO or 5 ng/ml TGF-β in the presence or absence of knockdown of E2 ligase proteins, SMAD3, or SMAD7 by RNA interference, and migration was determined as outlined in Materials and Methods. The error bars indicate SE (*, P < 0.05). Cells were transfected with siCtl (control) or oligonucleotides targeting E2 ligase proteins treated with 5 ng/ml TGF-β for 5 h, and whole-cell lysates were analyzed in a Western blot. (B and C) Cells were treated and/or transfected with various constructs, and the effects of the proteasome inhibitor MG132 on SMAD7 expression was determined in Western blots. (D to G) Cells were treated with various compounds and/or transfected with p38-derived constructs, whole-cell lysates were immunoprecipitated with SMAD7 antibodies, and the immunoprecipitates were solubilized and analyzed for ubiquitinated SMAD7 species by Western blotting.
Does NR4A1 play a role in TGF-β-induced β-catenin expression and EMT?
It has previously been reported that p38 induces expression of β-catenin (20), and the results illustrated in Fig. 6A show that low levels of β-catenin are expressed in MDA-MB-231 cells. However, treatment with TGF-β for 5 h induced β-catenin protein expression in MDA-MB-231 cells, and this response was decreased after cotreatment with the p38 inhibitor SB202190. TGF-β also induced expression of several EMT marker proteins, including Slug, Snail, ZEB-1, N-cadherin, and vimentin; cotreatment with SB202190 inhibited these responses, and this was accompanied by increased expression of the epithelial marker ZO-1. Surprisingly, cotreatment of MDA-MB-231 cells with TGF-β plus LMB or the C-DIM/NR4A1 antagonists also inhibited expression of the β-catenin-regulated protein markers of EMT (Slug, Snail, ZEB-1, and N-cadherin but not vimentin); however, the induced levels of β-catenin were not decreased. As a positive control, we also observed the TGF-β-induced Slug and Snail mRNA levels (Fig. 6B). The results shown in Fig. 6C demonstrate that low levels of β-catenin are expressed in MDA-MB-231 cells, TGF-β induces nuclear β-catenin, and cotreatment with SB202190 inhibits this response; however, cotreatment with LMB resulted in both nuclear and cytosolic β-catenin, whereas β-catenin is primarily cytosolic after cotreatment with TGF-β plus the C-DIM/NR4A1 antagonists. Transfection of p38α also induced expression of β-catenin and EMT markers, where C-DIM/NR4A1 antagonists, LMB, and SB202190 inhibited β-catenin nuclear localization (Fig. 6D) and inhibited p38-mediated induction of EMT markers (Fig. 6E). The induction of β-catenin was abrogated by SB202190 (Fig. 6D and E), indicating the necessity for p38 in induction of β-catenin, and this was confirmed in studies in which p38 and p38 upstream kinases (TAK1, MKK3, and MKK6) were knocked down by RNA interference (RNAi) (Fig. 6F). The partial nuclear export of β-catenin observed in cells cotreated with TGF-β plus LMB (an exportin-1 inhibitor) may be due, in part, to an equilibrium between nuclear and cytosolic β-catenin, and this is currently being investigated. In cells transfected with p38α(CA), p38α(KD), and p38α(DN) alone or in combination with TGF-β (Fig. 6G), the induced [by p38α(CA) with or without TGF-β] β-catenin was nuclear and consistent with activation of the EMT β-catenin, Slug, Snail, ZEB1, N-cadherin, and vimentin genes; however, in the other treatment groups, β-catenin expression in the nucleus or cytosol was minimal to nondetectable, and induction of EMT gene expression was minimal (Fig. 6H). Therefore, the difference in β-catenin expression observed in Fig. 6E and F may be partially due to the duration of the treatment (5 and 12 h, respectively), and the time-dependent effects were further investigated (see below).
FIG 6.

TGF-β/p38-induced β-catenin/EMT genes are affected by NR4A1 antagonists. (A) MDA-MB-231 cells were treated with various agents for 5 h, and β-catenin and EMT genes associated with mesenchymal (β-catenin, Slug, Snail, ZEB1, N-cadherin and vimentin) and epithelial (ZO1) cells were determined by Western blot analysis of whole-cell lysates. (B) MDA-MB-231 cells were treated with DMSO or 5 ng/ml TGF-β for 5 h, and changes in Slug and Snail mRNA levels (compared to DMSO) were determined by real-time PCR as outlined in Materials and Methods. The results are means and SE for at least 3 separate determinations, and significant (*, P < 0.05) induction is indicated. (C and D) Cells were treated with various compounds and their combinations for 5 h, and nuclear and cytosolic extracts were separated and analyzed by Western blotting. (E and F) The cells were transfected with p38(CA) and treated with various compounds (E) or with 5 ng/ml TGF-β (F) and transfected with different oligonucleotides in RNAi experiments, and whole-cell lysates were analyzed by Western blotting as outlined in Materials and Methods. (G and H) Cells were transfected with p38-derived expression plasmids alone or in combination with 5 ng/ml TGF-β, and nuclear and cytosolic (G) or whole-cell (H) lysates were analyzed by Western blotting. (I) Cells were transfected with different siRNAs, and whole-cell lysates were analyzed by Western blotting.
We also used immunostaining and confocal microscopy analysis to investigate treatment-related cytosolic and nuclear localization of β-catenin (Fig. 7). The results demonstrate that TGF-β treatment induced expression and nuclear localization of β-catenin; however, β-catenin was sequestered in the cytosol when cells were cotreated with TGF-β plus SB202190, leptomycin B, CDIM8, and CDIM14. In cells transfected with p38(CA), β-catenin was induced and localized in the nucleus, whereas after cotreatment with LMB, CDIM8, CDIM14, or SB202190, β-catenin was sequestered in the cytosol. Both p38(KD) and p38(DN) alone also resulted in cytosolic β-catenin, and cotreatment with TGF-β did not rescue this effect, corroborating results obtained previously showing that TGF-β cannot rescue cells expressing p38(KD) or p38(DN).
FIG 7.
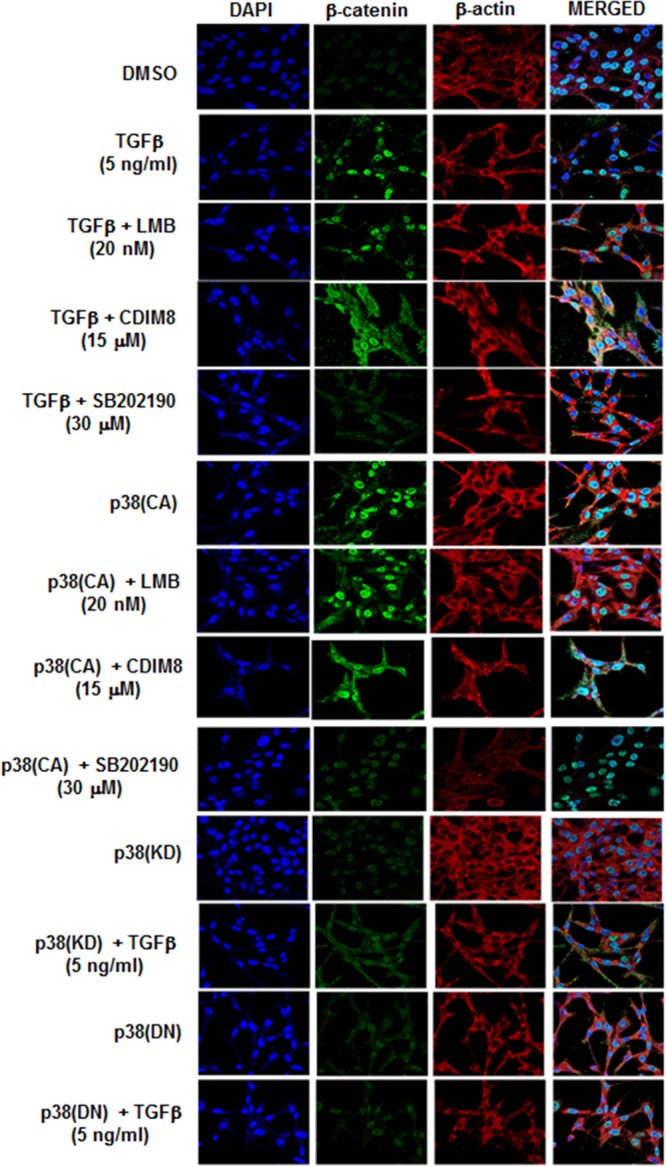
Analysis of β-catenin location by confocal microscopy. MDA-MB-231 cells were treated with DMSO or 5 ng/ml TGF-β or transfected with p38(CA), and after various treatments (5 h), the cells were fixed, immunostained, and analyzed by confocal microscopy as outlined in Materials and Methods.
Does NR4A1 play a role in regulation of TGF-β-induced β-catenin expression?
TGF-β-induced β-catenin was primarily nuclear after treatment for 5 h; however, after cotreatment with NR4A1 antagonists, β-catenin was cytosolic, and levels were decreased, suggesting a role for NR4A1 in regulating β-catenin expression (Fig. 6C). This was confirmed in RNAi studies, where knockdown of NR4A1 resulted in low to nondetectable levels of TGF-β-induced β-catenin or EMT genes in whole-cell lysates, whereas the epithelial marker ZO-1 was expressed after NR4A1 knockdown (Fig. 8A). β-Catenin levels in MDA-MB-231 cells are low (Fig. 5A and C); however, after treatment with TGF-β or transfection with p38(CA), β-catenin levels were induced and persisted for up to 24 h in the nucleus (Fig. 8B and C). In a parallel study, after treatment with the NR4A1 antagonist CDIM8, TGF-β- and p38(CA)-induced nuclear β-catenin was exported to the cytosol (Fig. 8D and E), as observed in Fig. 6C (5-h treatment with CDIM8); however, after treatment with CDIM8 for 24 h, the levels of β-catenin were undetectable. Since cytosolic β-catenin is subject to proteasome-dependent degradation, we repeated the studies outlined in Fig. 8D and E, but in the absence or presence of the proteasome inhibitor MG132. The results show that MG132 blocked the loss of β-catenin in cells treated with TGF-β or transfected with p38(CA) plus the NR4A1 antagonist CDIM8 (Fig. 8F and G). The mechanism of NR4A1 regulation of β-catenin is currently being investigated; however, our results demonstrate that NR4A1 antagonists that inhibit TGF-β-induced nuclear export of NR4A1 induce nuclear export of β-catenin. These results demonstrate that NR4A1 antagonists directly or indirectly inhibit accumulation of TGF-β-induced β-catenin in the nucleus, resulting in inhibition of EMT in breast cancer cells, and the mechanisms of NR4A1 ligands on the nuclear-cytosolic trafficking of NR4A1 and β-catenin are currently being investigated.
FIG 8.

NR4A1 regulation of β-catenin expression and degradation. (A) MDA-MB-231 cells were transfected with siCtl or siNR4A1, and whole-cell lysates were analyzed by Western blotting as outlined in Materials and Methods. (B to E) Cells were treated with TGF-β (B), transfected with p38(CA) (C) alone or in combination with the NR4A1 antagonist CDIM8 (D and E, respectively), and cytosolic and nuclear extracts were isolated and analyzed by Western blotting. (F and G) Cells were treated with CDIM8/TGF-β (F) or transfected with p38(CA)/CDIM8 (G) in the presence or absence of the proteasome inhibitor MG132, and whole-cell lysates were analyzed by Western blotting as outlined in Materials and Methods.
Roles of p38 and β-catenin in regulation of NR4A1 expression and subcellular localization.
TGF-β and p38 induce both β-catenin and NR4A1 expression and regulate their intracellular location in the nucleus and cytosol, respectively, and Fig. 9A illustrates that NR4A1 was nuclear after knockdown of β-catenin, p38, and upstream kinases (TAK1, MKK3, and MKK6) in MDA-MB-231 cells. The treatments decreased nuclear expression of NR4A1, but cytosolic NR4A1 levels were minimal and β-catenin was undetectable in either the nucleus or cytosol. TGF-β induced NR4A1 and its export to the cytosol, and this was accompanied by induction of nuclear β-catenin (Fig. 9B); knockdown of β-catenin, p38, and upstream kinases decreased expression of both NR4A1 and β-catenin and inhibited nuclear export of NR4A1. TGF-β-induced NR4A1 mRNA expression and induction were also inhibited in cells after knockdown of β-catenin, p38, and upstream kinases (Fig. 9C). Moreover, using the same treatment protocol, TGF-β-induced migration in MDA-MB-231 was also inhibited by knockdown of β-catenin, p38, and upstream kinases (Fig. 9D), and this correlated with the effects of these treatments as inhibitors of induction of β-catenin and inhibition of nuclear export of NR4A1 (Fig. 9B).
FIG 9.

β-Catenin regulates expression of NR4A1. (A and B) Cells were transfected with siCtl and various oligonucleotides targeting β-catenin or kinases alone (A) or after treatment with 5 ng/ml TGF-β (B), and nuclear and cytosolic extracts were isolated and analyzed by Western blotting. (C) Cells were treated with DMSO or 5 ng/ml TGF-β, and mRNA levels were determined by real-time PCR. The results are means and SE for at least 3 replicates per treatment group, and a significant (*, P < 0.05) decrease in TGF-β-induced NR4A1 mRNA levels (compared to DMSO) is indicated. (D) Cells were treated with DMSO or 5 ng/ml TGF-β and transfected with various oligonucleotides, and migration was determined as outlined in Materials and Methods. The results are means and SE of 3 replicate determinations, and a significant (*, P < 0.05) decrease in migration is indicated. (E) Schematic illustration of TCF/LEF sites on the NR4A1 promoter and primers used for ChIP analysis. (F and G) Cells were treated with 5 ng/ml TGF-β transfected with various oligonucleotides for knockdown by RNAi, and NR4A1 mRNA (F) and protein (G) levels were determined by real-time PCR and Western blotting, respectively. The results (F) are expressed as means and SE for 3 replicate determinations, and significant (*, P < 0.05) decreases are indicated. (H) Cells were treated with 5 ng/ml TGF-β or transfected with p38 variants alone or in combination with other agents, and interactions of various factors with the TCF/LEF region of the NR4A1 promoter were determined in a ChiP assay.
β-Catenin is a nuclear transcription factor that regulates gene expression through interactions with DNA-bound TCF/LEF sites, and the NR4A1 promoter contains two of these cis elements at −346 to −328 and −191 to −170 (Fig. 9E). The potential role of β-catenin/TCF/LEF in regulating β-NR4A1 expression was investigated by RNAi, and knockdown of β-catenin, TCF3 (siTCF3), TCF4 (siTCF4), and LEF1 (siLEF1) decreased TGF-β-induced NR4A1 mRNA (Fig. 9F) and protein (Fig. 9G) levels. siTCF1 only partially decreased NR4A1 mRNA, but not protein, levels. Further confirmation that β-catenin regulates NR4A1 gene expression was investigated in a chromatin immunoprecipitation (ChIP) assay on the NR4A1 promoter, which showed that treatment with TGF-β, TGF-β plus LMB, p38(CA), and p38(CA) plus TGF-β induced RNA polymerase II (Pol II) and enhanced TCF3, TCF4, and LEF1 binding to the region of the NR4A1 promoter containing the TCF/LEF motifs. In contrast, these promoter interactions were not observed in cells treated/transfected with p38(KD) and p38(DN) alone or in combination with TGF-β or with TGF-β plus the NR4A1 antagonists CDIM8 and CDIM14 (Fig. 9H). These data are consistent with results showing that β-catenin regulates TGF-β-induced NR4A1 expression (Fig. 9F and D) and that β-catenin acts as a nuclear cofactor, along with TCF3, TCF4, and LEF1. Figure 10 summarizes the role of NR4A1 in TGF-β/p38α-dependent MDA-MB-231 cell migration and EMT as determined in this study. TGF-β-inducible migration is dependent on activation of p38 and p38-dependent nuclear export of NR4A1 and its subsequent interactions with the Arkadia, axin2, RNF12 E3 ubiquitin ligase complex that targets SMAD7 for proteasome-dependent degradation. This process is NR4A1 and also p38 dependent and can be inhibited by SB202190 and C-DIM/NR4A1 antagonists. TGF-β-induced p38 is also critical for induction of β-catenin and EMT genes and for β-catenin–NR4A1 mutual coregulation, where β-catenin acts as a trans-acting transcription factor to induce NR4A1 gene expression. NR4A1 antagonists also regulate NR4A1–β-catenin interactions, and this includes facilitating β-catenin nuclear export and its subsequent proteasome-dependent degradation (Fig. 10).
FIG 10.

Model for the role of NR4A1 in mediating TGF-β/p38 induction of migration, β-catenin and EMT, and β-catenin regulation of NR4A1 gene expression and inhibitory effects of CDIM/NR4A1 antagonists. Ub, ubiquitin.
DISCUSSION
NR4A1 is a member of the NR4A orphan receptor subfamily, and although NR4A1 plays a role in multiple physiological and pathophysiological processes, the endogenous ligand for the receptor has not been identified (21, 22). Several studies have now characterized ligands that bind NR4A1 (18, 23–26), and research in our laboratory has investigated the binding of several C-DIM analogs to the receptor (18). NR4A1 is prooncogenic in most solid tumors, and both CDIM8 and CDIM14 act as receptor antagonists and inhibit nuclear NR4A1-dependent gene expression, resulting in decreased tumor growth, survival, and migration/invasion of breast and other cancer cells (12–19). Zhou and coworkers demonstrated that NR4A1 plays a key role in cytokine- and TGF-β-induced invasion and EMT in breast cancer by interacting with the axin2, RNF12, and Arkadia E3 ubiquitin ligase complex, which degrades SMAD7 (11). Studies in our laboratory demonstrated that NR4A1 regulation of β1-integrin gene expression is primarily responsible for basal migration of MDA-MB-231 cells. We also observed that TGF-β-induced MDA-MB-231 cell migration is dependent on nuclear export of NR4A1, which is inhibited by the NR4A1 ligands CDIM8 and CDIM14 (19). Thus, the C-DIM/NR4A1 antagonists effectively block TGF-β-induced migration of breast cancer cells, and this study was focused on delineating the mechanisms of TGF-β–NR4A1 interactions.
TGF-β induces multiple kinases, including p38 (1–3, 27–34), and based on initial kinase inhibitor studies in three triple-negative breast cancer cell lines (H5587T, MDA-MB-231, and SUM159), we identified p38 as the principal kinase required for nuclear export of NR4A1. Moreover, p38(CA), but not p38(KD) or p38(DN), overexpression induced nuclear export of NR4A1, and subsequent studies with MDA-MB-231 cells showed that TGF-β- and p38-induced migration were inhibited by C-DIM/NR4A1 antagonists and LMB (Fig. 2A) (19). Interestingly, we also observed in these initial experiments that TGF-β and p38(CA) alone or in combination induced the same magnitude of cell migration and other responses, and the lack of activity observed for p38(DN) and p38(KD) was not rescued by cotreatment with TGF-β for any response examined in our studies.
Immunoprecipitation studies with NR4A1 antibodies confirmed that full-length NR4A1 interacted with axin2, Arkadia, RNF12, and SMAD7, and the C-terminal (axin2, Arkadia, and RNF12) and N-terminal (axin2 and SMAD7) domains of NR4A1 differentially interacted with members of this complex (Fig. 4). Although the functional E3 ubiquitin ligase complex containing NR4A1 is responsible for degradation of SMAD7 (a cytosolic protein) (11), we observed that, like NR4A1, Arkadia was also a nuclear protein that underwent TGF-β/p38-dependent nuclear export (data not shown). The ultimate target of the E3 ubiquitin ligase complex is SMAD7 (11), and we also observed that TGF-β/p38 induced proteasome-dependent SMAD7 ubiquitination and subsequent degradation. Moreover, inhibitors of NR4A1 (and Arkadia) nuclear export or knockdown of one or more members of this complex also decreased SMAD7 ubiquitination (Fig. 5), and these observations are consistent with studies showing the importance of the complex for SMAD7 degradation (11).
Previous studies showed that TGF-β induces cancer cell migration and EMT, and activation of the latter pathway may also involve increased expression of β-catenin (35–38). In addition, it has also been reported that β-catenin not only interacts with NR4A1, but there is evidence for mutual functional responses and interprotein regulatory pathways (39–43). Knockdown or overexpression of NR4A1 in colon cancer cells decreases or increases β-catenin expression, respectively, and knockdown or overexpression of β-catenin decreases or increases NR4A1 expression (39–41). Hypoxia also enhances β-catenin/NR4A1-mediated invasion of colon cancer cells; however, the mechanism of this response and the intracellular location of NR4A1 are unclear (41). The role of NR4A1 in TGF-β-induced expression of β-catenin and downstream EMT genes in MDA-MB-231 cells is unique. Both TGF-β and p38(CA) induced expression of β-catenin and downstream genes in MDA-MB-231 cells, and this response was inhibited not only by p38(DN), p38(KD), and knockdown of kinases upstream from p38 but also by the NR4A1 antagonists CDIM8 and CDIM14 (Fig. 6 and 7). Treatment with TGF-β resulted in induced β-catenin expression in the nucleus and nuclear export of NR4A1, and the effects of NR4A1 antagonists (5-h treatment) reversed the subcellular location of both proteins, suggesting nuclear (antagonist) ligand-bound NR4A1 either directly or indirectly enhanced nuclear export of β-catenin. Moreover, CDIM8- and CDIM14-induced nuclear export of β-catenin ultimately resulted in proteasome-dependent degradation of the protein within 24 h after initial treatment (Fig. 8). Knockdown of NR4A1 by RNA also decreased β-catenin expression (Fig. 5A), suggesting that the loss or inactivation of NR4A1-regulated genes/pathways contributed to nuclear export of β-catenin and its subsequent degradation, and the mechanisms of liganded NR4A1-induced nuclear export of β-catenin are currently being investigated.
β-Catenin overexpression or induction in colon cancer cells induces NR4A1 expression, and this has been linked to activation of AP1 and subsequent interactions with AP1 cis elements in the proximal region (−200 to −2) of the NR4A1 gene promoter (40). In contrast, TGF-β induced β-catenin, and β-catenin-dependent induction of NR4A1 was due to nuclear β-catenin interactions with TCF3/TCF4 (but not TCF1) and LEF cis elements (−346 to −328 and −190 to −170) in the proximal region of the NR4A1 gene promoter (Fig. 9E). These interactions were TGF-β dependent and illustrate a unique cross talk between NR4A1 and β-catenin, which are critical elements in late-stage TGF-β-induced breast cancer cell migration/invasion.
Thus, the results of this study demonstrate that TGF-β-induced responses, including cell migration, induction of EMT, β-catenin, and NR4A1, are dependent on induction of p38 and genomic regulation of NR4A1 expression by cis-acting β-catenin/TCF/LEF complexes (Fig. 10). In contrast, TGF-β/p38-dependent induction of β-catenin results in nuclear accumulation of β-catenin and activation of downstream EMT genes. The precise role of NR4A1 in mediating TGF-β/p38-dependent induction of β-catenin is unknown; however, NR4A1 antagonists that retain NR4A1 in the nucleus also induce β-catenin nuclear export and degradation, demonstrating a novel pathway for inhibiting EMT in triple-negative breast cancer cells with NR4A1 ligands. These results demonstrate a pivotal role for NR4A1 in orchestrating TGF-β-induced triple-negative breast cancer cell migration and EMT (Fig. 10). Thus, NR4A1 antagonists, such as C-DIMs, represent a novel class of mechanism-based drugs for treating the relatively high percentage of breast cancer patients who overexpress this receptor (11), and this study now shows that these C-DIM/NR4A1 compounds may be particularly effective against TGF-β-induced migration/invasion in patients with late-stage tumors.
MATERIALS AND METHODS
Cell lines, reagents, and antibodies.
Breast cancer cells (MDA-MB-231, Hs587T, and SUM159) were purchased from the American Type Culture Collection (Manassas, VA). The cells were maintained at 37°C in the presence of 5% CO2 in Dulbecco's modified Eagle's medium (DMEM)–Ham's F-12 medium with 10% fetal bovine serum with antibiotic. NR4A1, Arkadia, and RNF12 antibodies were purchased from Novus Biologicals (Littleton, CO). TGF-β was purchased from BD Biosystems (Bedford, MA). β-Actin antibody, Dulbecco's modified Eagle's medium, M2 Flag antibody, MG132, and 36% formaldehyde were purchased from Sigma-Aldrich (St. Louis, MO). Hematoxylin was purchased from Vector Laboratories (Burlingame, CA). β-Catenin, GAPDH (glyceraldehyde-3-phosphate dehydrogenase), p38, p-p38, p-NR4A1, axin2, anti-rabbit IgG Fab2–Alexa Fluor 488, or anti-mouse IgG Fab2–Alexa Fluor, phalloidin (Alexa Fluor 555-phalloidin), Hoechst 33342, leptomycin B, SP600125, SB202190, LY294002, PD98059, and NR4A1 immunofluorescent antibody were purchased from Cell Signaling Technologies (Manassas, VA). SMAD 6/7 and ubiquitin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and p84 antibody from GeneTex (Irvine, CA).
Plasmids.
FLAG-NR4A1, FLAG-NR4A1-(A-B), and FLAG-NR4A1-(C-F) were synthesized in the laboratory using site-directed mutagenesis. DDK-Myc-p38CA-(D176A; F327S), DDK-Myc-p38KD-(K53A), DDK-Myc-p38DN-(T180A;Y182F), DDK-Myc-arkadia, DDK-Myc-arkadia-(Δ405–989), DDK-Myc-arkadia-(Δ1–404), DDK-Myc-arkadia-(241–404), and DDK-Myc-arkadia (C937A) were purchased from Origene Technologies (Rockville, MD). The plasmids were transfected as described previously (19); medium was removed, and then the cells were treated with the respective compound.
Boyden chamber assay.
MDA-MB-231, SUM159, and H5587T TNBC cells (3.0 × 105 per well) were seeded in Dulbecco's modified Eagle's medium–Ham's F-12 medium supplemented with 2.5% charcoal-stripped fetal bovine serum and allowed to attach for 24 h. The cells were seeded and subsequently treated with various concentrations of DIM-C-pPhOH or DIM-C-pPhCO2Me for 24 h or 1 h prior (with or without TGF-β [5 ng/ml]; 4-h cotreatment) or with 100 nM siAxin2, siArkadia, siRNF12, siβ-catenin, siTAK1, siMKK3, siMKK6, or sip38α for 48 h. The cells were trypsinized, counted, placed in 24-well 8.0-μm-pore-size ThinCerts from BD Biosciences (Bedford, MA), allowed to migrate for 24 h, fixed with formaldehyde, and then stained with hematoxylin. Cells that migrated through the pores were counted as described previously (19).
RT-PCR.
RNA was isolated using a Zymo Research (Irvine, CA) Quick-RNA MiniPrep kit. Quantification of mRNA (Slug, Snail, and NR4A1) was performed using a Bio-Rad (Richmond, CA) iTaq Universal SYBER Green 1-step kit according to the manufacturer's protocol with real-time PCR. TATA binding protein (TBP) mRNA was used as a control to determine relative mRNA expression.
Immunoprecipitation.
MDA-MB-231 cancer cells (3.0 × 105 per well) were seeded in Dulbecco's modified Eagle's medium–Ham's F-12 medium supplemented with 2.5% charcoal-stripped fetal bovine serum and allowed to attach for 24 h. The medium was then changed to DMEM–Ham's F-12 medium containing 2.5% charcoal-stripped fetal bovine serum, and either dimethyl sulfoxide (DMSO) or TGF-β (5 ng/ml) was added for 4 h (after pretreatment or not with leptomycin B [20 nM] for 24 h or pretreatment or not with or without DIM-C-pPhOH [20 μM], DIM-C-pPhCO2Me [15 μM], or SB202190 [30 μM] for 1 h) or transfection of DDK-Myc-p38CA, DDK-Myc-p38KD, DDK-Myc-p38DN, D-NR4A1, FLAG-NR4A1-(A-B), FLAG-NR4A1-(C-F), DDK-Myc-arkadia, DDK-Myc-arkadia (Δ405–989), DDK-Myc-arkadia (Δ1–404), DDK-Myc-arkadia (241–404), and DDK-Myc-arkadia (C937A) plasmids 6 h posttransfection. Protein A Dynabeads were prepared, and binding of antibody with protein and protein-protein interactions were isolated with a Life Technologies (Grand Island, NY) Immunoprecipitation kit using Dynabeads coated with protein A, following the manufacturer's protocol. Protein-protein interactions of interest were determined using Western blotting.
Chromatin immunoprecipitation.
The ChIP assay was performed using a ChIP-IT Express magnetic chromatin immunoprecipitation kit (Active Motif, Carlsbad, CA) according to the manufacturer's protocol. MDA-MB-231 cells were treated with DMSO or TGF-β (5 ng/ml) for 4 h (after pretreatment or not with leptomycin B [20 nM] for 24 h or pretreatment or not with or without DIM-C-pPhOH [20 μM], DIM-C-pPhCO2Me [15 μM], SB202190 [30 μM] for 1 h or transfection of p38CA, p38KD, or p38DN plasmids 6 h posttransfection). The cells were then fixed with 1% formaldehyde, and the cross-linking reaction was stopped by addition of 0.125 M glycine. After washing twice with phosphate-buffered saline, the cells were scraped and pelleted. The collected cells were hypotonically lysed, and nuclei were collected. The nuclei were then sonicated to the desired chromatin length (∼200 to 1,500 bp). The sonicated chromatin was immunoprecipitated with normal IgG, p300 (Santa Cruz), siSp1 (Abcam), NR4A1 (Novus Biologicals), or Pol II (Active Motif) antibody and protein A-conjugated magnetic beads at 4°C overnight. After the magnetic beads were extensively washed, protein-DNA cross-links were reversed and eluted. DNA was prepared by proteinase K digestion, followed by PCR amplification. The primers for detection of the NR4A1 promoter region were 5′-CGAGGAGCCTATTTATAG-3′ (sense) and 5′-TCGACGTTTGGCCATACAAGG-3′ (antisense). PCR products were resolved on a 2% agarose gel in the presence of RGB-4103 GelRed nucleic acid stain.
Nuclear/cytosolic extraction.
MDA-MB-231 cancer cells (3.0 × 105 per well) were seeded onto Dulbecco's modified Eagle's medium–Ham's F-12 medium supplemented with 2.5% charcoal-stripped fetal bovine serum and allowed to attach for 24 h. The medium was then changed to DMEM–Ham's F-12 medium containing 2.5% charcoal-stripped fetal bovine serum, and either DMSO or TGF-β (5 ng/ml) was added for 4 h [(after pretreatment or not with leptomycin B [20 nM) for 24 h or with or without DIM-C-pPhOH [20 μM], DIM-C-pPhCO2Me [15 μM], SP600125 [30 μM], SB202190 [30 μM], LY294002 [30 μM], or PD98095 [30 μM] for 1 h or transfection of p38CA, p38KD, or p38DN plasmid 6 h posttransfection). Nuclear and cytosolic fractions were then isolated using a Thermo Scientific (Rockford, IL) NE-PER nuclear and cytoplasmic extraction kit according to the manufacturer's protocol. The fractions were then analyzed by Western blotting. GAPDH and p84 were used as cytoplasmic and nuclear positive controls, respectively.
Immunofluorescence.
MDA-MB-231 cells (1.0 × 105 per well) were seeded in 2-well Nunc Lab-Tek chambered B number 1.0 borosilicate coverglass slides from Thermo Scientific and were allowed to attach for 24 h. The medium was then changed to DMEM–Ham's F-12 medium containing 2.5% charcoal-stripped fetal bovine serum, and either DMSO or TGF-β (5 ng/ml) was added for 4 h (after pretreatment or not with leptomycin B [20 nM] for 24 h or pretreatment or not with or without DIM-C-pPhOH [20 μM], DIM-C-pPhCO2Me [15 μM], or SB202190 [30 μM] for 1 h or transfection of DDK-Myc-p38CA, DDK-Myc-p38K,D or DDK-Myc-p38DN plasmid 6 h posttransfection). The cells were then fixed with 37% formalin, blocked, and treated with fluorescent NR4A1 primary antibody (Nur77 [D63C5] XP) or β-catenin primary antibody for 24 h. The cells were then washed with PBS and treated with anti-rabbit IgG Fab2–Alexa Fluor 488 secondary antibody for 3 h. The cells were then treated with Hoechst (Hoechst 33342) stain and phalloidin (Alexa Fluor 555–phalloidin) for 15 min in the dark following the manufacturer's protocol. After washing with ice-cold PBS, the cells were visualized by confocal microscopy (LSM 780 confocal microscope; Zeiss, Peabody, MA; 405 nm, 488 nm, and 561 nm for excitation; 458 nm [blue], 514 nm [green], and 633 nm [red] for emission,). NR4A1 or β-catenin localization was determined by green fluorescence (green). Hoechst 33342 was used to stain the nucleus (blue), and phalloidin stained β-actin (red).
Western blot analysis.
MDA-MB-231, SUM159, and H5587T cancer cells (3.0 × 105 per well) were seeded in Dulbecco's modified Eagle's medium–Ham's F-12 medium supplemented with 2.5% charcoal-stripped fetal bovine serum and allowed to attach for 24 h. The cells were transfected with 100 nM siAxin2, siArkadia, siRNF12, siβ-catenin, siTAK1, siMKK3, siMKK6, sip38α, siTCF1, siTCF3, siTCF4, or siLEF1 for 72 h or treated under various conditions as described above. The cells were analyzed by Western blotting as described previously (19).
siRNA interference assay.
Small interfering RNA (siRNA) experiments were conducted as described previously (19). The siRNA complexes used in the study were as follows: siGL2-5′, CGUACGCGGAAUACUUCGA; siNR4A1, SASI_Hs02_00333289; siAxin2, SASI_Hs01_00110148; siArkadia, SASI_Hs01_00064840; siRNF12, SASI_Hs01_00238255; siTAK1, SASI_Hs02_00335227; siMKK3, SASI_Hs01_00228024; siMKK6, SASI_Hs01_00162806; sip38α, SASI_Hs01_00018467; siβ-catenin, SASI_Hs02_00318698; siTCF1, SASI_Hs01_00018982; siTCF3, SASI_Hs01_00214771; siTCF4, SASI_Hs02_00317381; siLEF1, SASI_Hs02_00349169.
TNBC orthotopic xenograft studies.
Female BALB/c nude mice (6 to 8 weeks old) were obtained (Charles River Laboratory, Wilmington, MA), maintained, and treated as previously described (16). The mice were housed in the Laboratory Animal Resources and Research Facility approved by the Institutional Animal Care and Use Committee (IACUC) at Texas A&M University, College Station, TX. The animals were handled in accordance with the IACUC guidelines under an approved animal use protocol. Tumor volumes and tumor weights were determined, and tumor lysates were obtained and analyzed by Western blottings.
Statistical analysis.
The statistical significance of differences between the treatment groups was determined as previously described (19).
ACKNOWLEDGMENTS
The financial assistance of the National Institutes of Health (P30-ES023512 to S.S.), Texas AgriLife Research, and the Sid Kyle endowment is gratefully acknowledged.
The help of Rola Barhoumi in carrying out the confocal microscopy analysis is gratefully acknowledged.
E.H. and S.S. participated in research design; E.H. conducted experiments; S.S. contributed new reagents or analytic tools; E.H. performed data analysis; E.H. and S.S. wrote or contributed to the writing of the manuscript.
We declare that there are no competing financial interests.
REFERENCES
- 1.Ikushima H, Miyazono K. 2010. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer 10:415–424. doi: 10.1038/nrc2853. [DOI] [PubMed] [Google Scholar]
- 2.Wakefield LM, Hill CS. 2013. Beyond TGFbeta: roles of other TGFbeta superfamily members in cancer. Nat Rev Cancer 13:328–341. doi: 10.1038/nrc3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Principe DR, Doll JA, Bauer J, Jung B, Munshi HG, Bartholin L, Pasche B, Lee C, Grippo PJ. 2014. TGF-beta: duality of function between tumor prevention and carcinogenesis. J Natl Cancer Inst 106:djt369. doi: 10.1093/jnci/djt369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang B, Yoo N, Vu M, Mamura M, Nam JS, Ooshima A, Du Z, Desprez PY, Anver MR, Michalowska AM, Shih J, Parks WT, Wakefield LM. 2007. Transforming growth factor-beta can suppress tumorigenesis through effects on the putative cancer stem or early progenitor cell and committed progeny in a breast cancer xenograft model. Cancer Res 67:8643–8652. doi: 10.1158/0008-5472.CAN-07-0982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morrison CD, Parvani JG, Schiemann WP. 2013. The relevance of the TGF-beta paradox to EMT-MET programs. Cancer Lett 341:30–40. doi: 10.1016/j.canlet.2013.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gonzalez DM, Medici D. 2014. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal 7:re8. doi: 10.1126/scisignal.2005189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi Y, Massague J. 2003. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113:685–700. doi: 10.1016/S0092-8674(03)00432-X. [DOI] [PubMed] [Google Scholar]
- 8.Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL. 2000. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell 6:1365–1375. doi: 10.1016/S1097-2765(00)00134-9. [DOI] [PubMed] [Google Scholar]
- 9.Koinuma D, Shinozaki M, Komuro A, Goto K, Saitoh M, Hanyu A, Ebina M, Nukiwa T, Miyazawa K, Imamura T, Miyazono K. 2003. Arkadia amplifies TGF-beta superfamily signalling through degradation of Smad7. EMBO J 22:6458–6470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang L, Huang H, Zhou F, Schimmel J, Pardo CG, Zhang T, Barakat TS, Sheppard KA, Mickanin C, Porter JA, Vertegaal AC, van Dam H, Gribnau J, Lu CX, ten Dijke P. 2012. RNF12 controls embryonic stem cell fate and morphogenesis in zebrafish embryos by targeting Smad7 for degradation. Mol Cell 46:650–661. doi: 10.1016/j.molcel.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 11.Zhou F, Drabsch Y, Dekker TJ, de Vinuesa AG, Li Y, Hawinkels LJ, Sheppard KA, Goumans MJ, Luwor RB, de Vries CJ, Mesker WE, Tollenaar RA, Devilee P, Lu CX, Zhu H, Zhang L, Dijke PT. 2014. Nuclear receptor NR4A1 promotes breast cancer invasion and metastasis by activating TGF-beta signalling. Nat Commun 5:3388. doi: 10.1038/ncomms4388. [DOI] [PubMed] [Google Scholar]
- 12.Safe S, Jin UH, Hedrick E, Reeder A, Lee SO. 2014. Minireview: role of orphan nuclear receptors in cancer and potential as drug targets. Mol Endocrinol 28:157–172. doi: 10.1210/me.2013-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee SO, Abdelrahim M, Yoon K, Chintharlapalli S, Papineni S, Kim K, Wang H, Safe S. 2010. Inactivation of the orphan nuclear receptor TR3/Nur77 inhibits pancreatic cancer cell and tumor growth. Cancer Res 70:6824–6836. doi: 10.1158/0008-5472.CAN-10-1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee SO, Andey T, Jin UH, Kim K, Singh M, Safe S. 2012. The nuclear receptor TR3 regulates mTORC1 signaling in lung cancer cells expressing wild-type p53. Oncogene 31:3265–3276. doi: 10.1038/onc.2011.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hedrick E, Lee SO, Kim G, Abdelrahim M, Jin UH, Safe S, Abudayyeh A. 2015. Nuclear receptor 4A1 (NR4A1) as a drug target for renal cell adenocarcinoma. PLoS One 10:e0128308. doi: 10.1371/journal.pone.0128308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hedrick E, Lee SO, Doddapaneni R, Singh M, Safe S. 2015. Nuclear receptor 4A1 as a drug target for breast cancer chemotherapy. Endocr Relat Cancer 22:831–840. doi: 10.1530/ERC-15-0063. [DOI] [PubMed] [Google Scholar]
- 17.Lee SO, Jin UH, Kang JH, Kim SB, Guthrie AS, Sreevalsan S, Lee JS, Safe S. 2014. The orphan nuclear receptor NR4A1 (Nur77) regulates oxidative and endoplasmic reticulum stress in pancreatic cancer cells. Mol Cancer Res 12:527–538. doi: 10.1158/1541-7786.MCR-13-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee SO, Li X, Hedrick E, Jin UH, Tjalkens RB, Backos DS, Li L, Zhang Y, Wu Q, Safe S. 2014. Diindolylmethane analogs bind NR4A1 and are NR4A1 antagonists in colon cancer cells. Mol Endocrinol 28:1729–1739. doi: 10.1210/me.2014-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hedrick E, Lee SO, Doddapaneni R, Singh M, Safe S. 2016. NR4A1 antagonists inhibit β1-integrin-dependent breast cancer cell migration. Mol Cell Biol 36:1383–1394. doi: 10.1128/MCB.00912-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bikkavilli RK, Feigin ME, Malbon CC. 2008. p38 mitogen-activated protein kinase regulates canonical Wnt-beta-catenin signaling by inactivation of GSK3beta. J Cell Sci 121:3598–3607. doi: 10.1242/jcs.032854. [DOI] [PubMed] [Google Scholar]
- 21.Maxwell MA, Muscat GE. 2006. The NR4A subgroup: immediate early response genes with pleiotropic physiological roles. Nucl Recept Signal 4:e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhan YY, Chen Y, Zhang Q, Zhuang JJ, Tian M, Chen HZ, Zhang LR, Zhang HK, He JP, Wang WJ, Wu R, Wang Y, Shi C, Yang K, Li AZ, Xin YZ, Li TY, Yang JY, Zheng ZH, Yu CD, Lin SC, Chang C, Huang PQ, Lin T, Wu Q. 2012. The orphan nuclear receptor Nur77 regulates LKB1 localization and activates AMPK. Nat Chem Biol 8:897–904. doi: 10.1038/nchembio.1069. [DOI] [PubMed] [Google Scholar]
- 23.Zhan Y, Du X, Chen H, Liu J, Zhao B, Huang D, Li G, Xu Q, Zhang M, Weimer BC, Chen D, Cheng Z, Zhang L, Li Q, Li S, Zheng Z, Song S, Huang Y, Ye Z, Su W, Lin SC, Shen Y, Wu Q. 2008. Cytosporone B is an agonist for nuclear orphan receptor Nur77. Nat Chem Biol 4:548–556. doi: 10.1038/nchembio.106. [DOI] [PubMed] [Google Scholar]
- 24.Wang WJ, Wang Y, Chen HZ, Xing YZ, Li FW, Zhang Q, Zhou B, Zhang HK, Zhang J, Bian XL, Li L, Liu Y, Zhao BX, Chen Y, Wu R, Li AZ, Yao LM, Chen P, Zhang Y, Tian XY, Beermann F, Wu M, Han J, Huang PQ, Lin T, Wu Q. 2014. Orphan nuclear receptor TR3 acts in autophagic cell death via mitochondrial signaling pathway. Nat Chem Biol 10:133–140. doi: 10.1038/nchembio.1406. [DOI] [PubMed] [Google Scholar]
- 25.Liu JJ, Zeng HN, Zhang LR, Zhan YY, Chen Y, Wang Y, Wang J, Xiang SH, Liu WJ, Wang WJ, Chen HZ, Shen YM, Su WJ, Huang PQ, Zhang HK, Wu Q. 2010. A unique pharmacophore for activation of the nuclear orphan receptor Nur77 in vivo and in vitro. Cancer Res 70:3628–3637. doi: 10.1158/0008-5472.CAN-09-3160. [DOI] [PubMed] [Google Scholar]
- 26.Takekawa M, Tatebayashi K, Itoh F, Adachi M, Imai K, Saito H. 2002. Smad-dependent GADD45beta expression mediates delayed activation of p38 MAP kinase by TGF-beta. EMBO J 21:6473–6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL. 2002. p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migration. J Cell Sci 115:3193–3206. [DOI] [PubMed] [Google Scholar]
- 28.Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K. 1995. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science 270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- 29.Kim SI, Kwak JH, Na HJ, Kim JK, Ding Y, Choi ME. 2009. Transforming growth factor-beta (TGF-beta1) activates TAK1 via TAB1-mediated autophosphorylation, independent of TGF-beta receptor kinase activity in mesangial cells. J Biol Chem 284:22285–22296. doi: 10.1074/jbc.M109.007146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N, Zhang S, Heldin CH, Landstrom M. 2008. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol 10:1199–1207. doi: 10.1038/ncb1780. [DOI] [PubMed] [Google Scholar]
- 31.Kim KY, Kim BC, Xu Z, Kim SJ. 2004. Mixed lineage kinase 3 (MLK3)-activated p38 MAP kinase mediates transforming growth factor-beta-induced apoptosis in hepatoma cells. J Biol Chem 279:29478–29484. doi: 10.1074/jbc.M313947200. [DOI] [PubMed] [Google Scholar]
- 32.Yoo J, Ghiassi M, Jirmanova L, Balliet AG, Hoffman B, Fornace AJ Jr, Liebermann DA, Bottinger EP, Roberts AB. 2003. Transforming growth factor-beta-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. J Biol Chem 278:43001–43007. doi: 10.1074/jbc.M307869200. [DOI] [PubMed] [Google Scholar]
- 33.Yu L, Hebert MC, Zhang YE. 2002. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J 21:3749–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao BX, Chen HZ, Lei NZ, Li GD, Zhao WX, Zhan YY, Liu B, Lin SC, Wu Q. 2006. p53 mediates the negative regulation of MDM2 by orphan receptor TR3. EMBO J 25:5703–5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang C, Lu Y, Li Q, Mao J, Hou Z, Yu X, Fan S, Li J, Gao T, Yan B, Wang B, Song B, Li L. 2016. Salinomycin suppresses TGF-beta1-induced epithelial-to-mesenchymal transition in MCF-7 human breast cancer cells. Chem Biol Interact 248:74–81. doi: 10.1016/j.cbi.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 36.Lin LC, Hsu SL, Wu CL, Hsueh CM. 2014. TGFbeta can stimulate the p(38)/beta-catenin/PPARgamma signaling pathway to promote the EMT, invasion and migration of non-small cell lung cancer (H460 cells). Clin Exp Metastasis 31:881–895. doi: 10.1007/s10585-014-9677-y. [DOI] [PubMed] [Google Scholar]
- 37.Kumawat K, Menzen MH, Slegtenhorst RM, Halayko AJ, Schmidt M, Gosens R. 2014. TGF-beta-activated kinase 1 (TAK1) signaling regulates TGF-beta-induced WNT-5A expression in airway smooth muscle cells via Sp1 and beta-catenin. PLoS One 9:e94801. doi: 10.1371/journal.pone.0094801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun Z, Cao X, Jiang MM, Qiu Y, Zhou H, Chen L, Qin B, Wu H, Jiang F, Chen J, Liu J, Dai Y, Chen HF, Hu QY, Wu Z, Zeng JZ, Yao XS, Zhang XK. 2012. Inhibition of beta-catenin signaling by nongenomic action of orphan nuclear receptor Nur77. Oncogene 31:2653–2667. doi: 10.1038/onc.2011.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu H, Lin Y, Li W, Sun Z, Gao W, Zhang H, Xie L, Jiang F, Qin B, Yan T, Chen L, Zhao Y, Cao X, Wu Y, Lin B, Zhou H, Wong AS, Zhang XK, Zeng JZ. 2011. Regulation of Nur77 expression by beta-catenin and its mitogenic effect in colon cancer cells. FASEB J 25:192–205. doi: 10.1096/fj.10-166462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.To SK, Zeng WJ, Zeng JZ, Wong AS. 2014. Hypoxia triggers a Nur77-beta-catenin feed-forward loop to promote the invasive growth of colon cancer cells. Br J Cancer 110:935–945. doi: 10.1038/bjc.2013.816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen HZ, Liu QF, Li L, Wang WJ, Yao LM, Yang M, Liu B, Chen W, Zhan YY, Zhang MQ, Cai JC, Zheng ZH, Lin SC, Li BA, Wu Q. 2012. The orphan receptor TR3 suppresses intestinal tumorigenesis in mice by downregulating Wnt signalling. Gut 61:714–724. doi: 10.1136/gutjnl-2011-300783. [DOI] [PubMed] [Google Scholar]
- 42.Rajalin AM, Aarnisalo P. 2011. Cross-talk between NR4A orphan nuclear receptors and beta-catenin signaling pathway in osteoblasts. Arch Biochem Biophys 509:44–51. doi: 10.1016/j.abb.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 43.Lu WJ, Chua MS, Wei W, So SK. 2015. NDRG1 promotes growth of hepatocellular carcinoma cells by directly interacting with GSK-3beta and Nur77 to prevent beta-catenin degradation. Oncotarget 6:29847–29859. doi: 10.18632/oncotarget.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]