

Abstract
Although nicotine has a broad impact on both the central and peripheral nervous systems, the molecular mechanisms remain largely unknown, especially at the signaling pathway level. To investigate that aspect, we employed both conventional molecular techniques, such as quantitative real-time PCR and Western blotting analysis, and high-throughput microarray approach to identify the genes and signaling pathways that are modulated by nicotine. We found 14 pathways significantly altered in SH-SY5Y neuroblastoma cells. Of these, the Toll-like receptor pathway (TLR; p = 2.57 × 10−4) is one of the most important innate immune pathways. The death receptor pathway (DR; p = 8.71 × 10−4), whose transducers coordinate TLR signals and help conduct the host immune response to infection, was also significantly changed by nicotine. Furthermore, we found that several downstream pathways of TLR and DR signaling, such as PI3K/AKT signaling (p = 9.55 × 10−6), p38 signaling (p = 2.40 × 10−6), and ERK signaling (p = 1.70 × 10−4), were also significantly modulated by nicotine. Interestingly, most of the differentially expressed genes in these pathways leading to nuclear factor κB (NF-κB) activation and those important inhibitors of pathways leading to apoptosis, including FLIP and Bcl-2, were up-regulated by nicotine. Taken together, our findings demonstrate that nicotine can regulate multiple innate immune-related pathways, and our data thus provide new clues to the molecular mechanisms underlying nicotine's regulatory effects on neurons.
Keywords: Nicotine, Signaling, Immune system, SH-SY5Y
Introduction
In the USA, 20.8% of adults are smokers (CDC 2008). As the top cause of preventable death, cigarette smoking accounts for an estimated 438,000 deaths in the USA annually (Mokdad et al. 2004). Economically, smoking is responsible for about 7% of the total US health-care costs, an estimated $157.7 billion each year, of which $75.8 billion is direct medical costs. Nicotine is the primary addictive component of tobacco smoke and exerts its pharmacologic effects primarily through nicotinic acetylcholine receptors (nAChRs), resulting in broad effects on both the central and the peripheral nervous system. The influence of nicotine on the inflammatory process has received much attention recently. In the central nervous system, inflammatory responses are suggested to play a key role in the development of Alzheimer's disease (AD) and Parkinson's disease (PD). Human and animal studies have revealed an inverse correlation between nicotine intake and the onset and progression of some neurodegenerative diseases, such as PD and AD, an indicator of the neuroprotective and anti-apoptotic effects of nicotine (Picciotto and Zoli 2008; Zeidler et al. 2007). In the peripheral nervous system, nicotine can significantly suppress TLR4-mediated inflammation in both macrophages and a sepsis animal model (Wang et al. 2003, 2004).
Because nAChRs function as Ca2+ channels (Colquhoun 1987; Derkach et al. 1983) and nicotine affects intracellular Ca2+ (Dajas-Bailador et al. 2003), multiple Ca2+-dependent kinases can be induced by nicotine, including PI3K, ERK, PKC and PKA, all of which have been implicated as significant in innate immune responses (Dajas-Bailador et al. 2002; Damaj 2000; Fenster et al. 1999; Messing et al. 1989). By regulating the pathways triggered by these kinases, nicotine exerts broad effects in neurons. For example, the ERK pathway regulates cell survival (Hetman and Gozdz 2004) and is involved in neural plasticity (Brunzell et al. 2003). Activation of PI3K is related to neuron protection and promotes neuron survival (del Peso et al. 1997; Kihara et al. 2001). Acute administration of nicotine also activates nuclear transcription factor NF-κB (Barr et al. 2007) in rat mesencephalic cells in a dose-dependent manner. This protein, a key regulator of neuronal survival and involved in learning, memory formation, and neuron degeneration (Mattson and Camandola 2001; Meffert et al. 2003; Van Antwerp et al. 1996), can be activated by various innate immune pathways as well.
Although nicotine is immunosuppressive in the peripheral nervous system (Wang et al. 2003, 2004) and has positive effects in multiple neurodegenerative diseases involving inflammatory processes (Smith 1998), to date, only a few studies have investigated the effect of nicotine on immune-related signaling pathways (Parrish et al. 2008; Wang et al. 2003). Therefore, how nicotine regulates the innate immunity signaling pathways in neurons is largely unknown. In this study, we used the SH-SY5Y cell line as an in vitro model to study the effect of nicotine on the neuronal innate immune system. First, by measuring gene expression in SH-SY5Y cells treated with nicotine, we identified a number of innate immune-related genes and signaling pathways modulated significantly by nicotine. Second, we confirmed some of our key results using qRT-PCR and Western blotting analyses. On the basis of these results, we propose the first model of nicotine modulation of the neuronal innate immune system.
Materials and methods
Cell culture and nicotine treatment
Human neuroblastoma SH-SY5Y cells were purchased from the American Type Culture Collection (Manassas, VA) and cultured in a 1:1 mixture of ATCC-formulated Eagle's Minimal Essential Medium and F12 Medium supplemented with 10% fetal bovine serum (GIBCO Invitrogen, Grand Island, NY) at 37°C in a humidified atmosphere of 5% CO2. When the cells reached about 80% confluence at 24 h post-seeding, they were treated with either 1 mM nicotine (Sigma, St. Louis, MO) or PBS (used as the control) for 1 h, as described in a previous study (Dunckley and Lukas 2003). Considering the rapid ERK phosphorylation after nicotine treatment, we treated cells with 1 mM nicotine for 5, 15, 30, or 60 min in Western blotting analysis for the ERK phosphorylation experiments reported in this study.
RNA isolation and quantitative RT-PCR
Total RNA was isolated from SH-SY5Y cells using Trizol reagent (Invitrogen, Carlsbad, CA). To eliminate potential residual DNA contamination, the samples were treated with RNase-free DNase I at 37°C for 30 min, followed by inactivation at 65°C for 10 min. Two micrograms of total RNA was reverse-transcribed using Superscript II RT. The mixture was incubated at 25°C for 10 min, 42°C for 1.5 h, and 70°C for 15 min. Quantitative real-time PCR was performed in a total volume of 20 μl containing 10 μl of Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA), 250 nM primers, 2 μl of cDNA mixture, and water. All primer sequences shown in Table 1 were designed using Primer Express Software 3.0 (Applied Biosystems). The RT-PCRs were performed in a 96-well plate using an ABI Prism 7000 Sequence Detection System (Applied Biosystems) with the following thermal cycling conditions: 1 cycle at 50°C for 2 min, initial denaturation at 95°C for 10 min, 40 cycles of denaturation at 95°C for 15 s, and annealing/extension at 60°C for 1 min. Subsequent to the last cycle, a dissociation curve was generated to check for non-specific products. The expression of all the measured genes was normalized using β-actin or GAPDH as internal controls. Each sample was repeated with four independent replicates.
Table 1. Primer sequences used for quantitative RT-PCR.
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| DUSP | AGTGCGCTGAGCTACCTTCAG | CACCTCCCGTGGCCTTT |
| IL6ST | GCTGATTGCAAAGCAAAACG | CCAGACTTCAATGTTGACAAAATACA |
| TRAF6 | ATTCCATGCACATTCAGTACTTTTG | GGCGTGCCAAGTGATTCCT |
| TGFBR2 | CCCACCGCACGTTCAGA | TGCACCGTTGTTGTCAGTGA |
| ITGB1 | TGTAATTCAGTTGATCATTGATGCA | ACAATTTGCCGTTTTCCAAAA |
| HMGN1 | GAAGCGAAGCCGAAAAAGG | TTTGTTTGCACTTTTTTGTCTGAAG |
| DAXX | CCCGAAGCCTCCTTGGATT | ACCCCTGGGATGCCATTC |
| MAPK14 | GCCCGAGCGTTACCAGAAC | CGTAACCCCGTTTTTGTGTCA |
| RPS6KB | TGAGGACATGGCAGGAGTGTT | CAATGTTCCATGCCAAGTTCA |
| ARAF1 | GTCCCGCTGACCATGCA | ATGGAACAGAAACTTAAGGCAGAAG |
| GAPDH | ATGGAAATCCCATCACCATCTT | CGCCCCACTTGATTTTGG |
Microarray and microarray processing
A pathway-focused oligoarray designed specifically for brain-related research including drug addiction was used (Cao et al. 2011; Wei et al. 2011). Briefly, 3,565 genes implicated in the execution and regulation of CNS activities for the maintenance of neuronal homeostasis, as well as those associated with the neuron response to addictive substances such as nicotine, alcohol, or cocaine, were selected on the basis of an earlier version of a pathway-focused cDNA microarray developed by this laboratory (Konu et al. 2004; Li et al. 2004; Wang et al. 2007) and an extensive literature survey. These genes cover most of the major pathways related to cell metabolism, genetic information processing, cellular signaling transduction, neuron-related disease, and cell communication.
OligoWiz (http://www.cbs.dtu.dk/services/OligoWiz/) was used to design the oligonucleotide for each gene of interest. The final length of the selected oligonucleotides was 59.2 ± 3.8 bp (mean ± SD), with a GC content (fraction of total) of 0.53 ± 0.05 and a Tm of 76.4 ± 1.7°C. The oligonucleotides for the genes of interest and ten control oligos were synthesized and spotted at a concentration of 40 μM in 3 × SSC and 1.5 M betaine buffer onto CMT-GAPS II slides (Corning) using an OmniGrid MicroArrayer OGR-03 (Genomic Solutions, San Carlos, CA). To reduce the potential influence of gene-specific dye effect, a universal reference design was used in the microarray analysis. For each experimental group, six independent cultures were prepared. Another 12 independent control cultures were prepared, and these samples were referred to as “universal controls.” For each sample, 10 μg of total RNA was extracted for cDNA synthesis. Following purification with phenol:chloroform extraction and isopropanol precipitation, total RNA was dissolved in 28 μl of water, mixed with 4 μl of 10 × buffer, 4 μl of 10 mM dTTP-free dNTP, 1 μl of 10 mM dTTP, 2 μl of 1 mM cyanine 3-dUTP (for either nicotine-treated or control sample) or cyanine 5-dUTP (for universal control sample)(Enzo, Farmingdale, NY), and 1 μl of Klenow fragment (50 units/μl), then incubated at 37°C for 3 h prior to purification with a QIAquick PCR kit (Qiagen, Valencia, CA). Then, the cyanine 3-labeled sample (either nicotine treated or control) cDNA probes were mixed with the cyanine 5-labeled universal control cDNA probe and added to 7.5 μl of 20 × SSC, 3 μg of CotI DNA, 3 μg of polyA, and 0.5 μl of 10% SDS adjusted to a final volume of 50 μl. The mixture was applied to the pathway-focused oligonucleotide microarray and hybridized overnight at 60°C. Slides were then washed in 1 × SSC and 0.2% SDS at 60°C for 5 min followed by washing in 0.1 × SSC and 0.2% SDS and in 0.1 × SSC at room temperature for 10 min each. Hybridized slides were scanned using the ScanArray Gx microarray scanner, and the intensities of each probe were quantified with the ScanArray Express microarray analysis system (PerkinElmer, Waltham, CA).
Western blot analysis
The SH-SY5Y cells were washed with PBS before being digested with 500 μl of RIPA buffer (50 mM Tris Cl pH 7.5, 0.5% DOC, 0.1% SDS, 150 mM NaCl, 1% NP-40). After incubation on ice for 30 min, the samples were centrifuged at 12,000 rpm for 15 min, and the supernatant liquids were collected into new tubes. For each sample, 10 μg of total protein was electrophoresed on a 10% sodium dodecyl sulfate–polyacrylamide gel and then transferred electrophoretically to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA). After blocking with 1% BSA in TBST buffer at room temperature for 1 h, the filter was first incubated overnight with primary antibody (1:1000) at 4°C and then for 1.5 h at room temperature in the blocking buffer containing p38 or Phospho-p44/42 (Erk1/2) MAPK antibody (dilution 1:2000; Cell Signaling Technology, Inc., Danvers, MA). Immunoreactivity was detected using the SuperSignal West Pico Chemiluminescent Substract kit (PIERCE Inc., Rockford, IL), and the preparations were exposed to X-ray film. After the films were developed, they were scanned on a Microtek ScanMaker i800 with ScanWizard 5.5 at a resolution of 600 dpi for quantitative analysis with ImageQuant 5.1 (Molecular Dynamics, Sunnyvale, CA). The relative p38, ERK1 (p44) or ERK2 (p42) values were normalized to α-tubulin, and the significance of the difference between the nicotine-treated and control samples was determined by Student's t test via MATLAB (The Mathworks, Natick, MA). Four independent cell cultures were prepared under the same conditions used for the microarray experiments for both nicotine treatment and control groups (N = 3–4).
Bioinformatics analyses of microarray data
After scanning each array, the raw hybridization intensities of each element were obtained, and the background-subtracted median intensity of each spot was used for further statistical analysis. The two replicates of each gene on a chip were analyzed separately. To minimize spot variations and reduce experimental error, we discarded spots that were either oversaturated or poorly expressed (i.e., 5% of the weakest spots in each replicate of an array). We then used an intensity-dependent normalization method (locally weighted linear regress; Lowess) to normalize the data for each replicate (Yang et al. 2002). To minimize experimental error, genes with six or fewer valid measurements were removed from further analysis. The two technical replicates per array were averaged to obtain the measurement of each gene for a given sample. The normalized data from each drug treatment group were compared with the normalized data from the controls, and significantly regulated genes were identified by Student's t test. On the basis of the p values, the false discovery rate (FDR) was calculated by the method of Benjamini and Hochberg (1995) via MATLAB.
The genes significantly regulated by each treatment were analyzed by Ingenuity Pathway Analysis (IPA; https://analysis.ingenuity.com) with the goal of revealing the enriched biochemical pathways. The core of IPA is the Ingenuity Pathways Knowledge Base (IPKB), which contains the biological function, interaction, and other related information of a curated gene set and more than 330 biochemical pathways. This pathway-based software is designed to identify global canonical pathways, dynamically generated biological networks, and global functions from a given list of genes. Basically, the genes with their symbol, corresponding GenBank accession numbers, or both were uploaded into the IPA and compared with the genes included in each canonical pathway using the whole gene set of IPKB as the background. All the pathways with one or more genes overlapping the candidate genes were extracted. In IPA, each of these pathways was assigned a p value via Fisher's exact test, which denoted the probability of overlap between the pathway and input genes. Because a relatively large number of pathways were examined, multiple comparison correction for the individually calculated p values was necessary to make reliable statistical inferences. The FDR was also calculated with the method of Benjamini and Hochberg (1995).
Results
Identification of genes and pathways significantly modulated by nicotine
To study the acute effects of nicotine on signaling pathways, we treated SH-SY5Y cells under the paradigm reported previously (Dunckley and Lukas 2003) and then measured RNA expression for 3,565 genes that have been implicated in cell metabolism, genetic information processing, cellular signaling transduction, neuron-related disease, and cell communication using microarray techniques (Cao et al. 2011; Wei et al. 2011). Following a series of normalization and bioinformatics analyses, we identified 296 genes whose expression was significantly modulated by nicotine (p < 0.05; FDR ≤ 0.258) (Table S1). The FDR is the ratio of the false positives expected from multiple comparisons to total positives observed, which also represents the approximate probability for an identified gene to be a false positive (Blalock et al. 2005, 2004). The observed FDR of less than 0.258 was considered to be acceptable for most microarray studies; especially, it was the upper limit value for the indentified genes, with most genes having much lower FDRs, indicating an acceptable statistical power. To detect biochemical processes or pathways that were enriched in these differentially expressed genes, we analyzed them with Ingenuity Pathway Analysis (IPA) software and identified 14 pathways significantly enriched in the nicotine-treated group (Table 2), among which were included the critical innate immune pathway TLR (p = 2.57 × 10−4; FDR = 0.015) and its coordinate pathway DR (death receptor) signaling (p = 8.71 × 10−4; FDR = 0.035). Additionally, several important downstream pathways of TLR pathway and DR pathway, such as ERK signaling pathway (p = 1.70 × 10−4; FDR = 0.015), p38 signaling pathway (p = 2.40 × 10−6; FDR = 6.82 × 10−4), and PI3K/AKT signaling pathway (p = 9.55 × 10−4; FDR = 1.81 × 10−3), were significantly modulated by nicotine. Similarly, the inter-leukin-6 (IL-6) pathway, which is activated by the TLR pathway product IL-6, also reached a significant level (p = 5.13 × 10−4; FDR = 0.015). Together, these six pathways are designated as “innate immune-related pathways” in this report.
Table 2. Biological pathways significantly modulated by nicotine in SH-SY5Y cellsa.
| Pathway | p value | FDRb | Genes significantly regulated by nicotine |
|---|---|---|---|
| Glucocorticoid receptor signaling | 3.11 × 10−7 | 1.77 × 10−4 | AGT, AR, BCL2, CD3G, DUSP1, FGG, FOS, GTF2H2, IKKγ, JAK3, MAP3 K7IP1, MAPK14, MED14, NFATC3, PIK3CG, SMAD2, STAT5A, TGFB2, TGFBR2, TRAF6, YWHAH |
| p38 MAPK signaling | 2.40 × 10−6 | 6.82 × 10−4 | ATF2, DAXX, DUSP1, HMGN1, IRAK3, MAP3K7IP1, MAPK14, MEF2D, PLA2G4A, TGFB2, TGFBR2, TRAF6 |
| PI3K/AKT signaling | 9.55 × 10−6 | 1.81 × 10−3 | BCL2, EIF4EBP1, FOXO1, IKKγ, ITGB1, JAK3, PIK3CG, PPM1L, RPS6KB1, SFN, TRP53, YWHAH |
| PTEN signaling | 5.01 × 10−5 | 7.13 × 10−3 | BCL2, BCL2L11, CDC42, FOXO1, IKKγ, ITGB1, NGFR, PIK3CG, RPS6KB1, YWHAH |
| Acute-phase response signaling | 1.02 × 10−4 | 0.012 | AGT, APOA1, FGG, FOS, IKKγ, IL6R, IL6ST, MAP3K7IP1, MAPK14, NGFR, PIK3CG, TF, TRAF6 |
| ERK/MAPK signaling | 1.70 × 10−4 | 0.015 | ARAF, ATF2, DUSP1, EIF4EBP1, FOS, ITGB1, PIK3CG, PLA2G4A, PPM1L, PPP1CA, RAPGEF3, YWHAH |
| Mitochondrial dysfunction | 2.09 × 10−4 | 0.015 | COX6B1, COX7B, COX7C, NDUFA4, NDUFV1, OGDH, SDHB, SDHC, SNCA, UQCRFS1 |
| VEGF signaling | 2.14 × 10−4 | 0.015 | BCL2, EIF2B1, EIF2B5, FOXO1, PIK3CG, SFN, VCL, VEGFA |
| Toll-like receptor signaling | 2.57 × 10−4 | 0.015 | FOS, IKKγ, IRAK3, MAP3K7IP1, MAP4K4, MAPK14, TRAF6 |
| PPAR signaling | 2.75 × 10−4 | 0.015 | FOS, IKKγ, MAP3K7IP1, MAP4K4, NGFR, NR2F1, STAT5A, TRAF6 |
| IL-6 signaling | 2.95 × 10−4 | 0.015 | FOS, IKKγ, IL6R, IL6ST, MAP3K7IP1, MAP4K4, MAPK14, NGFR, TRAF6 |
| T-cell receptor signaling | 5.13 × 10−4 | 0.024 | CALM2, CD3G, CSK, FOS, IKKγ, NFATC3, PIK3CG, ZAP70 |
| Hepatic fibrosis/hepatic stellate cell activation | 7.24 × 10−4 | 0.032 | AGT, BCL2, IGF1R, IL6R, NGFR, SMAD2, TGFB2, TGFBR2, VEGFA |
| Death receptor signaling | 8.71 × 10−4 | 0.035 | BCL2, FLIP, DAXX, IKKγ, MAP4K4, TNFRSF10B |
The pathways and genes included in each pathway were based on the output of Ingenuity Pathway Analysis (IPA; http://www.ingenuity.com/). Some pathways may be partially overlapped with others. No action was taken to remove any possible redundancy
The FDR values were computed based on the p values of the pathways included in the IPA output via the method of Benjamini and Hochberg (1995). Pathways with FDR <0.05 are included in Table 2
Confirmation of mRNA expression of innate immune-related genes
Of the 296 genes identified as differentially expressed in response to acute nicotine treatment, there were 12 in each of the p38 MAPK signaling pathway, PI3K/AKT signaling pathway, and ERK MAPK signaling pathway, 9 in the IL-6 pathways, 5 in the TLR pathway, and 6 in the DR pathway that were identified by signaling pathway analysis using IPA (Table 3). Because a significant number of these genes are part of more than one biochemical pathway, in total, we found 32 uniquely affected genes within these six immune-related pathways. To confirm the microarray results, we performed quantitative real-time RT-PCR (qRT-PCR) analysis on ten randomly selected genes using RNA samples from an independent experiment under the same conditions used for the microarray experiment (Table 4). As shown in Fig. 1, the results from the two approaches were highly consistent, with a correlation coefficient of 0.92 (p < 0.01). All these genes identified in the microarray experiment, except RPS6KB1 and ARAF1, were confirmed by qRT-PCR analysis using independent samples. DUSP, HMGN1, DAXX, and MAPK14 showed significant up-regulation (p = 0.028–0.005) and IL6ST, TRAF6, TGFBR2, and ITGB1 demonstrated significant down-regulation (p = 0.027–0.004). Although the changes in RPS6KB1 and ARAF1 were not significant (p > 0.05) by qRT-PCR, they did show a trend in the same direction as that detected with the microarray approach. A detailed summary of these qRT-PCR results, including the average expression, standard deviation, fold change, and corresponding p value, is shown in Table 4.
Table 3. Genes significantly changed by nicotine at the mRNA level in the six immune-related pathways.
| Gene symbol | Gene | Ratio ± SD | p value | FDRa |
|---|---|---|---|---|
| ARAF1 | V-raf murine sarcoma 3611 viraloncogene homolog 1 | 1.24 ± 0.09 | 0.03 | 0.17 |
| ATF2 | Activating transcription factor 2 | 0.80 ± 0.04 | 0.02 | 0.14 |
| BCL2 | B cell leukemia/lymphoma 2 | 1.24 ± 0.16 | 0.03 | 0.17 |
| FLIP | CASP8 and FADD-like apoptosis regulator | 1.35 ± 0.10 | 0.01 | 0.12 |
| DAXX | Fas death domain-associated protein | 1.25 ± 0.09 | 0.03 | 0.17 |
| DUSP1 | Dual specificity phosphatase 1 | 1.17 ± 0.08 | 0.02 | 0.14 |
| EIF4EBP1 | Eukaryotic translation initiation factor 4E binding protein 1 | 0.65 ± 0.07 | 0.01 | 0.13 |
| FOS | Fos oncogene | 1.28 ± 0.16 | 0.03 | 0.17 |
| FOXO1 | Forkhead box 1 | 0.52 ± 0.10 | 0.00 | 0.05 |
| HMGN1 | High mobility group nucleosomal binding domain 1 | 1.42 ± 0.14 | 0.01 | 0.14 |
| IKKγ | Inhibitor of kappa light polypeptide gene enhancer in B cells, inase gamma | 1.27 ± 0.18 | 0.03 | 0.17 |
| IL6R | Interleukin 6 receptor | 1.17 ± 0.06 | 0.01 | 0.10 |
| IL6ST | Interleukin 6 signal transducer | 0.74 ± 0.06 | 0.01 | 0.12 |
| ITGB1 | Integrin beta 1 | 0.86 ± 0.05 | 0.04 | 0.18 |
| JAK3 | Janus kinase 3 | 0.80 ± 0.10 | 0.02 | 0.16 |
| MAP3K7IP1 | Mitogen-activated protein kinase kinase kinase 7 interacting protein 1 | 1.45 ± 0.17 | 0.01 | 0.11 |
| MAP4K4 | Mitogen-activated protein kinase kinase kinase kinase 4 | 0.69 ± 0.07 | 0.00 | 0.04 |
| MAPK14 | Mitogen-activated protein kinase 14 | 1.22 ± 0.19 | 0.05 | 0.18 |
| MEF2D | Myocyte enhancer factor 2d | 1.35 ± 0.08 | 0.04 | 0.18 |
| NGFR | Nerve growth factor receptor | 1.46 ± 0.18 | 0.05 | 0.18 |
| PIK3CG | Phosphoinositide-3-kinase, catalytic, gamma polypeptide | 1.30 ± 0.20 | 0.05 | 0.19 |
| PLA2G4A | Phospholipase A2, group IVA (cytosolic, calcium dependent) | 1.42 ± 0.16 | 0.03 | 0.17 |
| PPM1L | Protein phosphatase 1 (formerly 2C)-like | 1.35 ± 0.24 | 0.03 | 0.17 |
| PPP1CA | Protein phosphatase 1, catalytic subunit, alpha isoform | 1.21 ± 0.08 | 0.02 | 0.15 |
| RAPGEF3 | Rap guanine nucleotide exchange factor (GEF) 3 | 0.84 ± 0.04 | 0.03 | 0.17 |
| RPS6KB1 | Ribosomal protein S6 kinase, 70kD, polypeptide 1 | 1.28 ± 0.13 | 0.04 | 0.17 |
| SFN | Stratifin | 1.14 ± 0.04 | 0.02 | 0.16 |
| TGFB2 | Transforming growth factor, beta 2 | 0.74 ± 0.03 | 0.00 | 0.01 |
| TGFBR2 | Transforming growth factor, beta receptor 2 | 0.57 ± 0.13 | 0.01 | 0.10 |
| TNFRSF10B | Tumor necrosis factor receptor superfamily, member 10b | 1.43 ± 0.15 | 0.04 | 0.18 |
| TRAF6 | Tnf receptor-associated factor 6 | 0.75 ± 0.13 | 0.03 | 0.17 |
| YWHAH | Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, beta polypeptide | 1.21 ± 0.12 | 0.04 | 0.17 |
FDR is calculated from the p values of all the genes included in the microarray via the method of Benjamini and Hochberg (1995)
Table 4. Genes significantly changed by acute nicotine treatment as confirmed by real-time PCR.
| Gene symbol | Gene | Ratio ± SDa | p value | FDRb |
|---|---|---|---|---|
| DUSP | Dual specificity phosphatase 2 | 1.23 ± 0.04 | 0.005 | 0.025 |
| IL6ST | Interleukin 6 signal transducer | 0.81 ± 0.04 | 0.013 | 0.028 |
| TRAF6 | Tnf receptor-associated factor 6 | 0.90 ± 0.01 | 0.027 | 0.035 |
| TGFBR2 | Transforming growth factor, beta receptor 2 | 0.73 ± 0.07 | 0.009 | 0.028 |
| ITGB1 | Integrin beta 1 | 0.73 ± 0.10 | 0.004 | 0.025 |
| HMGN1 | High mobility group nucleosomal binding domain 1 | 1.37 ± 0.07 | 0.028 | 0.035 |
| DAXX | Fas death domain-associated protein | 1.12 ± 0.06 | 0.023 | 0.035 |
| MAPK14 | Mitogen-activated protein kinase 14 | 1.14 ± 0.05 | 0.014 | 0.028 |
The ratio shown in this column is between gene expression in the nicotine-treated and control groups. For the ten genes verified by real-time PCR, the expression of eight is significantly (p < 0.05) different in response to acute nicotine treatment. For the other two genes, i.e., ARAF1 and RPS6KB1, the p value is 0.197 and 0.519, respectively. Although the expression change of the two genes did not reach significance, they showed a trend in the same direction as that detected with the microarray approach
FDR is calculated from the p values of the ten genes verified by real-time PCR via the method of Benjamini and Hochberg (1995). For ARAF1 and RPS6KB1, the FDR is 0.219 and 0.519, respectively; the values are not shown in the table, because the expression difference in the two genes was not significant in real-time PCR tests
Fig. 1.

Comparison of results from qRT-PCR and microarrays for ten randomly selected genes significantly modulated by acute nicotine treatment. The mRNA expression of these genes was measured by qRT-PCR in SH-SY5Y cells either treated or untreated with nicotine for 1 h. Of the ten genes verified by real-time RT-PCR, eight showed significant changes in response to nicotine (p < 0.05), and the other two showed alteration in the same direction as detected with the microarray approach, although the changes did not reach significance. Data are presented as the mean ± SD (n = 5) for both microarray and qRT-PCR experiments
Regulation by nicotine of innate immune-related genes at protein expression level
Given that MAPK14 (p38) is one of the most critical kinases in the p38 pathway (Sanz et al. 2000; Tamura et al. 2000), we performed Western blotting analysis of SH-SY5Y cells that had or had not been exposed to nicotine to measure p38 protein expression, with the goal of using it as a key indicator of changes detected in all the immune-related pathways that contained 32 genes significantly altered by nicotine according to the microarray data. Our results clearly demonstrated that nicotine treatment caused a significant increase (45%; p = 0.04) in the expression of p38 in SH-SY5Y cells (Fig. 2).
Fig. 2.
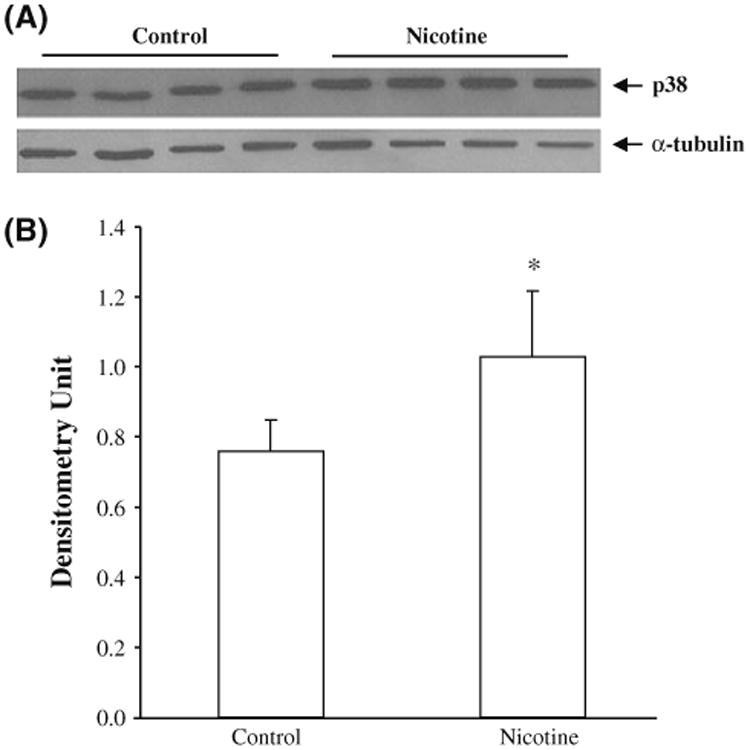
Expression of p38 (MAPK14) protein. The SH-SY5Y cells were treated with nicotine for 1 h, and then subjected to Western blotting with p38 antibody. Untreated cells were used as controls. a A representative blot for Western analysis; b the mean ± SD optical density measurements in the two experimental groups (n = 4). *p < 0.05
Activation of ERK pathways by nicotine in SH-SY5Y cells
The results from the microarray, qRT-PCR, and Western blotting analyses indicated that many genes in the six immune-related pathways were significantly modulated by nicotine; however, the activation of these pathways has not been demonstrated. To provide direct evidence of nicotine's activation of these pathways, Western blotting with phos-phor-p44/42 MAPK (Erk1/2) antibody in SH-SY5Y cells treated with nicotine was performed. Although there was no significant induction of ERK phosphorylation at 1 h of treatment, we found that phosphorylation was significantly increased at 5 min (Fig. 3; ERK1: 73%, p = 0.006; ERK2: 67%, p = 0.02), 15 min (ERK1: 36%, p = 0.04; ERK2: 43%, p = 0.04), and 30 min (ERK1: 46%, p = 0.02; ERK2: 51%, p = 0.02).
Fig. 3.
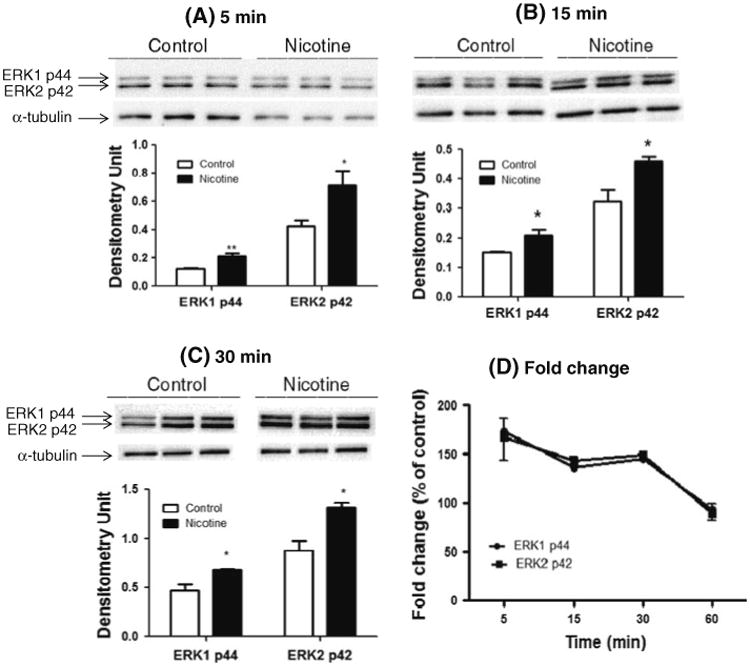
Activation of the ERK pathway by nicotine. Immunoblot analysis of whole-cell lysates from SH-SY5Y cells treated with nicotine for 5 min (a), 15 min (b), 30 min (c), and 1 h (data not shown) was performed to measure phosphorylation of ERK. Untreated cells were used as controls. In a, b and c, the upper panels are representative blots for Western analysis and the lower panel shows the mean ± SD of the optical density measurements (n = 3). The fold changes of p-ERK relative to untreated cells at each time point are shown in d as mean ± SD. **p < 0.01; *p < 0.05
Nicotine's effect on pro- and anti-apoptotic DR pathway
The death domain of the death receptor can trigger both pro-and anti-apoptosis pathways (Ashkenazi and Dixit 1998). Although we demonstrated that nicotine modulated the DR pathway, we were not sure which branch or even if both were affected by nicotine. As shown in Fig. 4, nicotine up-regulates the anti-apoptotic branch of the DR pathway by elevating the expression of genes such as TAB 1 (nicotine/control = 1.45; p = 0.01), IKKγ (nicotine/control = 1.27; p = 0.03) and its downstream p38 (p = 2.40 × 10−6; FDR = 6.82 × 10−4), and ERK (p = 1.70 × 10−4; FDR = 0.015) signaling pathway. Regarding the apoptotic pathway, considering that the expression of two key inhibitors of the pathway, BCL-2 (nicotine/control = 1.24; p = 0.03) and FLIP (nicotine/control = 1.35; p = 0.01), are significantly increased by nicotine, we suspect that overall trend of the pathway is down-regulated by nicotine even though we detected no significant changes of genes in the apoptotic branch, except for DR5 (nicotine/control = 1.43; p = 0.04) and DAXX (nicotine/control = 1.25; p = 0.03).
Fig. 4.
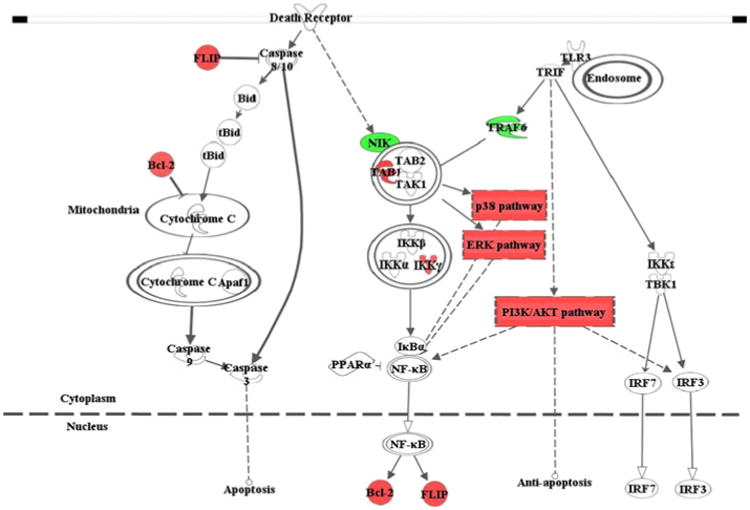
Schematic model of the effect of nicotine on the innate immune system. The TLR and death receptor pathways, as well as their downstream pathways including ERK, p38 and PI3K, may be modulated by acute nicotine exposure in SH-SY5Y neuroblastoma cells. The red (or dark gray in print)-highlighted genes are significantly up-regulated by nicotine, whereas the green (or light gray in print)-highlighted genes are down-regulated. The interaction between the pathways shown here is mainly based on the result from this study; the irrelevant details are not included. The figure was drawn by Ingenuity Pathway Analysis (IPA; http://www.ingenuity.com/)
Discussion
To determine the mechanism by which acute nicotine exposure exerts its effect on neurons, instead of investigating a few genes of interest, as is commonly described in the literature, in this study, we first employed a broad and powerful search strategy with the goal of identifying most, if not all, targets that might be affected. When we examined the expression profiles of almost all genes implicated in the involvement and regulation of CNS activities and responses to addictive substances such as nicotine in SH-SY5Y cells, we found that the expression of 296 genes was significantly modulated by nicotine. To better delineate the effect of nicotine on these genes, we then performed a series of pathway analyses, which indicated that these genes were overrepresented in 14 biological pathways. Of these pathways, six are innate immunity related, i.e., TLR, DR, p38, ERK, PI3K/AKT, and IL-6 signaling pathway. On the basis of the extent of change and the biological function of these genes, we selected ten representative genes for verification with qRT-PCR using independent samples and obtained consistent results. Furthermore, we showed the regulatory effect of nicotine on the innate immune system by demonstrating that both the expression of p38 and the activation of ERK pathway were significantly up-regulated. Consistent with reports from other research groups (Dajas-Bailador et al. 2002; Nakayama et al. 2001; Steiner et al. 2007), induction of ERK phosphorylation by nicotine was observed at earlier treatment time points. Such early activation may contribute to the changes in RNA expression in the overall pathway and downstream gene expression observed at 1 h of treatment.
SH-SY5Y cells express α3, α5, α7, β2, and β4 nAChR subunits that assemble to form various α3*-nAChR sub-types or homomeric α7-nAChRs. Experiments conducted in this study were blind about the role of specific α3*- or α7-nAChR that might mediate the nicotine-altered gene expression. No attempt was made to delineate which genes may be acting via α3*- or α7-nAChR. On its surface, the work reported here may have some degree of semblance to the work of Dunckley and Lukas (2003). The similarity between our study and the earlier one (Dunckley and Lukas (2003) is that we used the same type of cells, which were treated with the same concentration of nicotine for the same period of time. Their work clearly demonstrated that acute nicotine exposure could regulate the expression of genes with diverse functions in SH-SY5Y cells; at the same time, when treated with 1 mM nicotine for 1 h, most of the affected genes were regulated through nAChR-mediated mechanisms. On the other hand, we tried to obtain a more detailed and comprehensive profile of the genes and pathways regulated by acute nicotine exposure. Above and beyond that, significant technical differences exist between our work and the work by Dunckley and Lukas (2003): use of different arrays (array selected for brain-related research, especially for addiction research, vs. randomly selected genes); use of statistical and bioinformatics tools to draw conclusions (sophisticated and advanced tools vs. Student's t test); use of pathway analysis (IPA vs. none); selection of gene sets for further study (6 related but different pathways related to innate immunity vs. randomly selected 17 genes); and cross validation of results (expression analysis of key genes at both the mRNA and protein level vs. expression analysis of genes at the mRNA level only). Because of these quantitative and qualitative differences between the two studies, we identified a set of novel genes and pathways related to the cellular response to acute nicotine exposure in the SH-SY5Y cell line. Thus, the results and conclusions derived from our study should represent a significant update and enhancement of the field.
The genes that are differentially expressed in SH-SY5Y cells in response to nicotine treatment have been categorized broadly by Dunckley and Lukas (2003) into four groups, i.e., transcription factors, protein processing factors, RNA-binding proteins, and plasma membrane-associated proteins. Our work here did not endeavor to classify the differentially regulated genes in a similar manner. Rather, our results suggest that nicotinic activation of genes may have a broad role in altering cellular physiology by modulating gene expression and affecting biological pathways. The genes affected by nicotine treatment in SH-SY5Y cells belong to such diverse pathways as glucocorticoid receptor signaling, ERK/MAPK signaling, and T-cell receptor signaling. In this study, we mainly focused on the modulatory effect of nicotine on innate immune pathways and its influence on neuronal activities. It has been reported that nicotine exerts broad effects on the CNS, such as memory improvement (Rezvani and Levin 2001), release of neurotransmitters (Okuda et al. 1994; Serova and Sabban 2002) and neurotrophic factors (Belluardo et al. 2004), and protection of neurons from apoptosis (Garrido et al. 2001; Guan et al. 2003; Hejmadi et al. 2003). For example, cigarette smoking has a protective effect against AD and PD (Fratiglioni and Wang 2000; Morens et al. 1994) by affecting cellular inflammation. Further, improved cognitive performance has been identified in healthy non-smokers and AD patients after a short period of nicotine treatment. Indeed, nicotine has been considered a potential treatment for AD and PD (Mihailescu and Drucker-Colin 2000). Moreover, nicotine's anti-inflammatory effect has been demonstrated in multiple immune cell types (Ulloa 2005). However, it remains unclear how nicotine regulates the innate immune-related pathways in neurons.
Although neurons are not typical immune cells, innate immune-related pathways, such as TLR, are functional in these cells. Under normal conditions, TLRs play a key role in building up the first line of defense against infections. Previous studies have revealed the presence of TLR2, TLR3, TLR4, and TLR8 in neurons (Ma et al. 2006; Tang et al. 2007). Although lipopolysaccharide (LPS), a TLR4 ligand, does not trigger immune responses in SH-SY5Y cells (Monaghan et al. 2008), functional TLR3 has been found in this cell line and can induce the expression of interferon-lambda (Zhou et al. 2009). Human neurons also express IFN-β in response to poly(I:C), which specifically activates TLR3, but not LPS (Prehaud et al. 2005). Activation of TLRs can trigger multiple downstream signaling pathways, such as MAPK and PI3K/AKT.
The role of nicotine in the crosstalk between the immune pathways in neurons is still a mystery. By using a customized pathway-focused microarray, consisting of 3,565 genes essential to CNS activity, we identified six pathways significantly modulated by nicotine and closely related to innate immune responses, five of which showed a clear trend. Among these pathways, the expression of the genes in the TLR pathway and its downstream signaling pathways, including p38, ERK, and PI3K/AKT, was up-regulated; whereas the apoptosis branch of the DR pathway was down-regulated by nicotine, and there was no clear trend for the IL-6 pathway. To validate our microarray data, we measured the expression of p38, which is the most important mediator of the p38 MAPK pathway, and found it to be significantly increased by acute nicotine treatment at both the RNA and protein levels. Moreover, increased activation of the ERK MAPK pathway by acute nicotine treatment was detected in the current study. All these pathways are closely related and help to maintain immune homeostasis through a delicate balance. Death receptors, which belong to the tumor necrosis factor receptor superfamily, control cellular activities ranging from gene activation to apoptosis. Death receptor signal transducers participate in the regulation of pattern-recognition receptor (PRR) signaling pathways, including TLR (Chen et al. 2008; Ermolaeva et al. 2008; Imtiyaz et al. 2006; Ma et al. 2004; Pobezinskaya et al. 2008). The interaction between the TLR and DR pathways plays an important role in maintaining cellular homeostasis (Wilson et al. 2009). In this study, we found that TLR signaling was enhanced, whereas the apoptosis branch of DR signaling was suppressed. The ERK pathway regulates proliferation, differentiation, long-term memory, and synaptic plasticity in response to growth factors (Sweatt 2004); the PI3K pathway regulates neural survival and plasticity and axonal growth (Dudek et al. 1997; Lin et al. 2001); and phosphorylation of p38 is involved in both the proliferation of neuronal precursors (Dougherty et al. 2005) and neuronal death (Xia et al. 1995). By modulating these pathways, nicotine may influence a panel of neuron activities, including the plasticity associated with addiction. However, most significantly, the ERK, PI3K, and p38 pathways are all involved in the regulation of neuron survival.
Activated ERK can inhibit caspase-3 activation, leading to the blockade of the caspase cascade during apoptosis (Allan et al. 2003). Another MAPK pathway, JNK, promotes pro-apoptotic responses, but this pathway was not significantly changed by nicotine in our study, suggesting that nicotine administration may have a greater impact on the MAPK pathways involved in anti-apoptosis. Another downstream pathway, PI3K/AKT, which also was up-regulated by nicotine, is a major pathway mediating neuron survival (Dudek et al. 1997). Further, all the above-mentioned five pathways except the apoptosis branch of the DRs can lead to activation of NF-κB, which regulates multiple physiological functions in the brain (Meffert and Baltimore 2005). Activated NF-κB promotes cell survival by inducing caspase inhibitors (Chu et al. 1997; Karin and Lin 2002; Liu et al. 1996; Wang et al. 1996, 1998), including c-IAPs, A1, TRAF1, TRAF2, FLIP, and BCL-2, of which FLIP and BCL-2 were significantly up-regulated by nicotine in our microarray study. On the basis of our findings that nicotine affects both DR and TLR pathways through investigating regulatory mechanisms at the RNA, protein, and phosphorylation levels, we offer a model to explain how nicotine regulates innate immune pathways in neurons (Fig. 4). This model not only describes a mechanism by which nicotine can regulate the innate immune system in neurons through crosstalk between the pathways, but also provides new clues to the potential beneficial effects of nicotine treatment on neurons. Given that no functional TLR4 is expressed (Monaghan et al. 2008) and that functional TLR3, which is one of the most important and best known TLRs, was identified in SH-SY5Y cells (Zhou et al. 2009), our model is primarily based on the TLR3 pathway. For the DR pathway, there are two branches triggered from the death domain, with one leading to apoptosis and the other to anti-apoptosis (Ashkenazi and Dixit 1998). Although basal expression of caspase-8 is not detectable in SH-SY5Y cells (Casciano et al. 2004; Ribas et al. 2005), exposing these neuroblastoma cells to β-amyloid and IFN-γ can result in a rapid increase in cas-pase-8 levels that may mediate in part the toxic effects of these treatments (Cantarella et al. 2003; Tong et al. 2007). Thus, we included this pathway in our model. Activation of the anti-apoptosis branch of both the DR and the TLR pathways could trigger NF-κB, which suppresses the caspase cascade, leading to cell survival. Thus, we conclude that nicotine can modulate innate immune-related pathways in neurons, probably contributing to neuron survival.
Although we have demonstrated that SH-SY5Y neuroblastoma cells can be used to explore the innate immune response to nicotine exposure in nervous system and that a number of genes and pathways involved in this process have been identified, this study represents only a small piece of a much large puzzle on the relation between nicotine and the innate immune system. The current study is based on the assumption that in SH-SY5Y cells, most of the differentially expressed genes are modulated via nAChR-dependent approaches, which is derived from the results of an earlier study under the same conditions (Dunckley and Lukas 2003). For the time being, we could not provide a direct relation between the genes modulated by nicotine and the nAChR subtypes. Additionally, SH-SY5Y cells can synthesize multiple types of neurotransmitters such as noradrenaline and dopamine; it is also able to express receptors like Mu and delta opioid receptors, α2-adrenoceptors, and neuropeptide Y receptors. Thus, we cannot totally exclude the possibility that dopamine, noradrenaline, or other neurotransmitters may have effect on some of the detected genes. However, by identifying a relatively small number of genes and pathways potentially related to the innate immune system, further studies will be able to obtain a more detailed delineation of gene regulation and the action of specific nAChRs, as well as the potential role of other neurotransmitters.
Supplementary Material
Acknowledgments
The project was supported, in part, by NIH grants DA-013783 to MDL, DA-016149 to SLC, and DA-026356 to SLC and MDL, and China Scholarship Council to WYC. We also thank Drs. David L Bronson and Bhagirathi Dash for their editing and comments on the paper.
Footnotes
Electronic supplementary material: The online version of this article (doi:10.1007/s00726-011-1171-0) contains supplementary material, which is available to authorized users.
Contributor Information
Wen-Yan Cui, State Key Laboratory of Protein and Plant Gene Research, Peking University, 100871 Beijing, China; Department of Psychiatry and Neurobehavioral Sciences, University of Virginia, 1670 Discovery Drive, Suite 110, Charlottesville, VA 22911, USA.
Ju Wang, Department of Psychiatry and Neurobehavioral Sciences, University of Virginia, 1670 Discovery Drive, Suite 110, Charlottesville, VA 22911, USA.
Jinxue Wei, Department of Psychiatry and Neurobehavioral Sciences, University of Virginia, 1670 Discovery Drive, Suite 110, Charlottesville, VA 22911, USA.
Junran Cao, Department of Psychiatry and Neurobehavioral Sciences, University of Virginia, 1670 Discovery Drive, Suite 110, Charlottesville, VA 22911, USA.
Sulie L. Chang, Department of Biology, Institute of NeuroImmune Pharmacology, Seton Hall University, South Orange, NJ, USA
Jun Gu, State Key Laboratory of Protein and Plant Gene Research, Peking University, 100871 Beijing, China.
Ming D. Li, Department of Psychiatry and Neurobehavioral Sciences, University of Virginia, 1670 Discovery Drive, Suite 110, Charlottesville, VA 22911, USA
References
- Allan LA, Morrice N, Brady S, Magee G, Pathak S, Clarke PR. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nat Cell Biol. 2003;5:647–654. doi: 10.1038/ncb1005. [DOI] [PubMed] [Google Scholar]
- Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- Barr J, Sharma CS, Sarkar S, Wise K, Dong L, Periyakaruppan A, Ramesh GT. Nicotine induces oxidative stress and activates nuclear transcription factor kappa B in rat mesencephalic cells. Mol Cell Biochem. 2007;297:93–99. doi: 10.1007/s11010-006-9333-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belluardo N, Mudo G, Blum M, Itoh N, Agnati L, Fuxe K. Nicotine-induced FGF-2 mRNA in rat brain is preserved during aging. Neurobiol Aging. 2004;25:1333–1342. doi: 10.1016/j.neurobiolaging.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer's disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci USA. 2004;101:2173–2178. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock EM, Chen KC, Stromberg AJ, Norris CM, Kadish I, Kraner SD, Porter NM, Landfield PW. Harnessing the power of gene microarrays for the study of brain aging and Alzheimer's disease: statistical reliability and functional correlation. Ageing Res Rev. 2005;4:481–512. doi: 10.1016/j.arr.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Brunzell DH, Russell DS, Picciotto MR. In vivo nicotine treatment regulates mesocorticolimbic CREB and ERK signaling in C57Bl/6J mice. J Neurochem. 2003;84:1431–1441. doi: 10.1046/j.1471-4159.2003.01640.x. [DOI] [PubMed] [Google Scholar]
- Cantarella G, Uberti D, Carsana T, Lombardo G, Bernardini R, Memo M. Neutralization of TRAIL death pathway protects human neuronal cell line from beta-amyloid toxicity. Cell Death Differ. 2003;10:134–141. doi: 10.1038/sj.cdd.4401143. [DOI] [PubMed] [Google Scholar]
- Cao J, Dwyer JB, Mangold JE, Wang J, Wei J, Leslie FM, Li MD. Modulation of cell adhesion systems by prenatal nicotine exposure in limbic brain regions of adolescent female rats. Int J Neuropsychopharmacol. 2011;14:157–174. doi: 10.1017/S1461145710000179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casciano I, Banelli B, Croce M, De Ambrosis A, di Vinci A, Gelvi I, Pagnan G, Brignole C, Allemanni G, Ferrini S, Ponzoni M, Romani M. Caspase-8 gene expression in neuroblastoma. Ann N Y Acad Sci. 2004;1028:157–167. doi: 10.1196/annals.1322.017. [DOI] [PubMed] [Google Scholar]
- Chen NJ, Chio II, Lin WJ, Duncan G, Chau H, Katz D, Huang HL, Pike KA, Hao Z, Su YW, Yamamoto K, de Pooter RF, Zuniga-Pflucker JC, Wakeham A, Yeh WC, Mak TW. Beyond tumor necrosis factor receptor: TRADD signaling in toll-like receptors. Proc Natl Acad Sci USA. 2008;105:12429–12434. doi: 10.1073/pnas.0806585105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kappaB control. Proc Natl Acad Sci USA. 1997;94:10057–10062. doi: 10.1073/pnas.94.19.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D. Molecular neurobiology. A new type of ion-channel block. Nature. 1987;329:204–205. doi: 10.1038/329204a0. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador FA, Soliakov L, Wonnacott S. Nicotine activates the extracellular signal-regulated kinase 1/2 via the alpha7 nicotinic acetylcholine receptor and protein kinase A, in SH-SY5Y cells and hippocampal neurones. J Neurochem. 2002;80:520–530. doi: 10.1046/j.0022-3042.2001.00725.x. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador FA, Heimala K, Wonnacott S. The allosteric potentiation of nicotinic acetylcholine receptors by galantamine is transduced into cellular responses in neurons: Ca2+ signals and neurotransmitter release. Mol Pharmacol. 2003;64:1217–1226. doi: 10.1124/mol.64.5.1217. [DOI] [PubMed] [Google Scholar]
- Damaj MI. The involvement of spinal Ca(2+)/calmodulin-protein kinase II in nicotine-induced antinociception in mice. Eur J Pharmacol. 2000;404:103–110. doi: 10.1016/s0014-2999(00)00579-3. [DOI] [PubMed] [Google Scholar]
- DCC. Smoking-attributable mortality, years of potential life lost, and productivity losses–United States, 2000–2004. MMWR Morb Mortal Wkly Rep. 2008;57:1226–1228. [PubMed] [Google Scholar]
- del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- Derkach VA, Selyanko AA, Skok VI. Acetylcholine-induced current fluctuations and fast excitatory post-synaptic currents in rabbit sympathetic neurones. J Physiol. 1983;336:511–526. doi: 10.1113/jphysiol.1983.sp014595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty JD, Garcia AD, Nakano I, Livingstone M, Norris B, Polakiewicz R, Wexler EM, Sofroniew MV, Kornblum HI, Geschwind DH. PBK/TOPK, a proliferating neural progenitor-specific mitogen-activated protein kinase kinase. J Neurosci. 2005;25:10773–10785. doi: 10.1523/JNEUROSCI.3207-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Dunckley T, Lukas RJ. Nicotine modulates the expression of a diverse set of genes in the neuronal SH-SY5Y cell line. J Biol Chem. 2003;278:15633–15640. doi: 10.1074/jbc.M210389200. [DOI] [PubMed] [Google Scholar]
- Ermolaeva MA, Michallet MC, Papadopoulou N, Utermohlen O, Kranidioti K, Kollias G, Tschopp J, Pasparakis M. Function of TRADD in tumor necrosis factor receptor 1 signaling and in TRIF-dependent inflammatory responses. Nat Immunol. 2008;9:1037–1046. doi: 10.1038/ni.1638. [DOI] [PubMed] [Google Scholar]
- Fenster CP, Beckman ML, Parker JC, Sheffield EB, Whitworth TL, Quick MW, Lester RA. Regulation of alpha4beta2 nicotinic receptor desensitization by calcium and protein kinase C. Mol Pharmacol. 1999;55:432–443. [PubMed] [Google Scholar]
- Fratiglioni L, Wang HX. Smoking and Parkinson's and Alzheimer's disease: review of the epidemiological studies. Behav Brain Res. 2000;113:117–120. doi: 10.1016/s0166-4328(00)00206-0. [DOI] [PubMed] [Google Scholar]
- Garrido R, Mattson MP, Hennig B, Toborek M. Nicotine protects against arachidonic-acid-induced caspase activation, cytochrome c release and apoptosis of cultured spinal cord neurons. J Neurochem. 2001;76:1395–1403. doi: 10.1046/j.1471-4159.2001.00135.x. [DOI] [PubMed] [Google Scholar]
- Guan ZZ, Yu WF, Nordberg A. Dual effects of nicotine on oxidative stress and neuroprotection in PC12 cells. Neurochem Int. 2003;43:243–249. doi: 10.1016/s0197-0186(03)00009-3. [DOI] [PubMed] [Google Scholar]
- Hejmadi MV, Dajas-Bailador F, Barns SM, Jones B, Wonnacott S. Neuroprotection by nicotine against hypoxia-induced apoptosis in cortical cultures involves activation of multiple nicotinic acetylcholine receptor subtypes. Mol Cell Neurosci. 2003;24:779–786. doi: 10.1016/s1044-7431(03)00244-6. [DOI] [PubMed] [Google Scholar]
- Hetman M, Gozdz A. Role of extracellular signal regulated kinases 1 and 2 in neuronal survival. Eur J Biochem. 2004;271:2050–2055. doi: 10.1111/j.1432-1033.2004.04133.x. [DOI] [PubMed] [Google Scholar]
- Imtiyaz HZ, Rosenberg S, Zhang Y, Rahman ZS, Hou YJ, Manser T, Zhang J. The Fas-associated death domain protein is required in apoptosis and TLR-induced proliferative responses in B cells. J Immunol. 2006;176:6852–6861. doi: 10.4049/jimmunol.176.11.6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- Kihara T, Shimohama S, Sawada H, Honda K, Nakamizo T, Shibasaki H, Kume T, Akaike A. Alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J Biol Chem. 2001;276:13541–13546. doi: 10.1074/jbc.M008035200. [DOI] [PubMed] [Google Scholar]
- Konu O, Xu X, Ma JZ, Kane J, Wang J, Shi SJ, Li MD. Application of a customized pathway-focused microarray for gene expression profiling of cellular homeostasis upon exposure to nicotine in PC12 cells. Brain Res Mol Brain Res. 2004;121:102–113. doi: 10.1016/j.molbrainres.2003.11.012. [DOI] [PubMed] [Google Scholar]
- Li MD, Kane JK, Wang J, Ma JZ. Time-dependent changes in transcriptional profiles within five rat brain regions in response to nicotine treatment. Brain Res Mol Brain Res. 2004;132:168–180. doi: 10.1016/j.molbrainres.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Lu KT, Leu TH, Chang WC, Gean PW. A role for the PI-3 kinase signaling pathway in fear conditioning and synaptic plasticity in the amygdala. Neuron. 2001;31:841–851. doi: 10.1016/s0896-6273(01)00433-0. [DOI] [PubMed] [Google Scholar]
- Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- Ma Y, Liu H, Tu-Rapp H, Thiesen HJ, Ibrahim SM, Cole SM, Pope RM. Fas ligation on macrophages enhances IL-1R1-Toll-like receptor 4 signaling and promotes chronic inflammation. Nat Immunol. 2004;5:380–387. doi: 10.1038/ni1054. [DOI] [PubMed] [Google Scholar]
- Ma Y, Li J, Chiu I, Wang Y, Sloane JA, Lu J, Kosaras B, Sidman RL, Volpe JJ, Vartanian T. Toll-like receptor 8 functions as a negative regulator of neurite outgrowth and inducer of neuronal apoptosis. J Cell Biol. 2006;175:209–215. doi: 10.1083/jcb.200606016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Camandola S. NF-kappaB in neuronal plasticity and neurodegenerative disorders. J Clin Invest. 2001;107:247–254. doi: 10.1172/JCI11916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meffert MK, Baltimore D. Physiological functions for brain NF-kappaB. Trends Neurosci. 2005;28:37–43. doi: 10.1016/j.tins.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. NF-kappa B functions in synaptic signaling and behavior. Nat Neurosci. 2003;6:1072–1078. doi: 10.1038/nn1110. [DOI] [PubMed] [Google Scholar]
- Messing RO, Stevens AM, Kiyasu E, Sneade AB. Nicotinic and muscarinic agonists stimulate rapid protein kinase C translocation in PC12 cells. J Neurosci. 1989;9:507–512. doi: 10.1523/JNEUROSCI.09-02-00507.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihailescu S, Drucker-Colin R. Nicotine, brain nicotinic receptors, and neuropsychiatric disorders. Arch Med Res. 2000;31:131–144. doi: 10.1016/s0188-4409(99)00087-9. [DOI] [PubMed] [Google Scholar]
- Mokdad AH, Marks JS, Stroup DF, Gerberding JL. Actual causes of death in the United States, 2000. JAMA. 2004;291:1238–1245. doi: 10.1001/jama.291.10.1238. [DOI] [PubMed] [Google Scholar]
- Monaghan TK, Pou C, MacKenzie CJ, Plevin R, Lutz EM. Neurotrophic actions of PACAP-38 and LIF on human neuroblastoma SH-SY5Y cells. J Mol Neurosci. 2008;36:45–56. doi: 10.1007/s12031-008-9082-6. [DOI] [PubMed] [Google Scholar]
- Morens DM, Grandinetti A, Reed D, White LR. Smoking-associated protection from Alzheimer's and Parkinson's disease. Lancet. 1994;343:356–357. doi: 10.1016/s0140-6736(94)91194-0. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Numakawa T, Ikeuchi T, Hatanaka H. Nicotine-induced phosphorylation of extracellular signal-regulated protein kinase and CREB in PC12h cells. J Neurochem. 2001;79:489–498. doi: 10.1046/j.1471-4159.2001.00602.x. [DOI] [PubMed] [Google Scholar]
- Okuda S, Saito H, Katsuki H. Arachidonic acid: toxic and trophic effects on cultured hippocampal neurons. Neuroscience. 1994;63:691–699. doi: 10.1016/0306-4522(94)90515-0. [DOI] [PubMed] [Google Scholar]
- Parrish WR, Rosas-Ballina M, Gallowitsch-Puerta M, Ochani M, Ochani K, Yang LH, Hudson L, Lin X, Patel N, Johnson SM, Chavan S, Goldstein RS, Czura CJ, Miller EJ, Al-Abed Y, Tracey KJ, Pavlov VA. Modulation of TNF release by choline requires alpha7 subunit nicotinic acetylcholine receptor-mediated signaling. Mol Med. 2008;14:567–574. doi: 10.2119/2008-00079.Parrish. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M. Neuroprotection via nAChRs: the role of nAChRs in neurodegenerative disorders such as Alzheimer's and Parkinson's disease. Front Biosci. 2008;13:492–504. doi: 10.2741/2695. [DOI] [PubMed] [Google Scholar]
- Pobezinskaya YL, Kim YS, Choksi S, Morgan MJ, Li T, Liu C, Liu Z. The function of TRADD in signaling through tumor necrosis factor receptor 1 and TRIF-dependent Toll-like receptors. Nat Immunol. 2008;9:1047–1054. doi: 10.1038/ni.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prehaud C, Megret F, Lafage M, Lafon M. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J Virol. 2005;79:12893–12904. doi: 10.1128/JVI.79.20.12893-12904.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvani AH, Levin ED. Cognitive effects of nicotine. Biol Psychiatry. 2001;49:258–267. doi: 10.1016/s0006-3223(00)01094-5. [DOI] [PubMed] [Google Scholar]
- Ribas J, Gomez-Arbones X, Boix J. Caspase 8/10 are not mediating apoptosis in neuroblastoma cells treated with CDK inhibitory drugs. Eur J Pharmacol. 2005;524:49–52. doi: 10.1016/j.ejphar.2005.09.021. [DOI] [PubMed] [Google Scholar]
- Sanz V, Arozarena I, Crespo P. Distinct carboxy-termini confer divergent characteristics to the mitogen-activated protein kinase p38alpha and its splice isoform Mxi2. FEBS Lett. 2000;474:169–174. doi: 10.1016/s0014-5793(00)01598-2. [DOI] [PubMed] [Google Scholar]
- Serova L, Sabban EL. Involvement of alpha 7 nicotinic acetylcholine receptors in gene expression of dopamine biosynthetic enzymes in rat brain. J Pharmacol Exp Ther. 2002;303:896–903. doi: 10.1124/jpet.102.039198. [DOI] [PubMed] [Google Scholar]
- Smith MA. Alzheimer disease. Int Rev Neurobiol. 1998;42:1–54. doi: 10.1016/s0074-7742(08)60607-8. [DOI] [PubMed] [Google Scholar]
- Steiner RC, Heath CJ, Picciotto MR. Nicotine-induced phosphorylation of ERK in mouse primary cortical neurons: evidence for involvement of glutamatergic signaling and CaM-KII. J Neurochem. 2007;103:666–678. doi: 10.1111/j.1471-4159.2007.04799.x. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Tamura K, Sudo T, Senftleben U, Dadak AM, Johnson R, Karin M. Requirement for p38alpha in erythropoietin expression: a role for stress kinases in erythropoiesis. Cell. 2000;102:221–231. doi: 10.1016/s0092-8674(00)00027-1. [DOI] [PubMed] [Google Scholar]
- Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, Magnus T, Camandola S, Mattson MP. Pivotal role for neuronal Tolllike receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci USA. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong HX, Lu CW, Zhang JH, Ma L. Combination of gamma-interferon with TRAIL and cisplatin or etoposide induces apoptosis in human neuroblastoma cell line SH-SY5Y. Chin Med Sci J. 2007;22:38–43. [PubMed] [Google Scholar]
- Ulloa L. The vagus nerve and the nicotinic anti-inflammatory pathway. Nat Rev Drug Discov. 2005;4:673–684. doi: 10.1038/nrd1797. [DOI] [PubMed] [Google Scholar]
- Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Baldwin AS., Jr TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, Al-Abed Y, Metz C, Miller EJ, Tracey KJ, Ulloa L. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med. 2004;10:1216–1221. doi: 10.1038/nm1124. [DOI] [PubMed] [Google Scholar]
- Wang J, Gutala R, Sun D, Ma JZ, Sheela RC, Ticku MK, Li MD. Regulation of platelet-derived growth factor signaling pathway by ethanol, nicotine, or both in mouse cortical neurons. Alcohol Clin Exp Res. 2007;31:357–375. doi: 10.1111/j.1530-0277.2006.00331.x. [DOI] [PubMed] [Google Scholar]
- Wei J, Wang J, Dwyer JB, Mangold J, Cao J, Leslie FM, Li MD. Gestational nicotine treatment modulates cell death/survival-related pathways in the brains of adolescent female rats. Int J Neuropsychopharmacol. 2011;14:91–106. doi: 10.1017/S1461145710000416. [DOI] [PubMed] [Google Scholar]
- Wilson NS, Dixit V, Ashkenazi A. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol. 2009;10:348–355. doi: 10.1038/ni.1714. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002;30:e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidler R, Albermann K, Lang S. Nicotine and apoptosis. Apoptosis. 2007;12:1927–1943. doi: 10.1007/s10495-007-0102-8. [DOI] [PubMed] [Google Scholar]
- Zhou L, Wang X, Wang YJ, Zhou Y, Hu S, Ye L, Hou W, Li H, Ho WZ. Activation of toll-like receptor-3 induces interferon-lambda expression in human neuronal cells. Neuroscience. 2009;159:629–637. doi: 10.1016/j.neuroscience.2008.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.