

Abstract
Protein folding is governed by a variety of molecular forces including hydrophobic and ionic interactions. Less is known about the molecular determinants of protein stability. Here we used a recently developed computer algorithm (pHinder) to investigate the relationship between buried charge and thermostability. Our analysis revealed that charge networks in the protein core are generally smaller in thermophilic organisms as compared to mesophilic organisms. To experimentally test whether core network size influences protein thermostability, we purified 18 paralogous Ras superfamily GTPases from yeast and determined their melting temperatures (Tm, or temperature at which 50% of the protein is unfolded). This analysis revealed a wide range of Tm values (35–63 °C) that correlated significantly (R = 0.87) with core network size. These results suggest that thermostability depends in part on the arrangement of ionizable side chains within a protein core. An improved capacity to predict protein thermostability may be useful for selecting the best candidates for protein crystallography, the development of protein-based therapeutics, as well as for industrial enzyme applications.
Graphical abstract
A fundamental goal in biological chemistry is to understand the determinants of protein structure and function. A particularly important objective is to identify enzymes that remain active for long periods of time at high temperatures in vitro. Increasing the thermostability of enzymes can make biochemical processes more efficient and cost-effective; reactions that can be done at higher temperatures are often more resistant to chemical solvents and denaturants, and are less susceptible to microbial contamination. Accordingly, thermostable enzymes are widely used in industrial applications such as paper production and biofuel development. In the research laboratory, improvements in protein thermostability are almost always beneficial. Thermodynamic stabilization can increase the likelihood of obtaining crystals for protein structure determination, improve the effectiveness of protein-based biopharmaceuticals, and increase the efficiency of laboratory processes that use enzymes. 1,2
Historically, thermostable enzymes have been isolated from organisms that thrive at extreme temperatures and pressures.3 An example familiar to most molecular biologists is the DNA polymerase from Thermus aquaticus, commonly used in the polymerase chain reaction. Despite many successes, however, the reliance on thermophilic organisms has some drawbacks. Of the thermophilic organisms identified to date, nearly all are archaea and bacteria. Thus, the range of proteins from thermophilic organisms is restricted by the limited diversity of enzymes found in nature. Moreover, the function of proteins is often dependent on the host cellular environment,4 which for thermophiles can include high concentrations of salts, proteins, and protein stabilizers such as polyamines. Consequently, the function of a thermophilic protein is not necessarily preserved in vitro or in a heterologous expression system. These factors may account for the surprisingly weak correlation between protein thermostability, characterized by its Tm, and the optimal growth temperature of the host organism.5
Given these limitations, there is interest in alternative methods for identifying thermostable proteins, particularly from eukaryotes. Such methods could allow investigators to select the most promising candidates from newly expanded genome sequence data sets and protein three-dimensional structures. To that end, and as described here, we have implemented recently developed computational and experimental approaches to investigate the relationship between buried charge and protein thermostability.6,7 Using a structure-based algorithm called pHinder, we show that networks of buried charge residues, hereafter referred to as core networks, are smaller in hyperthermophilic proteins than in mesophilic proteins. This finding led us to predict that proteins having greater thermostability should contain smaller core networks. To control for variables such as protein size and disulfide bonding, we tested this prediction on a set of 18 Ras family paralogs from the yeast species Saccharomyces cerevisiae. By restricting our experiments and analysis to an evolutionarily related family of protein paralogs, we could demonstrate that thermostability is indeed correlated with core network size as well as with cysteine content.
EXPERIMENTAL PROCEDURES
Acquisition of Protein Structures
The PDB (Protein Data Bank) accession codes for the nonredundant protein chains used in this study (compiled on 3/11/2015) were downloaded from the National Center for Biotechnology Information (NCBI). Uniprot identifiers for hyperthermophilic proteins were obtained from the Uniprot database (www.uniprot.org) using the search term “hyperthermophile OR hyperthermophilic”. The resultant list of hyperthermophilic structures was cross-referenced against the nonredundant PDB accession codes to build a unique set of hyperthermophilic protein chains. PDB identifiers for structures of Ras superfamily GTPases were obtained from the Pfam database (Wellcome Trust Sanger Institute) using the accession code PF00071. The full set of PDB identifiers used in this study is listed in Supplementary Data 1.
Homology Modeling
Where indicated (Table 1 and Figure 5), homology models were generated for yeast Ras paralogs using the Swiss-Model server (http://swissmodel.expasy.org). The primary sequence of each Ras paralog was obtained from the Saccharomyces Genome Database (www.yeastgenome.org). Models were built for each Ras paralog using the default Swiss-model parameters and the five most similar structural templates.
Table 1.
| Ras paralog |
Tm °C fQCR |
Tm°C ThermoFluor |
core network size a,b |
number of cysteinesc |
model type |
|---|---|---|---|---|---|
| Arf1 | 58 | 54 | 2.1 | 1 | crystal structure |
| Arf3 | 63 | 62 | 1.4 | 1 | liomology model |
| Arl1 | 59 | 51 | 1.5 | 1 | crystal structure |
| Cdc42d | 41 | 35 | 8.8 | 7 | liomology model |
| Gsp1d | 48 | 46 | 4.6 | 3 | crystal structure |
| Gsp2 | 47 | 46 | 5.2 | 3 | liomology model |
| Ras1 | 52 | 53 | 4.0 | 3 | liomology model |
| Ras2 | 52 | 49 | 3.6 | 2 | liomology model |
| Rho2 | 43 | 39 | 6.2 | 6 | liomology model |
| Rho3 | 47 | 46 | 6.6 | 8 | liomology model |
| Rho4 | 49 | 46 | 6.4 | 6 | liomology model |
| Rsr1 | 54 | 51 | 4.4 | 2 | liomology model |
| Sarld | 62 | 56 | 2.0 | 1 | crystal structure |
| Sec4d | 53 | 52 | 3.7 | 3 | crystal structure |
| Vps21 | 56 | 54 | 2.0 | 2 | crystal structure |
| Ypt1d | 53 | 53 | 3.7 | 4 | crystal structure |
| Ypt31 | 59 | 48 | 4.0 | 3 | crystal structure |
| Ypt6 | 62 | 55 | 3.6 | 3 | liomology model |
Calculated using parameter set 1 (see Supplementary Figure 2).
Mean core network size for each set of paralog structural models.
None of the Ras paralogs contain disulfide bonds.
Ras paralogs essential for yeast viability.
Figure 5.

Thermostability of yeast Ras paralogs correlates with core network size and cysteine content. Tm values (fQCR method) for the 18 yeast Ras paralogs as a function of core network size (A, B), percentage of Cys residues per protein (C, D), and pH. Core network size as shown in panels A and B corresponds to the average core network size calculated for the crystal structures (black circles) or homology models (red circles) for each Ras paralog. Linear least-squares fit of the data (black lines) gives correlation coefficients (R values) of 0.87 (A), 0.86 (B), 0.77 (C), and 0.91 (D). (D) Thermal unfolding curves for the yeast paralog Cdc42 in complex with GDP (gray, Tm = 41 °C) and GTPγS (red, Tm= 48 °C) measured by fQCR. Each unfolding curve represents an average of three experimental replicates. Tm values were quantified by fitting a two-state model of thermal unfolding (eq 1) to the Cdc42-GDP (gray line) and Cdc42-GTPγS (red line) data. (F) pH dependence of thermostability for the 18 yeast Ras paralogs. Change in Tm value (ΔTm) is the difference in paralog thermostability at pH S.O and 7.0 (i.e., ΔTm = TmpHS − TmpH7 ) measured using the fQCR method.
pHinder Calculation
The pHinder algorithm was used to calculate the topology of ionizable groups in protein structures using a previously described two-step triangulation procedure.6,7 In step one, a Delaunay triangulation is calculated using the terminal side chain atoms of the ionizable residues Asp (centroid of OD1 and OD2 atoms), Glu (centroid of OE1 and OE2 atoms), His (centroid of ND1 and NE2 atoms), Cys (SG atom), Lys (NZ atom), and Arg (centroid of NH1 and NH2 atoms). The triangulation is then minimized by removing edges ≥10 Å (set by the pHinder parameter maxNetworkEdgeLength) and further simplified by removing redundant network connections. In step two, the Cα atoms of the protein are triangulated. A molecular surface is then calculated from the Cα triangulation by iteratively removing surface-exposed triangulation facets (i.e., tetrahedra) having circumspheres >6.5 Å. The surface facets are then regularized by iteratively subdividing facets having area >20 Å2, internal angles <45°, or edges 3.0 > Å The facets of the molecular surface are then used to classify each ionizable side chain as core (≥3.0 Å below the surface; set by the pHinder parameter coreCutoff), margin (<3.0 Å below and <1.05 Å above the surface; set by the pHinder parameter marginCutOff), or exposed (≥1.05 Å above the surface). Depth of side chain burial is determined by calculating the mean distance from the terminal side chain atom to the coordinates of the closest surface facet pair. Core networks are identified as contiguous runs of ionizable side chains classified as core or deep margin (defined as margin side chains located >2.0 Å below the molecular surface, set by the pHinder parameter marginCutoff CoreNetwork). Margin side chains that are connected to core and deep margin network nodes are also included in the core network result. The sensitivity of the core network versus Tm correlation (Figure 5A,B) is presented in Supplementary Figure 2 for pHinder parameters coreCutoff (Figure S2A), marginCutoff (Figure S2B), marginCutoff CoreNetwork (Figure S2C), and maxNetworkEdgeLength (Figure S2D).
Accurate structure-based pKa predictions are notoriously difficult for core and margin residues. pHinder offers an alternative approach that complements existing predictive methods. pHinder is an informatics-based algorithm that identifies topological arrangements of ionizable side chains likely to be important for protein structure and function. Topological arrangements of buried ionizable side chains (i.e., core networks) are relatively rare and are usually indicative of important structure-function relationships. The ionizable side chains included in the pHinder algorithm all have pKa values that can be shifted into the physiological pH range (pH 5–8) when positioned in the protein core or margin. Like Asp and Glu side chains, Cys side chains are neutral/charged at pH values below/above their pKa values. Dehydration of Cys by burial in the protein core would tend to promote the neutral form of the Cys side chain and cause an upshift in its pKa value. Thus, a Cys side chain is less likely than an Asp or Glu side chain to be charged in the protein core and margin. However, there is precedent for depressed Cys pKa values in Ras proteins.8 Furthermore, when Cys residues are removed from the analysis of Ras paralog structures, the correlation between core network size and thermostability is diminished (Supplementary Figure 3). On the basis of these observations, we conclude that the inclusion of Cys side chains in our analysis was justified.
Consensus Network Analysis
Consensus networks of buried ionizable residues were calculated for each Ras family GTPase using a five-step procedure, as described previously.7 First, the set of 885 GTPases in the PDB were aligned using the cealign function in Pymol (Schrödinger). Second, core networks for each GTPase were calculated using pHinder. Third, core network nodes from the individual pHinder calculations were combined and clustered using a distance constraint of 0.5 Å. Fourth, a Delaunay triangulation was calculated for the set of clustered nodes using a 10.0 Å distance constraint. Fifth, the triangulated cluster nodes were subjected to a second round of iterative clustering using a refined distance constraint of 0.5 Å and a minimum cluster size of 90 residues. This procedure was repeated for minimum cluster sizes of 45 and 20 residues to generate the data shown in Figure 2D.
Figure 2.

Core networks in Ras superfamily small GTPases. (A) pHinder results for core networks in 885 Ras family GTPase structures. The fractional occurrence of ionizable residue types buried within cores of (B) Ras family GTPases and (C) 14253 nonredundant protein chains. (D) Spatially conserved positions of core ionizable residues (inset) within the 885 Ras family GTPase structures calculated by consensus network analysis. Results are shown for high (90 residues), medium (45 residues), and low (20 residues) thresholds for minimum cluster size. The displayed crystal structure corresponds to PDB code 121P (chain A).
Protein Production and Purification
The 18 Ras paralogs examined in this study were cloned directly from yeast genomic DNA (strain BY4741) by standard polymerase chain reaction procedure and subcloned into pLIC-His vectors for heterologous overexpression in Escherichia coli. Sequences for the LIC-compatible cloning primers are listed in Supplementary Table 2.
Proteins were overexpressed in E. coli (BL21 RIPL strain) using the method of autoinduction.9 Starter cultures in LB medium (10 g/L tryptone, 5 g/L yeast extract, 10 g/L NaCl, 50 µg/µL carbenicillin) were inoculated with freshly transformed bacterial colonies and grown overnight at 37 °C. The following day the entire starter culture was used to inoculate autoinduction media: 800 mL of autoclaved ZY medium (10 g/L tryptone, 5 g/L yeast extract), 16 mL of sterile-filtered 50× 5052 solution (25% w/v glycerol, 2.5% w/v glucose, 10% w/v α-lactose), 16 mL of sterile filtered 50× M buffer (1.25 M Na2HPO4, 1.25 M KH2PO4, 2.5 M NH4Cl, 0.25 M Na2SO4), 1.6 mL of 1 M MgSO4, and 800 µL of 50 µg/µL carbenicillin in 50% EtOH. The cultures were grown at 37 °C for 8 h to A600nm between 2 and 4. The temperature was then reduced to 18 °C and the cultures were left to grow overnight. After 16 h of growth the bacteria were harvested by centrifugation and resuspended in 50 mL PBS (25 mM potassium phosphate, 100 mM KCl, pH 7.0). The resuspended bacteria were then frozen at −20 °C.
Overexpressed Ras paralogs containing C-terminal 6xHistags were batch purified on ice using HIS-Select nickel affinity gel (Sigma-Aldrich, P6611). Frozen bacteria were thawed, tris(2-carboxyethyl)phosphine was added (1 mM final), and cells were lysed using a Nano DeBEE homogenizer (Bee International, South Easton, MA) in PBS supplemented with 50 µM GDP, 50 µM MgCl2, and 1 mM tris(2-carboxyethyl)-phosphine (PBS-GMT). Samples were kept at 4 °C throughout the purification. Following centrifugation at 125g, the clarified lysate (13 mL) was combined with 50% affinity gel slurry (1 mL) in a 15 mL conical vial and gently mixed end-over-end for 15 min. The affinity gel was collected by centrifugation and washed in 13 mL of PBS-GMT. This procedure was repeated two more times for a total of three washes. Protein was eluted by transferring a 2 mL slurry of protein-bound affinity gel to a 3 mL fritted-spin column (Thermo Scientific, 89896), and excess buffer was drained by gravity flow. The spin column was then capped, 2 mL of elution buffer (PBS-GMT with 250 mM imidazole) was added, and the column was closed and gently mixed end-over-end for 15 min. The eluted protein sample was then transferred to a 3 mL Slide-A-Lyzer cassette (Thermo Scientific, 66330) and dialyzed against 4 L of PBS-GMT for 1 h. The dialysis cassette was then transferred to 4 L of fresh PBS-GMT and dialyzed overnight. Following dialysis, aliquots of the purified protein were frozen and stored at −80 °C. This procedure typically yielded 2 mL of protein at a concentration between 50 and 100 µM. Protein purity was evaluated by SDS-polyacrylamide gel electrophoresis.
Temperature Denaturation by Fast Quantitative Cysteine Reactivity (fQCR)
0.5 M stock solutions of the thiol-reactive probe 4-(aminosulfonyl)-7-fluoro-2,1,3-benzoxadiazole (ABD; TCI America; A5597) were prepared in dimethyl sulfoxide. All ABD stock concentrations were confirmed spectroscopically (ε313nm = 4200 M−1 cm−1) and stored in 30 µL aliquots at −20 °C. fQCR reaction mixtures were prepared by diluting Ras protein stocks to 1 µM in cold PBS containing 50 µM GDP or GTPγS, chilled on ice, and combined with ABD. For unfolding reactions at pH 7.0, 5 µL of a 26 mM ABD stock solution (1 mM final) was combined with 125 µL of ice-cold protein sample. For unfolding reactions at pH 5.0, 10 µL of a 26 mM ABD stock solution (2 mM final) was combined with 120 µL of ice-cold protein sample. Following ABD addition, the sample mixture was distributed in 10 µL aliquots across a 12-well PCR strip-tube (USA Scientific, 1402-2408), placed in a gradient thermocycler (Biometra Professional Thermocycler), heated for 3 min, immediately transferred to ice, and quenched by adding 2 µL of 0.1 N HCl. The ABD-labeled samples were then transferred to a 384-well plate (Greiner, 788076) and read in a BMG Labtech PHERAstar plate reader using excitation and emission bandpass filters of 400 and 500 nm. The resultant unfolding curves were fit with a two-state model of protein unfolding to quantify the midpoint of thermal denaturation (Tm). fQCR experiments for each Ras paralog were done in triplicate. Prior to fitting, the three data sets for each Ras paralog were used to calculate a mean fQCR-unfolding curve. The Tm values and denaturation profiles presented in Figure 3, Figure 5, and Supplementary Figure 1 correspond to the mean fQCR curve for each Ras paralog. Experimental errors for Tm values obtained by fQCR were derived from the uncertainty of each Tm fit parameter and in all cases were less than 1 °C.
Figure 3.

Thermostability of selected Ras family paralogs from yeast. (A, B) Representative thermal unfolding curves for the yeast paralogs Gsp1 and Vps21 measured independently by the fQCR (left) and ThermoFluor (right) methods. Each unfolding curve represents an average of three experimental replicates. Tm values were quantified by fitting a two-state model of thermal unfolding (eq 1) (black line) to the data. Insets show the purity of each paralog as assessed by SDS-PAGE. (C) Comparison of Tm values quantified by fQCR and ThermoFluor at pH 7.0. Linear least-squares fit of the data (black line) gives a correlation coefficient (R value) of 0.88. (D) Rank-order of the averaged Tm values measured for the set of 18 yeast Ras family paralogs using the fQCR method at pH 7.0.
Temperature Denaturation by ThermoFluor
Ras protein stocks were diluted to 5 µM in PBS containing 50 µM GDP and mixed with 1.5 µL of 500× SYPRO (Invitrogen, S-6650). This sample mixture was then distributed in 20 µL aliquots across a 384-well plate (Genesee 24-305W) and sealed with adhesive film (Bioexpress T-2417-8). An Applied Biosystems RT-PCR machine was programmed to record SYPRO fluorescence from 20 to 85 °C over the course of 45 min (which corresponds to a 3% temperature ramp speed). The resultant unfolding curves were fit with a two-state model of protein unfolding to quantify Tm. Thermofluor experiments for each Ras paralog were done in triplicate. Prior to fitting, the three data sets for each Ras paralog were used to calculate a mean Thermofluor unfolding curve. The Tm values and denaturation profiles presented in Figure 3 and Supplementary Figure 1 correspond to the mean ThermoFluor curve for each Ras paralog. Experimental errors for Tm values obtained by the ThermoFluor method were derived from the uncertainty of each Tm fit parameter, and in all cases were less than 1 °C.
Quantification of Tm Values
The midpoint of thermal denaturation (Tm value) for the fQCR and ThermoFluor unfolding curves was determined for each Ras paralog by fitting a two-state model of unfolding to the temperature dependence of normalized ABD or SYPRO fluorescence:
| (1) |
where fN and fD are linear relationships (e.g., fN = mNT + bN) that describe the temperature-dependent fluorescence of the native (N) and denatured (D) unfolding baselines, and ΔG(T) is the Gibbs-Helmholtz relationship:
| (2) |
where ΔHm is the unfolding enthalpy, ΔCp is the heat capacity, and Tm is the midpoint of thermal denaturation.
RESULTS
Many factors contribute to the stability of folded proteins. Prominent examples include hydrogen bonding, van der Waals interactions, and the hydrophobic effect. In addition, charged amino acid side chains (Asp, Glu, His, Cys, Lys, and Arg) tend to be disfavored within hydrophobic protein cores. However, networks of buried ionizable residues occur regularly in nature and are often required for protein function.11–15 Because they are sequestered from bulk water, buried charges tend to exhibit dramatically altered pKa values that in some cases approach neutrality.12,13 In these situations, proteins must expend free energy to maintain the shifted pKa values. Thus, proteins with numerous ionizable residues in their cores are expected to be relatively unstable. Here we examine how the arrangement of buried ionizable residues affect global protein stability.
To identify charge networks in protein cores we used an informatics-based method known as pHinder. The pHinder computation differs from other structure-based electrostatics calculations by considering only the spatial organization of ionizable groups. Previously we used pHinder to identify core networks of ionizable residues buried within G protein α subunits and showed that these networks are sensitive to physiological changes in intracellular pH.6,7 Furthermore, we identified a distinct core network that links the activated Gα subunit to the G protein-coupled receptor ligand-binding pocket, and showed that similar networks are present in 7-transmembrane receptors from archaea and bacteria.6 Building on these findings, we asked whether differences in core network size could account for differences in protein thermostability across species. We began by comparing a nonredundant set of 14253 mesophilic and 457 hyperthermophilic protein structures from the PDB, listed in Supplementary Data 1. As shown in Figure 1, pHinder analysis revealed a fairly strict upper limit to core network size within both mesophilic and hyperthermophilic protein chains. In mesophilic proteins (Figure 1A–C), this limit scales linearly with chain length and is capped at ~10% of protein size. For thermophilic proteins (Figure 1B–C), the core network size limit is smaller by half (~5%). On the basis of these findings we postulated that core network size is a prominent determinant of protein thermostability.
Figure 1.

Core networks in mesophilic and hyperthermophilic proteins. pHinder results for core networks in a nonredundant set of 14253 mesophilic (A, B, gray) and 457 hyperthermophilic (B, purple) protein chains. (C) Comparison of core networks in mesophilic (gray) and hyperthermophilic (purple) protein chains having less than 250 residues.
To further explore the relationship between core network size and thermostability, we focused on the Ras superfamily of small GTPases. By limiting our analysis to a single family of enzymes, with conserved sequence and structure, we were able to control for variations in protein size, ligand binding, and catalytic activity. As described in Figure 2, we assessed core network sizes for all 885 Ras family GTPases in the PDB. These calculations revealed GTPase core network sizes ranging from 0 to 17 residues, with most (95%) having core networks ≤10 residues (Figure 2A). Cysteine is the most frequently buried ionizable residue type in GTPases (52%), followed by Lys (7.4%), Asp (3.1%), Arg (2.0%), His (1.4%), and Glu (0.01%) (Figure 2B). Cysteine is also the most frequently buried ionizable residue in the set of 14253 nonredundant proteins (40.7%, Figure 2C). However, the occurrence of buried ionizable residues in the nonredundant protein set differs from that of Ras family GTPases, with His being the second most frequently buried ionizable residue (13.1%), followed by Asp (5.8%), Arg (5.4%), Glu (3.9%), and Lys (2.3%). Notably, the frequency of buried Lys residues in Ras family GTPases (7.4%) is more than 3.0-fold higher than in the nonredundant set of reference structures (2.3%). On the basis of these findings, we conclude that ionizable residues are often buried within Ras family GTPases.
We next examined the spatial conservation of buried ionizable residues within Ras family GTPases (Figure 2D). For this calculation we used an approach called consensus network analysis (CNA), which we previously developed to study the conservation of core charges in G protein-coupled receptors.6 Using CNA, we aligned the pHinder-calculated core networks for each of the 885 Ras family GTPases. We then clustered the resultant set of 4389 core residues into distinct spatial nodes (Figure 2D). By far, the most densely populated nodes corresponded to Lys (cluster size 744), Asp (cluster size 736), and Arg (cluster size 285) residues located within structural motifs important for GTPase function and regulation (i.e., the P-loop, switch I, and switch II regions). The second most densely populated nodes corresponded to clusters of Arg and Cys residues (cluster sizes between 100 and 250) located within the core of the GTPase fold. We then searched for smaller node clusters by reducing the threshold cluster size from 90 (occurring in >10% of structures) to 45 (>5% of structures) and 20 (>2% of structures). This procedure revealed additional clusters, most of which contained <50 residues. On the basis of these findings, we conclude that residues comprising GTPase core networks are spatially conserved throughout the Ras superfamily.
Our analysis in Figure 1 compared core network size between mesophilic and hyperthermophilic proteins. That analysis revealed that, on average, smaller core networks are correlated with increased thermostability. Our analysis in Figure 2 compared available Ras family structures and revealed a broad range of core network sizes. Our next objective was to experimentally determine if core network size predicts thermostability in Ras. To avoid any potential bias due to evolutionary history or phylogenetic diversity, we chose to limit our analysis to 18 Ras paralogs from a single organism, Saccharomyces cerevisiae. This set of paralogs was designed to include members from each of the five Ras subfamilies (Ras, Rho, Ran, Rab, and Arf) and includes examples that are essential and nonessential to yeast survival (i.e., gene deletions that are lethal and nonlethal respectively, as indicated in Table 1). All 18 Ras paralogs were cloned directly from the yeast genome, overexpressed in E. coli, and purified via nickel affinity chromatography.
We then measured the thermostability (Tm value) of each purified Ras paralog using two independent methods, fast quantitative cysteine reactivity (fQCR) and Thermo-Fluor.10,16,17 Both approaches are high-throughput and require less material than conventional methods. However, the labeling mechanism of each assay differs. In the fQCR experiment, Cys residues are labeled with a fluorogenic reagent by a mechanism that is analogous to hydrogen exchange.10,17 Most Cys residues in proteins are shielded from bulk solvent by protein structure and local sterics. Upon heating, these protected Cys residues become exposed and can be covalently labeled by cysteine-specific probes. In past studies we have shown that fQCR is applicable to proteins with one or more Cys residues and is largely independent of Cys microenvironment.7,10,17 In particular, the success of our previous studies on Gα protein subunits,7 which are similar to Ras proteins, indicated that fQCR could be used to systematically measure Ras thermo-stabilities. Furthermore, each Ras paralog contained at least one Cys residue (Table 1) and no disulfide bonds (which cannot be labeled by the fQCR probe). As a means of cross-validating the fQCR-derived Tm values, we also measured Tm values using the ThermoFluor approach. In the ThermoFluor assay, a proprietary fluorescent dye (SYPRO) binds to hydrophobic protein regions that become exposed by temperature-induced unfolding. The binding of the SYPRO dye to the protein leads to a fluorescence increase that serves as an indirect indicator of protein denaturation.
Representative thermostability profiles for the Ras paralogs Gsp1 and Vps21 are shown in Figure 3A,B. Unfolding curves for the full set of paralogs are available in Supplementary Figure 1. Using a two-state model of denaturation (eq 1), we analyzed each Ras paralog unfolding profile to quantify the midpoint of temperature unfolding (Tm value). The Tm values obtained from the fQCR and ThermoFluor methods were in excellent agreement (Figure 3C), having a correlation coefficient of 0.88. As shown in Table 1, the fQCR- and ThermoFluor-derived Tm values for the set of 18 Ras paralogs at pH 7.0 ranged from 35 to 63 °C. In the fQCR data set (Figure 3D), which was used to calculate the correlations reported below, Tm values ranged from 41 to 63 °C. When the fQCR- and ThermoFluor-derived Tm values were clustered by subfamily and averaged, the Rho/Cdc42 family had the lowest average Tm (46 °C), followed by Ran (47 °C), Ras (52 °C), Rab (55 °C), and finally Sar/Arf (57 °C). The wide range of values is noteworthy considering the structural and catalytic similarities shared by these protein family members.
We then examined the relationship between Tm values and core network size. Structures have been solved for 8 of the 18 Ras paralogs tested, two of which are shown in Figure 4A–C. For the 10 remaining GTPases, we used homology models obtained from Swiss Model18,19 (see Table 1). Our decision to focus on Ras family paralogs, which display a high degree of sequence and structural similarity, enabled us to generate reliable homology models. As shown in Figure 4B, nucleotides bind at a site that is proximal to the core networks. Network residues closest to the nucleotide-binding site (Figure 4C) include the conserved Asp (Asp67 in Gsp1, Asp62 in Vps21) and Lys (Lys25 in Gsp1, Lys20 in Vps21) located within the switch II and P-loop motifs (Figure 2D), respectively. As anticipated by our model, the experimentally determined Tm values of Ras family paralogs trended linearly with core network size (Figure 5A–B), having correlation coefficients of 0.87 and 0.86 at pH 7.0 and 5.0, respectively. As shown in Supplementary Figure 2, the correlation between core network size and Tm value is largely independent of pHinder parameter set. Thus, Ras paralogs with larger core networks tend to be less thermostable. These findings show that our structure-based approach can predict relative thermostabilities among protein family paralogs.
Figure 4.

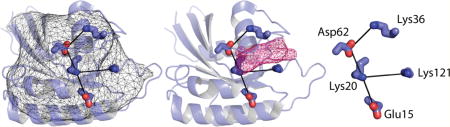
Representative core networks in Ras family paralogs from yeast. (A) Representative pHinder surfaces (thin mesh) and core networks (thick lines) for the yeast Ras family paralogs Gsp1 (3ICQ) and Vps21 (1EK0). (B) Core networks of Gspl and Vps21 in relation to their bound nucleotides (shown as pink mesh). (C) Detail of each core network.
The experimental data in Figure 5A,B demonstrate a strong correlation between buried charge networks and Ras paralog thermostability. This relationship was anticipated by our unbiased and comprehensive computational analysis of charged networks in protein structures. In order to uncover any unanticipated bias in our data, we attempted to correlate the observed Tm values to other protein parameters including amino acid composition, predicted pI, and contact order. In addition, yeast is the only organism for which there is comprehensive data on protein abundance and half-life, 20,21 and for which the identity of all essential genes is known. Of all these parameters (see Supplementary Table 1), only one correlated with the thermostability of the Ras paralogs. As shown in Figure 5C–D, the percentage of Cys residues is inversely proportional to Tm, having correlation coefficients of 0.77 and 0.91 at pH 7.0 and pH 5.0, respectively. We speculate that the lower correlation coefficient is related to a temperature-dependent increase in Cys oxidation at pH 7.0. Cysteine oxidation is pH-dependent because it is mediated primarily by the thiolate form of the Cys side chain. As a result, Cys oxidation is 2 orders of magnitude faster at pH 7.0 than at pH 5.0. Although speculative, the tendency for Cys residues to oxidize at higher temperatures may also explain why Cys residues are underrepresented in thermophilic proteins.22 The potential effects of increased Cys oxidation at pH 7.0 do not greatly affect the correlation between core network size and thermostability. As reported in Supplementary Figure 3, pHinder calculations with and without Cys side chains give correlation coefficients of 0.87 and 0.73, respectively. The findings reported in Figure 5C–D show that cysteine content, which can be quantified directly from protein sequence, is predictive of the relative thermostabilities of Ras paralogs.
Because the focus of this study was to assess the relationship between core network size and protein thermostability, and not binding or catalytic activity, we restricted our analysis to Ras paralogs in the GDP-bound state. This strategy was necessary because most Ras paralogs require specific guanine nucleotide exchange proteins to promote GDP release and GTP binding. Nucleotide exchange can also be enhanced by chelating-away the Mg2+ cofactor with EDTA. However, this approach could not guarantee stoichiometric nucleotide exchange for each of the 18 Ras paralogs studied. In one case, Cdc42, nucleotide exchange was spontaneous in the time frame of the fQCR experiment, which allowed us to assess the effects of stoichiometric nucleotide binding on Cdc42 thermostability. As show in Figure 5E, the thermostability of yeast Cdc42 bound to GTPγS (Tm = 48 °C) is seven degrees higher than Cdc42 bound to GDP (Tm = 41 °C). The magnitude of this difference was striking given that GDP and GTPγS differ by only one phosphate group. While we cannot say whether this is a common feature of all Ras paralogs from yeast, these differences mirror those we observed previously for yeast and mammalian Gα subunits bound to GDP and GTPγS.7
We next investigated Ras paralog thermostability as a function of pH. As noted earlier, buried ionizable residues often exhibit pKa shifts that are coupled to protein stability. The pHinder program was designed to predict pH-sensitive and pH-sensing proteins by identifying these buried residues. Thus, our pHinder calculations predicted that most Ras family paralogs would exhibit some degree of pH sensitivity. To test this prediction, we remeasured the thermostability of each Ras paralog at pH 5.0 and calculated the change in Tm value (ΔTm) (Figure 5F). As anticipated, 14 of 18 paralogs exhibited a pH-sensitive ΔTm (i.e., ΔTm values > 2), with values ranging from 0 to 12 degrees. With the exception of Sar1 and Ypt6, which lacked a pH-dependent Tm shift, Tm values were always higher at pH 5.0 than at pH 7.0. We speculate this dependence of thermostability on pH is due to pKa shifts in the Asp, Lys, or phosphate groups buried within the enzyme active site. There are no obvious structural features that explain the pH insensitivity of the four Ras paralogs Sar1, Ypt6, Arf1, and Rho3. We speculate that the pH insensitivity of these Ras paralogs may be explained by an increase in structural dynamics. Enhanced sampling of conformational space tends to dampen the strength of electrostatic interactions and reduce the magnitude of the pKa shifts that give rise to pH-dependent changes in stability.
DISCUSSION
Understanding the determinants of protein stability is of fundamental importance in biochemistry and biotechnology. In a cell, proteins must be stable enough to function, but not so stable as to escape eventual degradation. In the laboratory, improvements in protein thermostability can advance applications in molecular biology, such as increasing the likelihood of obtaining crystals for protein structure determination. 1,2 In the clinic, increasing protein stability can improve the effectiveness of biopharmaceuticals.23
The isolation of Taq DNA polymerase was a particular milestone in biomedical research. Since that time, considerable effort has gone toward isolating and characterizing other potentially useful proteins from thermophilic organisms. Such proteins are usually active at temperatures near the optimal growth temperature of the host organism, which could be as high as 121 °C.24 However, there has been relatively little progress in understanding the biophysical basis of protein thermostability.1,2 An early comparison of orthologous protein structures from thermophilic and mesophilic organisms revealed a correlation in the number of ion pairs and the optimal growth temperature of the host.25 A more recent analysis confirmed those findings and revealed an additional correlation with protein hydrophobicity.26 Apart from the expected phylogenetic variation, however, orthologous proteins from thermophiles and mesophiles are largely similar. They have similar catalytic mechanisms, and their three-dimensional structures are usually superimposable.1,2 Less attention has been paid to differences among paralogous proteins from a single organism.
In this study, our strategy was to examine networks of ionizable (acidic and basic) residues within protein cores. First, we observed striking differences in core network sizes when comparing protein structures from mesophilic and hyper-thermophilic organisms. Given that these proteins come from a wide range of species, have diverse structures, and have a broad array of biochemical functions, it was important to test whether the same relationship exists without these variables. To that end, we examined all available structures for a single protein superfamily and found that these proteins also contained a broad range of core network sizes. Finally, to control for potential bias due to evolutionary history or phylogenetic diversity, we tested the functional significance of these differences in 18 paralogous proteins derived from a single organism. This systematic comparison of Ras paralogs from yeast revealed that differences in core network size, calculated from protein structure, correlate with Tm values measured experimentally. It is unlikely that this correlation is limited to a single protein family. However, demonstrating the relationship between core network size and thermostability in other protein families will require future experimentation.
We selected the Ras superfamily for this study because it is ideally suited for computational and experimental studies of protein thermostability. More than 800 crystal structures of Ras proteins are available in the PDB. Furthermore, the sequence similarity of Ras family proteins allows for homology modeling of unknown Ras structures. Biochemically, Ras proteins function as compact (~200 residues) molecular switches that cycle between a GDP-bound unactivated state and a GTP-bound activated state.27,28 Biologically, Ras proteins are conserved from yeast to humans and are classified into subfamilies according to the cellular processes they regulate.29,30 The Ras subfamily transmits signals from cell surface receptors to intracellular protein kinases that control cell growth and differentiation. The Rho subfamily regulates cytoskeletal organization and polarized cell growth. The Ran subfamily participates in nucleo-cytoplasmic transport and microtubule organization. And last, the Sar/Arf and Rab subfamilies are involved in the priming of vesicle formation and vesicle docking, respectively. Here we show that yeast Ras paralogs derived from these different subfamilies display an unexpectedly wide range of thermostabilities. These findings imply that temperature-dependent inactivation of selected Ras paralogs might allow the cell to prioritize specific cellular functions in the face of high-temperature stress.
Our decision to focus on Ras family proteins from yeast was based on some important considerations. From a practical point of view, yeast is used commercially for the production of biopharmaceuticals such as human insulin as well as for industrial conversion of complex carbohydrates to alcohol and other biofuels. 1,2 These processes are typically done at high temperatures and under harsh solvent conditions. Moreover, yeast cells in the wild are exposed to a wide range of temperatures. Since they are nonmotile, yeast cannot evade these changes, and their constituent proteins will have evolved to tolerate high temperatures. In that light, we were surprised to observe such a wide range of Tm values among the 18 Ras paralogs tested here. If the low Tm values observed in vitro are recapitulated in vivo, some Ras family members may begin to denature at high (but physiological) growth temperatures and would require interactions with other proteins and chaperones to counteract unfolding. Assuming that these stability effects are the product of natural selection, such differences could be viewed as an ongoing experiment in directed evolution through random mutagenesis. 31,32
A challenge for the future is to consider the effects of protein-protein interactions on Ras stability. While our analysis focused on monomeric Ras proteins, Ras family proteins have numerous binding partners including exchange factors (promote GDP release and GTP binding) and GTPase activating proteins (accelerate GTP hydrolysis). Those protein-protein interactions are likely to influence protein conformation, core network architecture, and (consequently) protein thermostability. For example, we previously described a network of ionizable residues within G protein α subunits.7 When the G protein assembles with a receptor, a new core network is formed that links the GTP-binding pocket of Gα to the ligand binding pocket of the receptor.6
These findings show that the pHinder algorithm can predict relative thermostability among protein family paralogs. Prior to our analysis, investigators relied on a small group of bacterial and archaeal species as a source of thermostable enzymes. Despite considerable effort, little is known about the biophysical basis for protein stability in these organisms; even less in known about determinants of protein thermostability in mesophilic proteins from eukaryotes. By focusing on a newly recognized feature of protein architecture (core networks), 6,7 rather than on overall structure or sequence motifs, we have identified a novel determinant of protein thermostability. Using the Ras superfamily as a test case, we have demonstrated the predictive power of our approach.
In the longer term we hope to be able to design, and not just predict, proteins with increased thermostability. Although challenging, there have been some notable successes. Recently, computational methods were used to predict a series of point mutations that conferred thermostabilization, without loss of catalytic efficiency, on the enzyme cytosine deaminase from yeast.33 Although the authors of that work did not consider charge networks, they noted that future design efforts would likely benefit from modeling interactions involving buried polar and charged side chains, as we have done here. We anticipate that our approach will accelerate such efforts, as well as ongoing efforts to identify thermostable proteins for use as catalysts, biosensors, and therapeutics.
Supplementary Material
Acknowledgments
The authors would like to thank Roy Pourab for early contributions to this project, and the Sondek lab for providing the materials, protocols, and advice pertaining to the pLIC cloning system. The authors would also like to thank Richard Wolfenden for critical reading of the manuscript.
Funding
Supported by National Institutes of Health Grant GM101560 to H.G.D.
ABBREVIATIONS
- fQCR
fast quantitative cysteine reactivity
- ABD
4-(amino-sulfonyl)-7-fluoro-2,1,3-benzoxadiazole
- GTPγS
guanosine 5′-O-[gamma-thio]triphosphate
- PDB
Protein Data Bank
- PBS
phosphate buffered saline
- CNA
consensus network analysis
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00901.
Table of Tm value correlations at pH 7.0; supplementary figures (PDF)
PDB codes (XLSX)
The authors declare no competing financial interest.
References
- 1.Liszka MJ, Clark ME, Schneider E, Clark DS. Nature versus nurture: developing enzymes that function under extreme conditions. Annu. Rev. Chem. Biomol. Eng. 2012;3:77–102. doi: 10.1146/annurev-chembioeng-061010-114239. [DOI] [PubMed] [Google Scholar]
- 2.Vieille C, Zeikus GJ. Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol Mol. Biol. Rev. 2001;65:1–43. doi: 10.1128/MMBR.65.1.1-43.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pikuta EV, Hoover RB, Tang J. Microbial extremophiles at the limits of life. Crit. Rev. Microbiol. 2007;33:183–209. doi: 10.1080/10408410701451948. [DOI] [PubMed] [Google Scholar]
- 4.Monteith WB, Cohen RD, Smith AE, Guzman-Cisneros E, Pielak GJ. Quinary structure modulates protein stability in cells. Proc. Natl. Acad. Sci. U. S. A. 2015;112:1739–1742. doi: 10.1073/pnas.1417415112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dehouck Y, Folch B, Rooman M. Revisiting the correlation between proteins’ thermoresistance and organisms’ thermophilicity. Protein Eng., Des. Sel. 2008;21:275–278. doi: 10.1093/protein/gzn001. [DOI] [PubMed] [Google Scholar]
- 6.Isom DG, Dohlman HG. Buried ionizable networks are an ancient hallmark of G protein-coupled receptor activation. Proc. Natl. Acad. Sci. U. S. A. 2015;112:5702–5707. doi: 10.1073/pnas.1417888112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Isom DG, Sridharan V, Baker R, Clement ST, Smalley DM, Dohlman HG. Protons as second messenger regulators of G protein signaling. Mol. Cell. 2013;51:531–538. doi: 10.1016/j.molcel.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hobbs GA, Gunawardena HP, Campbell SL. Biophysical and proteomic characterization strategies for cysteine modifications in Ras GTPases. Methods Mol. Biol. 2014;1120:75–96. doi: 10.1007/978-1-62703-791-4_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expression Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 10.Isom DG, Marguet PR, Oas TG, Hellinga HW. A miniaturized technique for assessing protein thermodynamics and function using fast determination of quantitative cysteine reactivity. Proteins: Struct, Funct, Genet. 2011;79:1034–1047. doi: 10.1002/prot.22932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Isom DG, Cannon BR, Castaneda CA, Robinson A, Garcia-Moreno EB. High tol erance for ionizable residues in the hydrophobic interior of proteins. Proc. Natl. Acad. Sci. U. S. A. 2008;105:17784–17788. doi: 10.1073/pnas.0805113105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Isom DG, Castaneda CA, Cannon BR, Garcia-Moreno E B. Large shifts in pKa values of lysine residues buried inside a protein. Proc. Natl. Acad. Sci. U. S. A. 2011;108:5260–5265. doi: 10.1073/pnas.1010750108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isom DG, Castaneda CA, Cannon BR, Velu PD, Garcia-Moreno EB. Charges in the hydrophobic interior of proteins. Proc. Natl. Acad. Sci. U. S. A. 2010;107:16096–16100. doi: 10.1073/pnas.1004213107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perutz MF. Electrostatic effects in proteins. Science. 1978;201:1187–1191. doi: 10.1126/science.694508. [DOI] [PubMed] [Google Scholar]
- 15.Perutz MF. What are enzyme structures telling us? Faraday Discuss. 1992;93:1–11. doi: 10.1039/fd9929300001. [DOI] [PubMed] [Google Scholar]
- 16.Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 17.Isom DG, Vardy E, Oas TG, Hellinga HW. Picomole-scale characterization of protein stability and function by quantitative cysteine reactivity. Proc. Natl. Acad. Sci. U. S. A. 2010;107:4908–4913. doi: 10.1073/pnas.0910421107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 19.Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 2008;4:1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- 20.Belle A, Tanay A, Bitincka L, Shamir R, O’Shea EK. Quantification of protein half-lives in the budding yeast proteome. Proc. Natl. Acad. Sci. U. S. A. 2006;103:13004–13009. doi: 10.1073/pnas.0605420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 22.Kumar S, Tsai CJ, Nussinov R. Factors enhancing protein thermostability. Protein Eng., Des. Sel. 2000;13:179–191. doi: 10.1093/protein/13.3.179. [DOI] [PubMed] [Google Scholar]
- 23.Asial I, Cheng YX, Engman H, Dollhopf M, Wu B, Nordlund P, Cornvik T. Engineering protein thermostability using a generic activity-independent biophysical screen inside the cell. Nat. Commun. 2013;4:2901. doi: 10.1038/ncomms3901. [DOI] [PubMed] [Google Scholar]
- 24.Kashefi K, Lovley DR. Extending the upper temperature limit for life. Science. 2003;301:934. doi: 10.1126/science.1086823. [DOI] [PubMed] [Google Scholar]
- 25.Szilagyi A, Zavodszky P. Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: results of a comprehensive survey. Structure. 2000;8:493–504. doi: 10.1016/s0969-2126(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 26.Gromiha MM, Pathak MC, Saraboji K, Ortlund EA, Gaucher EA. Hydrophobic environment is a key factor for the stability of thermophilic proteins. Proteins: Struct, Funct, Genet. 2013;81:715–721. doi: 10.1002/prot.24232. [DOI] [PubMed] [Google Scholar]
- 27.Rojas AM, Fuentes G, Rausell A, Valencia A. The Ras protein superfamily: evolutionary tree and role of conserved amino acids. J. Cell Biol. 2012;196:189–201. doi: 10.1083/jcb.201103008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J. Cell Sci. 2005;118:843–846. doi: 10.1242/jcs.01660. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Ranea JA, Valencia A. Distribution and functional diversification of the ras superfamily in Saccharomyces cerevisiae. FEBS Lett. 1998;434:219–225. doi: 10.1016/s0014-5793(98)00967-3. [DOI] [PubMed] [Google Scholar]
- 30.Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol. Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- 31.Bloom JD, Labthavikul ST, Otey CR, Arnold FH. Protein stability promotes evolvability. Proc. Natl. Acad. Sci. U. S. A. 2006;103:5869–5874. doi: 10.1073/pnas.0510098103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Noort V, Bradatsch B, Arumugam M, Amlacher S, Bange G, Creevey G, Falk S, Mende DR, Sinning I, Hurt E, Bork P. Consistent mutational paths predict eukaryotic thermostability. BMC Evol. Biol. 2013;13:7. doi: 10.1186/1471-2148-13-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Korkegian A, Black ME, Baker D, Stoddard BL. Computational thermostabilization of an enzyme. Science. 2005;308:857–860. doi: 10.1126/science.1107387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.