

Abstract
Wilson Disease (WD), a copper transport disorder caused by a genetic defect in the ATP7B gene, has been a long time strong candidate for newborn screening (NBS), since early interventions can give better results by preventing irreversible neurological disability or liver cirrhosis. Several previous pilot studies measuring ceruloplasmin (CP) in infants or children showed that this marker alone was insufficient to meet the universal screening for WD. WD results from mutations that cause absent or markedly diminished levels of ATP7B. Therefore, ATP7B could serve as a marker for the screening of WD, if the protein can be detected from dried blood spots (DBS). This study demonstrates that the immuno-SRM platform can quantify ATP7B in DBS in the picomolar range, and that the assay readily distinguishes affected cases from normal controls (p < 0.0001). The assay precision was <10% CV, and the protein was stable for a week in DBS at room temperature. These promising proof-of-concept data open up the possibility of screening WD in newborns and the potential for a multiplexed assay for screening a variety of congenital disorders using proteins as biomarkers in DBS.
Keywords: Wilson’s disease, WD, newborn screening, NBS, ATP7B, Dried Blood Spots, DBS, Immuno-SRM, Peptide Immunoaffinity Enrichment, Mass Spectrometry
Introduction
Wilson Disease (WD) is an autosomal recessive disorder caused by mutations in ATP7B gene (OMIM *6O6882).1–3 ATP7B encodes a transmembrane protein ATPase (ATP7B), which is highly expressed in the liver and kidney and functions as a copper-dependent P-type ATPase. ATP7B is required for transmembrane transport of copper from hepatocytes into the biliary system. Absent or reduced function of ATP7B protein results in copper accumulation in the liver and subsequently in the brain, kidneys, and other organs. The impaired function of ATP7B protein also fails to incorporate copper into apoceruloplasmin, resulting in the decreased blood level of ceruloplasmin (CP) in the majority of patients with WD.4–6
The prevalence of WD is approximately 1 in 30,000 newborns, with a carrier frequency of 1 in 90 (higher in certain populations)7. However, regional variations exist. In particular, Costa Rica, Sardinia, the Canary Island and Crete have all reported to have increased incidence.8–11 WD is a slow, progressive disease. Although the biochemical defects are present from birth, patients with WD typically present with chronic hepatitis, cirrhosis or acute liver failure in the first or second decade of life. They may have tremors, ataxia, dysarthria and swallowing difficulty. WD was fatal until treatments were developed a half-century ago. In 1955, the identification of D-penicillamine by John Walshe dramatically improved the outcome of WD by increasing the urinary excretion of copper.12 Other treatments have since been introduced, including trientine and zinc salts, and have proven efficacious.13–17 Unfortunately, despite the fact that effective medical treatments have been available for over 50 years and can prevent a fatal outcome if patients are diagnosed early, clinically recognizing WD remains difficult because of its slow progression and the broad clinical spectrum of symptoms. Therefore, many patients still present with irreversible multi-organ damage at the time of diagnosis.
Ideally patients should be recognized in the presymptomatic stage. There has been a consensus that the best way to achieve early diagnosis of WD before the onset of serious symptoms is through NBS.18–20 The current gold standard for diagnosis of WD includes multiple laboratory tests, such as copper determination in the urine and liver tissue, followed by confirmation with genetic testing of the ATP7B gene. These current diagnostic tools, however, are not suitable for large-scale screening.
With the discovery that CP was reduced in the majority (~85%) of patients with WD5,6,20, several methods were developed to measure CP, a proposed marker for WD, in DBS or urine using different analytical platforms such as a sandwich ELISA assay and an LC-MS/MS assay.19,21–25 Unfortunately, from the few pilot studies published measuring CP in infants or children, CP determination alone was insufficient to screen for WD because of high false discovery rates resulting from: (1) CP is physiologically low in some unaffected newborn babies, and (2) some heterozygote carriers for WD have reduced levels of CP. Pediatric screening around 3 years of age has been proposed in Japan but it is practically difficult to screen the entire population.26 Despite a strong need for more reliable markers and/or methods in order to meet the requirement for the universal screening of WD, no further developments have been made in attempting to screen for WD in recent years.
Most WD mutations have been shown to disrupt ATP7B stability, resulting in absent or diminished levels of the protein; thus, quantifying ATP7B levels has enormous potential in the screening of WD. There are more than 370 mutations reported worldwide27,28 most are rare and infrequent, except the two most common mutations, p.H1069Q (~37–63% of the white population) and p.R778L (57% of the East Asian population).8,29–31 In line with previous observations for disease-causing missense mutations32–35, some WD-associated missense mutations including p.H1069Q and p.R778L resulted in a markedly decreased level of the ATP7B protein caused by enhanced degradation.31,36,37 Other prevalent mutations such as protein-truncating nonsense mutations (~13% of known point mutations)38 and frameshift mutations30 are predicted to result in the absence or decay of mRNA39,40 or a severely truncated protein, resulting in absent or diminished levels of the protein. Taken together, it is expected that most patients with WD would have absence or reduced levels of ATP7B. While these findings suggest absent or diminished ATP7B levels can be an indicator for WD, it has not been tested yet.
Targeted mass spectrometry, in particular Multiple/Selected Reaction Monitoring (M/SRM), has been used for rapid development of quantitative assays with high specificity, high-throughput, precision, robustness41–45, and cross-laboratory (including international) transferability of SRM-based assays has been achieved.45 The combination of DBS with SRM is the standard analytical approach in clinical and/or NBS laboratories for a variety of small metabolites that accumulate as a result of inborn errors of metabolism.46,47 Although SRM is capable of quantifying proteins present in μg/ml and higher concentrations, many potential protein markers of greatest interest are often in the low ng/mL range. Quantification of proteins and peptides in complex samples (e.g. plasma) in the ng/ml range by SRM is challenging because of the complexity and large dynamic range of the matrix. The sensitivity of SRM is not sufficient to measure low abundance protein markers directly from DBS without an enrichment process, due to interferences from more abundant analytes present in the matrix.
In recent years, peptide immunoaffinity enrichment coupled to SRM (immuno-SRM) has emerged as a promising technique for the quantification of low abundance proteins in complex matrices.48–57 The benefits of immunoaffinity enrichment of the target peptide analyte from digests of complex samples are to greatly enrich the peptide of interest before LC-MS/MS, reducing ion suppression from background components and greatly enhancing the sensitivity of the method. Immuno-SRM has been successfully implemented to address the detection-limit challenges associated with measuring low-abundance protein biomarkers in the low- and sub-μg/L range in a wide array of studies.48–51,58–62
While the immuno-SRM technology has been demonstrated on clinical samples63,64, it has not yet been adapted for measuring low-abundance proteins from dried blood spots on filter paper. In this study, we proposed to evaluate ATP7B as a marker for WD. As proof-of-concept, we investigated the immuno-SRM methodology and applied this assay to determine the concentrations of ATP7B in DBS from unaffected and WD-affected individuals. The results demonstrate that immuno-SRM is a high-sensitivity platform for DBS analysis of proteins in the low-picomolar range and ATP7B is a potential marker for screening WD in newborns.
Experimental Procedures
Study approval
Blood samples involved in this study included a total of 13 WD patients and 12 healthy volunteers. Of these, 10 WD and 12 healthy subjects were from Seattle Children’s Hospital and 3 WD subjects were from Asan Medical Center in Seoul, Korea. All patients signed informed consent and all research procedures were approved by the institutional review boards of afore-mentioned institutions. Whole blood from each subject was collected in ACD (acid citrate dextrose) tubes. Dried blood spots were prepared by pipetting 70 μL blood/spot onto filter paper card (Protein Saver™ 903® Card, Whatman Inc, Piscayaway, NJ), and allowed to dry at room temperature overnight, and then stored in sealed plastic bags at −80 °C until use. Fifteen year-old DBS samples from proven carriers and affected patient from a previous study21 were retrieved from −20°C and tested as described.
Materials
ProteaseMAX™ Surfactant (#V2072) was purchased from Promega Corporation (Madison, WI). Proteomics grade trypsin, bovine serum albumin protein standard (200 mg/mL), and (3-[3-cholamidopropyl) dimethylammonio]-1-propanesulfonate)(CHAPS, #PI28300) detergent, ammonium bicarbonate (XX) were obtained from Sigma Life Science (St. Louis, MO). Acetonitrile (#A955) and water (#W6, LCMS optima grade), and formic acid (#PI28905), phosphate buffered saline (PBS, #10010-023) were obtained from Thermo Fisher Scientific (Waltham, MA). HepG2 cell line was obtained from the ATCC (Manassas, VA).
Generation of Immuno-SRM Assay Reagents
Rabbit polyclonal antibodies were produced by Pacific Immunology (Ramona, CA). Polyclonal antibodies were affinity purified from 25 mL of antiserum. Purified (>95% by HPLC) heavy stable isotope labeled peptides were obtained from Anaspec (Fremont, CA). For stable isotope-labeled peptides, the C-terminal arginine or lysine was labeled with [13C and 15N] labeled atoms, resulting in a mass shift of +8 or +10 Da, respectively. Aliquots were stored in 5% acetonitrile/0.1% formic acid at −20°C until use. The antibody was coupled and immobilized to 2.8 μm Protein G magnetic beads (#100-04D, Invitrogen, Carlsbad, CA) in a 1 μg antibody-to-2.5 μL of beads ratio. Briefly, 250 μL of the beads were added to 1.6 mL Eppendorf tubes and washed once with 250 μL of 1X PBS, followed by addition of 100 μg of antibody and 1X PBS + 0.03% CHAPS (#28300, Thermo Scientific) to yield a total 250 μL volume. The antibodies were allowed to couple to the beads overnight with tumbling at 4°C. The next day, the antibodies were immobilized onto the beads as follows (the work was performed in a fume hood). The supernatant was discarded and 250 μL of freshly prepared 20 mM DMP (dimethyl pimelimidate dihydrochloride, #D8388, Sigma) in 200 mM triethanolamine, pH 8.5 (#T1377, Sigma) was added. The samples were tumbled for 30 minutes at room temperature and the DMP in triethanolamine was discarded. To quench the reaction, 250 μL of 150 mM monoethanolamine (#411000, Sigma) was added and the beads were tumbled at room temperature for 30 minutes. The antibody-beads were washed twice using 250 μL of 5% acetic acid + 0.03% CHAPS (5 minutes tumbling at room temperate each time), and washed once more using 250 μL of 1X PBS + 0.03% CHAPS. The antibody-beads suspension was finally resuspended in 250 μL of 1X PBS and stored at 4°C until use.
Trypsin digestion
From each DBS sample, twenty 3 mm punches (containing ~3.7 μl of blood per disc) were obtained with a standard leather punch, placed into a 1.7 mL microcentrifuge, followed by addition of 500 μL of 0.1% ProteaseMax in 50mM ammonium biocarbonate (pH 8). Tubes were vortex-mixed for 1 h on Eppendorf MixMate® (Eppendorf, Hamburg, Germany). At this point, aliquots were reserved for a Bradford assay. Disulfide bond reduction and trypsin digestion were performed in a single step with 2 M DTT and acetonitrile added to final concentrations of 5 mM and 15%, respectively. Trypsin was used in a 1:50 enzyme to protein ratio (w:w). The mixture was incubated in a 37°C water bath overnight. After a 10-min centrifugation, the supernatant was transferred to a new tube, dried under a nitrogen stream, and stored at −80°C until use.
Peptide Immunoaffinity Enrichment and Liquid Chromatography-Mass Spectrometry
The DBS digests were resuspended in 1X PBS + 0.03% CHAPS to yield a 1 μg/μL nominal protein digest concentration. Next, approximately 2 mg of protein digest was combined with 4.8 μg of the antibody (immobilized on beads) in each tube and tubes were incubated overnight at 4°C with tumbling. (The total capture volume was 500 μL.) The beads with immobilized antibodies and captured peptides were washed twice in PBS buffer + 0.03% CHAPS, washed once in PBS diluted 1:10, and peptides were eluted in 30 μL of 5% acetic acid/3% acetonitrile. The elution was frozen at −80°C until analysis. An Eksigent Ultra nanoLC 2D system (Eksigent Technologies, Dublin, CA) with a nano autosampler was used for liquid chromatography. The peptides were loaded on a trap column (0.3×5mm, C18, LCPackings, Dionex) at 10 μL/min and the LC gradient was delivered at 300 nL/minute and consisted of a linear gradient of mobile phase B developed from 2–40% B in 18 minutes on a 10 cm × 75 μm column (Reprosil AQ C18 particles, 3 μm; Dr. Maisch, Germany). The nanoLC system was connected to a hybrid triple quadrupole/ion trap mass spectrometer (6500 QTRAP, ABSciex, Foster City, CA) equipped with a nanoelectrospray interface operated in the positive ion SRM mode. Parameters for declustering potential (DP) and collision energy (CE) were taken from a linear regression of previously optimized values in Skyline.65 SRM transitions were acquired at unit/unit resolution in both the Q1 and Q3 quadrupoles with 5 ms dwell time and 3 ms pause between mass ranges, resulting in a cycle time of 1.5 seconds. All samples were run in a blinded fashion.
Data analysis
All SRM data were analyzed using Skyline. The presence of multiple transitions and retention time alignment with standard peptides were manually reviewed to verify detection of the correct peptide analyte. Data were exported from Skyline for analysis and plotting. The amount of the peptide in each DBS sample was determined by calculating the ratio of the peak areas for the signature peptide to that of its labeled IS present at a known concentration.
Method Assessment
Response curves were generated in a DBS matrix. The heavy stable isotope -labeled peptides were added to the tryptic digests of DBS covering the following concentrations: 0.03, 0.13, 0.67, 3.35, and 16.77 fmol/uL. Three process replicates were prepared and analyzed at all concentration points. Repeatability was determined using two samples: (i) DBS from normal control and (ii) DBS pooled from two WD affected siblings. Complete process triplicates were prepared and analyzed on three independent days. Intraassay variation was calculated as the average CV obtained within each day. Interassay variation was the CV calculated from the average values of the three days. The stability of analytes in DBS was tested by using the same DBS card stored at room temperature and at −20°C for 0, 3, and 7 days. For each DBS sample, samples were prepared in triplicate.
Results
Selection of target peptide
To develop a quantitative method for the quantification of ATP7B in DBS, candidate peptides for ATP7B were screened by in silico trypsin digestion, using criteria previously described44, followed by BLAST searching to ensure that the sequences are unique within the human genome. These peptides were screened using the tryptic digests of HepG2 cells to empirically determine which ATP7B peptides could be best detected and quantified by LC-MS/MS. Several peptides were chosen based on the intensity of the extracted ion chromatogram and their fragmentation pattern in SRM mode. Data for the 4 most abundant peptides are presented in Table 1, and an example of full-MS and MS/MS spectra of ATP7B 1056-1077 are shown in Figure 1. Affinity-purified, rabbit polyclonal antibodies were generated against all 4 peptides. Because the sequence for ATP7B 1056 is highly hydrophobic in the N-terminal region (the first five amino acids in particular), the shortened sequence, ATP7B 1061-1077, was used as a target for polyclonal antibody generation. While polyclonal antibodies for ATP7B 301 and 887 allowed very weak recovery of the peptides, ATP7B 325 and 1056 peptides showed recovery efficiencies ranged from ~50% to 70%. Of these two peptides, the ATP7B 1056 peptide was pursued further as a target peptide to quantify ATP7B in subsequent human samples because: (i) there was no background signal resulting from carrier peptides copurified with the antibodies49,50,66, and (ii) the most common mutation, p.H1069Q, occurs in this peptide, taking advantage of absence of this peptide in WD patient with p.H1069Q.
Table 1.
List of candidate peptides.
| Peptide | Sequence | Molecular weight | Parent ion | Daughter ions |
|---|---|---|---|---|
| ATP7B 301–313 | YDPSCTSPVALQR | 1435.7 | 718.8 | 579.8 (y11+2), 683.4 (y6), 871.5 (y8), 974.5 (y9) |
| ATP7B 325–339 | VSLPDGAEGSGTDHR | 1496.7 | 499.9 | 599.8 (y12+2), 656.30 (y13+2), 729.33 (y7), 858.37 (y8) |
| ATP7B 887–901 | ATHVGNDTTLAQIVK | 1566.8 | 523.3 | 558.4 (y5), 671.4 (y6), 772.5 (y7), 897.4 (b9) |
| ATP7B 1056–1077 | VLAWGTAEASSE HPLGVAVTK | 2134.2 | 712.4 | 827.4 (y17+2), 876.9 (y18+2), 926.5 (y19+2), 966.0 (y20+2) |
The ion type for daughter ions are in parenthesis.
Figure 1.
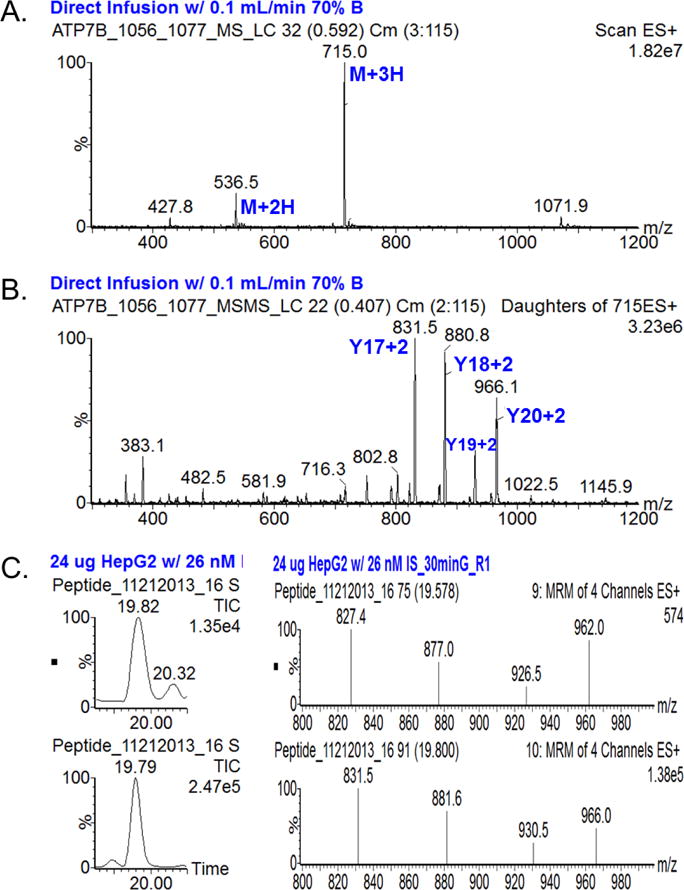
A, Mass Spectrum of heavy peptide 1056 for ATP7B and B, Tandem mass spectrum of the most abundant parent ion (M+3H). Abundant fragments are selected and optimized for SRM analysis. C, Total ion chromatogram (TIC) and SRM spectra of endogenous (top) and heavy (bottom) peptide 1056 observed in HepG2 cell extract. Chromatographic peaks overlap and SRM patterns are compatible.
Quantification of target peptide by immunoaffinity peptide enrichment and LC-MS/MS
While ATP7B is highly abundant in several tissues including liver, kidney, and placenta67, it is known to present at very low abundance in white blood cells and it has also been observed in blood including lymphocytes and erythrocytes (gpmdb.thegpm.org). Thus, ATP7B 1056 peptide was first analyzed in peripheral blood mononuclear cells (PBMC) to see if we could identify the peptide. Isolated PBMCs were lysed, digested by trypsin, and the ATP7B 1056 immuno-SRM assay was used to enrich the peptide upstream of LC-SRM. A chromatogram of the sample eluate showing the ATP7B 1056 peptide identified in PBMC is shown in Figure 2. We next tested the feasibility of measuring ATP7B 1056 peptide in DBS. These results show the feasibility of developing an assay in DBS, we then characterized and assessed the assay for use in DBS samples.
Figure 2. Extracted ion chromatograms for ATP7B 1056 peptide after peptide capture in normal PBMC.
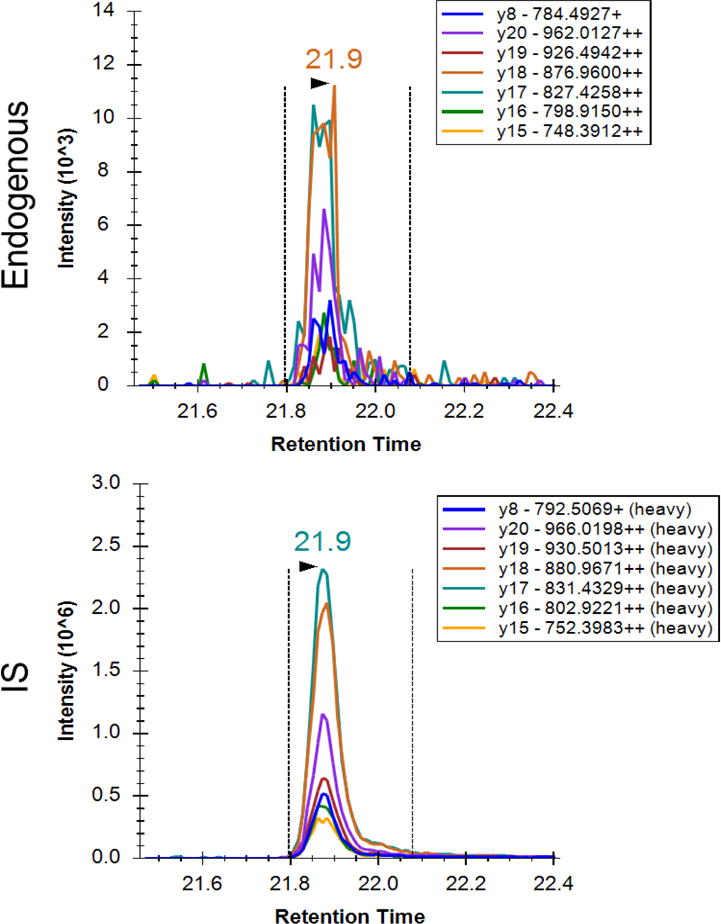
Top panel is a signature peptide found in the PBMC. Bottom panel is the isotopically labeled internal standard. Chromatographic peaks overlap, and SRM patterns are comparable. Transition labels refer to the precursor charge, fragment ion, fragment m/z, and fragment charge state.
Assay Assessment
The linearity, imprecision, and stability of the assay was assessed by following fit-for-purpose guidelines (detailed in the Experimental Section).68 The linear dynamic range was determined by generating a 5-point response curve using synthetic standard peptides. Three DBS samples for each concentration level were prepared to account for any variation in protein extraction from the DBS card. These samples were analyzed in the order of increasing concentration with one blank injection between different sample concentrations. The assay showed a linear response (r2 = 0.99) for all peptide amounts tested, spanning the peptide concentration of 27 to 16,765 pmol/L (0.7 to 417 femtomoles) (Figure 3). Results with a signal-to-noise ratio (S/N) < 10 were considered unreliable data. The low limit of quantification (LOQ) was estimated to be approximately 27 pmol/L based on the lowest concentration of the response curve (27 pmol/L) and patient sample #1 (29.5 pmol/L). The average CV was < 12% for the three replicates of DBS samples prepared and analyzed at each concentration level. The linear response with the high reproducibility shows constant protein recovery and protein digestion efficiency for the target peptide. We assessed intra- and inter-assay imprecision by the LC-SRM analysis of complete process triplicates prepared from a healthy subject and a pooled patient sample and analyzed on three independent days. The results are summarized in Table 2. Intra- and interassay imprecision from the normal control were 8.4% CV and 2.9% CV, respectively, suggesting high reproducibility of both sample preparation and the method of analysis. The assay imprecision from the pooled WD patients was not calculated because most peaks observed were below the LOQ. The stability data for the ATP7B peptide is presented in Figure 4. The ATP7B peptide was stable for at least 7 days in DBS at RT and −20°C.
Figure 3. Response curve for ATP7B 1056 peptide.
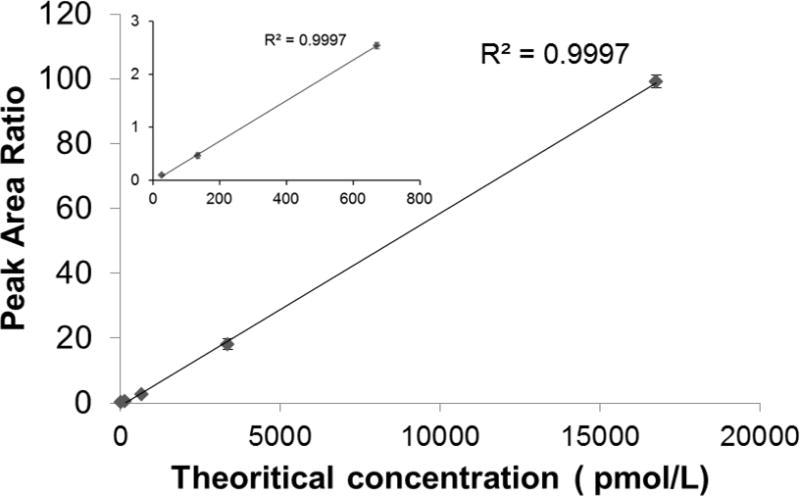
Curves are plotted for the sum of all transitions. The inset plot shows more detail of lower end of the concentration range. Error bars are the standard deviation of three process replicates.
Table 2.
Intra-assay and inter-assay imprecision of immuno-SRM assay for ATP7B 1056 peptide.
| Sample | Intra-assay, % | Inter-assay, % |
|---|---|---|
| normal control | 8.42 | 2.9 |
| WD patient | NA | NA |
NA, not applicable.
Figure 4. Stability of ATP7B 1056 peptide in normal control DBS at room temperature and −20°C for 0, 3, and 7 days.
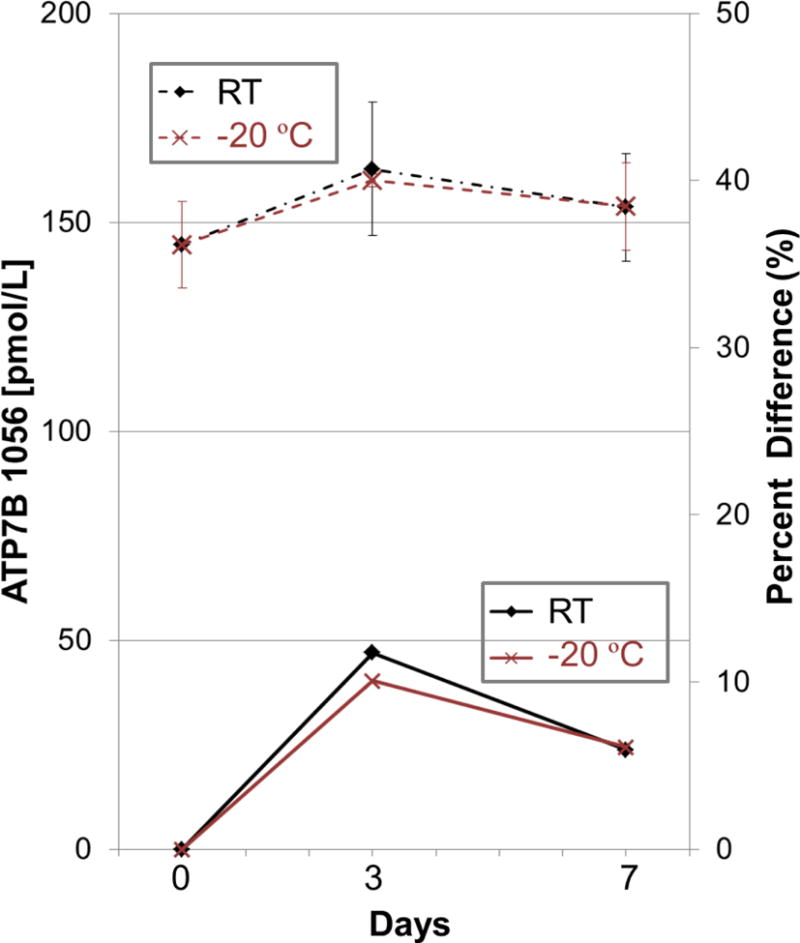
The data represent the average of three replicates. Dashed and solid lines represent ATP7B concentrations and percent difference, respectively. Error bars are the standard deviation of three process replicates.
Evaluation of ATP7B as a marker for early screening of WD
To determine if the chosen target peptide could be used as a screening marker for WD, a total of 25 DBS samples from 12 controls and 13 confirmed WD patients were analyzed in a blinded fashion for ATP7B. Representative SRM traces obtained from a healthy control and a WD patient are presented in Figure 5. The results are summarized in Table 3 and Figure 6. As shown, the assay readily distinguished affected from controls (p < 0.0001). While we were able to reliably detect endogenous ATP7B ranging from 105 to 708 pmol/L in normal controls, the analyte response from WD patients was either not detected or below 29.5 pmol/L except a case, WD08. Of note, there was no ATP7B 1056 peptide detected in either of the WD patients who carried p.H1069Q mutation, as predicted. The case 8 presented with presumably alcoholic liver cirrhosis at the age of 56 years with history of chronic jaundice, ascites and hepatosplenomegaly. The copper content in the liver biopsy was marginally elevated at 116 mcg/g dry weight tissue (control <35 mcg/g), which prompted genetic test for ATP7B gene. She was found to carry one known pathogenic variant, p.Thr974Met and one variant of unknown significance, p.Ser391Leu in trans. Her 24 hour urine copper was normal at 17 mcg/24 hours, serum ceruloplasmin was within the normal range, and no Kayser-Fleischer ring was detected in her eyes. Given her clinical and biochemical evaluations, she was suspected presumably a carrier for Wilson disease, and the VUS was predicted to be benign in nature. Her mildly elevated copper content in the liver tissue was considered most likely secondary to long-standing liver disease.
Figure 5. Extracted ion chromatograms for ATP7B 1056 peptide after peptide capture in DBS from (A) normal control and (B) WD patient.
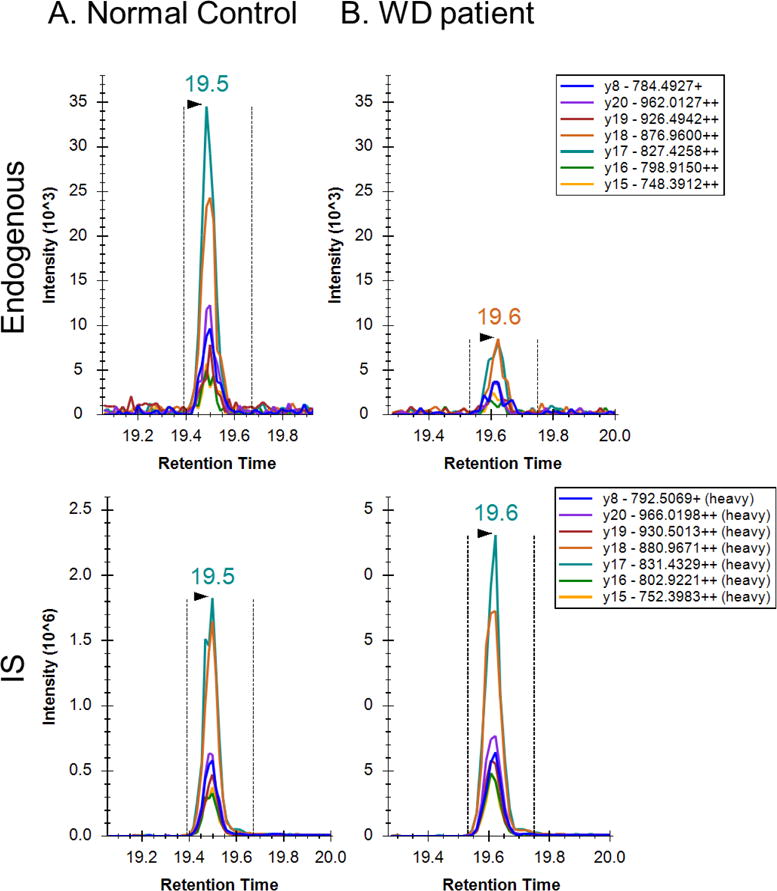
Top panel is a signature peptide found in DBS. Bottom panel is the isotopically labeled internal standard. Chromatographic peaks overlap and SRM patterns are comparable. Transition labels refer to the precursor charge, fragment ion, fragment m/z, and fragment charge state.
Table 3.
ATP7B 1056 peptide concentration in 13 DBS samples from WD patients.
| Sample | ATP7B 1056 (pmol/L) |
Mutation |
|---|---|---|
| WD01 | 29.5 | p.R778W and p.T977M |
| WD02 | NDa | p.H1069Q and p.R1319* |
| WD03 | ND | p.H1069Q and p.R1319* |
| WD04 | ND | not available at this time |
| WD05 | ND | p.R778L Homozygote |
| WD06 | ND | p.C2304_2305insc and p.L1083F |
| WD07 | NAb | p.R778L and p.A874V |
| WD08 | 244.8 | p.Thr974Met and p.Ser391 Leu |
| WD09 | ND | p.R778G and p.K175S_fs/p.Q260P_fs |
| WD10 | ND | p.R778G and p.K175S_fs/p.Q260P_fs |
| WD11 | ND | p.R778L and p.E1064A |
| WD12 | ND | p.R778L and p.E1064A |
| WD13 | ND | p.H1069Q and p.Y1331Tfs*61 |
|
| ||
| Controls (N=12) | 320.9 ± 147.4 | |
ND, not detected
NA, not applicable due to S/N <10
Figure 6. Distribution of the levels of ATP7B in DBS from 13 WD patients and 12 normal controls.
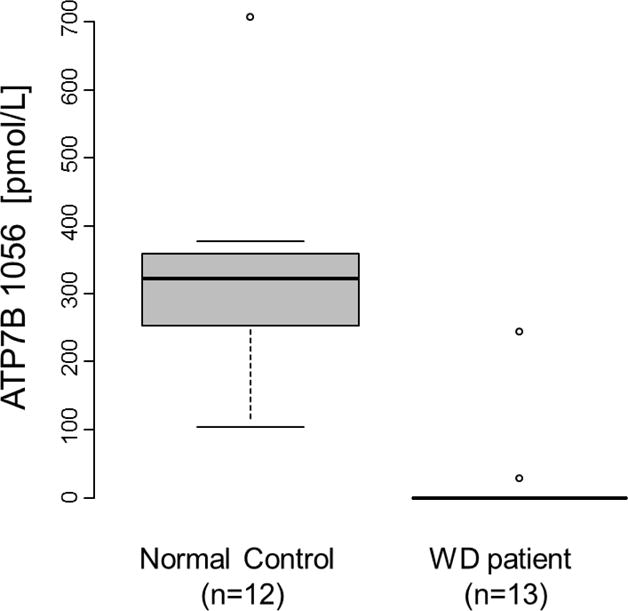
The bold black line indicates the median, the inner quartiles are represented by boxes, and the whiskers show 95% of the data.
The levels of ATP7B 1056 peptide were determined to see if the assay can distinct between WD patients and proven carriers. Because of the high prevalence of carriers, it is important that a screening test should be able to limit or avoid carrier detections. A set of DBS specimens used for this test included a WD patient, two proven carriers (the WD patient’s mother and father), and two age-matched normal controls. These DBS had been stored at −20°C for approximately 15 years. The results are shown in Table 4. ATP7B 1056 peptide in the WD patient was markedly reduced compared to the two carriers and two age-matched controls. The levels of ATP7B 1056 peptide did not differ between the carriers and age-matched controls. Of note, the levels of ATP7B from the WD patient and the four controls were in the range of the WD patients and the control groups tested in this study, indicating the protein in DBS could be stable for many years.
Table 4.
ATP7B 1056 peptide concentration in 15 year-old DBS.
| Sample | ATP7B 1056 (pmol/L) |
|---|---|
| WD patient | 60.3 |
| Carrier 1 (mother) | 270.9 |
| Carrier 2 (father) | 251.4 |
| age-matched NC 1 | 369.5 |
| age-matched NC 2 | 452.6 |
Discussion
WD is a progressive and fatal disorder that is treatable, where early detection can make a significant impact on disease outcome and even be life-saving. However, there is currently no suitable marker and method available for population screening. In this study, we: (i) propose ATP7B as a potential marker to screen WD, taking advantage of an absence or decrease in the amount of the ATP7B protein in most WD patients; (ii) provide a sensitive immuno-SRM assay for the quantification of ATP7B in DBS, demonstrating the feasibility of DBS/immuno-SRM formats for screening congenital disorders lacking marker proteins; and (iii) suggest our approach can be further applied to aid diagnostics in conjunction with clinical and other biochemical test results.
We were able to reliably detect endogenous ATP7B in DBS from normal controls in the range of 105–708 pmol/L. To our knowledge, the detection and quantification of ATP7B protein in DBS has never been achieved with any method before. In 12 out of 13WD patients, the amount of the deficient ATP7B was below the control range, suggesting feasibility of the use of ATP7B as a potential marker for WD. This approach is unique as most studies are looking for accumulated metabolites or markers, whereas this assay directly analyzes the affected protein. Although the number of samples tested in this study is quite limited, the results on the patients carrying two most common mutations (p.R778L and p.H1069Q) are promising and demonstrate feasibility of the immuno-SRM approach for mass screening.
The result on the case 8 highlights the use of this assay for those patients with ambiguous genetic or biochemical test results, which is not uncommon situation in the clinic. The variant of uncertain significance is not uncommon in ATP7B gene while there is no definite diagnostic test available. The diagnosis for WD could be challenging especially with those ambiguous or borderline results which could potentially lead to unnecessary treatment. Our result seems very promising in aiding the appropriate and accurate diagnosis in conjunction with other genetic and biochemical test results although further studies on many clinical samples are necessary.
While the data presented in this study indicate the possible use of ATP7B in DBS for screening WD, we acknowledge that the findings are preliminary. Larger studies including both controls with proven carriers and patient samples with a broad mutational spectrum will be required to determine more accurate reference, disease ranges and cut-off. In addition, patient samples tested in this study are limited to children or adults, due to the difficulty of identifying newborn samples from affected patients. Although we expect no significant age-dependence of its abundance, the effect of age on the level of ATP7B protein (in particular for newborns) is not known. As with all NBS assays, the implementation of this assay will need to be tested in the newborns and/or infants on whom the testing would be carried out.
As anticipated, there was no detectable ATP7B 1056 peptide in either of the WD patients who carried p.H1069Q mutation that occurs within this peptide. However, it can be argued that the absence of this peptide could be due to sequence variations (mutations/polymorphisms) from presumed healthy subjects. This could be resolved by monitoring multiple peptides for each target protein, which will help ensure that negative results are truly negative. The reduced/absence of multiple peptides in a SRM assay could increase confidence of a negative result. This applies to the quantification of any protein to prevent underestimation due to single nucleotide polymorphisms and posttranslational modifications.
Due to the nature of the individual variants, measurement of a target protein, ATP7B, may not be sensitive enough to identify all affected individuals. For example, some patients with mutations that affect protein function/structure but not quantity may not be detected. One approach to address this limitation is the use of additional/secondary markers such as CP. When the primary marker shows ambiguous or undetermined result, the application of secondary markers can help improve both the sensitivity and specificity of the assay. It is important to note that the ability to measure multiple analytes in the same analysis would not require additional sample process and/or sample collection.
DBS offers many advantages such as a less invasive sampling, a simpler storage and an easier transfer. However, there are several variables that may affect how uniformly blood and analytes spread across the filter paper, influencing precision in quantitative analysis. These include the hematocrit values, blood volume spotted, and chromatographic effects. There are contradicting reports in the literature as to whether or not these variables have a direct impact on the precision of the analytical result.69,70 Note that, in our proof of principle study, we chose to reduce the effects of these variables by spotting the same volume of blood for all DBS samples used in experiments in this study and using nearly the entire blood spots for analysis.
The performance characteristics of our immuno-SRM assay were found to be compatible with expectations for clinical sample analysis. The inter-assay and intra-assay CV of ATP7B assay using DBS from a healthy control specimen were <10%, demonstrating that the assay is precise. The response curve was linear, with an R2 of 0.99 and the dynamic range from 27 pmol/L to 16,765 pmol/L. The use of immuno-SRM also offers other compelling advantages with respect to NBS. These advantages include: (i) this assay can be multiplexed without loss of specificity and sensitivity, enabling simultaneous analysis of multiple proteins from a single sample and a single injection, permitting greater statistical power to be achieved and robust cross-correlations to be made, and (ii) screening programs already have been using MS/MS technology, particularly SRM.
DBS are an attractive alternative to the collection of plasma or serum, particularly for NBS. Analytes are known to be more stable in DBS compared to those in plasma, blood, or other solutions71, likely because of the dehydration of the sample on the card and consequent minimization of chemical and enzymatic hydrolysis of the analytes71. Note that, in our bottom-up approach to quantify the protein, we focused on the integrity of the targeted peptide that serves as surrogates for the protein and not the intact protein itself. Our stability test showed that the ATP7B peptide is fairly stable in DBS for a week at room temperature and −20°C. Additionally, results from the ATP7B peptide from DBS stored at −20°C for ~15 years were comparable to fresh samples, suggesting good long-term stability. Our data strongly suggest that peptides in DBS may be stable, opening up the possibility of application if immuno-SRM to NBS for testing for other genetic conditions such as primary immunodeficiencies or cystinosis.
While screening programs already utilize MS/MS technology for small molecules, immuno-SRM for quantification of proteins is a novel platform in the clinical and NBS laboratories. The method for measuring proteins uses quite different procedures from measuring small metabolites. The assay requires a relatively large number of steps before LC-MS/MS: (i) proteolytic digestion of proteins in the DBS; (ii) immunoaffinity enrichment of target peptides; and (iii) MS/MS coupled with liquid chromatography. Although this is a relatively complex assay format, the implementation of robotic sample preparation for trypsin digestion and peptide enrichment and a robust chromatography configuration45 should enable this technology to advance into routine clinical analysis for thousands of samples.
A limitation for setting up an immuno-SRM assay may be the time required and likelihood of success in generating an anti-peptide antibody.66 Monoclonal antibodies (mAb) are preferable for clinical or screening assays, since they provide a renewable resource for long-term supply with acceptable consistency and reproducibility. Some monoclonal antibodies outperform the polyclonal antibodies with increased recovery efficiency and sensitivity of the immuno-SRM assay.
Conclusion
We developed a novel, sensitive immunoaffinity LC-MS/MS assay for quantitative measurement of ATP7B levels in DBS. This study demonstrates the feasibility of the use of immuno-SRM to quantify ATP7B in DBS to screen for WD. To the best of our knowledge, this is the first published report of employing immuno-SRM strategy for measuring a clinically important, low-abundance protein in DBS. The described method opens up future opportunities for the analysis of other protein markers in DBS for many other life-threatening congenital disorders that are currently not a part of the NBS.
Acknowledgments
We thank the patients and their parents for their generous blood donations for this study. S.J. and S.H. were funded by NIH R21HD069890-01A1, and J.R.W, L.Z., and A.G.P were supported by the National Cancer Institute of the National Institutes of Health (NIH) Clinical Proteomics Tumor Analysis Consortium Initiative (U24CA160034).
Footnotes
Conflict of interest statement: The authors have declared that no conflict of interest exists.
References
- 1.Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5:327. doi: 10.1038/ng1293-327. [DOI] [PubMed] [Google Scholar]
- 2.Petrukhin K, Fischer SG, Pirastu M, Tanzi RE, Chernov I, Devoto M, Brzustowicz LM, Cayanis E, Vitale E, Russo JJ, et al. Mapping, cloning and genetic characterization of the region containing the Wilson disease gene. Nat Genet. 1993;5:338. doi: 10.1038/ng1293-338. [DOI] [PubMed] [Google Scholar]
- 3.Tanzi RE, Petrukhin K, Chernov I, Pellequer JL, Wasco W, Ross B, Romano DM, Parano E, Pavone L, Brzustowicz LM, et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet. 1993;5:344. doi: 10.1038/ng1293-344. [DOI] [PubMed] [Google Scholar]
- 4.Scheinberg IH, Gitlin D. Deficiency of ceruloplasmin in patients with hepatolenticular degeneration (Wilson’s disease) Science. 1952;116:484. doi: 10.1126/science.116.3018.484. [DOI] [PubMed] [Google Scholar]
- 5.Lutsenko S, Efremov RG, Tsivkovskii R, Walker JM. Human copper-transporting ATPase ATP7B (the Wilson’s disease protein): biochemical properties and regulation. J Bioenerg Biomembr. 2002;34:351. doi: 10.1023/a:1021297919034. [DOI] [PubMed] [Google Scholar]
- 6.Das SK, Ray K. Wilson’s disease: an update. Nat Clin Pract Neurol. 2006;2:482. doi: 10.1038/ncpneuro0291. [DOI] [PubMed] [Google Scholar]
- 7.Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson’s disease. Lancet. 2007;369:397. doi: 10.1016/S0140-6736(07)60196-2. [DOI] [PubMed] [Google Scholar]
- 8.Shah AB, Chernov I, Zhang HT, Ross BM, Das K, Lutsenko S, Parano E, Pavone L, Evgrafov O, Ivanova-Smolenskaya IA, Anneren G, Westermark K, Urrutia FH, Penchaszadeh GK, Sternlieb I, Scheinberg IH, Gilliam TC, Petrukhin K. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet. 1997;61:317. doi: 10.1086/514864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giagheddu A, Demelia L, Puggioni G, Nurchi AM, Contu L, Pirari G, Deplano A, Rachele MG. Epidemiologic study of hepatolenticular degeneration (Wilson’s disease) in Sardinia (1902–1983) Acta Neurol Scand. 1985;72:43. doi: 10.1111/j.1600-0404.1985.tb01546.x. [DOI] [PubMed] [Google Scholar]
- 10.Loudianos G, Dessi V, Lovicu M, Angius A, Figus A, Lilliu F, De Virgiliis S, Nurchi AM, Deplano A, Moi P, Pirastu M, Cao A. Molecular characterization of wilson disease in the Sardinian population–evidence of a founder effect. Hum Mutat. 1999;14:294. doi: 10.1002/(SICI)1098-1004(199910)14:4<294::AID-HUMU4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 11.Dedoussis GV, Genschel J, Sialvera TE, Bochow B, Manolaki N, Manios Y, Tsafantakis E, Schmidt H. Wilson disease: high prevalence in a mountainous area of Crete. Ann Hum Genet. 2005;69:268. doi: 10.1046/j.1529-8817.2005.00171.x. [DOI] [PubMed] [Google Scholar]
- 12.Walshe JM. Wilson’s disease; new oral therapy. Lancet. 1956;270:25. doi: 10.1016/s0140-6736(56)91859-1. [DOI] [PubMed] [Google Scholar]
- 13.Scheinberg IH, Jaffe ME, Sternlieb I. The use of trientine in preventing the effects of interrupting penicillamine therapy in Wilson’s disease. N Engl J Med. 1987;317:209. doi: 10.1056/NEJM198707233170405. [DOI] [PubMed] [Google Scholar]
- 14.Walshe JM. The management of Wilson’s disease with trienthylene tetramine 2HC1 (Trien 2HC1) Prog Clin Biol Res. 1979;34:271. [PubMed] [Google Scholar]
- 15.Hoogenraad TU, Koevoet R, de Ruyter Korver EG. Oral zinc sulphate as long-term treatment in Wilson’s disease (hepatolenticular degeneration) Eur Neurol. 1979;18:205. doi: 10.1159/000115077. [DOI] [PubMed] [Google Scholar]
- 16.Hoogenraad TU, Van Hattum J, Van den Hamer CJ. Management of Wilson’s disease with zinc sulphate. Experience in a series of 27 patients. J Neurol Sci. 1987;77:137. doi: 10.1016/0022-510x(87)90116-x. [DOI] [PubMed] [Google Scholar]
- 17.Brewer GJ, Dick RD, Johnson VD, Brunberg JA, Kluin KJ, Fink JK. Treatment of Wilson’s disease with zinc: XV long-term follow-up studies. J Lab Clin Med. 1998;132:264. doi: 10.1016/s0022-2143(98)90039-7. [DOI] [PubMed] [Google Scholar]
- 18.Kroll CA, Ferber MJ, Dawson BD, Jacobson RM, Mensink KA, Lorey F, Sherwin J, Cunningham G, Rinaldo P, Matern D, Hahn SH. Retrospective determination of ceruloplasmin in newborn screening blood spots of patients with Wilson disease. Mol Genet Metab. 2006;89:134. doi: 10.1016/j.ymgme.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 19.deWilde A, Sadilkova K, Sadilek M, Vasta V, Hahn SH. Tryptic peptide analysis of ceruloplasmin in dried blood spots using liquid chromatography-tandem mass spectrometry: application to newborn screening. Clin Chem. 2008;54:1961. doi: 10.1373/clinchem.2008.111989. [DOI] [PubMed] [Google Scholar]
- 20.Hahn SH. Population screening for Wilson’s disease. Ann N Y Acad Sci. 2014;1315:64. doi: 10.1111/nyas.12423. [DOI] [PubMed] [Google Scholar]
- 21.Hahn SH, Lee SY, Jang YJ, Kim SN, Shin HC, Park SY, Han HS, Yu ES, Yoo HW, Lee JS, Chung CS, Lee DH. Pilot study of mass screening for Wilson’s disease in Korea. Mol Genet Metab. 2002;76:133. doi: 10.1016/s1096-7192(02)00026-4. [DOI] [PubMed] [Google Scholar]
- 22.Endo F, Taketa K, Nakamura K, Awata H, Tanoue A, Eda Y, Matsuda I. Measurement of blood holoceruloplasmin by EIA using a mouse monoclonal antibody directed to holoceruloplasmin. Implication for mass screening of Wilson disease. J Inherit Metab Dis. 1994;17:616. doi: 10.1007/BF00711601. [DOI] [PubMed] [Google Scholar]
- 23.Ohura T, Abukawa D, Shiraishi H, Yamaguchi A, Arashima S, Hiyamuta S, Tada K, Iinuma K. Pilot study of screening for Wilson disease using dried blood spots obtained from children seen at outpatient clinics. J Inherit Metab Dis. 1999;22:74. doi: 10.1023/a:1005455401076. [DOI] [PubMed] [Google Scholar]
- 24.Yamaguchi Y, Aoki T, Arashima S, Ooura T, Takada G, Kitagawa T, Shigematsu Y, Shimada M, Kobayashi M, Itou M, Endo F. Mass screening for Wilson’s disease: results and recommendations. Pediatr Int. 1999;41:405. doi: 10.1046/j.1442-200x.1999.01096.x. [DOI] [PubMed] [Google Scholar]
- 25.Owada M, Suzuki K, Fukushi M, Yamauchi K, Kitagawa T. Mass screening for Wilson’s disease by measuring urinary holoceruloplasmin. J Pediatr. 2002;140:614. doi: 10.1067/mpd.2002.122731. [DOI] [PubMed] [Google Scholar]
- 26.Nakayama K, Kubota M, Katoh Y, Sawada Y, Saito A, Nishimura K, Katsura E, Ichihara N, Suzuki T, Kouguchi H, Tamura M, Honma H, Kanzaki S, Itami H, Ohtake A, Kobayashi K, Ariga T, Fujieda K, Shimizu N, Aoki T. Early and presymptomatic detection of Wilson’s disease at the mandatory 3-year-old medical health care examination in Hokkaido Prefecture with the use of a novel automated urinary ceruloplasmin assay. Mol Genet Metab. 2008;94:363. doi: 10.1016/j.ymgme.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 27.Schilsky ML. Wilson disease: current status and the future. Biochimie. 2009;91:1278. doi: 10.1016/j.biochi.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 28.Kenney SM, Cox DW. Sequence variation database for the Wilson disease copper transporter, ATP7B. Hum Mutat. 2007;28:1171. doi: 10.1002/humu.20586. [DOI] [PubMed] [Google Scholar]
- 29.Caca K, Ferenci P, Kuhn HJ, Polli C, Willgerodt H, Kunath B, Hermann W, Mossner J, Berr F. High prevalence of the H1069Q mutation in East German patients with Wilson disease: rapid detection of mutations by limited sequencing and phenotype-genotype analysis. J Hepatol. 2001;35:575. doi: 10.1016/s0168-8278(01)00219-7. [DOI] [PubMed] [Google Scholar]
- 30.Vrabelova S, Letocha O, Borsky M, Kozak L. Mutation analysis of the ATP7B gene and genotype/phenotype correlation in 227 patients with Wilson disease. Mol Genet Metab. 2005;86:277. doi: 10.1016/j.ymgme.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 31.van den Berghe PV, Stapelbroek JM, Krieger E, de Bie P, van de Graaf SF, de Groot RE, van Beurden E, Spijker E, Houwen RH, Berger R, Klomp LW. Reduced expression of ATP7B affected by Wilson disease-causing mutations is rescued by pharmacological folding chaperones 4-phenylbutyrate and curcumin. Hepatology. 2009;50:1783. doi: 10.1002/hep.23209. [DOI] [PubMed] [Google Scholar]
- 32.Waters PJ. Degradation of mutant proteins, underlying “loss of function” phenotypes, plays a major role in genetic disease. Curr Issues Mol Biol. 2001;3:57. [PubMed] [Google Scholar]
- 33.Lappalainen I, Thusberg J, Shen B, Vihinen M. Genome wide analysis of pathogenic SH2 domain mutations. Proteins. 2008;72:779. doi: 10.1002/prot.21970. [DOI] [PubMed] [Google Scholar]
- 34.Arredondo L, Nelson HB, Beckingham K, Stern M. Increased transmitter release and aberrant synapse morphology in a Drosophila calmodulin mutant. Genetics. 1998;150:265. doi: 10.1093/genetics/150.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Voskoboinik I, Thia MC, Trapani JA. A functional analysis of the putative polymorphisms A91V and N252S and 22 missense perforin mutations associated with familial hemophagocytic lymphohistiocytosis. Blood. 2005;105:4700. doi: 10.1182/blood-2004-12-4935. [DOI] [PubMed] [Google Scholar]
- 36.Payne AS, Kelly EJ, Gitlin JD. Functional expression of the Wilson disease protein reveals mislocalization and impaired copper-dependent trafficking of the common H1069Q mutation. Proc Natl Acad Sci USA. 1998;95:10854. doi: 10.1073/pnas.95.18.10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Bie P, van de Sluis B, Burstein E, van de Berghe PV, Muller P, Berger R, Gitlin JD, Wijmenga C, Klomp LW. Distinct Wilson’s disease mutations in ATP7B are associated with enhanced binding to COMMD1 and reduced stability of ATP7B. Gastroenterology. 2007;133:1316. doi: 10.1053/j.gastro.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merle U, Weiss KH, Eisenbach C, Tuma S, Ferenci P, Stremmel W. Truncating mutations in the Wilson disease gene ATP7B are associated with very low serum ceruloplasmin oxidase activity and an early onset of Wilson disease. BMC Gastroenterol. 2010;10:8. doi: 10.1186/1471-230X-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, Dietz HC. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet. 2004;36:1073. doi: 10.1038/ng1429. [DOI] [PubMed] [Google Scholar]
- 40.Chang YF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem. 2007;76:51. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 41.Lawson AM. The scope of mass spectrometry in clinical chemistry. Clin Chem. 1975;21:803. [PubMed] [Google Scholar]
- 42.Grebe SK, Singh RJ. LC-MS/MS in the Clinical Laboratory - Where to From Here? Clin Biochem Rev. 2011;32:5. [PMC free article] [PubMed] [Google Scholar]
- 43.Gillette MA, Carr SA. Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nat Methods. 2013;10:28. doi: 10.1038/nmeth.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kerfoot SA, Jung S, Golob K, Torgerson TR, Hahn SH. Tryptic peptide screening for primary immunodeficiency disease by LC/MS-MS. Proteomics Clin Appl. 2012;6:394. doi: 10.1002/prca.201100096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kennedy JJ, Abbatiello SE, Kim K, Yan P, Whiteaker JR, Lin C, Kim JS, Zhang Y, Wang X, Ivey RG, Zhao L, Min H, Lee Y, Yu MH, Yang EG, Lee C, Wang P, Rodriguez H, Kim Y, Carr SA, Paulovich AG. Demonstrating the feasibility of large-scale development of standardized assays to quantify human proteins. Nat Methods. 2014;11:149. doi: 10.1038/nmeth.2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Want EJ, Cravatt BF, Siuzdak G. The expanding role of mass spectrometry in metabolite profiling and characterization. ChemBioChem. 2005;6:1941. doi: 10.1002/cbic.200500151. [DOI] [PubMed] [Google Scholar]
- 47.Chace DH, Kalas TA. A biochemical perspective on the use of tandem mass spectrometry for newborn screening and clinical testing. Clin Biochem. 2005;38:296. doi: 10.1016/j.clinbiochem.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 48.Whiteaker JR, Zhao L, Anderson L, Paulovich AG. An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol Cell Proteomics. 2010;9:184. doi: 10.1074/mcp.M900254-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuhn E, Addona T, Keshishian H, Burgess M, Mani DR, Lee RT, Sabatine MS, Gerszten RE, Carr SA. Developing multiplexed assays for troponin I and interleukin-33 in plasma by peptide immunoaffinity enrichment and targeted mass spectrometry. Clin Chem. 2009;55:1108. doi: 10.1373/clinchem.2009.123935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoofnagle AN, Becker JO, Wener MH, Heinecke JW. Quantification of thyroglobulin, a low-abundance serum protein, by immunoaffinity peptide enrichment and tandem mass spectrometry. Clin Chem. 2008;54:1796. doi: 10.1373/clinchem.2008.109652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Whiteaker JR, Lin C, Kennedy J, Hou L, Trute M, Sokal I, Yan P, Schoenherr RM, Zhao L, Voytovich UJ, Kelly-Spratt KS, Krasnoselsky A, Gafken PR, Hogan JM, Jones LA, Wang P, Amon L, Chodosh LA, Nelson PS, McIntosh MW, Kemp CJ, Paulovich AG. A targeted proteomics-based pipeline for verification of biomarkers in plasma. Nat Biotechnol. 2011;29:625. doi: 10.1038/nbt.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anderson NL, Anderson NG, Haines LR, Hardie DB, Olafson RW, Pearson TW. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA) J Proteome Res. 2004;3:235. doi: 10.1021/pr034086h. [DOI] [PubMed] [Google Scholar]
- 53.Whiteaker JR, Paulovich AG. Peptide immunoaffinity enrichment coupled with mass spectrometry for peptide and protein quantification. Clin Lab Med. 2011;31:385. doi: 10.1016/j.cll.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ackermann BL, Berna MJ. Coupling immunoaffinity techniques with MS for quantitative analysis of low-abundance protein biomarkers. Expert Rev Proteomics. 2007;4:175. doi: 10.1586/14789450.4.2.175. [DOI] [PubMed] [Google Scholar]
- 55.Madian AG, Rochelle NS, Regnier FE. Mass-linked immuno-selective assays in targeted proteomics. Anal Chem. 2013;85:737. doi: 10.1021/ac302071k. [DOI] [PubMed] [Google Scholar]
- 56.Bostrom T, Takanen JO, Hober S. Antibodies as means for selective mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2015 doi: 10.1016/j.jchromb.2015.10.042. [DOI] [PubMed] [Google Scholar]
- 57.Kuhn E, Whiteaker JR, Mani DR, Jackson AM, Zhao L, Pope ME, Smith D, Rivera KD, Anderson NL, Skates SJ, Pearson TW, Paulovich AG, Carr SA. Interlaboratory evaluation of automated, multiplexed peptide immunoaffinity enrichment coupled to multiple reaction monitoring mass spectrometry for quantifying proteins in plasma. Mol Cell Proteomics. 2012;11:M111 013854. doi: 10.1074/mcp.M111.013854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schoenherr RM, Whiteaker JR, Zhao L, Ivey RG, Trute M, Kennedy J, Voytovich UJ, Yan P, Lin C, Paulovich AG. Multiplexed quantification of estrogen receptor and HER2/Neu in tissue and cell lysates by peptide immunoaffinity enrichment mass spectrometry. Proteomics. 2012;12:1253. doi: 10.1002/pmic.201100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van den Broek I, Nouta J, Razavi M, Yip R, Bladergroen MR, Romijn FP, Smit NP, Drews O, Paape R, Suckau D, Deelder AM, van der Burgt YE, Pearson TW, Anderson NL, Cobbaert CM. Quantification of serum apolipoproteins A-I and B-100 in clinical samples using an automated SISCAPA-MALDI-TOF-MS workflow. Methods. 2015;81:74. doi: 10.1016/j.ymeth.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 60.Ahn YH, Lee JY, Lee JY, Kim YS, Ko JH, Yoo JS. Quantitative analysis of an aberrant glycoform of TIMP1 from colon cancer serum by L-PHA-enrichment and SISCAPA with MRM mass spectrometry. J Proteome Res. 2009;8:4216. doi: 10.1021/pr900269s. [DOI] [PubMed] [Google Scholar]
- 61.Neubert H, Gale J, Muirhead D. Online high-flow peptide immunoaffinity enrichment and nanoflow LC-MS/MS: assay development for total salivary pepsin/pepsinogen. Clin Chem. 2010;56:1413. doi: 10.1373/clinchem.2010.144576. [DOI] [PubMed] [Google Scholar]
- 62.Whiteaker JR, Zhao L, Yan P, Ivey RG, Voytovich UJ, Moore HD, Lin C, Paulovich AG. Peptide Immunoaffinity Enrichment and Targeted Mass Spectrometry Enables Multiplex, Quantitative Pharmacodynamic Studies of Phospho-Signaling. Mol Cell Proteomics. 2015;14:2261. doi: 10.1074/mcp.O115.050351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Netzel BC, Grant RP, Hoofnagle AN, Rockwood AL, Shuford CM, Grebe SK. First Steps toward Harmonization of LC-MS/MS Thyroglobulin Assays. Clin Chem. 2016;62:297. doi: 10.1373/clinchem.2015.245266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Razavi M, Anderson NL, Yip R, Pope ME, Pearson TW. Multiplexed longitudinal measurement of protein biomarkers in DBS using an automated SISCAPA workflow. Bioanalysis. 2016 doi: 10.4155/bio-2016-0059. [DOI] [PubMed] [Google Scholar]
- 65.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Whiteaker JR, Zhao L, Abbatiello SE, Burgess M, Kuhn E, Lin C, Pope ME, Razavi M, Anderson NL, Pearson TW, Carr SA, Paulovich AG. Evaluation of large scale quantitative proteomic assay development using peptide affinity-based mass spectrometry. Mol Cell Proteomics. 2011;10:M110 005645. doi: 10.1074/mcp.M110.005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu F, Wang J, Pu C, Qiao L, Jiang C. Wilson’s disease: a comprehensive review of the molecular mechanisms. Int J Mol Sci. 2015;16:6419. doi: 10.3390/ijms16036419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carr SA, Abbatiello SE, Ackermann BL, Borchers C, Domon B, Deutsch EW, Grant RP, Hoofnagle AN, Huttenhain R, Koomen JM, Liebler DC, Liu T, MacLean B, Mani DR, Mansfield E, Neubert H, Paulovich AG, Reiter L, Vitek O, Aebersold R, Anderson L, Bethem R, Blonder J, Boja E, Botelho J, Boyne M, Bradshaw RA, Burlingame AL, Chan D, Keshishian H, Kuhn E, Kinsinger C, Lee JS, Lee SW, Moritz R, Oses-Prieto J, Rifai N, Ritchie J, Rodriguez H, Srinivas PR, Townsend RR, Van Eyk J, Whiteley G, Wiita A, Weintraub S. Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol Cell Proteomics. 2014;13:907. doi: 10.1074/mcp.M113.036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peng M, Liu L, Peng L. Evaluation of factors influencing accuracy in the analysis of succinylacetone in dried blood spots. Clin Chim Acta. 2012;413:1265. doi: 10.1016/j.cca.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 70.Mei JV, Alexander JR, Adam BW, Hannon WH. Use of filter paper for the collection and analysis of human whole blood specimens. J Nutr. 2001;131:1631S. doi: 10.1093/jn/131.5.1631S. [DOI] [PubMed] [Google Scholar]
- 71.Jung S, Tran NT, Gospe SM, Jr, Hahn SH. Preliminary investigation of the use of newborn dried blood spots for screening pyridoxine-dependent epilepsy by LC-MS/MS. Mol Genet Metab. 2013;110:237. doi: 10.1016/j.ymgme.2013.07.017. [DOI] [PubMed] [Google Scholar]