

Abstract
Objective
Elastin deficiency due to heterozygous loss of an ELN allele in Williams syndrome causes obstructive aortopathy characterized by medial thickening and fibrosis and consequent aortic stiffening. Previous work in Eln-null mice with a severe arterial phenotype showed that inhibition of mechanistic target of rapamycin (mTOR), a key regulator of cell growth, lessened the aortic obstruction but did not prevent early postnatal death. We investigated effects of mTOR inhibition in Eln-null mice partially rescued by human ELN that manifest a less severe arterial phenotype and survive long-term.
Approach and Results
Thoracic aortas of neonatal and juvenile mice with graded elastin deficiency exhibited increased signaling through both mTOR complex 1 and complex 2. Despite lower predicted wall stress, there was increased phosphorylation of focal adhesion kinase, suggestive of greater integrin activation, and increased transforming growth factor-β signaling mediators, associated with increased collagen expression. Pharmacologic blockade of mTOR by rapalogs did not improve luminal stenosis, but reduced mechanosignaling (in delayed fashion following mTOR complex 1 inhibition), medial collagen accumulation, and stiffening of the aorta. Rapalog administration also retarded somatic growth, however, and precipitated neonatal deaths. Complementary, less toxic strategies to inhibit mTOR via altered growth factor and nutrient responses were not effective.
Conclusions
In addition to previously demonstrated therapeutic benefits of rapalogs decreasing smooth muscle cell proliferation in the absence of elastin, we find that rapalogs also prevent aortic fibrosis and stiffening attributable to partial elastin deficiency. Our findings suggest that mTOR-sensitive perturbation of smooth muscle cell mechanosensing contributes to elastin aortopathy.
Keywords: elastin, collagen, smooth muscle cell, aorta, mechanistic target of rapamycin
Subject Codes: [118] Cardiovascular Pharmacology, [130] Animal models of human disease, [97] Other Vascular biology
Graphical abstract
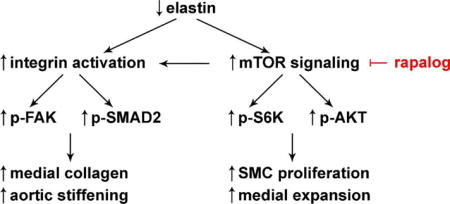
Introduction
Elastin deficiency due to heterozygous loss of ELN from chromosomal microdeletion in Williams syndrome (WS) causes obstructive aortic disease characterized by smooth muscle cell (SMC) proliferation, medial thickening, and decreased aortic diameter [1]. In addition to luminal stenosis, most individuals with WS have stiffer aortas associated with an increased incidence of hypertension [2–4]. Histological analysis of operative and post-mortem specimens demonstrate disorganized extracellular matrix (ECM) in the media with increased collagen that may explain the increased aortic stiffness [5,6]. This aortopathy of WS likely contributes to an increased risk of cardiovascular events and premature death [7]. Surgical patch enlargement is successful in treating discrete aortic obstruction, but there are no effective treatments for diffuse obstructive lesions or aortic stiffening.
We have pursued an experimental approach to prevent elastin-related aortopathy by targeting mechanistic target of rapamycin (mTOR), a key regulator of cell growth. mTOR is a serine/threonine protein kinase that interacts with several proteins to form two distinct complexes having different substrates and functions [8]. mTOR complex 1 (mTORC1) integrates multiple input signals from nutrients, energy level, mechanical stress, and growth factors and acts to switch from catabolic to anabolic processes, such as protein synthesis, and to drive cell-cycle progression. In contrast, mTOR complex 2 (mTORC2) responds to growth factors and regulates the cytoskeleton as well as cell survival. mTORC1 and mTORC2 were identified based on their differential sensitivity to rapamycin. Acute treatment with rapamycin, and related compounds such as everolimus, inhibits mTORC1, but not mTORC2, activity [9] while prolonged treatment for at least several days also indirectly inhibits mTORC2 signaling in some, though not all, cell types by sequestering free mTOR and thereby suppressing de novo mTORC2 assembly [10]. Rapamycin and its analogs, collectively termed rapalogs, inhibit mTOR kinase activity through an allosteric mechanism by binding to the FRB domain that lies upstream to the catalytic domain. A new class of ATP-competitive mTOR kinase inhibitors directly inhibits mTORC2 as well as mTORC1 activity. Both mTOR complexes may regulate the size of cells, organs, and organisms; the effects of mTORC1 are universal whereas those of mTORC2 are cell type-specific [11].
Our previous work demonstrated that mTORC1 signaling is activated in the aorta of Eln-null mice [12]. These animals without elastin develop severe stenosis of the aorta due to SMC proliferation, medial thickening, and aortic hypoplasia. They die soon after birth [13]. Treatment of the mothers by intraperitoneal administration of rapamycin during late pregnancy and lactation inhibits mTOR activity in the aorta of Eln-null pups, diminishes SMC proliferation, and reduces aortic obstruction, but does not prevent perinatal mortality [12]. This extreme phenotype prevents further investigation in juvenile and adult animals. A model with complete absence of elastin expression is also not directly comparable to WS where partial elastin expression results from the remaining ELN allele, albeit at levels less than the expected 50% of normal and with disordered ECM architecture [14]. Conversely, the mild aortic obstruction in mice with heterozygous loss of Eln and around 50% expression of elastin provides limited information on the effects of rapamycin treatment on aortic size and properties [12]. We therefore tested mTOR inhibitors in a more clinically applicable model in which Eln-null mice are partially rescued by transgenic human ELN leading to approximately 30% elastin expression with an arterial phenotype similar to that seen in typical WS patients, namely, moderate, diffuse luminal obstruction and aortic stiffening [15]. We hypothesized that mTOR inhibition would reduce both luminal stenosis and aortic stiffening in hBAC-mNULL mice by inhibiting SMC proliferation and medial thickening. We find that rapalogs prevent aortic stiffening in this model of partial elastin deficiency, but do not reduce aortic obstruction.
Materials and Methods
Available in the online-only Data Supplement.
Results
mTORC1, mTORC2, FAK, and SMAD2 Activity are Increased in Elastin-Deficient Aortas
To gain insight into molecular mechanisms underlying elastin aortopathy, we examined intracellular signaling within thoracic aortas of mice with graded deficiency of elastin, namely wild-type (mWT), heterozygous (mHET), or homozygous null (mNULL) for murine Eln and partially rescued with human ELN in a bacterial artificial chromosome (hBAC) [15]. The animals were fasted overnight before euthanasia and western blot analysis to minimize the impact of nutritional intake on signaling activity. Consistent with our previous observation of mTOR activation in aortas of neonatal Eln-null mice [12], there was greater mTORC1 activity, as indicated by increased phosphorylation of S6K and its target S6, in the thoracic aortas of hBAC-mNULL mice at 3 weeks of age (Fig. 1A,B). Similarly, there was greater mTORC2 activity, as indicated by increased phosphorylation of AKT at serine 473. Since the integrity of the ECM may influence the sensing of hemodynamically induced loads by medial SMCs [16], we examined mechanosignaling in terms of the integrin intracellular signaling effector, focal adhesion kinase (FAK). There was increased phosphorylation of FAK at the activating tyrosine 397 residue in hBAC-mNULL mice. There was also increased phosphorylation of SMAD2, a canonical signaling pathway of transforming growth factor-β that can be released from its extracellular latent complexes via activated integrins in response to mechanical loads [17]. In addition to differences when normalized to GAPDH, activation of S6K, S6, AKT, FAK, and SMAD2 remained significant when calculated as a fraction of total protein levels (Supplemental Fig. I). The activity of other stress-sensitive kinases, such as ERK1/2, p38, PKCα, and MLC, were not significantly altered by elastin deficiency and were not investigated further (data not shown). In agreement with biochemical assays suggesting fibrosis of aortas in hBAC-mNULL mice [15], there was increased levels of matrix proteins, including type III collagen, but no change in contractile proteins, such as smooth muscle α-actin (Fig. 1A,B). Thus, dysregulated mTOR signaling presents a target for pharmacotherapy and assessing consequent effects on the anomalous morphology, mechanosignaling, and fibrosis of elastin-deficient aortas.
Figure 1. mTOR Activity and Mechanosignaling in Elastin-Deficient Aortas.
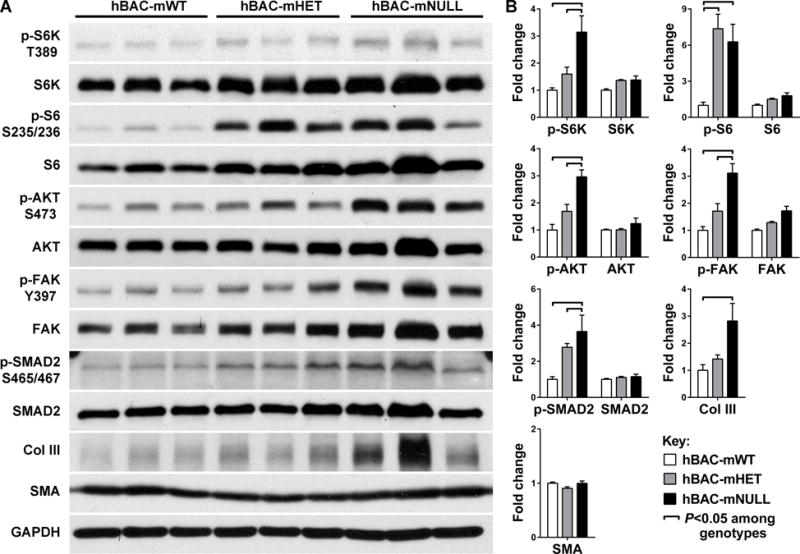
(A) Western blotting for phosphorylated and total levels of S6K, S6, AKT, FAK, SMAD2, type III collagen (Col III), smooth muscle α-actin (SMA), and GAPDH in 3 thoracic aortas of hBAC-mWT, hBAC-mHET, and hBAC-mNULL mice at 3 week of age. (B) Protein band densities were normalized to the corresponding GAPDH bands and expressed as a fraction of the mean control value, n = 3 per group, ͆ P < 0.05 among genotypes, one- or two-way ANOVA.
mTOR Inhibition Reduces Medial Collagen and Aortic Stiffening in Neonatal Elastin-Deficient Mice
We tested the effects of mTOR inhibitors since this class of drugs prevented stenosis of the aorta (but not lethality) in 1–3 day old Eln-null pups [12]. First, neonatal hBAC-mNULL mice were treated with rapamycin or everolimus at 3 mg/kg/d orally in divided doses given twice daily beginning on the day of birth and continued for 3 weeks. Both drugs were poorly tolerated, however, for the pups failed to thrive and the majority died between 8–18 days postnatally (Fig. 2A). In contrast, all vehicle-treated animals survived suggesting that mortality resulted from common effects of rapamycin and everolimus rather than the inherent aortopathy of hBAC-mNULL mice. Everolimus at the full dose resulted in uniform lethality and even one-third of this dose had similar effects on pup survival as the full dose of rapamycin. Analysis of the 4 runts that survived to 3 weeks of age revealed decreased aorta size and mass in response to mTOR inhibition, although these differences may be explained by the correspondingly smaller body size and mass (Table 1 and Supplemental Fig. II). Interestingly, everolimus and rapamycin reduced absolute and relative levels of fibrillar collagen within the media, but did not affect medial cell density or elastin concentration (Fig. 2B–D). In association with diminished medial fibrosis, mTOR inhibition increased aortic distensibility both in absolute terms and when indexed to aortic size (Fig. 2C). Second, we confirmed that a briefer, 5-day, non-lethal course of drugs initiated in 3 week old weanlings inhibited mTOR signaling in the thoracic aorta, with everolimus demonstrating greater efficacy than the same dose of rapamycin (Fig. 2E). Consistent mTOR inhibition in target tissues by oral everolimus may have resulted from efficient absorption of the drug in its proprietary microemulsion solution, although both drugs displayed significant toxicity.
Figure 2. mTOR Inhibition in Neonatal Elastin-Deficient Mice.
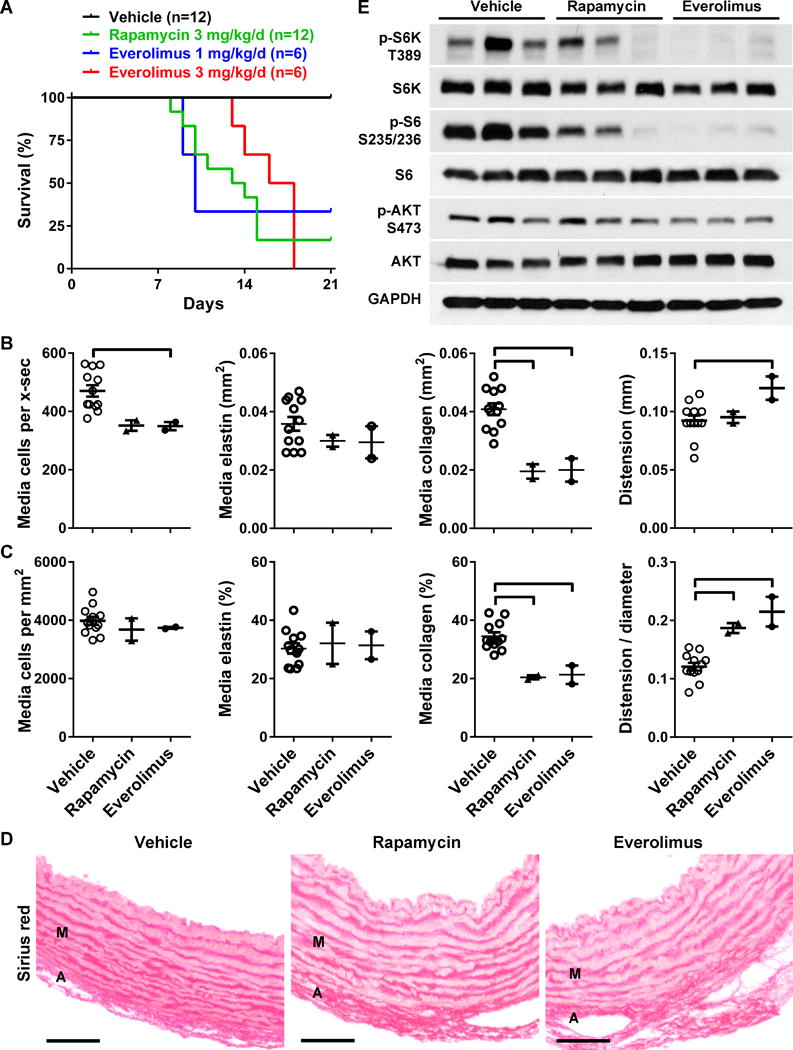
(A) Survival of hBAC-mNULL mice treated with vehicle (n = 12), rapamycin at 3 mg/kg/d (n = 12), everolimus at 3 mg/kg/d (n = 6), or everolimus at 1 mg/kg/d (n = 6) p.o. in divided doses twice daily from 0 to 3 weeks of age. (B) The ascending aortas of the surviving animals were analyzed for number of medial cells per cross-section (x-sec), media staining positive for elastin per x-sec, media staining positive for collagen per x-sec, and distension from end-diastole to end-systole. (C) The same measurements were indexed to medial area or end-diastolic aortic diameter. Scatter dot plots of data with mean ± SEM, n = 2–12, ͆ P < 0.05 vs. vehicle, one-way ANOVA. (D) Representative images of sirius red stain detecting collagen in the media (M) and adventitia (A), bars = 50 μm. (E) An additional group of 3 week old mice were treated for 5 days and the thoracic aortas (pooled from 2 mice for each sample) were immunoblotted for phosphorylated and total levels of S6K, S6, AKT, and GAPDH; n = 3 per group.
Table 1.
Effects of mTOR Inhibitors in Neonatal Elastin-Deficient Mice.
| Vehicle | Rapamycin | Everolimus | |
|---|---|---|---|
| n = 12 | n = 2 | n = 2 | |
| Aorta width (mm) | 0.657±0.024 | 0.498±0.014* | 0.501±0.001* |
| Aorta length (mm) | 17.2±0.2 | 14.3±0.4* | 14.9±1.0* |
| Medial thickness (μm) | 74.2±2.4 | 84.0±12.2 | 79.9±8.0 |
| Medial area (mm2) | 0.119±0.004 | 0.097±0.015 | 0.094±0.005 |
| Lumen area (mm2) | 0.124±0.005 | 0.062±0.005* | 0.066±0.007* |
| Aorta mass (mg) | 3.21±0.07 | 2.15±0.15* | 2.10±0.20* |
| Cardiac mass (mg) | 57.0±1.9 | 43.2±0.2* | 42.0±5.9* |
| Body mass (g) | 8.73±0.33 | 5.33±0.08* | 5.61±0.65* |
| Aorta/body mass × 10−3 | 0.372±0.014 | 0.403±0.022 | 0.376±0.008 |
hBAC-mNULL mice were treated with vehicle, rapamycin at 3 mg/kg/d, or everolimus at 3 or 1 mg/kg/d p.o. in two divided doses per day from 0 to 3 weeks of age. All of the higher dose and the majority of the lower dose everolimus-treated mice and most of the rapamycin-treated mice died prematurely and were lost to analysis. Following euthanasia of surviving animals at 3 weeks of age, the thoracic aortas were analyzed by stereomicroscopy in situ (width, length) and weight ex vivo (mass). Following standard fixation and processing, the ascending aortas were also analyzed by histomorphometry of transverse sections (thickness, area). Hence, all size dimensions are unloaded. Ascending aorta parameters are shown except for the length and mass of the entire thoracic aorta. Blood pressure was not measured in neonates as the tail-cuff apparatus was not of appropriate size. Data represent mean ± SEM,
P < 0.05 vs. vehicle, one-way ANOVA.
mTOR Inhibition Reduces Medial Collagen and Aortic Stiffening in Juvenile Elastin-Deficient Mice
To avoid drug toxicity, we next delayed the onset of therapy until after weaning at 3 weeks of age. Rapamycin was not used in these experiments as its oral preparation had achieved suboptimal mTOR inhibition in aortas of neonatal mice. Although everolimus did not result in lethality of juvenile mice treated at 3 mg/kg/d orally from 3 to 9 weeks of age, significant growth retardation persisted. To assess possible synergy with other drugs having lower toxicity profiles, we also compared treatment during this time frame with imatinib at 200 mg/kg/d and metformin at 300 mg/kg/d, which reduce mTOR signaling indirectly via PDGF β-receptor inhibition and AMPK activation, respectively. Drug doses were selected on the basis of documented efficacy in other murine disease models (cf. Materials and Methods) and we verified drug bioactivity in hBAC-mNULL mice (Supplemental Fig. III). Despite decreased medial thickening in response to everolimus, none of the drugs reduced luminal stenosis of elastin-deficient aortas (Table 2 and Supplemental Fig. IV). The drugs showed common toxicity in that all resulted in 10–20% lower body mass. Notably, no synergistic effects on aortic size were seen for half doses of everolimus in combination with either imatinib or metformin (Supplemental Table I). Histological analysis revealed that the total number of medial cells per cross-section and the absolute expression levels of medial collagen, but not elastin, were reduced by everolimus and imatinib treatment (Fig. 3A). When accounting for the corresponding changes in medial area, cell density was similar and only the reduction in relative collagen content by everolimus reached statistical significance (Fig. 3B,C). As a functional correlate to decreased medial collagen, ultrasound examination revealed improved distension and compliance of the ascending aorta in everolimus-treated animals (Fig. 3A,B and Table 2). Their blood pressure, as measured by tail-cuff, was not affected.
Table 2.
Effects of mTOR Inhibitors in Juvenile Elastin-Deficient Mice.
| Vehicle | Everolimus | Imatinib | Metformin | |
|---|---|---|---|---|
| n = 12 | n = 6 | n = 6 | n = 6 | |
| Aorta width (mm) | 0.796±0.022 | 0.775±0.033 | 0.796±0.029 | 0.856±0.056 |
| Aorta length (mm) | 20.9±0.5 | 18.6±0.6* | 19.3±0.4 | 19.5±0.4 |
| Medial thickness (μm) | 97.9±3.1 | 86.3±3.1* | 92.1±1.8 | 94.1±1.4 |
| Medial area (mm2) | 0.179±0.011 | 0.141±0.008* | 0.156±0.006 | 0.175±0.010 |
| Lumen area (mm2) | 0.152±0.010 | 0.139±0.019 | 0.139±0.007 | 0.135±0.009 |
| Aorta mass (mg) | 5.68±0.23 | 4.97±0.36 | 5.10±0.17 | 5.33±0.39 |
| Cardiac mass (mg) | 117.6±5.5 | 87.2±7.1* | 111.6±8.7 | 107.1±5.3 |
| Body mass (g) | 20.3±0.4 | 15.7±0.68* | 17.8±0.2* | 17.1±0.4* |
| Aorta/body mass × 10−3 | 0.280±0.010 | 0.321±0.030 | 0.288±0.011 | 0.313±0.023 |
| Aorta compliance (μm/mmHg) | 3.47±0.18 | 4.55±0.36* | 4.13±0.38 | 3.73±0.17 |
| Mean blood pressure (mmHg) | 104.8±3.5 | 100.0±4.5 | 97.5±3.2 | 114.5±4.2 |
hBAC-mNULL mice were treated with vehicle, everolimus at 3 mg/kg/d, imatinib at 200 mg/kg/d, or metformin at 300 mg/kg/d p.o. in two divided doses per day from 3 to 9 weeks of age. All of the animals survived. Blood pressure was measured by tail-cuff while awake. Aortas were analyzed under anesthesia by ultrasound in vivo (distension), then following euthanasia by stereomicroscopy in situ (width, length), weight ex vivo (mass), and histomorphometry of transverse sections (thickness, area). Ascending aorta parameters are shown except for the length and mass of the entire thoracic aorta. Data represent mean ± SEM,
P < 0.05 vs. vehicle, one-way ANOVA.
Figure 3. mTOR Inhibition in Juvenile Elastin-Deficient Mice.
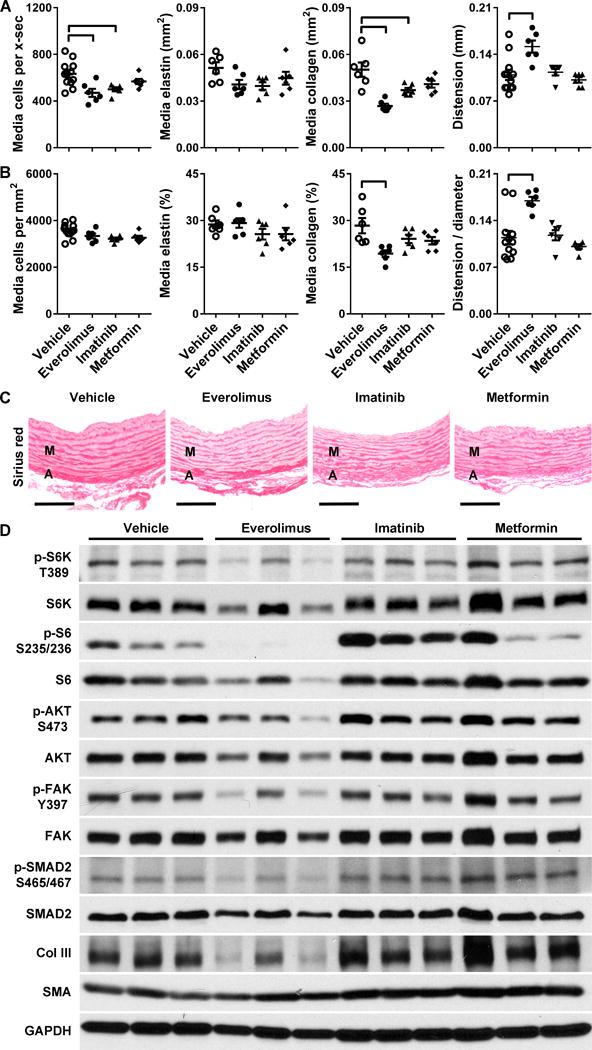
hBAC-mNULL mice were treated with vehicle, everolimus at 3 mg/kg/d, imatinib at 200 mg/kg/d, or metformin at 300 mg/kg/d p.o. in two divided doses per day from 3 to 9 weeks of age. (A) The ascending aortas were analyzed for number of medial cells per cross-section (x-sec), media staining positive for elastin per x-sec, media staining positive for collagen per x-sec, and distension from end-diastole to end-systole. (B) The same measurements were indexed to medial area or end-diastolic aortic diameter. Scatter dot plots of data with mean ± SEM, n = 6–12, ͆ P < 0.05 vs. vehicle, one-way ANOVA. (C) Representative images of sirius red stain detecting collagen in the media (M) and adventitia (A), bars = 100 μm. (D) Alternatively, the animals were treated for 7 days from 4.5 to 5.5 weeks of age and the thoracic aortas were immunoblotted for phosphorylated and total levels of S6K, S6, AKT, FAK, SMAD2, type III collagen (Col III), smooth muscle α-actin (SMA), and GAPDH; n = 3 per group.
Everolimus Rapidly Reduces mTORC1 Activity Followed by Partial Inhibition of mTORC2, FAK, and SMAD2 Signaling in Elastin-Deficient Aortas
We examined how the drug treatments affected signaling in the thoracic aorta of juvenile hBAC-mNULL mice treated for 7 days beginning at 5 weeks of age. Everolimus, but not imatinib or metformin, led to a marked reduction in mTORC1 activity and a lesser reduction in mTORC2, FAK, and SMAD2 signaling at the doses tested (Fig. 3D). We also followed the time course of everolimus effects in hBAC-mNULL mice that had been treated orally twice daily for 1 to 7 days from 5 to 6 weeks of age (Fig. 4A). After 1–3 doses over 1 to 25 hours, everolimus fully suppressed S6K and S6 phosphorylation indicative of blocked mTORC1 activity. In contrast, AKT phosphorylation at serine 473, indicative of mTORC2 activity, was only partially inhibited after 7–15 doses over 3 to 7 days, consistent with an indirect action of free mTOR sequestration [10]. Phosphorylation of FAK and SMAD2 showed a similar delayed and partial suppression by everolimus, with inhibition of the former manifesting slightly earlier by 5 doses at 2 days of treatment. Quantification of combined results at the common time points of 7 days among different experiments on juvenile hBAC-mNULL mice substantiated the inhibitory effects of everolimus on mTOR activity and mechanosignaling (Supplementary Fig. V). In this time frame, everolimus also moderately decreased type III collagen and slightly, but significantly, increased smooth muscle α-actin expression.
Figure 4. Temporal Effects of mTOR Inhibition on Mechanosignaling.
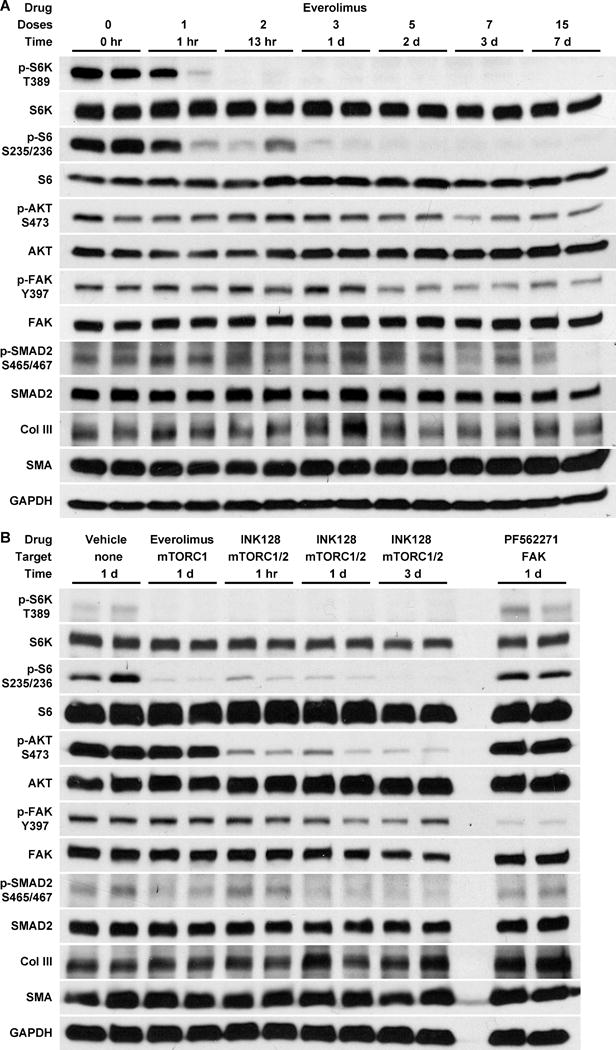
(A) hBAC-mNULL mice were treated with everolimus at 1.5 mg/kg p.o. every 12 hours (i.e., at 3 mg/kg/d) for 0–7 days, staggering the initial doses such that all treatments were completed at 6 weeks of age. (B) Alternatively, 4 week old hBAC-mNULL mice were treated with INK128 at 1.5 mg/kg p.o. every 12 hours for 1, 3, or 7 doses (dual mTORC1 and mTORC2 inhibition at all time points) and compared with vehicle alone for 3 doses, everolimus at 1.5 mg/kg p.o. every 12 hours for 3 doses (selective mTORC1 inhibitor at this treatment duration), and PF562271 at 25 mg/kg p.o. every 12 hours for 3 doses (selective FAK inhibitor). The animals were euthanized 1 hour after the last dose of drug, the thoracic aortas were rapidly procured, and extracted protein was immunoblotted for phosphorylated and total levels of S6K, S6, AKT, FAK, SMAD2, type III collagen (Col III), smooth muscle α-actin (SMA), and GAPDH; n = 2 per group.
To differentiate between delayed (including possible transcriptional and translational) effects of mTORC1 activity and contemporaneous signaling effects of mTORC2 activity on mechanosignaling, we tested additional pharmacological agents. INK128, an ATP-competitive mTOR kinase inhibitor with equal affinity for mTORC2 and mTORC1, markedly decreased activation of AKT in addition to S6K and S6 as early as 1 hour after the first dose of treatment, but did not prevent FAK and SMAD2 phosphorylation until 1 to 3 days later (Fig. 4B). Yet, inhibition of FAK activity by PF562271 did not affect mTOR or SMAD2 signaling. Levels of type III collagen and smooth muscle α-actin were not significantly altered by this short duration of drug therapy that modulated protein phosphorylation.
Discussion
We find increased mTORC1 and mTORC2 signaling in thoracic aortas with partial elastin deficiency and associated increases in FAK and SMAD2 activation, collagen accumulation, and medial thickening, which together increase aortic stiffness. We postulate that this mTOR signaling is activated in vivo by an increased sensation of mechanical stress by medial SMCs similar to responses observed in cultured, mechanically loaded smooth muscle and skeletal muscle cells [18,19]. Rapalog inhibition of mTOR reduces the aberrant mechanosignaling and attendant medial fibrosis and aortic stiffening, but does not reduce the luminal stenosis in hBAC-mNULL mice.
To determine the role of mTOR signaling in elastin-associated aortopathy, we tested the effects of several pharmacological inhibitors. The drugs were administered orally as injection of neonates is problematic due to their delicate tissues. Everolimus appears to be more effective than an equal dose of rapamycin, notwithstanding possible differences in pharmaceutical formulation or pharmacokinetics. In contrast to rapid effects on mTORC1, inhibition of mTORC2 activity by everolimus is delayed for 3 days similar to reports for other cell lines and tissues [10]. The induction of mTOR signaling by elastin deficiency is not affected by imatinib or metformin, which suggests that the pathological pathway does not depend on growth factor or nutrient availability (although not all growth factors or nutrients were investigated). mTORC2 activity is rapidly and profoundly inhibited by INK128, but effects of prolonged treatment in hBAC-mNULL mice are unknown. It is possible that complete loss of mTORC2 activity may be counterproductive by further limiting the growth of the aorta as occurs in some organs [11]. Although western blot analyses were performed with limited replicates, due to the number of experimental variables tested, confidence in the reliability of the results accrues from analyzing pooled results and considering the consistent effects on multiple mTOR signaling effectors, by several drugs of the same class, at different animal ages, and at varying times of administration.
Consistent with the SMCs sensing an increased mechanical stress, we find increased activation of FAK, a key intracellular effector for activated integrins that have engaged their ECM ligands [20]. Increased SMAD2 phosphorylation may similarly reflect an extracellular effect of activated integrins whereby transforming growth factor-β is released from ECM-bound latent complexes [21]. Notably, both FAK and SMAD2 are activated within aortas subjected to increased mechanical loads in vivo [22,23]. Mechanosensing of elevated arterial stresses typically elicits ECM production and wall thickening, as is common in hypertension. Increased FAK and SMAD2 activation in intramural cells of elastin-deficient aortas is thus consistent with the medial accumulation of collagen, but was initially unexpected from a mechanical perspective. Circumferential wall stress is predicted to be much lower in thoracic aortas of hBAC-mNULL mice than controls due to an ~50% smaller luminal diameter and ~100% thicker wall, but only ~50% higher mean blood pressure [15]; recall the Laplace equation that mean circumferential stress σθ=Pa/h, where P is pressure, a is inner radius, and h is wall thickness. Axial wall stress is also predicted to be lower in hBAC-mNULL mice given a lengthening of the ascending aorta and expected reduction in axial stretch [24]. These findings suggest either altered mechanosensing or a resetting of the target value of stress. In support of the former, a normal ECM appears to shield intramural cells from typical hemodynamically-induced intramural stresses of 100 to 200 kPa such that non-contracted cells experience stresses of only 3 to 5 kPa at focal adhesions [16]. Assuming that total wall stress , where elastin typically carries most of the stress in a normal elastic artery so as to store elastic energy [25], a markedly reduced load bearing capability by elastin should initially increase the stress seen by the SMCs even in the case of an overall net lower wall stress. Subsequent mechano-mediated production of collagen could then restore the normal stresses borne by the SMCs despite an over-compensative wall thickening. A related possibility is that an initial deficiency in elastin would alter the ratio of elastin:collagen integrin binding (and similarly for elastin-associated glycoproteins, such as fibrillin and fibulin), which in turn might stimulate collagen production. Of course, FAK can also be stimulated by growth factors, as cross-talk exists between growth factor and integrin signaling [26], and reduced elastin could affect other signaling pathways as well.
Importantly, mTOR inhibition decreased both FAK and SMAD2 signaling, though the interaction was not reciprocal at the times studied. FAK and SMAD2 activation were similarly reduced in a delayed fashion by everolimus and INK128, thus suggesting that they are downstream of mTORC1 signaling. A previous study using a panel of tumor cell lines similarly showed that rapamycin inhibits FAK signaling via an mTORC1 pathway [27]. Precise mechanisms linking mechanosensing to mTOR as well as mTOR to mechanoresponses in the aortic wall remain unknown. One possibility is that mechanical stress activates mTOR signaling through a unique PI3K- and nutrient-independent pathway that involves phospholipase D and phosphatidic acid [19]. Another is that mTOR triggers polymerization of F-actin, which promotes phosphorylation of focal adhesion proteins or activates integrins via IRS-1 [27–29]. The ability of mTOR inhibitors to modulate mechanoresponses in SMCs may relate to its beneficial effects on pathological aortic remodeling and reinforce the concepts that vascular cell activation and integrin-mediated mechanotransduction play important roles in elastin aortopathy [30,31]. Additionally, mTOR inhibition increased contractile protein expression in differentiated SMCs in vivo, though to a lesser extent than previously reported for de-differentiated cultured SMCs [28] and aortas with loss of SMC differentiation due to disruption of TGF-β signaling [32,33].
We also find that mTOR inhibition reduces medial collagen accumulation and structural stiffening of the aorta in hBAC-mNULL mice using different drugs (rapamycin and everolimus) for various durations (3 weeks in neonates and 6 weeks in juveniles). Absolute and relative levels of collagen are decreased, whereas the amount of elastin is not affected, sparing any worsening of the elastopathy. Changes in sirius red staining within the tunica media localize alongside elastic laminae, consistent with previous descriptions of type III collagen distribution [34]. Collagen levels are not significantly altered by mTOR inhibitors administered for less than 1 week, suggesting that changes in collagen are not responsible for the reduction in FAK signaling. Importantly, the increased aortic distension of treated hBAC-mNULL mice equals that of hBAC-mWT controls when normalized to vessel size. Our novel mechanistic finding of mTOR inhibition improving aortic compliance is consistent with limited clinical observations; use of rapamycin in the immunosuppression regimen of kidney transplant recipients associates with less aortic stiffening when compared with cyclosporine [35]. It is possible that the common abnormalities of elastin degradation and aortic stiffening of the elderly may also be susceptible to mTOR inhibition. Although mTOR inhibition for 6 weeks did not alter blood pressure in our hBAC-mNULL mice, beneficial effects could manifest over longer periods similar to a beneficial effect of rapamycin on blood pressure in renal transplant patients that has been observed after 40, but not 14, weeks of treatment [35]. Aortic stiffening without or with hypertension is well described in children and adults with WS [2–4]. Independent of blood pressure, aortic stiffening predicts cardiovascular complications and mortality in the general population [36] and is likely to be a similar prognosticator in WS patients.
The current study, together with our previous findings [12], suggests that mTOR inhibition prevents multiple maladaptive responses in elastin aortopathy (Supplemental Fig. VI). Treatment of Eln-null fetuses and pups reduces SMC proliferation and aortic obstruction resulting from absent elastin expression. Treatment of hBAC-mNULL neonates and weanlings decreases aortic fibrosis and stiffening despite not improving aortic hypoplasia associated with partial elastin deficiency. The limitation of mTOR inhibition in the latter may relate to the lack of SMC proliferation causing aortic obstruction in this model [37] as well as a smaller aorta associated with retarded somatic growth. Beneficial effects of mTOR inhibitors have to be weighed against their side effects, including reduced weight gain in growing animals. We believe, however, that the promising results of mTOR inhibitors in our experimental models support the rationale for a clinical trial in patients with WS. Treatment early in life (effects on somatic growth notwithstanding) to alleviate severe obstructive lesions and continuation of the treatment in older patients to prevent aortic stiffening may represent complementary therapeutic strategies.
Supplementary Material
Highlights.
Deficient elastin expression leads to mTOR complex 1 and complex 2 activation and greater integrin and TGF-β signaling in smooth muscle cells of the thoracic aorta, despite lower predicted wall stresses.
mTOR inhibition by rapalogs reduces smooth muscle cell mechanosignaling, medial collagen accumulation, and aortic stiffening, but does not reduce stenosis in mice with partial elastin deficiency.
Modulation of FAK and SMAD2 phosphorylation correlates with a 1–3 day delay following changes in mTOR complex 1 activity suggesting transcriptional and translational links.
Alteration of growth factor and nutrient responses by imatinib and metformin does not affect mTOR activity or pathological aortic remodeling due to partial elastin deficiency.
Acknowledgments
Everolimus and imatinib were a generous gift of Novartis (Basel, Switzerland).
Sources of Funding
This work was supported by the Kiev Foundation and the Williams Syndrome Association (G.T.), NIH HL086418 and HL105297 (J.D.H.), NIH HL134712 (G.T. and J.D.H.), NIH HL116705 and Connecticut Regenerative Medicine Research 12-SCB-YALE-06 and 15-RMB-YALE-08 (Y.Q.), and NIH HL125815 and American Heart Association 14GRNT19990019 (D.M.G.).
Abbreviations
- ECM
extracellular matrix
- FAK
focal adhesion kinase
- hBAC
bacterial artificial chromosome coding human ELN
- mHET
heterozygous for murine Eln
- mNULL
null for murine Eln
- mTOR
mechanistic target of rapamycin
- mTORC1
mTOR complex 1
- mTORC2
mTOR complex 2
- mWT
wild-type for murine Eln
- SMC
smooth muscle cell
- WS
Williams syndrome
Footnotes
Disclosures
There are no conflicts of interest.
References
- 1.Pober BR, Johnson M, Urban Z. Mechanisms and treatment of cardiovascular disease in Williams-Beuren syndrome. J Clin Invest. 2008;118:1606–15. doi: 10.1172/JCI35309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salaymeh KJ, Banerjee A. Evaluation of arterial stiffness in children with Williams syndrome: Does it play a role in evolving hypertension? Am Heart J. 2001;142:549–55. doi: 10.1067/mhj.2001.116763. [DOI] [PubMed] [Google Scholar]
- 3.Bassareo PP, Mercuro G. Increased arterial stiffness in children with Williams syndrome and normal blood pressure. Blood Press Monit. 2010;15:257–61. doi: 10.1097/MBP.0b013e32833e4f7d. [DOI] [PubMed] [Google Scholar]
- 4.Kozel BA, Danback JR, Waxler JL, Knutsen RH, de Las Fuentes L, Reusz GS, Kis E, Bhatt AB, Pober BR. Williams syndrome predisposes to vascular stiffness modified by antihypertensive use and copy number changes in NCF1. Hypertension. 2014;63:74–9. doi: 10.1161/HYPERTENSIONAHA.113.02087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Connor WN, Davis JB, Jr, Geissler R, Cottrill CM, Noonan JA, Todd EP. Supravalvular aortic stenosis. Clinical and pathologic observations in six patients. Arch Pathol Lab Med. 1985;109:179–85. [PubMed] [Google Scholar]
- 6.van Son JA, Edwards WD, Danielson GK. Pathology of coronary arteries, myocardium, and great arteries in supravalvular aortic stenosis. Report of five cases with implications for surgical treatment. J Thorac Cardiovasc Surg. 1994;108:21–8. [PubMed] [Google Scholar]
- 7.Wessel A, Gravenhorst V, Buchhorn R, Gosch A, Partsch CJ, Pankau R. Risk of sudden death in the Williams-Beuren syndrome. Am J Med Genet A. 2004;127A:234–7. doi: 10.1002/ajmg.a.30012. [DOI] [PubMed] [Google Scholar]
- 8.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–8. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 10.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–68. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 11.Thomanetz V, Angliker N, Cloëtta D, Lustenberger RM, Schweighauser M, Oliveri F, Suzuki N, Rüegg MA. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J Cell Biol. 2013;201:293–308. doi: 10.1083/jcb.201205030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li W, Li Q, Qin L, Ali R, Qyang Y, Tassabehji M, Pober BR, Sessa WC, Giordano FJ, Tellides G. Rapamycin inhibits smooth muscle cell proliferation and obstructive arteriopathy attributable to elastin deficiency. Arterioscler Thromb Vasc Biol. 2013;33:1028–35. doi: 10.1161/ATVBAHA.112.300407. [DOI] [PubMed] [Google Scholar]
- 13.Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998;393:276–80. doi: 10.1038/30522. [DOI] [PubMed] [Google Scholar]
- 14.Urbán Z, Riazi S, Seidl TL, Katahira J, Smoot LB, Chitayat D, Boyd CD, Hinek A. Connection between elastin haploinsufficiency and increased cell proliferation in patients with supravalvular aortic stenosis and Williams-Beuren syndrome. Am J Hum Genet. 2002;71:30–44. doi: 10.1086/341035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirano E, Knutsen RH, Sugitani H, Ciliberto CH, Mecham RP. Functional rescue of elastin insufficiency in mice by the human elastin gene: implications for mouse models of human disease. Circ Res. 2007;101:523–31. doi: 10.1161/CIRCRESAHA.107.153510. [DOI] [PubMed] [Google Scholar]
- 16.Humphrey JD, Milewicz DM, Tellides G, Schwartz MA. Cell biology. Dysfunctional mechanosensing in aneurysms. Science. 2014;344:477–9. doi: 10.1126/science.1253026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–28. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 18.Li W, Chen Q, Mills I, Sumpio BE. Involvement of S6 kinase and p38 mitogen activated protein kinase pathways in strain-induced alignment and proliferation of bovine aortic smooth muscle cells. J Cell Physiol. 2003;195:202–9. doi: 10.1002/jcp.10230. [DOI] [PubMed] [Google Scholar]
- 19.Hornberger TA, Chu WK, Mak YW, Hsiung JW, Huang SA, Chien S. The role of phospholipase D and phosphatidic acid in the mechanical activation of mTOR signaling in skeletal muscle. Proc Natl Acad Sci U S A. 2006;103:4741–6. doi: 10.1073/pnas.0600678103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–23. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 21.Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, Springer TA. Latent TGF-β structure and activation. Nature. 2011;474:343–9. doi: 10.1038/nature10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuang SQ, Geng L, Prakash SK, Cao JM, Guo S, Villamizar C, Kwartler CS, Peters AM, Brasier AR, Milewicz DM. Aortic remodeling after transverse aortic constriction in mice is attenuated with AT1 receptor blockade. Arterioscler Thromb Vasc Biol. 2013;33:2172–9. doi: 10.1161/ATVBAHA.113.301624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Louis H, Kakou A, Regnault V, Labat C, Bressenot A, Gao-Li J, Gardner H, Thornton SN, Challande P, Li Z, Lacolley P. Role of α1β1-integrin in arterial stiffness and angiotensin-induced arterial wall hypertrophy in mice. Am J Physiol Heart Circ Physiol. 2007;293:H2597–604. doi: 10.1152/ajpheart.00299.2007. [DOI] [PubMed] [Google Scholar]
- 24.Wagenseil JE, Nerurkar NL, Knutsen RH, Okamoto RJ, Li DY, Mecham RP. Effects of elastin haploinsufficiency on the mechanical behavior of mouse arteries. Am J Physiol Heart Circ Physiol. 2005;289:H1209–17. doi: 10.1152/ajpheart.00046.2005. [DOI] [PubMed] [Google Scholar]
- 25.Bellini C, Ferruzzi J, Roccabianca S, Di Martino ES, Humphrey JD. A microstructurally motivated model of arterial wall mechanics with mechanobiological implications. Ann Biomed Eng. 2014;42:488–502. doi: 10.1007/s10439-013-0928-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eliceiri BP. Integrin and growth factor receptor crosstalk. Circ Res. 2001;89:1104–10. doi: 10.1161/hh2401.101084. [DOI] [PubMed] [Google Scholar]
- 27.Liu L, Chen L, Chung J, Huang S. Rapamycin inhibits F-actin reorganization and phosphorylation of focal adhesion proteins. Oncogene. 2008;27:4998–5010. doi: 10.1038/onc.2008.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin KA, Merenick BL, Ding M, Fetalvero KM, Rzucidlo EM, Kozul CD, Brown DJ, Chiu HY, Shyu M, Drapeau BL, Wagner RJ, Powell RJ. Rapamycin promotes vascular smooth muscle cell differentiation through insulin receptor substrate-1/phosphatidylinositol 3-kinase/Akt2 feedback signaling. J Biol Chem. 2007;282:36112–20. doi: 10.1074/jbc.M703914200. [DOI] [PubMed] [Google Scholar]
- 29.Tai YT, Podar K, Catley L, Tseng YH, Akiyama M, Shringarpure R, Burger R, Hideshima T, Chauhan D, Mitsiades N, Richardson P, Munshi NC, Kahn CR, Mitsiades C, Anderson KC. Insulin-like growth factor-1 induces adhesion and migration in human multiple myeloma cells via activation of beta1-integrin and phosphatidylinositol 3′-kinase/AKT signaling. Cancer Res. 2003;63:5850–8. [PubMed] [Google Scholar]
- 30.Campuzano V, Segura-Puimedon M, Terrado V, Sánchez-Rodríguez C, Coustets M, Menacho-Márquez M, Nevado J, Bustelo XR, Francke U, Pérez-Jurado LA. Reduction of NADPH-oxidase activity ameliorates the cardiovascular phenotype in a mouse model of Williams-Beuren Syndrome. PLoS Genet. 2012;8:e1002458. doi: 10.1371/journal.pgen.1002458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Misra A, Sheikh AQ, Kumar A, Luo J, Zhang J, Hinton RB, Smoot L, Kaplan P, Urban Z, Qyang Y, Tellides G, Greif DM. Integrin β3 inhibition is a therapeutic strategy for supravalvular aortic stenosis. J Exp Med. 2016;213:451–63. doi: 10.1084/jem.20150688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li W, Li Q, Jiao Y, Qin L, Ali R, Zhou J, Ferruzzi J, Kim RW, Geirsson A, Dietz HC, Offermanns S, Humphrey JD, Tellides G. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest. 2014;124:755–67. doi: 10.1172/JCI69942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferruzzi J, Murtada SI, Li G, Jiao Y, Uman S, Ting MY, Tellides G, Humphrey JD. Pharmacologically improved contractility protects against aortic dissection in mice with disrupted transforming growth factor-β signaling despite compromised extracellular matrix properties. Arterioscler Thromb Vasc Biol. 2016;36:919–27. doi: 10.1161/ATVBAHA.116.307436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCullagh KG, Duance VC, Bishop KA. The distribution of collagen types I, III and V (AB) in normal and atherosclerotic human aorta. J Pathol. 1980;130:45–55. doi: 10.1002/path.1711300107. [DOI] [PubMed] [Google Scholar]
- 35.Joannidès R, Monteil C, de Ligny BH, Westeel PF, Iacob M, Thervet E, Barbier S, Bellien J, Lebranchu Y, Seguin SG, Thuillez C, Godin M, Etienne I. Immunosuppressant regimen based on sirolimus decreases aortic stiffness in renal transplant recipients in comparison to cyclosporine. Am J Transplant. 2011;11:2414–22. doi: 10.1111/j.1600-6143.2011.03697.x. [DOI] [PubMed] [Google Scholar]
- 36.Laurent S, Cockcroft J, Van Bortel L, Boutouyrie P, Giannattasio C, Hayoz D, Pannier B, Vlachopoulos C, Wilkinson I, Struijker-Boudier H, European Network for Non-invasive Investigation of Large Arteries Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur Heart J. 2006;27:2588–605. doi: 10.1093/eurheartj/ehl254. [DOI] [PubMed] [Google Scholar]
- 37.Jiao Y, Li G, Korneva A, Caulk AW, Qin L, Bersi MR, Li Q, Li W, Mecham RP, Humphrey JD, Tellides G. Deficient Circumferential Growth is the Primary Determinant of Aortic Obstruction Attributable to Partial Elastin Deficiency. Arterioscler Thromb Vasc Biol. 2017;37:930–41. doi: 10.1161/ATVBAHA.117.309079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.