

Abstract
One direct route for the discovery of therapeutic human monoclonal antibodies (mAbs) involves the isolation of peripheral B cells from survivors/sero-positive individuals after exposure to an infectious reagent or disease etiology followed by single-cell sequencing or hybridoma generation. Peripheral B cells, however, are not always easy to obtain and only represent a small percentage of the total B cell population across all bodily tissues. Although it has been demonstrated that tandem mass spectrometry (MS/MS) techniques can interrogate the full polyclonal antibody (pAb) response to an antigen in vivo, all current approaches identify MS/MS spectra against databases derived from genetic sequencing of B cells from the same patient. In this proof-of-concept study, we demonstrate the feasibility of a novel MS/MS antibody discovery approach in which only serum antibodies are required, without the need for sequencing of genetic material. Peripheral pAbs from a CMV exposed individual were purified by glycoprotein B antigen-affinity and de novo sequenced from MS/MS data. Purely MS-derived mAbs were then manufactured in mammalian cells to validate potency via antigen-binding ELISA. Interestingly, we found that these mAbs accounted for 1–2% of total donor IgG but were not detected in parallel sequencing of memory B cells from the same patient.
Introduction
Monoclonal antibodies (mAbs) are a well-validated biotherapeutic platform with very high specificity and potency. MAbs are a low-risk class of drug for development through licensure1,2 and offer great potential as therapies for cancer3 in addition to addressing emerging and re-emerging infectious diseases4,5 and other pathologies. One of the most directly translatable methods for the discovery and therapeutic development of potent human mAbs is via isolation of peripheral B cells from survivors and/or seropositive individuals post exposure to a pathogen or pathology followed by single cell sequencing6 or hybridoma generation7 using B-cell cloning. To date, these methods have proved invaluable towards understanding the immune system and producing therapeutic drug candidates, including development of broadly-neutralizing HIV-1 mAbs, such as VRC018, that have exceptional breadth and potency, many of which are currently undergoing clinical trials. Unfortunately, antibody discovery approaches that rely on B cells from peripheral blood remain limited by (1) clone amplification biases, (2) very limited sampling depth that cannot encompass a complete B cell repertoire, and (3) absence of sampling of B cells from spleen, lymph nodes, mucosal surfaces, and bone marrow9.
Immunity research that aims to identify novel antibodies from living human donors currently relies on genetic sequencing or resource-intensive hybridoma cloning of peripheral B cells. However, it is unknown how much of the diversity of peripheral antibodies is represented by peripheral B cells. Although clonal B-cell expansion occurs in peripheral blood immediately following infection, long lived plasma B cells (LLPCs) predominantly reside in bone marrow10 and lymphoid tissues. The LLPCs are capable of producing a diverse population of high-affinity antibodies for years after infection. In some special cases LLPCs may be obtained from tonsils or bone marrow extraction but, in most cases, researches are limited to analyzing peripheral blood where B cells are estimated to account for at most 2% of the total B cell population across all bodily tissues9,11.
State-of-the-art Next-Generation Sequencing (NGS) techniques can thus sample only a small fraction of a B cell repertoire even if that repertoire was readily available. The sheer complexity of a full B cell repertoire (estimates as high as 1013)9 is a serious obstacle for DNA sequencing and can only be addressed with single-cell techniques that introduce preferential cloning biases9 (e.g., cells expressing high affinity antibodies can have adverse growth effects during cloning). In contrast, polyclonal antibodies (pAbs) produced by B cells throughout different bodily tissues are present in peripheral blood (IgG isotype antibodies typically have a half-life of ~20 days in circulation12) and can be enriched by affinity purification to the antigen of interest. This classical procedure involves specific binding of antibodies to the antigen displayed on a solid support. High affinity pAbs can readily be extracted, yielding a dramatically reduced complexity of pAbs that are accessible to tandem mass spectrometry (MS/MS) analysis13,14.
Due to limitations associated with MS/MS acquisition15, current MS-based polyclonal antibody sequencing approaches must cross reference peptides against a collection of sequences derived from genetic sequencing of peripheral B cells from the same patient13,14. In contrast, de novo MS/MS sequencing offers a strategy to sequence pAbs directly from donor plasma without the need to sequence any B cells. The difficulty lies in navigating the complexity of polyclonal mixtures, where hundreds of pAbs can be present at detectable abundance following affinity purification13,14. De novo sequencing tools have only recently achieved the capability of sequencing >100 amino acid (AA)-long segments of simple protein mixtures at 99% sequencing accuracy16, with many more approaches that utilize homology to known antibody framework domains for the specialized task of sequencing purified hybridoma-produced mAbs17–19 (in cases where the mAb sequence is unknown due to unavailability of genetic material or failure to correctly sequence DNA of the source hybridoma cell line).
In this work we have extended the Meta-Shotgun Protein Sequencing (Meta-SPS)16,20 approach with semi-automated customized tools to sequence pAbs that were purified by gB antigen-affinity from peripheral blood of a live human donor exposed to Cytomegalovirus (CMV). Individual mAbs having the most supporting proteomic evidence were manufactured in mammalian cells, from de novo sequence, to functionally validate their potency by antigen-binding ELISA. Parallel Next-Generation Sequencing (NGS) of peripheral memory B cells from the same patient yielded no support for these mAbs, even though they were estimated to account for ~1–2% of total IgG, meaning they were not encoded in peripheral B cells or not detectable.
Experimental Procedures
Figure 1 illustrates the overall antibody discovery approach. Circulating plasma was collected from a live donor and pAbs were purified by Protein A and antigen affinity. MS and MS/MS analysis was utilized to de novo sequence the most abundant mAbs from the pAb mixture, which were then expressed in mammalian cells from de novo sequence. MAb potency was validated by downstream ELISA functional assays.
Figure 1.
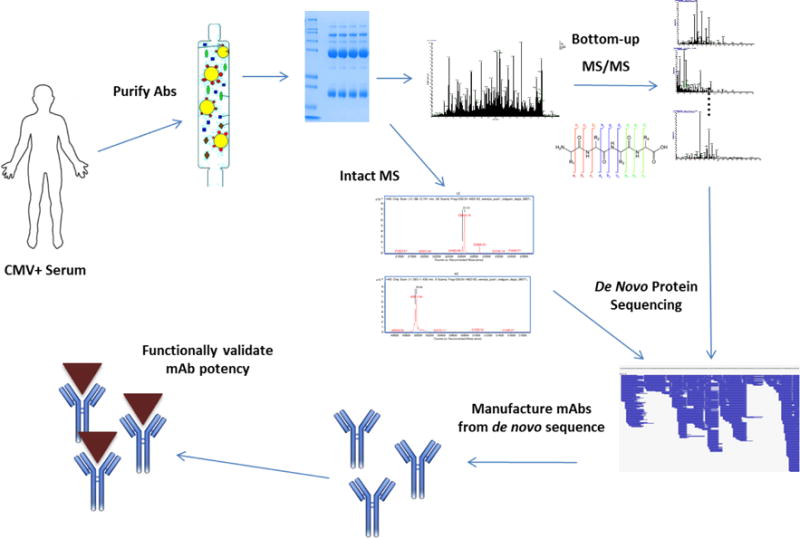
Overview of novel antibody discovery approach. mAbs are sequenced directly from circulating pAbs using MS/MS de novo analysis, circumventing the need to sequence B cells from peripheral plasma. The main benefit is the ability to capture antibodies that are not produced by peripheral B cells, which account for at most 2% of B cells across all tissues9.
Sample Preparation
200 mL of whole blood was collected from a cytomegalovirus (CMV) infected donor. After centrifugation, 50mL of serum IgGs were isolated using affinity chromatography with Protein A (MabSelectSuRe, GE Healthcare, Piscataway, NJ). Fractions showing highest absorbence by UV were pooled and passed over a column composed of the CMV glycoprotein B antigen coupled to resin (Affi-gel10, BioRad, Hercules, CA) via primary amines to isolate antigen- specific monoclonal antibodies (mAbs), which were eluted with a pH 3 elution. The most abundant mAbs were further purified by ion exchange chromatography. This yielded a mix of 2–3 abundant mAbs (~1mL eluate at 1.7 mg/mL) among a background of pAbs (Figure 2) that accounted for 1–2% of total IgG by weight.
Figure 2.
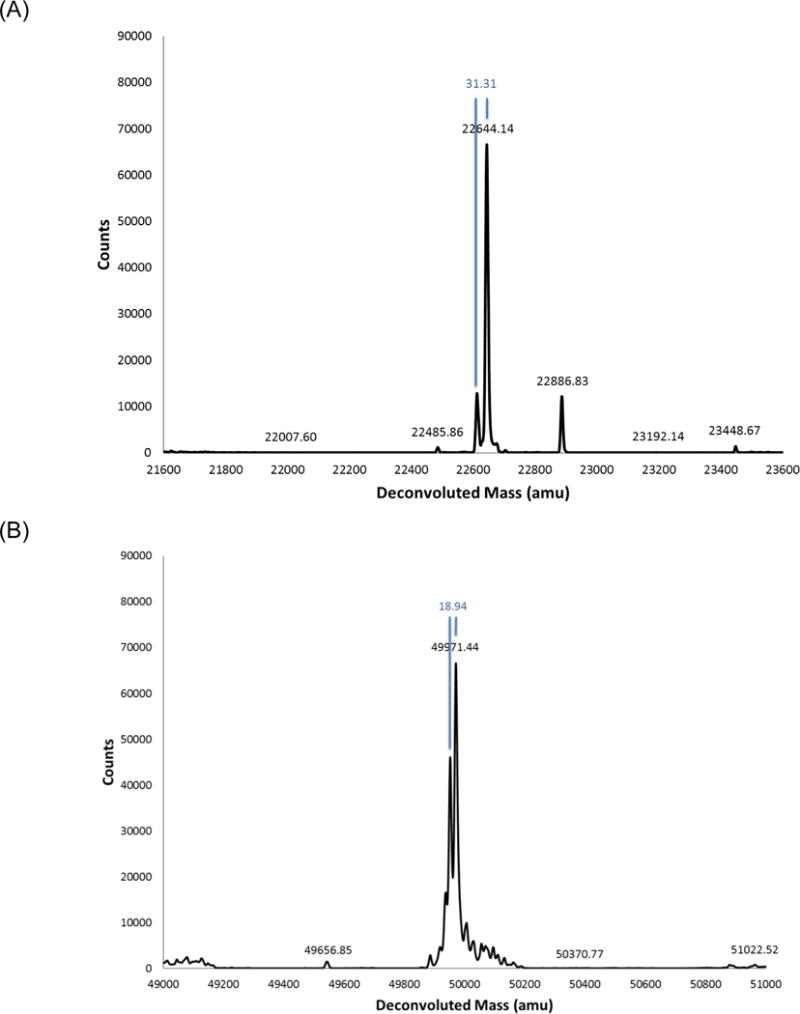
Deconvoluted intact mass spectra of reduced and deglycosylated light chain (A) and heavy chain (B) anti-CMV pAb material that was purified from donor plasma. The most abundant heavy chains weighed approximately 49971 and 49953 Da while the most abundant light chains had weights of 22644 Da, 22613 Da, and 22887 Da.
Tandem Mass Spectrometry
After extraction, samples were alkylated with NIPIA, the heavy and light chains were denatured and separated via SDS-page. Resultant gel bands corresponding to heavy and light chains were separately digested with one of eight enzymes (trypsin, chymotrypsin, AspN, GluC, ArgC, LysC, LysN, or pepsin). MS/MS spectra were acquired on a LTQ Orbitrap Velos mass spectrometer with a 2-hour chromatography gradient and three CID/HCD/ETD high resolution MS/MS spectra triplets acquired per peptide precursor as previously described16. All MS/MS spectra can be downloaded at the MassIVE data repository21.
Intact Mass Spectrometry
Intact molecular weight analysis was performed on a Q-TOF instrument (Agilent, Santa Clara, CA) on the deglycosylated and reduced protein level pAb mixture to determine the intact masses and relative abundance of the most prominent mAbs. These measurements not only provided a useful snapshot of the complexity of antigen-specific pAbs, but yielded the exact intact masses of the most abundant mAbs to approximately 1 Da resolution (Figure 2). Given that pAbs were purified by antigen affinity prior to intact molecular weight analysis, protein abundance at this stage likely corresponds to some combination of (1) mAb abundance in host plasma and (2) affinity of each mAb to the source antigen. Ultimately, mAb sequences were filtered against these intact masses and ranked by the number of supporting MS/MS spectra. Full-length de novo mAb sequences were excluded from downstream manufacture/testing if they did not match an abundant intact mass or contained gaps in MS/MS coverage.
De Novo Sequencing
Thermo RAW files were converted to mzXML with ProteoWizard22 (version 3.0.3324). MS/MS spectra were then processed through the Meta-SPS pipeline16, which involves the following steps:
All MS/MS peaks were first converted to charge one via fragment charge deconvolution20.
(PepNovo+)23 was used to interpret known CID/HCD/ETD MS/MS ion offsets and convert each MS/MS spectrum into a PRM (Prefix Residue Mass) spectrum where peak intensities are replaced with CID-, HCD- or ETD-specific log-likelihood scores and where peak masses correspond to cumulative amino acid masses of N-terminal prefixes of the peptide sequence24.
Each triplet of CID, HCD, and ETD PRM spectra from the same precursor was merged into one scored PRM spectrum as previously described16. This procedure boosts the merged score of PRMs independently observed in CID/ETD and HCD/ETD spectrum pairs due to the decreased likelihood of observing random b/c and y/z ion pairs from noise25,26. We refer to this set of merged PRM spectra as scored spectra.
Scored spectra were aligned and converted into star spectra27, which are near noise-free versions of scored spectra where PRMs that are not supported by aligned peaks from overlapping peptides are removed.
Star spectra were then re-aligned and assembled into contigs (sets of spectra from overlapping peptides)28 which were further connected to form meta-contigs (sets of overlapping contigs)20. Figure 3A illustrates a resulting de novo sequence automatically extracted from a meta-contig covering the variable region of a putative polyclonal antibody.
Figure 3.
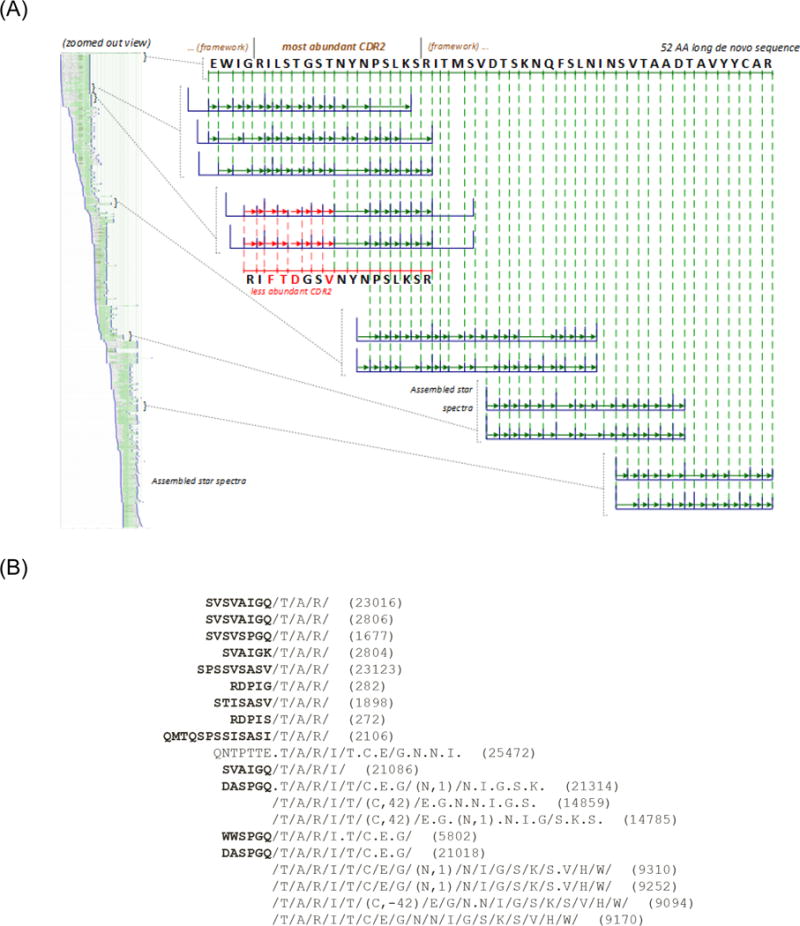
(A) Assembled meta-contig covering 52 AA of the variable region of a putative antibody. The top-most de novo sequence is the highest-scoring interpretation of all 536 star spectra from this meta-contig (seen on left in zoomed out view). Each blue spectrum denotes a star spectrum where noise peaks (non-PRM masses) are removed based on alignment to neighboring spectra27. The sequence in red denotes a de novo sequence covering a less-abundant clonal derivative that was manually extracted using the PolyExtend tool. (B) Example output of PolyExtend while extending a partial protein sequence “TARITCEGNNIGSKSVHW” towards the N-term, suggesting that “GQ” is the most abundant extension.
As shown in Figure 3A, Meta-SPS is designed to extract a single sequence (supported by the highest number of star spectra) from each meta-contig as it was not developed to sequence mixtures of highly similar proteins. Thus, we developed a semi-automated approach, PolyExtend, to sequence polyclonal sequence variations supported by less spectra than the highest abundance clone. PolyExtend takes as input (A) partial protein sequence P (e.g., from previous de novo sequencing or from a detected framework region), (B) a set of PRM spectra {Si}, and (C) a binary option e = Preff/Suff to extend the root protein sequence from the N- or C-terminus, respectively. For both the heavy chain (HC) and light chain (LC), the partial protein sequence used for input (A) began as a known constant region identified by database search and was iteratively extended towards the N-terminus. We then used peptide IDs against known variable regions to capture remaining sequences (extending towards both N- and C-termini). We mainly used star spectra as input (B) due to their high signal-to-noise ratio while scored spectra were used in rare cases to sequence areas with low MS/MS coverage from overlapping peptides. Rather than manually de novo sequencing individual unfiltered MS/MS spectra, PolyExtend first finds spectra with tags that match P and aligns these spectra to P allowing for partial matches between each spectrum and prefixes of P (if e = Suff, then suffixes are considered). For each spectrum that extends past the N- or C-terminus of P, the user is displayed the highest scoring de novo interpretation that can extend P in the desired direction. Figure 3B illustrates such an output while trying to extend a putative framework de novo sequence towards the N-terminus.
A more detailed explanation of the algorithm is given below while a full Python implementation of PolyExtend (with source code) can be downloaded at the MassIVE data repository21. Define each PRM spectrum Si as a set of m PRM masses .
The partial protein sequence P is converted into a PRM spectrum containing all n PRM masses of the sequence.
A pre-computed set of 3-mer amino acid tags was extracted from each PRM spectrum and was used to match spectra to P. Each set of tags is defined as the set of all possible combinations of three amino acids that are supported by four consecutive PRMs from a spectrum. A spectrum Si is matched if and only if there exists a PRM peak pair (s ∈ Si, sP ∈ SP) such that a tag starting at peak s equals the 3-mer substring of P following the prefix with mass sP.
The dynamic programming algorithm for spectral alignment described by Bandeira et al28 was used to find the maximum scoring alignment between all pairs of spectra (Si, SP) allowing for up to two post-translational modifications (PTMs). A spectral alignment is defined on the set of all matching peaks (si ∈ Si, sP ∈ SP) while the maximum scoring spectral alignment maximizes the score of matched peaks28. We modified this approach to only allow for known PTMs (such as oxidized Met, or M+16, and deamidated Asp, or N+1) rather than allowing for any PTM of unknown mass. Alignments were discarded if they i) matched <4 PRMs or ii) matched <30% of summed PRM scores in scored spectra. These criteria do not correspond to any particular false-positive rate, but were chosen because they significantly improved the quality of alignments over less stringent thresholds while being sensitive enough to enable sequencing of all mAbs presented here without additional adjustment of thresholds.
Given a spectral alignment over peaks (si ∈ Si, sP ∈ SP), consider the matched peak pair such that is the matched peak from spectrum Si with minimal mass. We compute the maximum scoring de novo sequence from the set of peaks that can extend P past its N-terminus (if e = Pref). If e = Suff, we consider the matched peak pair such that is the matched peak from spectrum Si with maximum mass and we compute the de novo sequence over peaks .
Extension alignments were first sorted by decreasing number of matched peaks and then by decreasing log likelihood score. Finally, display to the user each alignment (Si, SP) followed by the de novo sequence extension extracted from Si. By default, the top 75 alignments (with de novo extensions) are displayed to the user but this parameter can be adjusted.
Once the list of alignments is displayed, to the user can choose the next de novo sequence extension based on the number and quality of corresponding alignments to the tag sequence (we only considered de novo extensions supported by at least two spectra, and only extended the root sequence by 1–2 AA at a time). The decision of which de novo extensions to choose is ultimately up to the user, where those with the highest ranking alignments should be the most reliable (see Figure S1 for a more detailed use case). After appending the de novo extension to the root sequence, the process is repeated until all possible sequence extensions have been explored or until reaching the N- or C-terminus of the putative antibody sequence, which can be determined by BLAST29,30 searching the putative sequence against known N-/C-terminal IgG framework domains.
Selecting full-length sequences for in silico mAb manufacture
Rather than working with thousands of full-length antibody sequences individually, all sequences and their corresponding mutations were manually organized into the following format: Given a protein sequence PEPTIDE with mutations PESTIDE and PEPTALE, they were encoded succinctly as PE[P,S]T[ID,AL]E. Custom Python scripts were developed to parse sequences in this format and output the subset of antibody sequences that match observed intact masses shown in Figure 2. Given the high complexity (possibly leading to isotope deconvolution errors) and unknown presence of abundant post-translational modifications in human polyclonal antibody samples, a slightly wider tolerance of 4 Da was allowed. All MS/MS spectra were then searched against a combined database encoding these sequences plus known contaminants31 using MS-GFDB32 and filtered to 1% False-Discovery Rate (FDR). Although FDR cannot be accurately estimated when searching against de novo sequences, resulting peptide-spectrum matches (PSMs) were useful for estimating what percentage of residues in each full-length de novo sequence was supported by MS/MS spectra and how many spectra supported each sequence. Sequences matching observed intact masses with 100% MS/MS coverage were ultimately ranked by numbers of supporting MS/MS spectra, where the top four LC and seven HC sequences were chosen.
Expression of antibody derived from de novo sequencing
All pairwise combinations of the four LC and seven HC candidate sequences were expressed in mammalian cells to validate antigen binding. Antibody variable light and heavy sequences were synthesized (Genewiz, NJ, USA) and subcloned into mammalian expression plasmids encoding human kappa/lambda, and IgG1 constant region, respectively33. Recombinant antibodies were expressed in Expi293 (Thermo Fisher Scientific) and purified using Protein A affinity resins. Different combinations of heavy and light chain were co-expressed.
Evaluation of recombinant antibody binding to target by ELISA
gBCterm-His6 (2 μg ml−1) in PBS, pH 7.4, was coated on ELISA plates (Nunc Maxisorp) at 4 °C overnight. Plates were blocked with casein blocker in PBS (Pierce) for 1 h at room temperature. Serial threefold dilutions of antibody IgGs in PBST (PBS with 0.05% Tween-20) buffer were added to the plates and incubated for 1 h at room temperature. The plates were then washed with PBST and bound antibodies were detected with peroxidase-conjugated goat anti-human Fab specific IgG (Sigma). TMB substrate (3,3′,5,5′-tetramethylbenzidine, BioFx) was used and the reactions were stopped with 100 μl stop solution (BioFx) before absorbance at 650 nM was read using a standard ELISA plate reader. Absorbance was plotted against concentrations of IgGs using KaleidaGraph (Synergy Software).
Memory B-cell Sequencing
To test if de novo sequenced pAbs were detectable in peripheral B cells using standard sequencing protocols, peripheral B-cell sequencing was performed. Memory B cells were isolated from the heparinized plasma of the same CMV positive individual using a RosetteSep™ Human B cell Enrichment Cocktail kit according to the manufacturer’s protocol (#15064, STEMCELL Technologies, Vancouver, BC, Canada) that included purification over a Ficoll-Paque PLUS (GE Healthcare Life Sciences, Pittsburg, PA) density gradient. B cells were washed in phosphate buffered saline, pelleted by centrifugation and then prepared for total RNA isolation (RNeasy, QIAGEN, Redwood City, CA). First strand cDNA synthesis was performed using oligo d(T) and Supercript III Reverse Transcriptase (ThermoFisher, Waltham, MA) according to the manufacturer’s recommendations. PCR amplicons for human IgG, IgM, IgK and IgL were generated using 5′ degenerate framework 1 oligonucleotides with gene specific 3′ constant region oligonucleotides (Platinum PCR SuperMix High Fidelity, Thermo Fisher). PCR products were analyzed visually by E-Gel electrophoresis (Thermo Fisher) and then purified (QIAquick PCR Purification, QIAGEN) prior to sequencing. A database encoding all sequencing reads was constructed to enable error-tolerant search34 of de novo protein sequences obtained using PolyExtend.
Results
Assuming that intact molecular weight analysis was not confounded by undetected PTMs and/or MS deconvolution artifacts, the donor’s detectable immune response to the CMV gB antigen consisted of 2–3 abundant mAbs among a background of pAbs (Figure 2). The most abundant heavy chains weighed approximately 49971 and 49953 Da while the most abundant light chains had weights of 22644 Da, 22613 Da, and 22887 Da, with a putative HC/LC pairing of 49971/22644 Da and 49953/22644 Da based on MS intensity (Figure 2). The HC/LC de novo sequence pair, named POS1, matched the molecular weights 49971/22644 Da and was ultimately shown to have positive binding to the CMV gB antigen (Figure 4), although the HC intact mass was roughly 4 Da heavier than expected, which is either the result of a sequencing error or unanticipated post-translational modification(s). The HC/LC de novo sequence pair named POS2, matched the molecular weights 49953/22644 Da and also showed positive binding to the CMV gB antigen (Figure 4), with both HC and LC intact masses within 1–2 Da of expected values.
Figure 4.
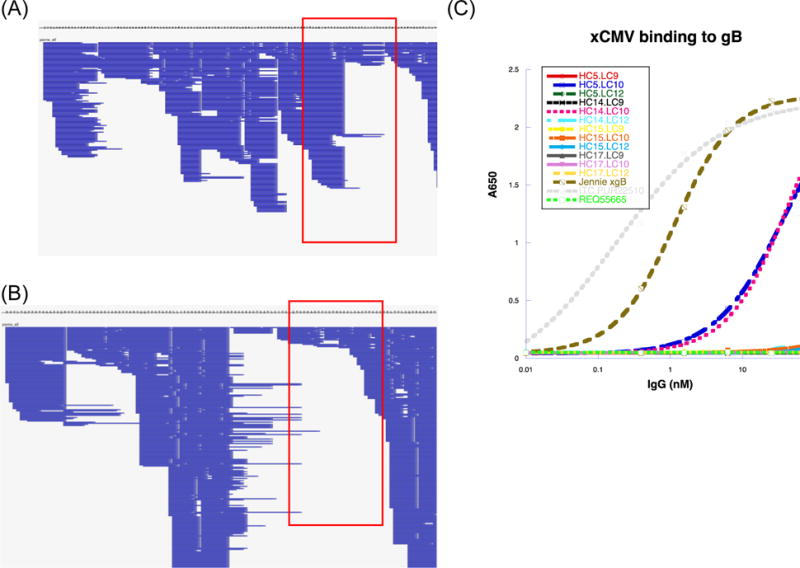
Complete MS/MS coverage of the heavy chain (A) and light chain (B) variable regions. The LC matches the most abundant intact molecular weight of 22644 Da and the HC matches the most abundant intact molecular weight of 49971 Da (Figure 2). Panels show a zoomed-out view of coverage from the N-terminus to the C-terminus of the variable regions. Each horizontal blue line denotes a spectrum matched by MSGFDB database search against all de novo sequences. Areas boxed in red denote coverage of the CDR3 domain. (C) ELISA binding assay for expressed mAbs (different combinations of de novo LC and HC sequences) against the same CMV gB antigen used to purify pAbs from donor plasma. The grey dotted line is a positive control murine mAb against gB; the brown dashed line is anti-CMV pAb material from the donor; the blue dashed line indicating positive binding is a MS-derived mAb with the same variable region shown on the right (POS1); and the pink dotted line indicating positive binding refers to POS2, a mAb similar to POS1 with the same light chain but mutations in HC CDR2. The bright green dotted line is a negative control mAb. Remaining lines indicating negative binding correspond to other mAbs manufactured from de novo sequences.
Bottom-up de novo sequencing revealed IgG1/IgG2 heavy chain isotypes as well as lambda/kappa light chain isotypes. Multiple variable IgG framework sequences were detected with the majority of mutations localized to CDR domains. Table 1 exhibits how many unique CDR domains were sequenced from available MS/MS data. Without applying intact molecular weight filters, 864 unique HC and 19,524 LC sequences exhibited 100% coverage of MS/MS spectra. Since the detection of even single AA variants multiplies the number of possible full-length sequences (ie. 3 unique CDR1, 4 unique CDR2, and 5 unique CDR3 sequences would result in up to 3×4×5=60 full-length sequences representing all combinations of unique CDR1/2/3 variants), the high number of full-length sequences should not be used to approximate the number of polyclonal antibodies present in this sample. This is a result of polyclonal diversity and the inability to phase mutations that are not spanned by a single peptide, regardless of the computational approach being used. This represents the extent of de novo sequencing achieved with PolyExtend over two weeks of analysis on a quad-core desktop computer with 24Gb available RAM.
Table 1.
Number of CDR domains obtained from de novo sequencing of bottom-up MS/MS spectra. This does not include the number of framework mutations.
| CDR1 | CDR2 | CDR3 | Constant Regions | |
|---|---|---|---|---|
|
| ||||
| Heavy Chain | 2 | 5 | 2 | IgG1/2 |
|
| ||||
| Light Chain | 15 | 108 | 9 | Lambda/kappa |
See the PolyExtend download package for a compact representation of all framework sequences obtained from de novo sequencing as well as those chosen for downstream manufacture. Ultimately, four full-length LC and seven full-length HC candidate sequences were chosen for manufacture in mammalian cells (all sequences were made as IgG1) based on these criteria: 2 LC sequences matched the intact mass 22644 Da (Figure 2) with maximal supporting MS/MS spectra and 2 LC sequences had maximal supporting MS/MS spectra while matching no observed intact masses; 4 HC sequences matched the intact mass 49971 Da with maximal supporting MS/MS spectra and 3 HC sequences had maximal supporting MS/MS spectra while matching no observed intact masses. Some mAbs were chosen for manufacture without supporting intact molecular weights to test if the deconvoluted intact mass spectra acquired from the complex polyclonal mixture was accurate enough to aid in mAb selection.
Of all combinations of manufactured heavy/light chains, two, named POS1 and POS2, exhibited positive binding to the CMV gB antigen by ELISA (Figure 4C). POS1 did not closely match 49971/22644 Da (Figure 2) while POS2 closely matched the observed masses of 49953/22644 Da. This underscores a limitation of relying on bottom-up MS/MS analysis to sequence full-length antibodies from a polyclonal mixture: it is impossible to phase (i.e., to determine the protein-level combinations of) polyclonal mutations that are not spanned by at least one peptide. Without peptides from middle-down digestions which span multiple CDRs (and/or top-down spectra covering entire variable heavy/light chains), multiple unique combinations of mutations can match observed intact molecular weights with 100% coverage of MS/MS spectra. This necessitated the use of heuristics, such as spectral counts, to rank candidate full-length sequences prior to downstream selection by ELISA, which may have led to inaccurate full-length de novo sequencing.
It is unclear why the binding potency of POS1 and POS2 did not match that of polyclonal serum from donor plasma (Figure 4C). They could have been incorrectly sequenced, or they could have had very weak potency in the absence of other less abundant pAbs from serum. The same can be said as to why other candidates did not bind. Another possibility is that pAbs could have had affinity for gB-bound solid support streptavidin agarose during affinity purification. Future projects should seek to remove such broadly active pAbs from plasma prior to affinity purification via agarose negative purification.
Conclusions and Discussion
An important question regarding this novel antibody discovery approach is whether or not it can detect antibodies that are missed by traditional peripheral B-cell technologies. Although we were not able to detect any of the proteomic-sequenced pAbs from parallel memory B-cell sequencing of this donor, it is entirely possible that our methods failed to detect peripheral B cells encoding POS1 and POS2. More exhaustive sampling of B cells, particularly the isolation of B cells displaying antibodies with affinity to the targeted antigen6, should be tested in parallel with this approach. In fact, this proteomic-centric approach is very complementary to all peripheral B-cell technologies because they typically discard pAbs during B-cell purification and MS/MS data can be searched against B-cell genetic sequencing data13,14,35.
De novo protein sequencing approaches are traditionally limited by MS/MS peptide sampling bias as a result of hydrophobicity, ionizability, and locations of basic amino acids, which leads to incomplete MS/MS coverage. We addressed these concerns with 16 total MS/MS runs (eight separate enzyme digests of each heavy/light chain), each with a 2-hour chromatography gradient and high-resolution CID/HCD/ETD MS/MS fragmentation of each precursor, to acquire enough MS/MS spectra from overlapping peptides to cover full-length pAbs. Compared to traditional CID or HCD MS/MS analysis13,14, high resolution CID/HCD/ETD is particularly effective at improving MS/MS fragmentation of peptides that are poorly fragmented by CID or HCD alone, such as long, highly charged peptides containing basic residues26. In fact, a prior study comparing triplet CID/HCD/ETD MS/MS analysis of a six protein mixture16 with just CID and HCD alone20 revealed that even with the decreased scan rate of ETD allowing for analysis of 1/3 as many peptide precursors, more accurate de novo sequencing could be achieved from CID/HCD/ETD with greater sequence coverage. The scan rate trade-off of CID/HCD/ETD versus CID or HCD becomes even less of a concern when considering the latest generation of Orbitrap Fusion mass spectrometers (ThermoFisher), which feature a ~5× faster scan rate than the Orbitrap Velos used in this study and allow for greater flexibility in separately optimizing CID/HCD/ETD fragmentation settings.
Other sample preparation procedures can impact the accuracy and sensitivity of this approach independent of available MS/MS instrumentation. We employed ion exchange chromatography to purify abundant mAbs upstream of MS/MS analysis. Any step that can reduce the complexity of pAbs in the sample will almost always improve MS/MS sensitivity for the most abundant mAbs. We also suggest digesting serum pAbs with IdeS36 to allow purification of F(ab)2 fragments, which removes much of the constant region on the HC, thereby increasing available MS/MS signal on the HC variable region. Absence of this step could explain why we detected fewer CDRs and overall sequence diversity on the HC compared to the LC, although high diversity on the LC can be expected if antigen binding is mediated by the HC.
Any approach that relies upon MS/MS identification at the protein level faces difficulty in accurately differentiating between isobaric amino acids with similar mass, such as I/L, K/Q, and SV/W. High-resolution MS/MS acquisition enables separation when the difference in mass is resolvable at 10–30ppm, such as for K/Q, but not for amino acid combinations having the same mass, such I/L or GG/N and AG/GA (when there is incomplete fragmentation). One can use homology to known sequences when possible, but this is not always reliable, especially when sequencing novel CDRs with a tool such as PolyExtend. However, the presence or absence of prefix masses should enable resolution of multi-isobaric amino acid jumps such as GG/N and AG/GA when sufficient MS/MS fragmentation is observed. Other ambiguities such as I/L must be called with caution, and in some cases may necessitate expression of different I/L variants followed by in vitro assays to determine the optimal sequence.
Although Meta-SPS was originally designed to sequence simple mixtures of only a few unknown proteins, we were able to extract sequences for hundreds of putative proteins because of shared sequence along constant and framework antibody domains (many unique antibodies diverged by just a few AA). But shared sequence homology created the problem of phasing polyclonal mutations within the same singular mAb, which we addressed with customized software to (1) sequence less-abundant mutations in a semi-automated fashion and (2) rank full-length sequences by numbers of supporting MS/MS spectra and how well their expected molecular weight matches observed weights of the most abundant mAbs within the pAb mixture. One possible avenue towards improving the accuracy of this approach would be to apply middle-down and/or top-down protocols37–40 to acquire MS/MS spectra covering entire variable regions of the most abundant pAbs. Intact molecular weight analysis (Figure 2) suggests that purification by antigen affinity can dramatically reduce the complexity of pAbs from circulating plasma, which may facilitate targeted top-down analysis of the most abundant mAbs. Top-down analysis may also be crucial to addressing the possibility that the strong peaks observed in intact molecular weight analysis correspond to multiple polymorphic pAbs that happen to have the same molecular weight, rather than singular mAbs at high abundance.
The restrictions of this proof-of-concept study are reflected in the limited activity of anti-CMV mAbs compared to donor plasma (Figure 4C). Nevertheless, we demonstrate that overlapping enzyme digestions coupled with CID/HCD/ETD analysis has the potential to enable full-length de novo sequencing of potent mAbs from donor plasma. This demonstrates the feasibility of de novo polyclonal discovery and illustrates how it could advance the field of immunological research by, for the first time, interrogating the expressed repertoire of peripheral antibodies produced within a live host in response to a foreign antigen.
Supplementary Material
References
- 1.Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC. Monoclonal antibody successes in the clinic. Nat Biotechnol. 2005;23(9):1073–1078. doi: 10.1038/nbt0905-1073. [DOI] [PubMed] [Google Scholar]
- 2.Reichert JM. Trends in the development and approval of monoclonal antibodies for viral infections. BioDrugs. 2007;21(1):1–7. doi: 10.2165/00063030-200721010-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drake CG, Lipson EJ, Brahmer JR. Breathing new life into immunotherapy: review of melanoma, lung and kidney cancer. Nat Rev Clin Oncol. 2014;11(1):24–37. doi: 10.1038/nrclinonc.2013.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qiu X, Wong G, Audet J, Bello A, Fernando L, Alimonti JB, Fausther-Bovendo H, Wei H, Aviles J, Hiatt E, et al. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature. 2014;514(7520):47–53. doi: 10.1038/nature13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Furuyama W, Marzi A, Nanbo A, Haddock E, Maruyama J, Miyamoto H, Igarashi M, Yoshida R, Noyori O, Feldmann H, et al. Discovery of an antibody for pan-ebolavirus therapy. Sci Rep. 2016;6:20514. doi: 10.1038/srep20514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bornholdt ZA, Turner HL, Murin CD, Li W, Sok D, Souders CA, Piper AE, Goff A, Shamblin JD, Wollen SE, et al. Isolation of potent neutralizing antibodies from a survivor of the 2014 Ebola virus outbreak. Science (80−) 2016;351(6277):1078–1083. doi: 10.1126/science.aad5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith SA, de Alwis R, Kose N, Durbin AP, Whitehead SS, de Silva AM, Crowe JE. Human monoclonal antibodies derived from memory B cells following live attenuated dengue virus vaccination or natural infection exhibit similar characteristics. J Infect Dis. 2013;207(12):1898–1908. doi: 10.1093/infdis/jit119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou T, Georgiev I, Wu X, Yang Z-Y, Dai K, Finzi A, Kwon Y Do, Scheid JF, Shi W, Xu L, et al. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science. 2010;329(5993):811–817. doi: 10.1126/science.1192819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Georgiou G, Ippolito GC, Beausang J, Busse CE, Wardemann H, Quake SR. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat Biotechnol. 2014;32(2):158–168. doi: 10.1038/nbt.2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hibi T, Dosch HM, Ig T. Limiting dilution analysis of the B cell compartment in human bone marrow. Eur J Immunol. 1986;16(2):139–145. doi: 10.1002/eji.1830160206. [DOI] [PubMed] [Google Scholar]
- 11.Apostoaei IA, Trabalka JR. Review, Synthesis, and Application of Information on the Human Lymphatic System to Radiation Dosimetry for Chronic Lymphocytic Leukemia. Oak Ridge, TN: 2010. [Google Scholar]
- 12.Brekke OH, Sandlie I. Therapeutic antibodies for human diseases at the dawn of the twenty-first century. Nat Rev Drug Discov. 2003;2(1):52–62. doi: 10.1038/nrd984. [DOI] [PubMed] [Google Scholar]
- 13.Cheung WC, Beausoleil SA, Zhang X, Sato S, Schieferl SM, Wieler JS, Beaudet JG, Ramenani RK, Popova L, Comb MJ, et al. A proteomics approach for the identification and cloning of monoclonal antibodies from serum. Nature Biotechnology. 2012:447–452. doi: 10.1038/nbt.2167. [DOI] [PubMed] [Google Scholar]
- 14.Sato S, Beausoleil Sa, Popova L, Beaudet JG, Ramenani RK, Zhang X, Wieler JS, Schieferl SM, Cheung WC, Polakiewicz RD. Proteomics-directed cloning of circulating antiviral human monoclonal antibodies. Nat Biotechnol. 2012;30(11):1039–1043. doi: 10.1038/nbt.2406. [DOI] [PubMed] [Google Scholar]
- 15.Duncan MW, Aebersold R, Caprioli RM. The pros and cons of peptide-centric proteomics. Nat Biotechnol. 2010;28(7):659–664. doi: 10.1038/nbt0710-659. [DOI] [PubMed] [Google Scholar]
- 16.Guthals A, Clauser KR, Frank AM, Bandeira N. Sequencing-Grade De novo Analysis of MS/MS Triplets (CID/HCD/ETD) From Overlapping Peptides. J Proteome Res. 2013;12(6):2846–2857. doi: 10.1021/pr400173d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bandeira N, Pham V, Pevzner P, Arnott D, Lill JR. Automated de novo protein sequencing of monoclonal antibodies. Nat Biotechnol. 2008;26(12):1336–1338. doi: 10.1038/nbt1208-1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castellana NE, Pham V, Arnott D, Lill JR, Bafna V. Template proteogenomics: sequencing whole proteins using an imperfect database. Mol Cell Proteomics. 2010;9(6):1260–1270. doi: 10.1074/mcp.M900504-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X, Han Y, Yuen D, Ma B. Automated protein (re)sequencing with MS/MS and a homologous database yields almost full coverage and accuracy. Bioinformatics. 2009;25(17):2174–2180. doi: 10.1093/bioinformatics/btp366. [DOI] [PubMed] [Google Scholar]
- 20.Guthals A, Clauser KR, Bandeira N. Shotgun protein sequencing with meta-contig assembly. Mol Cell Proteomics. 2012;10(11):1084–1096. doi: 10.1074/mcp.M111.015768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MassIVE. ftp://massive.ucsd.edu/MSV000080039.
- 22.Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24(21):2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frank AM. Predicting intensity ranks of peptide fragment ions. J Proteome Res. 2009;8(5):2226–2240. doi: 10.1021/pr800677f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dancík V, Addona TA, Clauser KR, Vath JE, Pevzner PA. De novo peptide sequencing via tandem mass spectrometry. J Comput Biol. 1999;6(3–4):327–342. doi: 10.1089/106652799318300. [DOI] [PubMed] [Google Scholar]
- 25.Datta R, Bern M. Spectrum Fusion: Using Multiple Mass Spectra for De Novo Peptide Sequencing. J Comput Biol. 2009;16(8):1169–1182. doi: 10.1089/cmb.2009.0122. [DOI] [PubMed] [Google Scholar]
- 26.Guthals A, Bandeira N. Peptide identification by tandem mass spectrometry with alternate fragmentation modes. Mol Cell Proteomics. 2012;11(9):550–557. doi: 10.1074/mcp.R112.018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bandeira N, Tsur D, Frank A, Pevzner PA. Protein identification by spectral networks analysis. Proc Natl Acad Sci USA. 2007;104(15):6140–6145. doi: 10.1073/pnas.0701130104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bandeira N, Clauser KR, Pevzner PA. Shotgun protein sequencing: assembly of peptide tandem mass spectra from mixtures of modified proteins. Mol Cell Proteomics. 2007;6(7):1123–1134. doi: 10.1074/mcp.M700001-MCP200. [DOI] [PubMed] [Google Scholar]
- 29.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 30.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.The Global Proteome Machine. cRAP protein sequences. http://www.thegpm.org/crap/
- 32.Kim S, Mischerikow N, Bandeira N, Navarro JD, Wich L, Mohammed S, Heck AJR, Pevzner PA. The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: applications to database search. Mol Cell Proteomics. 2010;9(12):2840–2852. doi: 10.1074/mcp.M110.003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, et al. High Resolution Mapping of the Binding Site on Human IgG1 for Fcgamma RI, Fcgamma RII, Fcgamma RIII, and FcRn and Design of IgG1 Variants with Improved Binding to the Fcgamma R. J Biol Chem. 2001;276(9):6591–6604. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- 34.Altschul S, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Safonova Y, Bonissone S, Kurpilyansky E, Starostina E, Lapidus A, Stinson J, DePalatis L, Sandoval W, Lill J, Pevzner PA. IgRepertoireConstructor: a novel algorithm for antibody repertoire construction and immunoproteogenomics analysis. Bioinformatics. 2015;31(12):i53–61. doi: 10.1093/bioinformatics/btv238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.von Pawel-Rammingen U, Johansson BP, Björck L. IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin G. EMBO J. 2002;21(7):1607–1615. doi: 10.1093/emboj/21.7.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frank AM, Pesavento JJ, Mizzen CA, Kelleher NL, Pevzner PA. Interpreting top-down mass spectra using spectral alignment. Anal Chem. 2008;80(7):2499–2505. doi: 10.1021/ac702324u. [DOI] [PubMed] [Google Scholar]
- 38.Zabrouskov V, Senko MW, Du Y, Leduc RD, Kelleher NL. New and automated MSn approaches for top-down identification of modified proteins. J Am Soc Mass Spectrom. 2005;16(12):2027–2038. doi: 10.1016/j.jasms.2005.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dekker L, Wu S, Vanduijn M, Tolić N, Stingl C, Zhao R, Luider T, Paša-Tolić L. An integrated top-down and bottom-up proteomic approach to characterize the antigen-binding fragment of antibodies. Proteomics. 2014;14:1239–1248. doi: 10.1002/pmic.201300366. [DOI] [PubMed] [Google Scholar]
- 40.Durbin KR, Fornelli L, Fellers RT, Doubleday PF, Narita M, Kelleher NL. Quantitation and Identification of Thousands of Human Proteoforms below 30 kDa. J Proteome Res. 2016;15(3):976–982. doi: 10.1021/acs.jproteome.5b00997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.