

 This work is licensed under a
This work is licensed under a Abstract
Chronic ACTH exposure is associated with adrenal hypertrophy and steroidogenesis. The underlying molecular processes in mice have been analysed by microarray, histological and immunohistochemical techniques. Synacthen infused for 2 weeks markedly increased adrenal mass and plasma corticosterone levels. Microarray analysis found greater than 2-fold changes in expression of 928 genes (P < 0.001; 397 up, 531 down). These clustered in pathways involved in signalling, sterol/lipid metabolism, cell proliferation/hypertrophy and apoptosis. Signalling genes included some implicated in adrenal adenomas but also upregulated genes associated with cyclic AMP and downregulated genes associated with aldosterone synthesis. Sterol metabolism genes were those promoting cholesterol supply (Scarb1, Sqle, Apoa1) and disposal (Cyp27a1, Cyp7b1). Oil red O staining showed lipid depletion consistent with reduced expression of genes involved in lipid synthesis. Genes involved in steroidogenesis (Star, Cyp11a1, Cyp11b1) were modestly affected (P < 0.05; <1.3-fold). Increased Ki67, Ccna2, Ccnb2 and Tk1 expression complemented immunohistochemical evidence of a 3-fold change in cell proliferation. Growth arrest genes, Cdkn1a and Cdkn1c, which are known to be active in hypertrophied cells, were increased >4-fold and cross-sectional area of fasciculata cells was 2-fold greater. In contrast, genes associated with apoptosis (eg Casp12, Clu,) were downregulated and apoptotic cells (Tunel staining) were fewer (P < 0.001) and more widely distributed throughout the cortex. In summary, long-term steroidogenesis with ACTH excess is sustained by genes controlling cholesterol supply and adrenal mass. ACTH effects on adrenal morphology and genes controlling cell hypertrophy, proliferation and apoptosis suggest the involvement of different cell types and separate molecular pathways.
Keywords: ACTH, cholesterol, adrenal hyperplasia, adrenal hypertrophy
Introduction
Temporal control of adrenocortical responses to ACTH involves several processes, mediated by a common signalling system. In vivo and in vitro studies show that within five minutes, stress and ACTH cause increased adrenal corticosteroid release (1, 2). Closer analysis of acute responses has demonstrated that activation of melanocortin receptors trigger cyclic AMP synthesis, leading to the synthesis of StaR, which promotes the uptake of cholesterol into mitochondria (3, 4, 5). As steroid hormones are synthesised on demand, the availability of intramitochondrial cholesterol initiates steroidogenesis by providing substrate for the first rate-limiting enzyme in the steroidogenic pathway (6). Long-term exposure to ACTH requires changes in cholesterol supply, steroidogenic enzyme expression and adrenocortical cell hypertrophy and hyperplasia, which take place over hours, days and weeks to maintain steroid output at a continuous high level. Although many of these key processes involve non-genomic enzyme activation, transcriptional control is also important.
Long-term supply of cholesterol substrate is maintained by de novo synthesis, by uptake from the circulation and by release from stored intracellular lipid droplets. Key steps in de novo synthesis are hydroxymethyglutaryl CoA reductase (HMGCR), squalene epoxidase (SQLE) and various post-lanosterol reductase and dehydrogenase steps (7). Plasma LDL and HDL cholesterol (8, 9) are also available for steroidogenesis via LDL (LDLR) and scavenger receptor (SCARB1) respectively. Within the adrenal cortex, cholesterol is stored as an ester in lipid droplets or utilised for steroidogenesis depending on the balance between lipase and esterifying enzyme activity (10, 11, 12).
Although prolonged stress or ACTH treatment causes adrenal gland hypertrophy, effects on the expression of genes encoding steroidogenic enzymes are less profound (13, 14). In fact, it may be that induction of intra-adrenal steroid hormone-metabolising enzymes help mitigate the effects of excess ACTH. In sheep, for example, there is a marked increase in 20 hydroxylation of corticosteroid intermediates, which have no clearly defined biological activity (15). Similarly, there are reports that adrenal 5α reductase and sulfotransferase activities may affect the secretion of biologically active hormones (16, 17).
Genomic and somatic mutations of various genes have been identified that explain excess steroid production in cortisol- and aldosterone-producing adenomas (18, 19). These involve gain/loss of function that affect adrenocortical signalling processes. Although these genes are required, physiological control of steroidogenesis is not necessarily mediated by regulation of their expression. Moreover, there is a need to determine that signalling elements responsible for acute changes in steroid output are the same as those mediating adrenocortical adaptation to chronic stress or prolonged ACTH exposure.
In this study, we have used microarray analysis of mouse adrenal tissue to gain a comprehensive picture of transcriptional control processes affecting cell signalling, cholesterol supply and cell turnover in response to chronic stimulation of corticosterone synthesis in mice infused with an ACTH analogue, Synacthen. We found modest changes in genes encoding steroidogenic enzymes. Our data suggest that enhanced steroidogenic capacity reflects increases in cell size and a shift in the balance between proliferation and apoptosis that increases cell number. Sterol/lipid metabolic pathways are also changed in several ways to allow cholesterol to be channelled towards steroidogenesis.
Materials and methods
All experiments involving animals were approved by the University of Edinburgh Animal Welfare and Ethical Review Body and were carried out in strict accord with accepted standards of humane animal care under the auspices of the Animal (Scientific Procedures) Act UK 1986. Groups (n = 5/6) of age-matched male C57BL6 mice (Harlan Olac) weighing approximately 25 g were fed a diet containing 0.3% Na (SDS Diets, Witham, Essex, UK) with free access to water in a temperature and light-controlled (12 h light/dark cycle; 07:00 h lights on) room. Mice were infused sc via miniosmotic infusion pumps (Alzet Cupertino, Model 2002) with either vehicle (0.154 M NaCl) or ACTH (Synacthen Ciba-Geigy, UK; 3 μg/day). At the end of the study, mice were killed by carbon dioxide at the nadir of the circadian cycle. Pairs of adrenal glands were collected into a solution of RNeasy (Qiagen). Each adrenal was carefully trimmed free of fat under a dissecting microscope and weighed.
Histology and immunohistochemistry
Additional groups of mice were infused with ACTH or saline as above to assess the effects on adrenal morphology and cell proliferation. Blood was collected by cardiac puncture into lithium heparin tubes for corticosterone measurements by ELISA (20). In one experimental cohort, bromodeoxyuridine (1 mg/mL) was added to the minipump infusates to monitor cell proliferation. After a two-week infusion, mice were killed by decapitation and tissues collected for fixation in buffered formalin and embedded in paraffin wax. BrdU-positive and Ki67-positive nuclei in mid-adrenal sections were located by immunohistochemistry as previously described (21). Cells with Ki67-positive nuclei were counted in the zona glomerulosa/outer zona fasciculata region of the cortex and numbers were normalised to length of capsule perimeter. Haematoxylin and eosin-stained sections were used to estimate cell size in different regions of the adrenal cortex (21).
To assess the cellular storage of cholesterol, adrenals were collected without fixation and stored at −80°C before cryosectioning for oil red O staining of lipid droplets. Images were captured with a Niko Coolpix colour camera with a Zeiss Axioskop 2 compound microscope and MCID imaging software (Imaging Research Inc, St Catharines, Ontario, Canada).
Apoptosis was analysed using a Roche Tunel staining kit (Sigma-Aldrich).
Microarray processing
RNA for microarray analysis was prepared from individual adrenal glands (n = 5 and 6 for saline and ACTH-treated mice respectively) using TRIzol (ThermoFisher Scientific) and then processed through standard Affymetrix protocols, with one round of cDNA amplification (22). Processed RNAs from individual adrenal glands were hybridised to Affymetrix Mouse Genome 430 2.0 GeneChip. RNA processing and microarray analyses were carried out by Ark Genomics (Roslin, Edinburgh). Data were analysed as previously described (22). Microarray data have been archived in the ArrayExpress data repository with the accession number E-MTAB-5704. Differentially regulated transcripts were analysed with DAVID Bioinformatic Resources (23). Cluster analysis of genes involved in signalling, sterol and lipid metabolism and cell turnover was carried out with Miru software (24). Genes of interest that are discussed are listed in Supplementary Table 1 (see section on supplementary data given at the end of this article).
Real-time RT-PCR
Based on initial findings from microarrays, selected genes were quantified by pre-optimised RT-PCR assays in RNA from individual adrenals of separate cohorts of saline and ACTH-treated mice that were killed with CO2. Total RNA was extracted from tissue samples using the Qiagen RNeasy system and reverse transcribed into cDNA with random primers using the QuantiTect DNase/reverse transcription kit (Qiagen Ltd). cDNA (equivalent to 1ng total RNA) was incubated in triplicate with gene-specific primers and fluorescent probes (using pre-designed assays from Applied Biosystems, Warrington, UK) in 1× Roche LightCyclerR480 Probes mastermix. PCR cycling and detection of fluorescent signal were carried out using a Roche LightCyclerR480. A standard curve was constructed for each primer probe set using a serial dilution of cDNA pooled from all samples. Results were corrected for the mean of expression of beta-actin and 18S ribosomal RNA. Neither 18S RNA nor beta-actin was affected by ACTH treatment.
Statistics
Data are presented as mean ± s.e. After tests for Gaussian distribution, comparisons were made using either unpaired t-test or ANOVA with Bonferroni post hoc testing; P ≤ 0.05 were considered statistically significant. Gene expression profiles with Pearson correlation coefficients ≥0.9 were analysed for clustering.
Results
Adrenal response to ACTH treatment
Mice infused with ACTH had larger adrenals and higher plasma corticosterone levels (Fig. 1A and B). Previous studies of urinary corticosterone excretion in this model indicate that ACTH treatment produces a sustained increase in steroidogenesis (22). Body weight gain was not affected. After normalisation and correction for multiple testing, microarray analysis indicated that ACTH significantly affected the expression of approximately 9000 gene transcripts (P < 0.05; Fig. 1C); 397 and 531 annotated genes were upregulated and downregulated respectively by ≥ two-fold (P < 0.001). To validate the microarray results, a parallel analysis was carried out using PCR methods to quantify mRNA of representative genes that were upregulated and downregulated in the microarray. The choice of genes reflected a range of responses and their possible involvement in processes controlling signalling and sterol metabolism (Lpl, Srd5a1, Scarb1, Ren1, Mrap), cell proliferation (Fgfrl1, Impdh1) and apoptosis (Cidea, Casp12, Clu, Trib3, Elmo1). The patterns of change for the selected genes were broadly similar for microarray and qPCR methods (Fig. 1D).
Figure 1.

ACTH infusion causes adrenal hypertrophy (A), increased corticosterone levels (B) and altered expression of gene transcripts (C and D). Values shown in A and B are mean values ± s.e.m., n = 6. Microarray values (grey bars) are compared with RT-PCR values (black bars) for a range of up and downregulated genes (D).
Signalling genes
Figure 2A and B show heat maps of genes implicated in signal transduction that are upregulated and downregulated by ACTH treatment (P < 0.01). A cluster analysis of signalling genes with Pearson correlation coefficients ≥ 0.9 is shown in Supplementary Fig. 1. In general, the expression patterns of upregulated genes involved in signalling (Fig. 2A) were similar to that of the melanocortin receptor accessory protein, Mrap. Cross-tabulation of Pearson correlation coefficients for the upregulated cluster show all values ranged between 0.87 and 0.98. The cluster includes GTPase genes (Rab2, Rab10, Rhod) and genes involved in protein kinase A activity (Prkara1, Prkar2b) and localisation (Akap2). Paradoxically, Pde8b was also upregulated. Other genes in the upregulated cluster were those for factors controlling transcription (Creb312, Nr5a1), cell proliferation and hypertrophy (Rras2, Cdkn1a, Rcc2, Igflr, Shmt1) and several genes like Prkcd, Srxn1, Stx11 and Inha with known but ill-defined links to steroidogenesis. The function of others (Gucalb, Fam161a) has yet to be defined.
Figure 2.

Heat maps showing expression genes involved in cell signalling that are upregulated (A) and downregulated (B) by ACTH. Each square represents gene expression of a single sample. Shades of blue and red indicate levels of expression below and above normalised values for individual genes.
Analysis of downregulated signalling genes (Fig. 2B) suggest secondary events linked to aldosterone/zona glomerulosa functions (Rgs4, Kcnk1, Camkk1, Shh and Prkg2), neuronal/ adrenal medulla tissue (Insm1, Rgs11 and Kcnq2) or glucocorticoid activity (Irs1, Atp2b1 and Scn3b).
Cholesterol supply
Figure 3A shows the effects of ACTH on oil red O staining suggesting a depletion of cholesterol ester droplets in the zona fasciculata. Microarray data indicated that genes involved in (i) de novo cholesterol synthesis from acetyl CoA (Fig. 3B); (ii) promoting cholesterol uptake from the circulation (Fig. 3C); (iii) intracellular cholesterol mobilisation and trafficking and uptake of cholesterol into mitochondria (Fig. 3D); (iv) disposal of free cholesterol (Fig. 3E), were all affected by ACTH treatment. Genes involved in cholesterol synthesis included those encoding HMG-CoA reductase (Hmgcr) and also squalene epoxidase (Sqle) and post-lanosterol enzymes. In keeping with studies demonstrating that cholesterol for steroidogenesis is also derived from the circulation, genes encoding lipoprotein receptors and related proteins that are involved in cholesterol uptake from the circulation were increased. Intracellular cholesterol is trafficked to mitochondria (Npc1, Osbpl6 are upregulated). Genes implicated in the mitochondrial uptake of cholesterol (a rate-limiting step in steroidogenesis) were significantly upregulated too although changes in the expression of the key gene, Star (1.3 fold), was modest. Other than for immediate steroidogenesis, cholesterol may be stored or disposed. Storage as esters is mediated by transferase enzymes and requires a supply of lipid as well as cholesterol. Acat1 and Acat2 (Fig. 3E) and a wide range of genes involved in fatty acid synthesis (Supplementary Fig. 2) were downregulated. These changes are consistent with reduced oil red O staining.
Figure 3.
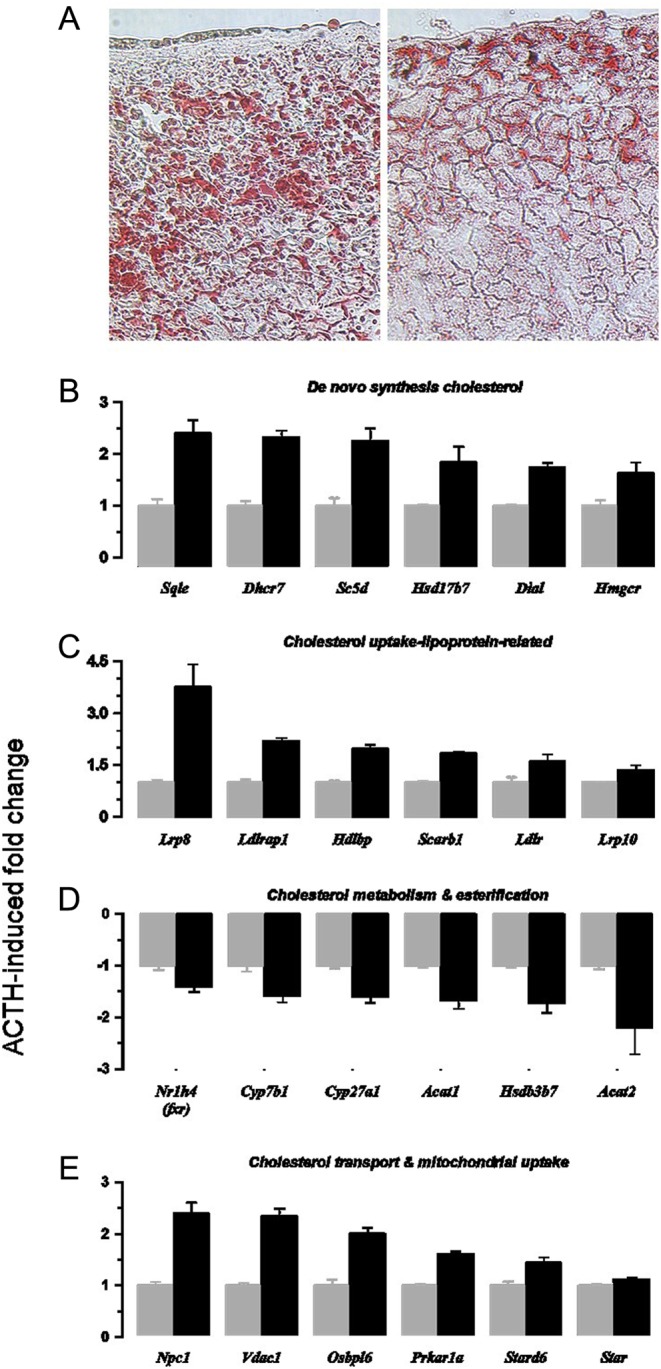
ACTH infusion causes depletion of lipid droplets in adrenal cortex (A) and upregulation of gene transcripts associated with cholesterol biosynthesis (B), the cellular uptake of cholesterol (C) and the intracellular distribution of cholesterol (E). Downregulated transcripts associated with non-steroidogenic routes of cholesterol metabolism are shown (D). Values are means ± s.e.m. of n = 5 (control) and 6 (ACTH) adrenal glands.
Genes affecting cholesterol disposal but not directly involved in steroidogenesis were also downregulated by ACTH. Cyp27a1, Cyp7b1 and Hsd3b7 genes mediate cholesterol hydroxylation and subsequent isomerase reactions. Overall, control of the enzymes and receptors involved in cholesterol synthesis, uptake and metabolism is normally mediated by transcription factors FXR and SREBF1, which in turn are regulated by cholesterol-sensing Insig proteins and intracellular sterol levels. Nr1h4 (Fxr), Serbf1 and Insig1 genes were all downregulated. Furthermore, upregulated genes implicated in cholesterol synthesis and uptake cluster together (Supplementary Fig. 3). Conversely, downregulated genes associated with cholesterol disposal and lipid biosynthesis form a separate larger cluster.
It is notable that genes encoding enzymes that metabolise steroids were also affected. Akr1c18, which encodes a progesterone 20α hydroxysteroid dehydrogenase enzyme was switched on in ACTH-treated adrenals (>80 fold) and expression of Srd5a1 and 2, which encode 5α steroid reductase enzymes, were reduced (1.6 and 5.4-fold respectively).
Adrenal size
Cell Hypertrophy: Part of the ACTH-induced increase in adrenal mass is due to adrenocortical cell hypertrophy. The cross-sectional area of zona fasciculata cells, the main cell type of the cortex, was >2 fold greater but cells of other cortical zones were also increased (Fig. 4A and B). Medullary cell size was not affected. The expression of representative genes potentially implicated in cell hypertrophy are shown in Fig. 4C and include factors affecting growth, cell cycle, transcription and protein synthesis.
Figure 4.
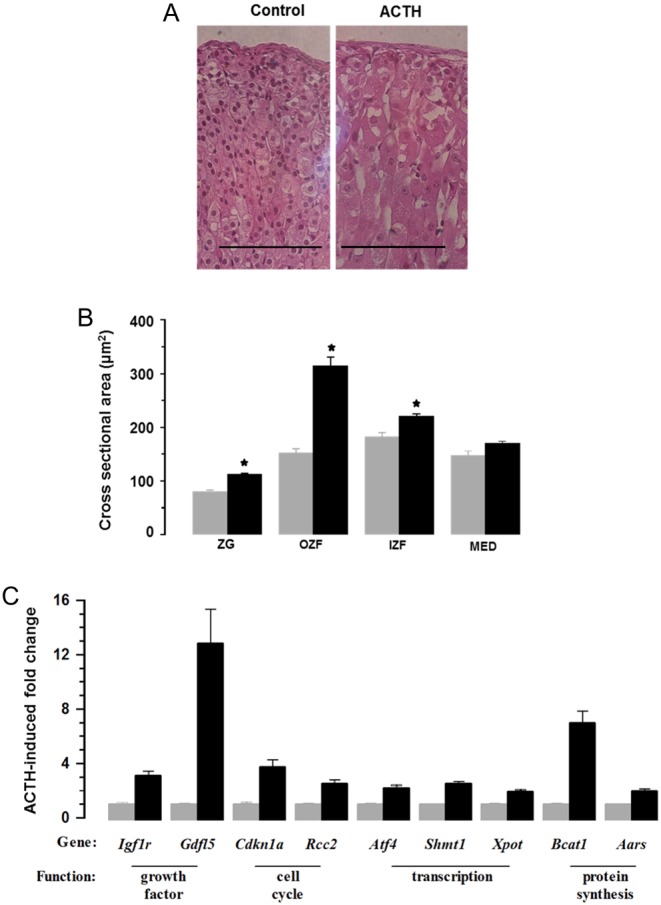
ACTH infusion caused adrenocortical cell hypertrophy (A and B) and increased expression of transcripts associated with cell growth (C). (A) Shows representative H&E-stained sections with bar indicating magnification (100 µm). (B) Shows cross-sectional area of cells in zona glomerulosa (ZG), outer zona fasciculata (OZF), inner zona fasciculata (IZF) and medulla (MED) regions of the gland. Values are means ± s.e.m. of n = 6 (control) and 6 (ACTH) adrenal glands. (C) Shows increased expression of genes associated with cell size.
Cell Hyperplasia: Fig. 5A and B show the effects of ACTH on BrdU incorporation and Ki67-positive cells in the adrenal cortex. Labelled cells are located predominantly in the outermost region of the gland. BrdU was incorporated cumulatively over the entire period of infusion, whereas Ki67-labelling identified only those cells in S-phase at the time of sacrifice. Immunohistochemistry identified approximately ten times more BrdU-positive than Ki67-positive cells (Fig. 5B). Interestingly, ACTH increased the numbers of BrdU- and Ki67-positive cells to a similar degree indicating that proliferative effects were sustained throughout the period of treatment. Increases in cell proliferation were matched by expression of genes associated with various aspects of cell division (Fig. 5C).
Figure 5.

ACTH infusion increased adrenocortical cell proliferation (A and B) and the expression of gene transcripts associated with cell division (C). Sections of adrenal gland from control and ACTH were dual immunostained (A) for Ki67 (brown nuclei) and bromodeoxyuridine (purple nuclei) and analysed for numbers of positive nuclei (B). Values are means ± s.e.m. of n = 6 (control) and 6 (ACTH) adrenal glands; *P < 0.01, **P < 0.001). ACTH-induced fold changes in gene transcripts associated with cell proliferation are shown in (C).
Apoptosis: The net effect of ACTH on adrenocortical volume may involve a decrease in cell death as well as increases in cell hyperplasia and hypertrophy (Fig. 6A and B). Figure 6C and Supplementary Fig. 4 show the decreased expression of genes with a pattern similar to that of a key apoptotic gene, Casp12, and include histocompatibility factors and components of the complement system.
Figure 6.

ACTH treatment reduced Tunel staining in the adrenal cortex (A). Numbers of apoptotic cells corrected for cross-sectional area of cortex (B) were significantly reduced (P < 0.001). Values shown are means ± s.e.m. of n = 4 control and ACTH-treated adrenals. ACTH decreased expression (fold change) associated with apoptosis are shown in (C) (see also Supplementary Fig. 2).
Cluster analysis of genes linked to cell size and turnover (Supplementary Fig. 5) shows upregulated and downregulated genes. Within the upregulated cluster were overlapping genes associated with hypertrophy and hyperplasia.
It is notable that genes associated with cell proliferation were more tightly clustered (Tk1, Mki67, Cdc2a). Genes in the downregulated cluster are associated with apoptosis
Discussion
An infusion of ACTH was used to model molecular and morphological changes in the mouse adrenal gland that contribute to long-term steroidogenic control. Previously we have reported pathophysiological responses to this treatment that suggest it represents a model of ACTH-dependent Cushings. These responses include raised urinary corticosterone to levels which are outwith normal circadian rhythms, thymic involution, hypertension and fluid and electrolyte imbalance (22). The central part of the current study is a microarray analysis of adrenal mRNA expression, which established that large numbers of genes were upregulated and downregulated indicating that adaptive genomic responses were far wider than might be anticipated from known acute changes in cell signalling and steroidogenesis. Closer analysis of clusters of changes in gene expression indicate that mechanisms are invoked with significant consequences for the continuing supply of steroid hormone substrate (cholesterol), for adrenocortical cell kinetics, for adrenocortical cell hypertrophy and for processes that might compensate excess glucocorticoid hormone. Genes encoding signalling factors that are responsible for these functional changes were also noted.
The obligatory role for cholesterol in steroidogenesis and the factors and genetic diseases involving cholesterol metabolism, which affect steroidogenesis are well recognised and have been reviewed extensively (11, 6). Sustained high corticosterone output requires continuing cholesterol supply rather than mobilisation of extant stores. This need is met by de novo synthesis and by uptake from the circulation. Genes encoding enzymes involved in cholesterol synthesis, including Hmgcr, Sqle, Scd5, Hsd17b7 and Dhcr7, are upregulated. It should be noted however that enzymes encoded by these genes and other enzymes in cholesterol biosynthesis are also regulated non-genomically and are subject to post-translational modification with activities controlled by kinases and various sterols (25). It is perhaps significant that Insig1 that encodes a cholesterol sensor is downregulated more than two-fold by ACTH. Insig1 reduces transcriptional activity and promotes degradation of key enzymes involved in cholesterol biosynthesis like HMG-CoA reductase (26).
Adrenal cholesterol is also provided by circulating lipoproteins, predominantly LDL in humans via LDL receptors (Ldlr) and HDL in rodents via scavenger receptors (Scarb1) (9, 11, 27). ACTH increased expression of both Ldlr and Scarb1 as well as Ldlrap1, which facilitates LDL uptake. Apoe, which is also known to affect LDL receptor activity and is highly expressed specifically in the adrenal cortex, was not affected. This contrasts with previous reports on apolipoprotein E protein and mRNA levels (28, 29). However, the gene Lrp8, which encodes a receptor for ApoE was upregulated as was Vldlr; both genes have been associated with the reelin signalling system, which controls neural development and plasticity (30) suggesting an alternative adrenal function. Similarly, Hdlbp, although increased by ACTH treatment and with an affinity for HDL (31), is also known as vigilin, an RNA-binding protein with wider functions that might be independent of steroidogenesis (32).
Cholesterol from circulating lipoproteins is taken up in the form of esters requiring hydrolysis to render free cholesterol for steroidogenesis. Two genes encoding adrenal cholesterol esterases have been identified: hormone-sensitive lipase (Lipe) (11) and more recently neutral cholesterol ester hydrolase (Nceh1) (33). Control of hormone-sensitive lipase activity may be post-translational (34); Lipe expression is not significantly affected in the present study (−1.67, P = 0.1). In contrast, Nceh1 is modestly increased by ACTH treatment (1.82, P < 0.0001). It is notable that genes encoding factors required for intracellular cholesterol trafficking (Npc1, Stx11) (35, 36) were also upregulated.
Under normal conditions, excess cholesterol is stored as esters in lipid droplets. As evidenced previously (28, 12) and here by oil red O staining, the zona fasciculata cells of ACTH-treated mice are deplete of lipid. A pattern of reduced expression of genes involved in cholesterol and lipid metabolism is consistent with this depletion. Plin4 (perilipin 4, a component of the droplet coat) (37) is downregulated as are Acat1 and 2, the transferases that esterify cholesterol. However, these transferases are also known to be allosterically activated by sterols (38). In addition, many genes implicated in the biosynthesis of the lipids, which are co-substrates in cholesterol ester synthesis are downregulated, implying that triglycerides are normally produced locally to maintain cholesterol ester reserves (Supplementary Fig. 2).
A further way of optimising cholesterol supply is to limit non-steroidogenic routes of metabolism. Excretion of cholesterol is via hepatic bile acid synthesis and although the adrenal does not produce significant amounts of bile, genes encoding three genes in bile synthesis are downregulated as is Fxr, a key transcription factor regulating the bile pathway (39). Stard10 that binds phosphatidyl choline and is involved in bile acid metabolism is also downregulated (40). The work of Schroeder and coworkers (41) has implicated sterol carrier protein-2 (Scp2) and caveolin-1 (Cav1) as factors in liver controlling the intracellular distribution, efflux and esterification of cholesterol. It is significant therefore that ACTH downregulated both Scp2 (−1.62-fold, P < 0.001) and Cav1 (−2.37-fold, P < 0.0001) expression.
The first step in steroidogenesis is the mitochondrial side chain cleavage of cholesterol to produce pregnenolone. Cleavage activity is determined by cholesterol uptake across the mitochondrial membrane. Papadopoulos and coworkers have suggested that this involves a complex of five proteins termed a transduceosome (42). Although this hypothesis is controversial (43), of the genes encoding these five proteins, Vdac1 and Prkar1a are upregulated twofold (P < 0.001); Star and Acbd3 are slightly increased (1.2-fold, P < 0.01) and Tspo (peripheral benzodiazepine receptor) expression is unaffected.
The relative change in genes encoding steroidogenic enzymes is modest (<1.3-fold) reaffirming studies that show cholesterol supply is critical in maintaining high corticosteroid output when enzyme expression is non-limiting. However, it is notable that ACTH has profound effects on other genes affecting steroid metabolism. Akr1c18, which encodes a 20α hydroxysteroid dehydrogenase enzyme, was switched on in ACTH-treated adrenals (>80-fold). This is consistent with observations in sheep where circulating levels of dihydroxyprogesterone were increased in ACTH-induced hypertension (15). Previous studies in mouse indicate that adrenal Akrc18 expression is normally high in prepubertal and low in adult males (44). In contrast, expression of Srd5a2 that encodes a 5α steroid reductase, which is normally high in the male mouse adrenal gland (17), was decreased more than 5-fold by ACTH. In general, the activity of these enzymes controls the availability of intermediates in the corticosteroid pathway thereby affecting throughput to biologically active hormonal end products.
A major contribution to steroidogenic capacity is the marked threefold increase in adrenal mass. The morphology of adrenals from ACTH-treated mice indicates that this is due to expansion of the cortex rather than the medulla and is a function of both the size and numbers of parenchymal cells. Histology shows that cell hypertrophy is predominantly a feature of the zona fasciculata. The twofold increase in cross-sectional area with ACTH treatment is perhaps an underestimate since volume expansion is offset by depletion of lipid droplets. Increased expression of Cdkn1a, a marker of cell cycle arrest, suggested cell cycle progression beyond the G1 stage is inhibited in some cells. The close correlation of Cdkn1a expression with other genes (correlation coefficient >0.9, P < 0.0001) reflects changes in growth and de novo protein. Cdkn1c, a gene known to affect adrenal cell turnover in embryonic life (45), was also increased (4.12-fold) but with a pattern of change that did not strongly correlate with that of Cdkn1a (r = 0.62).
BrdU and Ki67 immunostaining demonstrated ACTH caused a sustained stimulation of cell division, which appeared to be initiated in a subcapsular region of the gland with displacement of newly divided cells inwards (21). The majority of hypertrophied cells throughout the cortex were not immunostained suggesting that increased cell size is independent of mitosis. In line with Ki67 staining, expression of Mki67 was also increased as were a number of genes linked to cell proliferation and the cell cycle. Tk1, encoding thymidine kinase, another established marker of proliferation, was increased 2.25-fold (P < 0.0001). As with genes controlling cell hypertrophy, expression of genes associated with proliferation were closely clustered.
Cell number is determined by cell death (apoptosis) as well as division. The term apoptosis was first used in the seminal work of Kerr and coworkers (46) to describe programmed cell death in several experimental models including the effects of ACTH withdrawal on the rat adrenal cortex. Not surprisingly, ACTH infusion in the present experiment had anti-apoptotic effects, which were linked to decreased expression of genes like Casp12. These genes include histocompatibility factors, which previous studies have shown are expressed in inner regions of the cortex with a role in apoptosis (47). Similarly, various components of complement activation that have been shown to play a role in the clearance of apoptotic cells (albeit in non-adrenal cells (48)) were decreased. It should be stressed, however, that the level of apoptosis in a normal gland is low so that an ACTH-induced reduction in apoptosis may be difficult to detect, particularly if cell clearance is efficient (49). Nevertheless, Tunel staining in the present study showed a fivefold decrease with ACTH treatment. This is perhaps an overestimate given that the number of apoptotic cells is expressed relative to cross-sectional area of the cortex, and the cross-sectional area of cells from ACTH-treated adrenals is up to two times greater than controls.
The diversity of cellular effects underlying the response to chronic ACTH treatment is reflected in the range of genes involved in signalling. ACTH receptor regulation of adrenal activity is mediated through the cyclic AMP cascade (50) but few of the genes involved are affected. Adenylate cyclase isoforms are generally downregulated and Pde8b, which is negatively associated with steroidogenic activity (51), was upregulated by ACTH treatment. Genes involved in protein kinase A activity, which are mutated in some patients with ACTH-independent Cushings (18) were upregulated as was the protein kinase A anchor protein Akap2. The upregulated cluster was enriched with genes implicated in processes downstream of the cyclic AMP cascade including genes for transcription factors (Nr5a1, Creb3l2), for cell proliferation and hypertrophy (Rras2, Cdkn1a, Rrcc2, Igflr, Shmt1) and for snare proteins (Stx11). Genes like Prkcd, Srxn1 and Inha are known to be involved in steroidogenic control (52, 53, 54), whereas the function of others (Gucalb, Fam161a) are yet to be defined.
Analysis of downregulated signalling genes suggest secondary events. For example, Rgs4, Kcnk1, Camkk1, Shh and Prkg2 have been linked to aldosterone/zona glomerulosa functions (55, 56, 57, 58, 59, 60). Insm1, Rgs11 and Kcnq2 are associated with neuronal/adrenal medulla tissue (61, 62, 63) and expression of Irs1, Atp2b1 and Scn3b are controlled by glucocorticoids (64, 65, 66, 67).
In summary, a large number of adrenal genes are differentially affected by chronic ACTH treatment. Upregulated genes include those implicated in cell division and cell hypertrophy as well as those promoting cholesterol supply from de novo synthesis and uptake from the circulation. Downregulated genes are those involved in apoptosis, intracellular storage of cholesterol esters and non-steroidogenic metabolism of cholesterol. Preliminary observations indicate that genes controlling each of these different processes are co-ordinately regulated. Theoretically, this information could be used to target genes to manipulate glucocorticoid output or ACTH responsiveness. For example, the adverse effects of glucocorticoid hormones could be ameliorated in patients with Cushing’s disease due to excess pituitary ACTH or Cushing’s syndrome caused by ectopic ACTH production. It is important, however, to reiterate that many genes that are transcriptionally regulated by ACTH encode factors which are subject to non-genomic control. Finally, excessively high glucocorticoid hormone levels may, secondarily, affect gene expression in the zona glomerulosa and the adrenal medulla with consequences for aldosterone and catecholamine synthesis.
Supplementary Material
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research.
Funding
This work was supported by the Medical Research Council (Programme Grants G05000026, G0701528). The British Heart Foundation provided additional support.
Author contribution statement
Conception: C J K, J J M, L J M, D R D; Execution: R I M, D R D, L J M, X Z, N W, C C, C J K; Interpretation: D R D, M A B, R I M, J J M, L J M, C J K; Manuscript preparation: R I M, L J M, M A B, J J M, D R D, C J K.
Acknowledgments
The authors thank Mike Millar and Melanie McMillan for expert help with histological techniques.
References
- 1.Pearlmutter AF, Rapino E, Saffran M. Comparison of steroidogenic effects of cAMP and dbcAMP in the rat adrenal gland. Endocrinology 1973. 92 679–686. ( 10.1210/endo-92-3-679) [DOI] [PubMed] [Google Scholar]
- 2.Walker JJ, Spiga F, Gupta R, Zhao Z, Lightman SL, Terry JR. Rapid intra-adrenal feedback regulation of glucocorticoid synthesis. Journal of the Royal Society Interface 2015. 12 20140875 ( 10.1098/rsif.2014.0875) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Artemenko IP, Zhao D, Hales DB, Hales KH, Jefcoate CR. Mitochondrial processing of newly synthesized steroidogenic acute regulatory protein (StAR), but not total StAR, mediates cholesterol transfer to cytochrome P450 side chain cleavage enzyme in adrenal cells. Journal of Biological Chemistry 2001. 276 46583–46596. ( 10.1074/jbc.M107815200) [DOI] [PubMed] [Google Scholar]
- 4.Clark BJ. ACTH action on star biology. Frontiers in Neuroscience 2016. 10 547 ( 10.3389/fnins.2016.00547) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simpson ER, Waterman MR. Regulation by ACTH of steroid hormone biosynthesis in the adrenal cortex. Canadian Journal of Biochemistry and Cell Biology 1983. 61 692–707. ( 10.1139/o83-088) [DOI] [PubMed] [Google Scholar]
- 6.Miller WL, Bose HS. Early steps in steroidogenesis: intracellular cholesterol trafficking. Journal of Lipid Research 2011. 52 2111–2135. ( 10.1194/jlr.R016675) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ness GC. Physiological feedback regulation of cholesterol biosynthesis: Role of translational control of hepatic HMG-CoA reductase and possible involvement of oxylanosterols. Biochimica et Biophysica Acta 2015. 1851 667–673. ( 10.1016/j.bbalip.2015.02.008) [DOI] [PubMed] [Google Scholar]
- 8.Brown MS, Kovanen PT, Goldstein JL. Receptor-mediated uptake of lipoprotein-cholesterol and its utilization for steroid synthesis in the adrenal cortex. Recent Progress in Hormone Research 1979. 35 215–257. [DOI] [PubMed] [Google Scholar]
- 9.Gwynne JT, Strauss JF., 3rd. The role of lipoproteins in steroidogenesis and cholesterol metabolism in steroidogenic glands. Endocrine Reviews 1982. 3 299–329. ( 10.1210/edrv-3-3-299) [DOI] [PubMed] [Google Scholar]
- 10.Beckett GJ, Boyd GS. Purification and control of bovine adrenal cortical cholesterol ester hydrolase and evidence for the activation of the enzyme by a phosphorylation. European Journal of Biochemistry 1977. 72 223–233. ( 10.1111/j.1432-1033.1977.tb11243.x) [DOI] [PubMed] [Google Scholar]
- 11.Kraemer FB. Adrenal cholesterol utilization. Molecular and Cellular Endocrinology 2007. 265–266 42–45. ( 10.1016/j.mce.2006.12.001) [DOI] [PubMed] [Google Scholar]
- 12.Thorngate FE, Strockbine PA, Erickson SK, Williams DL. Altered adrenal gland cholesterol metabolism in the apoE-deficient mouse. Journal of Lipid Research 2002. 43 1920–1926. ( 10.1194/jlr.M200205-JLR200) [DOI] [PubMed] [Google Scholar]
- 13.Lehoux JG, Fleury A, Ducharme L. The acute and chronic effects of adrenocorticotropin on the levels of messenger ribonucleic acid and protein of steroidogenic enzymes in rat adrenal in vivo. Endocrinology 1998. 139 3913–3922. ( 10.1210/endo.139.9.6196) [DOI] [PubMed] [Google Scholar]
- 14.Ye P, Kenyon CJ, Mackenzie SM, Nichol K, Seckl JR, Fraser R, Connell JM, Davies E. Effects of ACTH, dexamethasone, and adrenalectomy on 11beta-hydroxylase (CYP11B1) and aldosterone synthase (CYP11B2) gene expression in the rat central nervous system. Journal of Endocrinology 2008. 196 305–311. ( 10.1677/JOE-07-0439) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butkus A, Coghlan JP, Denton DA, Scoggins BA. The effect of ACTH on the plasma concentrations of the ‘hypertensinogenic’ steroids, 17 alpha-hydroxyprogesterone and 17 alpha,20 alpha-dihydroxy-4-pregnen-3-one in sheep. Journal of Steroid Biochemistry 1985. 22 321–323. ( 10.1016/0022-4731(85)90433-9) [DOI] [PubMed] [Google Scholar]
- 16.Al-Dujaili EA, Kenyon CJ, Nicol MR, Mason JI. Liquorice and glycyrrhetinic acid increase DHEA and deoxycorticosterone levels in vivo and in vitro by inhibiting adrenal SULT2A1 activity. Molecular and Cellular Endocrinology 2011. 336 102–109. ( 10.1016/j.mce.2010.12.011) [DOI] [PubMed] [Google Scholar]
- 17.Luu-The V, Pelletier G, Labrie F. Quantitative appreciation of steroidogenic gene expression in mouse tissues: new roles for type 2 5alpha-reductase, 20alpha-hydroxysteroid dehydrogenase and estrogen sulfotransferase. Journal of Steroid Biochemistry and Molecular Biology 2005. 93 269–276. ( 10.1016/j.jsbmb.2005.01.003) [DOI] [PubMed] [Google Scholar]
- 18.Calebiro D, Di Dalmazi G, Bathon K, Ronchi CL, Beuschlein F. cAMP signaling in cortisol-producing adrenal adenoma. European Journal of Endocrinology 2015. 173 M99–M106. ( 10.1530/EJE-15-0353) [DOI] [PubMed] [Google Scholar]
- 19.Zennaro MC, Boulkroun S, Fernandes-Rosa F. An update on novel mechanisms of primary aldosteronism. Journal of Endocrinology 2015. 224 R63–R77. ( 10.1530/JOE-14-0597) [DOI] [PubMed] [Google Scholar]
- 20.Al-Dujaili EA, Mullins LJ, Bailey MA, Andrew R, Kenyon CJ. Physiological and pathophysiological applications of sensitive ELISA methods for urinary deoxycorticosterone and corticosterone in rodents. Steroids 2009. 74 938–944. ( 10.1016/j.steroids.2009.06.009) [DOI] [PubMed] [Google Scholar]
- 21.Chang SP, Morrison HD, Nilsson F, Kenyon CJ, West JD, Morley SD. Cell proliferation, movement and differentiation during maintenance of the adult mouse adrenal cortex. PLoS ONE 2013. 8 e81865 ( 10.1371/journal.pone.0081865) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dunbar DR, Khaled H, Evans LC, Al-Dujaili EA, Mullins LJ, Mullins JJ, Kenyon CJ, Bailey MA. Transcriptional and physiological responses to chronic ACTH treatment by the mouse kidney. Physiological Genomics 2010. 40 158–166. ( 10.1152/physiolgenomics.00088.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Research 2009. 37 1–13. ( 10.1093/nar/gkn923) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freeman TC, Goldovsky L, Brosch M, van Dongen S, Maziere P, Grocock RJ, Freilich S, Thornton J, Enright AJ. Construction, visualisation, and clustering of transcription networks from microarray expression data. PLOS Computational Biology 2007. 3 2032–2042. ( 10.1371/journal.pcbi.0030206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharpe LJ, Brown AJ. Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). Journal of Biological Chemistry 2013. 288 18707–18715. ( 10.1074/jbc.R113.479808) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong XY, Tang SQ, Chen JD. Dual functions of Insig proteins in cholesterol homeostasis. Lipids in Health and Disease 2012. 11 173 ( 10.1186/1476-511X-11-173) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rigotti A, Edelman ER, Seifert P, Iqbal SN, DeMattos RB, Temel RE, Krieger M, Williams DL. Regulation by adrenocorticotropic hormone of the in vivo expression of scavenger receptor class B type I (SR-BI), a high density lipoprotein receptor, in steroidogenic cells of the murine adrenal gland. Journal of Biological Chemistry 1996. 271 33545–33549. ( 10.1074/jbc.271.52.33545) [DOI] [PubMed] [Google Scholar]
- 28.Cheng B, Chou SC, Abraham S, Kowal J. Effects of prolonged ACTH-stimulation on adrenocortical cholesterol reserve and apolipoprotein E concentration in young and aged Fischer 344 male rats. Journal of Steroid Biochemistry and Molecular Biology 1998. 66 335–345. ( 10.1016/S0960-0760(98)00062-4) [DOI] [PubMed] [Google Scholar]
- 29.Prack MM, Nicosia M, Williams DL, Gwynne J. Relationship between apolipoprotein E mRNA expression and tissue cholesterol content in rat adrenal gland. Journal of Lipid Research 1991. 32 1611–1618. [PubMed] [Google Scholar]
- 30.Herz J, Chen Y. Reelin, lipoprotein receptors and synaptic plasticity. Nature Reviews Neuroscience 2006. 7 850–859. ( 10.1038/nrn2009) [DOI] [PubMed] [Google Scholar]
- 31.Fidge NH. High density lipoprotein receptors, binding proteins, and ligands. Journal of Lipid Research 1999. 40 187–201. [PubMed] [Google Scholar]
- 32.Mobin MB, Gerstberger S, Teupser D, Campana B, Charisse K, Heim MH, Manoharan M, Tuschl T, Stoffel M. The RNA-binding protein vigilin regulates VLDL secretion through modulation of Apob mRNA translation. Nature Communications 2016. 7 12848 ( 10.1038/ncomms12848) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohta K, Sekiya M, Uozaki H, Igarashi M, Takase S, Kumagai M, Takanashi M, Takeuchi Y, Izumida Y, Kubota M, et al. Abrogation of neutral cholesterol ester hydrolytic activity causes adrenal enlargement. Biochemical and Biophysical Research Communications 2011. 404 254–260. ( 10.1016/j.bbrc.2010.11.103) [DOI] [PubMed] [Google Scholar]
- 34.Yeaman SJ. Hormone-sensitive lipase – new roles for an old enzyme. Biochemical Journal 2004. 379 11–22. ( 10.1042/bj20031811) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X, Wang J, Coutavas E, Shi H, Hao Q, Blobel G. Structure of human Niemann-Pick C1 protein. PNAS 2016. 113 8212–8217. ( 10.1073/pnas.1607795113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin Y, Hou X, Shen WJ, Hanssen R, Khor VK, Cortez Y, Roseman AN, Azhar S, Kraemer FB. SNARE-Mediated Cholesterol Movement to Mitochondria Supports Steroidogenesis in Rodent Cells. Molecular Endocrinology 2016. 30 234–247. ( 10.1210/me.2015-1281) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kraemer FB, Khor VK, Shen WJ, Azhar S. Cholesterol ester droplets and steroidogenesis. Molecular and Cellular Endocrinology 2013. 371 15–19. ( 10.1016/j.mce.2012.10.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rogers MA, Liu J, Song BL, Li BL, Chang CC, Chang TY. Acyl-CoA:cholesterol acyltransferases (ACATs/SOATs): Enzymes with multiple sterols as substrates and as activators. Journal of Steroid Biochemistry and Molecular Biology 2015. 151 102–107. ( 10.1016/j.jsbmb.2014.09.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Russell DW. Fifty years of advances in bile acid synthesis and metabolism. Journal of Lipid Research 2009. 50 (Supplement) S120–S125. ( 10.1194/jlr.R800026-JLR200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alpy F, Tomasetto C. START ships lipids across interorganelle space. Biochimie 2014. 96 85–95. ( 10.1016/j.biochi.2013.09.015) [DOI] [PubMed] [Google Scholar]
- 41.Schroeder F, Huang H, McIntosh AL, Atshaves BP, Martin GG, Kier AB. Caveolin, sterol carrier protein-2, membrane cholesterol-rich microdomains and intracellular cholesterol trafficking. Subcellular Biochemistry 2010. 51 279–318. ( 10.1007/978-90-481-8622-8_10) [DOI] [PubMed] [Google Scholar]
- 42.Liu J, Rone MB, Papadopoulos V. Protein-protein interactions mediate mitochondrial cholesterol transport and steroid biosynthesis. Journal of Biological Chemistry 2006. 281 38879–38893. ( 10.1074/jbc.M608820200) [DOI] [PubMed] [Google Scholar]
- 43.Selvaraj V, Tu LN, Stocco DM. Crucial role reported for TSPO in viability and steroidogenesis is a misconception. Commentary: conditional steroidogenic cell-targeted deletion of TSPO unveils a crucial role in viability and hormone-dependent steroid formation. Frontiers in Endocrinology 2016. 7 91 ( 10.3389/fendo.2016.00091) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hershkovitz L, Beuschlein F, Klammer S, Krup M, Weinstein Y. Adrenal 20alpha-hydroxysteroid dehydrogenase in the mouse catabolizes progesterone and 11-deoxycorticosterone and is restricted to the X-zone. Endocrinology 2007. 148 976–988. ( 10.1210/en.2006-1100) [DOI] [PubMed] [Google Scholar]
- 45.Nagahama H, Hatakeyama S, Nakayama K, Nagata M, Tomita K, Nakayama K. Spatial and temporal expression patterns of the cyclin-dependent kinase (CDK) inhibitors p27Kip1 and p57Kip2 during mouse development. Anatomy and Embryology 2001. 203 77–87. ( 10.1007/s004290000146) [DOI] [PubMed] [Google Scholar]
- 46.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. British Journal of Cancer 1972. 26 239–257. ( 10.1038/bjc.1972.33) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wolkersdorfer GW, Ehrhart-Bornstein M, Brauer S, Marx C, Scherbaum WA, Bornstein SR. Differential regulation of apoptosis in the normal human adrenal gland. Journal of Clinical Endocrinology and Metabolism 1996. 81 4129–4136. ( 10.1210/jc.81.11.4129) [DOI] [PubMed] [Google Scholar]
- 48.Ogden CA, Elkon KB. Role of complement and other innate immune mechanisms in the removal of apoptotic cells. Current Directions in Autoimmunity 2006. 9 120–142. ( 10.1159/000090776) [DOI] [PubMed] [Google Scholar]
- 49.Carsia RV, Macdonald GJ, Gibney JA, Tilly KI, Tilly JL. Apoptotic cell death in the rat adrenal gland: an in vivo and in vitro investigation. Cell and Tissue Research 1996. 283 247–254. ( 10.1007/s004410050535) [DOI] [PubMed] [Google Scholar]
- 50.Gallo-Payet N. 60 YEARS OF POMC: adrenal and extra-adrenal functions of ACTH. Journal of Molecular Endocrinology 2016. 56 T135–T156. ( 10.1530/JME-15-0257) [DOI] [PubMed] [Google Scholar]
- 51.Horvath A, Giatzakis C, Tsang K, Greene E, Osorio P, Boikos S, Libe R, Patronas Y, Robinson-White A, Remmers E, et al. A cAMP-specific phosphodiesterase (PDE8B) that is mutated in adrenal hyperplasia is expressed widely in human and mouse tissues: a novel PDE8B isoform in human adrenal cortex. European Journal of Human Genetics 2008. 16 1245–1253. ( 10.1038/ejhg.2008.85) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brizuela L, Rabano M, Pena A, Gangoiti P, Macarulla JM, Trueba M, Gomez-Munoz A. Sphingosine 1-phosphate: a novel stimulator of aldosterone secretion. Journal of Lipid Research 2006. 47 1238–1249. ( 10.1194/jlr.M500510-JLR200) [DOI] [PubMed] [Google Scholar]
- 53.Hofland J, de Jong FH. Inhibins and activins: their roles in the adrenal gland and the development of adrenocortical tumors. Molecular and Cellular Endocrinology 2012. 359 92–100. ( 10.1016/j.mce.2011.06.005) [DOI] [PubMed] [Google Scholar]
- 54.Kil IS, Lee SK, Ryu KW, Woo HA, Hu MC, Bae SH, Rhee SG. Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Molecular Cell 2012. 46 584–594. ( 10.1016/j.molcel.2012.05.030) [DOI] [PubMed] [Google Scholar]
- 55.Azizan EA, Lam BY, Newhouse SJ, Zhou J, Kuc RE, Clarke J, Happerfield L, Marker A, Hoffman GJ, Brown MJ. Microarray, qPCR, and KCNJ5 sequencing of aldosterone-producing adenomas reveal differences in genotype and phenotype between zona glomerulosa- and zona fasciculata-like tumors. Journal of Clinical Endocrinology and Metabolism 2012. 97 E819–E829. ( 10.1210/jc.2011-2965) [DOI] [PubMed] [Google Scholar]
- 56.Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, Penton D, Schack VR, Amar L, Fischer E, et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nature Genetics 2013. 45 440–444. ( 10.1038/ng.2550) [DOI] [PubMed] [Google Scholar]
- 57.Nanba K, Chen A, Nishimoto K, Rainey WE. Role of Ca(2+)/calmodulin-dependent protein kinase kinase in adrenal aldosterone production. Endocrinology 2015. 156 1750–1756. ( 10.1210/en.2014-1782) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Romero DG, Zhou MY, Yanes LL, Plonczynski MW, Washington TR, Gomez-Sanchez CE, Gomez-Sanchez EP. Regulators of G-protein signaling 4 in adrenal gland: localization, regulation, and role in aldosterone secretion. Journal of Endocrinology 2007. 194 429–440. ( 10.1677/JOE-07-0153) [DOI] [PubMed] [Google Scholar]
- 59.Spiessberger B, Bernhard D, Herrmann S, Feil S, Werner C, Luppa PB, Hofmann F. cGMP-dependent protein kinase II and aldosterone secretion. FEBS Journal 2009. 276 1007–1013. ( 10.1111/j.1742-4658.2008.06839.x) [DOI] [PubMed] [Google Scholar]
- 60.Walczak EM, Kuick R, Finco I, Bohin N, Hrycaj SM, Wellik DM, Hammer GD. Wnt signaling inhibits adrenal steroidogenesis by cell-autonomous and non-cell-autonomous mechanisms. Molecular Endocrinology 2014. 28 1471–1486. ( 10.1210/me.2014-1060) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anderson GR, Posokhova E, Martemyanov KA. The R7 RGS protein family: multi-subunit regulators of neuronal G protein signaling. Cell Biochemistry and Biophysics 2009. 54 33–46. ( 10.1007/s12013-009-9052-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Etxeberria A, Aivar P, Rodriguez-Alfaro JA, Alaimo A, Villace P, Gomez-Posada JC, Areso P, Villarroel A. Calmodulin regulates the trafficking of KCNQ2 potassium channels. FASEB Journal 2008. 22 1135–1143. ( 10.1096/fj.07-9712com) [DOI] [PubMed] [Google Scholar]
- 63.Wildner H, Gierl MS, Strehle M, Pla P, Birchmeier C. Insm1 (IA-1) is a crucial component of the transcriptional network that controls differentiation of the sympatho-adrenal lineage. Development 2008. 135 473–481. ( 10.1242/dev.011783) [DOI] [PubMed] [Google Scholar]
- 64.Bhargava A, Mathias RS, McCormick JA, Dallman MF, Pearce D. Glucocorticoids prolong Ca(2+) transients in hippocampal-derived H19-7 neurons by repressing the plasma membrane Ca(2+)-ATPase-1. Molecular Endocrinology 2002. 16 1629–1637. ( 10.1210/mend.16.7.0861) [DOI] [PubMed] [Google Scholar]
- 65.Fahmi AI, Forhead AJ, Fowden AL, Vandenberg JI. Cortisol influences the ontogeny of both alpha- and beta-subunits of the cardiac sodium channel in fetal sheep. Journal of Endocrinology 2004. 180 449–455. ( 10.1677/joe.0.1800449) [DOI] [PubMed] [Google Scholar]
- 66.Giorgino F, Almahfouz A, Goodyear LJ, Smith RJ. Glucocorticoid regulation of insulin receptor and substrate IRS-1 tyrosine phosphorylation in rat skeletal muscle in vivo. Journal of Clinical Investigation 1993. 91 2020–2030. ( 10.1172/JCI116424) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Turnbow MA, Keller SR, Rice KM, Garner CW. Dexamethasone down-regulation of insulin receptor substrate-1 in 3T3-L1 adipocytes. Journal of Biological Chemistry 1994. 269 2516–2520. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.