

Abstract
Oxidative stress is a principal mechanism underlying the pathophysiology of neurodegeneration. Therefore, nutritional enhancement of endogenous antioxidant defenses may represent a viable treatment option. We investigated the neuroprotective properties of a unique whey protein supplement (Immunocal®) that provides an essential precursor (cystine) for synthesis of the endogenous antioxidant, glutathione (GSH). Primary cultures of rat cerebellar granule neurons (CGNs), NSC34 motor neuronal cells, or HT22 hippocampal cells were preincubated in medium containing Immunocal and then subsequently treated with agents known to induce oxidative stress. Immunocal protected CGNs against neurotoxicity induced by the Bcl-2 inhibitor, HA14-1, the nitric oxide donor, sodium nitroprusside, CuCl2, and AlCl3. Immunocal also significantly reduced NSC34 cell death due to either H2O2 or glutamate and mitigated toxicity in HT22 cells overexpressing β-amyloid1-42. The neuroprotective effects of Immunocal were blocked by inhibition of γ-glutamyl-cysteine ligase, demonstrating dependence on de novo GSH synthesis. These findings indicate that sustaining GSH with Immunocal significantly protects neurons against diverse inducers of oxidative stress. Thus, Immunocal is a nutritional supplement worthy of testing in preclinical animal models of neurodegeneration and in future clinical trials of patients afflicted by these diseases.
1. Introduction
Oxidative stress and mitochondrial dysfunction are major factors underlying the pathophysiology of several neurodegenerative disorders including Parkinson's disease, Alzheimer's disease, and amyotrophic lateral sclerosis (ALS) [1–4]. For instance, complex I deficiency and the consequent increase in mitochondrial reactive oxygen species (ROS) play a critical role in the death of dopaminergic neurons in Parkinson's disease [5, 6]. In models of Alzheimer's disease, evidence of mitochondrial dysfunction and oxidative stress precedes the deposition of characteristic amyloid beta-plaques during disease progression [7, 8]. In the case of ALS, mutant forms of copper-zinc superoxide dismutase (SOD1), which are collectively responsible for approximately 20% of cases of familial ALS, accumulate at mitochondria and trigger a shift in the redox state of these organelles [9]. The above findings strongly indicate that oxidative stress, particularly at the level of the mitochondria, plays a central role in the neuronal death that underlies a diverse group of neurodegenerative diseases.
Glutathione (GSH) is an endogenous tripeptide antioxidant that plays a key role in preventing oxidative stress, thereby preserving mitochondrial function and averting cellular apoptosis [10]. In many neurodegenerative disorders, GSH levels have been shown to be significantly depleted in patients suffering from these diseases, resulting in a diminished capacity to cope with increases in cellular ROS [11–13]. Indeed, decreases in GSH are often observed to precede other hallmarks of disease pathology, such as complex I deficiency and loss of dopaminergic neurons in Parkinson's disease [14]. Intriguingly, in vitro studies on GSH depletion have demonstrated that decreases in total cellular GSH levels can recapitulate disease pathology. For instance, in a dopaminergic PC12 cell line, deficiencies in GSH synthesis that led to an overall decrease in cellular GSH resulted in complex I inhibition, increased indices of oxidative stress, and deficits in mitochondrial respiration, as seen in cases of Parkinsonism [15]. Similarly, NSC34 motor neuron-like cells stably expressing the human G93A mutant form of SOD1 displayed a significant and selective depletion of mitochondrial GSH content in comparison to parental cells, reminiscent of some forms of familial ALS [16]. GSH depletion in vitro has also been shown to sensitize neurons to oxidative stress and mitochondrial dysfunction, leading to subsequent increases in ROS and apoptotic cell death. This was clearly demonstrated by a study in which primary cortical neurons treated with subtoxic levels of the GSH-depleting agent, buthionine sulfoximine (BSO), underwent apoptosis in the presence of trace amounts of extracellular copper [17]. Similarly, TAR DNA-binding protein-43 (TDP-43) forms cytoplasmic inclusions, which are a hallmark pathology observed in sporadic ALS patients, in cultured neurons subjected to GSH depletion [18]. Collectively, these studies demonstrate a critical role for GSH depletion in disease progression and pathology in multiple neurodegenerative disease states.
Given the prominent relationship between GSH depletion and neurodegeneration, it is not surprising that many studies have been undertaken to determine the neuroprotective effects of bolstering GSH levels through various treatment paradigms. Such treatments include administration of the GSH precursor, N-acetylcysteine (NAC), and GSH-monoethylester (GSH-MEE), a cell permeable form of GSH, and induction of the transcription factor, nuclear factor erythroid 2-related factor-2 (Nrf2), which is involved in transcriptional regulation of γ-glutamyl-cysteine ligase, the rate-limiting enzyme necessary for GSH synthesis [19]. Studies with NAC are extensive and indicate that NAC treatment offers a number of benefits across numerous disease models. For example, NAC demonstrated a significant protective capacity in a rotenone (complex I inhibition) rat model of Parkinson's disease by decreasing ROS generation, sustaining normal GSH levels, and ultimately preventing dopaminergic cell death [20]. In the G93A mutant SOD1 mouse model of familial ALS, NAC delayed the onset of disease-associated motor deficits and significantly extended survival, possibly due to its ability to elevate GSH levels in these animals [21]. Lastly, SAMP8 senescence-accelerated mice, which display many of the pathological features of Alzheimer's disease, demonstrated an increased cognitive performance with NAC treatment as compared to vehicle-treated controls [22]. Another study utilizing GSH-MEE in an MPTP rat model of Parkinson's disease demonstrated that GSH-MEE supplementation is capable of raising GSH levels in the brain when centrally delivered, and this increase in GSH corresponded to partial preservation of striatal dopamine levels [23]. Studies such as this have led to recent clinical trials testing the safety and tolerability of intranasal delivery of GSH to patients with PD [24]. Finally, Nrf2 induction or overexpression has shown similar promise in animal models of Parkinson's, ALS, and Alzheimer's disease. In the MPTP mouse model of Parkinson's disease, overexpression of Nrf2 in astrocytes attenuated the development of a Parkinsonian phenotype [25]. Likewise, astrocytic overexpression of Nrf2 in a mouse model of ALS both delayed onset and increased survival, as did treatment with chemical Nrf2 inducers [26, 27]. Comparatively, lentiviral Nrf2 overexpression caused significant improvements in observed learning deficits in a mouse model of Alzheimer's disease, accompanied by decreased amyloid plaque burden [28]. Cumulatively, these data indicate that treatments aimed at increasing GSH levels in the brain may be a viable option for treatment and prevention of neurodegenerative disease.
However, while existing treatment strategies have shown some promise in this capacity, the efficacy of such treatments is significantly limited by the relatively low stability and bioavailability of compounds such as GSH-MEE and NAC [23, 29]. Moreover, GSH-MEE requires direct injection into the brain for significant effects to be observed, further limiting its efficacy for treatment in human patients [23]. In the current study, we investigated the neuroprotective potential of a nondenatured whey protein supplement, Immunocal, in vitro in several models of oxidative stress. Immunocal has previously been shown to substantially increase blood or lymphocyte GSH levels in patients with HIV infection or cystic fibrosis, respectively, owing to its high concentration of nondenatured whey proteins containing the cysteine precursor, cystine (see Table 1 for composition) [30–32]. Cystine is resistant to trypsin proteolysis and able to travel through the bloodstream to the target cell where it is then readily reduced to two cysteine molecules which can serve as essential precursors for de novo GSH synthesis. In this manner, the stability of Immunocal lends itself to increased bioavailability, such that it can act as a cysteine delivery system. This is significant, as cysteine is spontaneously catabolized in the GI tract and bloodstream, and its supplementation alone can produce toxicity [33]. Additionally, because of its superior stability, the effects of Immunocal are not dependent upon an invasive administration system as is needed for GSH-MEE and have been observed with standard oral dosing regimens. These unique characteristics spurred us to examine the neuroprotective potential of Immunocal.
Table 1.
Immunocal constituents by mass per one packet of supplement (one packet of Immunocal contains approximately 10 g of protein supplement (one serving) in fine powder form and 40 calories per serving).
| Component | Supplement content | Percent of total supplement |
|---|---|---|
| Whey proteins (β-lactoglobulin, immunoglobulin, serum albumin, α-lactalbumin, and lactoferrin) |
8.8–9.2 g | 88–92% |
| Fat | ~0.05 g | <0.5% |
| Lactose | ~0.15 g | <1.5% |
| Minerals (Ca, Na) | ~0.30 g | <3.0% |
| Moisture | 0.5 g | ~5% |
2. Materials and Methods
2.1. Materials
Immunocal was provided by Immunotec Inc. (Quebec, Canada; Table 1). 2-Amino-6-bromo-α-cyano-3-(ethoxycarbonyl)-4H-1-benzopyran-4-acetic acid ethyl ester (HA14-1) and sodium nitroprusside (SNP) were obtained from Calbiochem (San Diego, CA). DL-buthionine-sulfoximine (BSO), 4, 6-diamidino-2-phenylindole (DAPI), Hoechst dye 33258, and a monoclonal antibody against β-tubulin (clone AA2; used at a dilution of 1 : 250) were from Sigma Aldrich Co. LLC (St Louis, MO). FITC-conjugated secondary antibodies were from Jackson Immunoresearch Laboratories (West Grove, PA).
2.2. Cell Culture and Treatment
Rat cerebellar granule neurons (CGNs) were isolated as previously described from 7-day-old Sprague-Dawley rat pups of both sexes [34]. CGNs were seeded on 35 mm diameter plastic dishes coated with poly-L-lysine at an average density of 2.0 × 106 cells/mL in basal modified Eagle's medium containing 10% fetal bovine serum, 25 mM KCl, 2 mM L-glutamine, and penicillin (100 units/mL)/streptomycin (100 μg/mL). Cytosine arabinoside (10 μM) was added to the culture medium 24 h after plating. Experiments were performed after 6 days in culture. In general, cells were pretreated with Immunocal at a concentration of 3.3%, w/v (unless otherwise noted) in serum-free medium for 24 h prior to treatment with the specified insult (i.e., SNP, HA14-1, etc.) for an additional 24 h.
NSC34 cells were maintained in DMEM with high glucose containing 10% fetal bovine serum, 2 mM L-glutamine, and penicillin (100 units/mL)/streptomycin (100 μg/mL). NSC34 cells were preincubated with Immunocal for 24 h prior to exposure to H2O2 or glutamate. For glutamate experiments, NSC34 cells were differentiated by withdrawing serum for 7 days prior to experimentation.
For transient transfection, HT22 mouse hippocampal cells were seeded in 6-well plates at an approximate confluency of 1.0 × 106 cells/mL and then cultured for 24 h in DMEM with low glucose containing 10% fetal bovine serum, 2 mM L-glutamine, and penicillin (100 units/mL)/streptomycin (100 μg/mL). Cells were transfected using lipofection (5 μg DNA/mL, 5 μL lipofectamine/mL) in OptiMEM medium for 4 h with either empty pIRES 2DsRed-Express2 bicistronic vector (Clontech, Mountain View, CA) or vector containing the sequence for amyloid-beta 1-42 (Aβ1-42). Following transfection, OptiMEM medium was replaced with DMEM culture medium, and cells were treated with Immunocal for 24 h. Percent apoptosis was then determined for only transfected (DSRed-positive) cells based on nuclear morphology.
2.3. Cell Viability, Lipid Peroxidation, and Cellular GSH Assay
All assays were performed according to commercially available manufacturer's instructions. GSH/GSSG assay kit was purchased from Oxford Biomedical Research (Oxford, MI). MTT cell viability assay was from BioAssay Systems (Hayward, CA). Malondialdehyde (MDA) lipid peroxidation assay was obtained from OXIS Research Inc. (Foster City, CA).
2.4. Immunofluorescence Microscopy
After treatment, cells were fixed in 4% paraformaldehyde for 1 h, washed once in PBS, and then permeabilized and blocked in 0.2% Triton X-100 and 5% bovine serum albumin (BSA) in PBS. Primary antibody (monoclonal antibody against β-tubulin; clone AA2; used at a dilution of 1 : 250; Sigma Aldrich Co. LLC, St Louis, MO) was diluted in 2% BSA and 0.2% Triton X-100 in PBS, and cells were incubated with primary antibodies for 24 h at 4°C. They were then washed 5 times in PBS and then incubated for 1 h in FITC-conjugated secondary antibody diluted in 2% BSA and 0.2% Triton-X 100 in PBS with DAPI. The cells were washed 5 times with PBS before the addition of antiquench (0.1% p-phenylenediamine in PBS). Images were captured using a Zeiss Axiovert 200 M epifluorescence microscope equipped with Zeiss Axiovision software.
2.5. Statistical Analysis
Each experiment was done in duplicate and repeated a minimum of three times; data are reported as mean ± SEM. Statistical significance was analyzed with a one-way analysis of variance (ANOVA) followed by post hoc Tukey's test.
3. Results
3.1. Immunocal Preserves Cellular GSH and Prevents Apoptosis in CGNs Exposed to the Bcl-2 Inhibitor, HA14-1
Initially, primary CGNs were incubated with 3.3% (w/v) Immunocal for 24 h to assess any potential toxicity that this supplement might induce. Immunocal is composed of five primary cystine- and glutamylcysteine-containing proteins, β-lactoglobulin, immunoglobulin, α-lactalbumin, serum albumin, and lactoferrin (Table 2) [35, 36]. Based upon the relative percentages for each of these four proteins within the whey protein fraction and the number of cystine or glutamylcysteine residues contained within each protein, we calculated the approximate concentration of each of these GSH precursors with which CGNs were treated (Table 3). In general, a 3.3% solution of Immunocal in culture medium contains 85.3 mM cystine and 30 mM glutamylcysteine, both of which have the potential to act as GSH precursors; however, it should be noted that since both precursors are contained within much larger proteins it is unlikely that all cystine and glutamylcysteine molecules are freely available to be utilized in GSH synthesis. Thus, the values calculated in Table 3 for these precursors should be considered as concentrations that could potentially be achieved rather than absolute concentrations.
Table 2.
Cystine [(Cys)2] and glutamylcysteine [Glu-(Cys)2] content of Immunocal whey proteins.
| Whey protein | Molecular mass (kDa) | Percent of protein fraction | Cystine (Cys)2 per molecule | Glu-(Cys)2 per molecule |
|---|---|---|---|---|
| β-Lactoglobulin | 18,400 | 56.3% | 2 | 0 |
| Immunoglobulin | 166,000 | 9.2% | 4 | 0 |
| α-Lactalbumin | 14,200 | 22.8% | 4 | 0 |
| Serum albumin | 66,000 | 11.1% | 17 | 6 |
| Lactoferrin | 77,000 | 0.7% | 17 | 4 |
Table 3.
Cystine [(Cys)2] and glutamylcysteine [Glu-(Cys)2] content of Immunocal in preincubation culture medium (3.3%, w/v final concentration).
| Whey protein | Total molecules per mL | Total number of (Cys)2 per mL | Total number of Glu-(Cys)2 per mL |
|---|---|---|---|
| β-Lactoglobulin | 5.44 × 1014 | 1.09 × 1015 | 0 |
| Immunoglobulin | 9.91 × 1012 | 3.96 × 1013 | 0 |
| α-Lactalbumin | 2.91 × 1014 | 1.17 × 1015 | 0 |
| Serum albumin | 3.01 × 1018 | 5.11 × 1019 | 1.80 × 1019 |
| Lactoferrin | 1.63 × 1016 | 2.76 × 1017 | 6.50 × 1016 |
| Final concentration | — | 85.3 mM | 30.0 mM |
Following Immunocal treatment, cells were fixed and stained with DAPI to analyze nuclear morphology. Cells treated with Immunocal alone displayed nuclear morphology comparable to that of untreated control cells (Figure 1). Moreover, observation under brightfield demonstrated that cells treated with Immunocal maintained a healthy neuronal morphology with intact processes and large somas, comparable to cells that were not supplemented with Immunocal (Figure 1).
Figure 1.
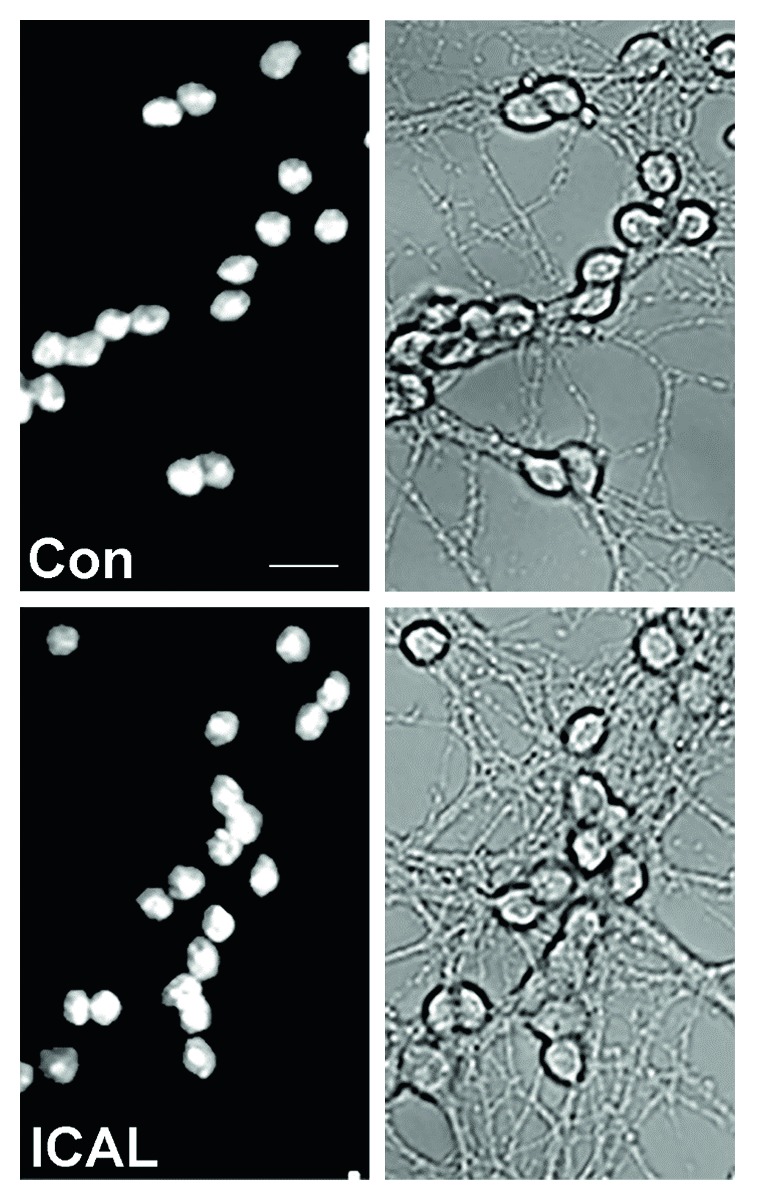
Cells treated with Immunocal display healthy neuronal morphology. Cells were left untreated (a) or treated with Immunocal alone (b) and assessed for overall health and appearance. Left-hand panels are representative images of cell nuclei stained with DAPI. Right-hand panels depict the same fields as viewed under brightfield to assess the state of neuronal processes and soma. Con: control; ICAL: Immunocal. Scale bar, 10 μm.
Having established that Immunocal displayed no overt toxicity to CGNs, cells were next treated with Immunocal and then exposed to the Bcl-2 homology-3 domain (BH3) mimetic, HA14-1. We have previously shown this Bcl-2 inhibitor to induce GSH-sensitive mitochondrial oxidative stress and intrinsic apoptosis in CGNs [37, 38]. HA14-1 induced marked nuclear condensation and microtubule disruption (Figure 2(a)) indicative of apoptosis (Figure 2(b)), while also causing significant depletion of GSH (Figure 2(c)). Immunocal significantly protected CGNs from apoptosis induced by HA14-1 and significantly preserved GSH levels. To confirm that the mechanism of protection was dependent, at least in part, on enhanced GSH synthesis, CGNs were cotreated with Immunocal and the γ-glutamyl-cysteine ligase inhibitor, BSO, which prevents GSH synthesis [39]. Coincubation with Immunocal and BSO for 24 h before HA14-1 treatment completely prevented any protective effect that Immunocal alone displayed against the Bcl-2 inhibitor (Figure 2(b)). Moreover, the capacity of Immunocal to preserve cellular GSH levels upon HA14-1 exposure was eliminated by BSO cotreatment (Figure 2(c)).
Figure 2.

Immunocal preserves cellular GSH and prevents apoptosis in CGNs exposed to the Bcl-2 inhibitor, HA14-1. (a) Representative images of CGNs left untreated (control), treated with HA14-1 (15 μM), or preincubated for 24 h with Immunocal before HA14-1 treatment for further 24 h. Panels from left to right, DAPI (nuclei), β-tubulin, merged images showing β-tubulin (green), and DAPI (blue). Scale bar, 10 μm. (b) Quantification of apoptosis for 4 independent experiments performed as in (a) except some cultures were preincubated with 200 μM BSO as well. Apoptotic cells were those with condensed or fragmented nuclei. Results are shown as mean ± SEM, n = 4. ∗∗∗ indicates p < 0.001 compared to control, ††† indicates p < 0.001 compared to HA14-1, ‡‡‡ indicates p < 0.001 compared to ICAL + HA14-1. (c) CGNs were treated exactly as described in (b). Total cellular GSH was measured as described in Materials and Methods. Data shown represent the percent of control cellular GSH concentration, mean ± SEM, n = 4. ∗∗∗ indicates p < 0.001 compared to control, † indicates p < 0.05 compared to HA14-1, and ‡‡ indicates p < 0.01 compared to ICAL + HA14-1. Significant differences were determined by one-way ANOVA with a post hoc Tukey's test. Con: control; ICAL: Immunocal; BSO: buthionine sulfoximine.
3.2. Immunocal Protects CGNs from CuCl2-Induced Oxidative Damage and Decreases Cellular Lipid Peroxidation
To further investigate the neuroprotective potential of Immunocal in primary neurons, we used copper chloride (CuCl2) as a model of oxidative stress. Copper overload is associated with free radical-induced lipid peroxidation and disruption of mitochondrial complex activity [40, 41]. Immunofluorescence analysis of the microtubule network revealed robust protection from this transition metal in CGNs pretreated with Immunocal (Figure 3(a)). Quantification of apoptotic cells revealed that there was a significant reduction in CGN apoptosis with Immunocal pretreatment compared to CGNs treated with CuCl2 alone (Figure 3(b)). The antioxidant effect of Immunocal was confirmed with a lipid peroxidation assay which revealed a significant decrease in malondialdehyde content in CGNs pretreated with Immunocal (Figure 3(c)).
Figure 3.

Immunocal decreases CuCl2-induced apoptosis and lipid peroxidation in CGNs. (a) Representative images of CGNs left untreated (control), treated with CuCl2 (50 μM), or preincubated with Immunocal for 24 h before CuCl2 treatment for further 24 h. Immunofluorescence shows β-tubulin (green) and DAPI (blue). Scale bar, 10 μm. (b) Quantification of apoptosis for 4 independent experiments performed as in (a). Results are shown as mean ± SEM, n = 4. ∗∗ indicates p < 0.01 compared to control and †† indicates p < 0.01 compared to CuCl2. (c) Cellular lipid peroxidation (malondialdehyde (MDA)) was measured as described in Materials and Methods. Results are shown as mean ± SEM, n = 5. ∗∗ indicates p < 0.01 compared to control, ††† indicates p < 0.001 compared to CuCl2. Con: control; ICAL: Immunocal.
3.3. Immunocal Protects CGNs Exposed to Sodium Nitroprusside- (SNP-) Generated Nitric Oxide Species and from AlCl3-Induced Neurotoxicity
SNP is a nitric oxide donor that causes dissipation of the mitochondrial membrane potential and enhanced generation of mitochondrial ROS in cortical neurons and CGNs [42, 43]. As expected, nitric oxide species generated by SNP caused overt apoptotic cell death in CGNs which was significantly mitigated by pretreatment with Immunocal (Figure 4(a)). Apoptotic cell counts confirmed that there was significant neuroprotection in CGNs pretreated with Immunocal, decreasing apoptosis by approximately 80% (Figure 4(b)). An MTT cell viability assay demonstrated similar results and showed that mitochondrial viability was also significantly preserved in Immunocal-pretreated cells, compared to CGNs treated with SNP alone (Figure 4(c)).
Figure 4.

Immunocal preserves CGN viability and protects from apoptosis after exposure to SNP. (a) Representative images of CGNs left untreated (control), treated with SNP (100 μM), or preincubated with Immunocal for 24 h before SNP treatment for further 24 h. Immunofluorescence shows β-tubulin (green) and DAPI (blue). Scale bar, 10 μm. (b) Quantification of apoptosis for 5 independent experiments performed as in (a). Results are shown as mean ± SEM, n = 5. (c) MTT cell viability was measured as described in Materials and Methods. Results are shown as mean ± SEM, n = 3. For (b) and (c), ∗∗∗ indicates p < 0.001 compared to control, and ††† indicates p < 0.001 compared to SNP. Con: control; ICAL: Immunocal.
Aluminum is a neurotoxic metal that impairs mitochondrial structure and function in neural cells exposed in vitro and in vivo [44, 45]. Aluminum chloride- (AlCl3-) induced toxicity in CGNs was characterized by nuclear condensation and marked disruption of the microtubule network; these effects were markedly decreased in CGNs pretreated with Immunocal (Figure 5(a)). To confirm that this protection was due to cysteine supplementation, and not metal chelation, we removed the Immunocal after the pretreatment period and washed the CGNs with serum-free media before treating with AlCl3. Under these conditions, we still observed a significant reduction in apoptosis compared to CGNs treated with AlCl3 alone (Figure 5(b)).
Figure 5.

Immunocal protects CGNs from AlCl3-induced toxicity. (a) Representative images of CGNs left untreated (control), treated with AlCl3 (10 μM), or preincubated with Immunocal for 24 h before AlCl3 treatment for further 48 h. Panels from left to right, DAPI (nuclei), β-tubulin, and merged image showing β-tubulin (green), and DAPI (blue). Scale bar, 10 μm. (b) CGN apoptosis was quantified for 4 independent experiments as described in (a). Results are shown as mean ± SEM, n = 4. ∗∗∗ indicates p < 0.001 compared to control, and †† indicates p < 0.01 compared to AlCl3. Con: control; ICAL: Immunocal.
3.4. Immunocal Protects NSC34 Motor Neuron-Like Cells from H2O2 and Glutamate/Glycine-Induced Excitotoxicity
H2O2-mediated cell death is a classic model of ROS toxicity in neuronal systems, as it generates free radicals that are implicated in neurodegeneration [46]. As expected, ROS generated by H2O2 caused an overt loss of viability in NSC34 cells, which was significantly mitigated by pretreatment with Immunocal. An MTT cell viability assay demonstrated that mitochondrial viability was preserved in Immunocal-pretreated cells in a dose-dependent manner, compared to NSC34 cells treated with H2O2 alone (Figure 6(a)). Incubation with Immunocal alone had no significant adverse effect on NSC34 cell viability assessed by MTT assay (data not shown).
Figure 6.

Immunocal protects NSC34 cells from H2O2 and glutamate/glycine-induced excitotoxicity. (a) Cell survival was quantified with MTT cell viability assay for 5 independent experiments in undifferentiated NSC34 left untreated (control), treated with H2O2 (250 μM), or preincubated with Immunocal for 24 h before H2O2 treatment for further 24 h. Results are shown as mean ± SEM, n = 5. ∗∗ indicates p < 0.01 compared to control, † indicates p < 0.05 compared to H2O2, and †† indicates p < 0.01 compared to H2O2. (b) Representative images showing morphological differences between undifferentiated (wildtype (WT)) and differentiated (DIFF) NSC34 cells, β-tubulin (green), and DAPI (blue). Scale bar, 10 μm. (c) Cell survival was quantified for 5 independent experiments with an MTT cell viability assay in differentiated NSC34 cells left untreated (control), treated with glutamate/glycine (1 mM/100 μM), or preincubated with Immunocal for 24 h before glutamate/glycine treatment for further 24 h. ∗ indicates p < 0.05 compared to control, and † indicates p < 0.05 compared to glutamate/glycine. Con: control; ICAL: Immunocal; GG: glutamate/glycine.
Glutamate excitotoxicity is thought to play a significant role in several forms of neurodegenerative disease, leading to neuronal damage and cell death through both apoptotic and nonapoptotic mechanisms. NSC34 motor neuron-like cells do not typically express functional glutamate receptors, which are the primary mediators of excitotoxicity. However, if they are exposed to serum withdrawal for 7 days, then they attain a semi-differentiated state and express functional N-methyl-D-aspartate (NMDA) receptors (Figure 6(b)). After this, point cells become sensitive to glutamate excitotoxicity [47]. We observed that exposure to glutamate/glycine caused a significant loss of viability in NSC34 cells differentiated by serum withdrawal. An MTT cell viability assay demonstrated that mitochondrial viability was significantly preserved in Immunocal-pretreated cells in a dose-dependent manner, compared to NSC34 cells treated with glutamate/glycine alone (Figure 6(c)).
3.5. Immunocal Protects HT22 Mouse Hippocampal Cells from Toxicity Induced by Overexpression of Amyloid-Beta Peptide (Aβ1-42)
Aβ1-42 is the major constituent of senile plaques, which form in the brains of Alzheimer's patients, leading to the hypothesis that increased production of this protein from aberrant processing of amyloid precursor protein is a major contributor to neuronal death and disease pathogenesis [48]. HT22 mouse hippocampal cells transfected with Aβ1-42 displayed a marked increase in apoptosis compared to controls transfected with empty vector, indicated by the presence of condensed and fragmented nuclei (Figure 7(a)). Strikingly, this effect was entirely mitigated by treatment with Immunocal, which preserved neuronal viability to an extent similar to that of empty vector controls (Figure 7(b)).
Figure 7.

Immunocal protects HT22 cells from toxicity induced by overexpression of Aβ1-42. (a) Representative images of HT22 cells transfected with either empty vector (IRES) or Aβ1-42. Top panels display colored images showing successful transfection of the cells, and bottom panels display decolorized images of cell nuclei to visualize nuclear condensation. Arrows indicate transfected cells. (b) Quantification of apoptosis for 4 independent experiments performed as in (a). Results are shown as mean ± SEM, n = 4. ∗∗∗ indicates p < 0.001 compared to control, and ††† indicates p < 0.001 compared to cells transfected with Aβ1-42 without Immunocal preincubation. Aβ: amyloid-beta; ICAL: Immunocal.
4. Discussion
Strategies aimed at scavenging ROS, including those that enhance the capacity of endogenous antioxidant defenses like GSH, are actively being investigated as therapeutic approaches for neurodegenerative diseases. In the present study, we assessed the neuroprotective potential of Immunocal, a cystine-rich whey protein supplement, against oxidative stress in vitro. This supplement contains high concentrations of proteins such as serum albumin, alpha-lactalbumin, and lactoferrin, which possess a substantial number of cystine residues in the unique nondenatured preparation. In addition, the direct GSH precursor, glutamylcysteine, is also present in the serum albumin fraction of this supplement. Due to these unique features, Immunocal has been used as a cysteine delivery system to boost GSH levels in individuals diagnosed with diseases for which oxidative stress is a prominent underlying factor [31, 32, 49]. Therefore, Immunocal may be an effective approach to elevate GSH in cases of neurodegeneration for which oxidative stress plays a significant role. To this end, we studied the potential of Immunocal to protect neurons in vitro from a diverse array of oxidative insults, which are not only known to cause oxidative damage and mitochondrial dysfunction but also to imitate some pathogenic factors in neurodegeneration such as diminished Bcl-2 function, increased levels of nitric oxide, or metal ion toxicity (Figure 8).
Figure 8.

Proposed neuroprotective mechanism of Immunocal. Immunocal provides the essential GSH precursor, cystine, which is transported into cerebellar granule neurons via the system xc− antiporter (Sxc−). Upon entry into the cell, cystine is rapidly hydrolyzed to form two cysteine molecules, which are then utilized in the de novo synthesis of GSH by γ-glutamylcysteine ligase (γ-GCL) and glutathione synthase (GSS). Newly synthesized glutathione inhibits oxidation caused by a variety of insults, thereby preventing mitochondrial oxidative stress (MOS) and subsequent induction of apoptosis.
GSH depletion is a widely studied phenomenon in cases of neurodegeneration. Although there are multiple mechanisms by which GSH may be depleted, one involves the downregulation of Bcl-2 expression or function. Increased expression of Bcl-2 leads to enhanced GSH synthesis and decreased GSH efflux from the cell [50, 51]. On the other hand, Bcl-2 knockdown leads to decreased levels of tissue GSH [52]. In the current study, we utilized the Bcl-2 inhibitor, HA14-1, to mimic loss of Bcl-2 function and assess the neuroprotective potential of Immunocal. We have previously shown HA14-1 to decrease the cellular GSH pool with a propensity to affect the mitochondrial GSH pool first and induce mitochondrial oxidative stress and intrinsic apoptosis in CGNs [37, 38]. Under these conditions, Immunocal displayed robust neuroprotection, indicating a capacity to counter the effects of mitochondrial GSH depletion and oxidative stress induced by loss of Bcl-2 function. Moreover, the protective effect of Immunocal against Bcl-2 inhibition is dependent upon de novo GSH synthesis as coincubation of Immunocal with BSO blocked neuroprotection.
Another factor implicated in the pathogenesis of several neurodegenerative diseases is copper toxicity. GSH is known to play a significant role in mitigating copper toxicity by facilitating the transport of copper to proteins that can safely store this toxic metal in the intracellular environment [53]. Depletion of GSH disrupts this important process and sensitizes neuronal cells to copper toxicity through copper-associated ROS generation, even when exposed to only trace amounts of copper [17, 54, 55]. Thus, copper toxicity may be a process that is dependent on GSH depletion, and indeed, increased concentrations of copper and dysregulation of copper homeostasis are observed in several neurodegenerative diseases in which GSH status is reduced, including Alzheimer's disease and models of ALS [54, 56]. In our study, elevation of GSH levels in cultured primary neurons with Immunocal proved to be an effective way to ameliorate the toxic effects of copper treatment by attenuating copper-induced lipid peroxidation, resulting in reduced cell death.
Neuroinflammation, in which microglia and astrocytes take on an inflammatory phenotype and secrete toxic factors such as cytokines and nitric oxide, is another major component of neurodegenerative disease [57, 58]. Induction of nitric oxide synthase (NOS) and subsequent production of nitric oxide is a well-established mechanism by which inflammatory cells trigger neuronal cell death [57]. Markers of nitrosative stress are prevalent in tissues from both Parkinson's and Alzheimer's disease patients, indicating a significant role for nitric oxide in disease pathogenesis [59, 60]. Reactive nitrogen species (RNS) such as nitric oxide promote damage to mitochondrial components, leading to dissipation of mitochondrial membrane potential and further increases in ROS and RNS production [42, 43]. This feed forward cycle ultimately exacerbates inflammatory responses and eventually results in neuronal death. GSH is known to detoxify both ROS and RNS, making it an essential antioxidant and key neuroprotective molecule. Consistent with this, preincubation with Immunocal significantly protected CGNs from toxicity induced by the nitric oxide donor SNP.
The neurotoxic effects of aluminum exposure are well documented, and recently, environmental aluminum and aluminum-containing vaccines have garnered attention as potential causes of neurodegeneration. In general, in vitro exposure of neural cells to aluminum has been shown to result in pronounced alterations in mitochondrial structure and function, leading to marked increases in ROS, reduction of mitochondrial enzyme activity, and cell death [45]. Aluminum also interferes with the activity of NADP-isocitrate at the mitochondria, decreasing the pool of NADPH that is available and necessary for the regeneration of GSH, and thereby decreasing GSH levels [61]. In vivo examination of aluminum neurotoxicity has demonstrated that healthy mice treated with aluminum hydroxide display significant motor deficits and develop pathological features similar to those observed in ALS [62]. These results are notable in that Veterans of the 1990-1991 Gulf War who received vaccines containing aluminum hydroxide adjuvant demonstrate a significant increase in the incidence of ALS, implicating aluminum toxicity as one potential environmental factor in some forms of sporadic ALS [62, 63]. Our experiments clearly demonstrate that Immunocal pretreatment is capable of significantly reducing the degree of neurotoxicity observed with aluminum in CGN cultures. We further confirmed that the protective effects of Immunocal were not due to metal chelation by removing Immunocal-containing media prior to the addition of AlCl3.
To determine if the protective action of Immunocal observed in CGNs was reproducible in other neuronal cell types bearing relevance to neurodegenerative disease, we examined the capacity of this supplement to protect NSC34 motor neuron-like cells from oxidative stress and excitotoxicity. NSC34 cells are a hybrid cell line consisting of spinal cord motor neurons fused with mouse neuroblastoma cells [64]. We first analyzed the ability of Immunocal to protect NSC34 cells from H2O2-induced oxidative stress. Immunocal pretreatment of NSC34 cells dose-dependently attenuated H2O2-induced cell death. We next examined the potential of Immunocal to ameliorate damage induced by excitotoxic insult in NSC34 cells, which were differentiated by prolonged serum withdrawal to induce the expression of NMDA receptors [47]. Excitotoxicity is known to play a prevalent role in the pathogenesis of multiple neurodegenerative diseases, including ALS, and is intimately linked with both oxidative and nitrosative stress [65]. Immunocal pretreatment of differentiated NSC34 motor neuron-like cells significantly reduced the injurious effects of glutamate excitotoxicity in a dose-dependent manner.
Lastly, we evaluated the ability of Immunocal to defend HT22 mouse hippocampal cells from toxicity induced by the overexpression of Aβ1-42. As previously discussed, Aβ1-42 is the primary constituent of senile plaques, one of the hallmarks of Alzheimer's disease pathology. In addition, this protein is also known to accumulate with amyloid precursor protein at mitochondria, leading to significant mitochondrial dysfunction [48]. Indeed, Aβ1-42 accumulation at the mitochondria has been shown to occur both in transgenic mouse models of the disease and in the brains of Alzheimer's patients [66–68]. Our data indicate that pretreatment with Immunocal was able to preserve HT22 hippocampal cell viability to a significant degree, indicating that GSH supplementation may be an effective way to mitigate cell death caused by Aβ1-42-induced toxicity.
5. Conclusions
Immunocal was initially studied for application to clinical disorders of immune system deficiency and cancer as an approach to augment the available GSH pool and increase cellular antioxidant and immune system defenses. More recently, Immunocal has been investigated as a potential treatment for disorders involving the CNS. Oral administration of Immunocal for 45 days has been shown to elevate GSH levels in the brains of healthy, nontransgenic mice by up to 300% compared to casein-treated controls, demonstrating that this supplement is able to directly affect the antioxidant status of tissues in the CNS [69]. Furthermore, we recently demonstrated that oral administration of Immunocal in the G93A mutant SOD1 mouse model of ALS delayed disease onset and preserved grip strength to a significant degree, in comparison to untreated transgenic mice [70]. These therapeutic effects correlated with preservation of both blood and spinal cord GSH levels in comparison to untreated transgenic controls, indicating that Immunocal is able to act directly on the CNS to preserve GSH status in the context of neurodegenerative disease. Based on the above studies and the data shown here, we suggest that Immunocal might hold significant potential as a novel therapeutic approach to bolster GSH levels in neurodegenerative disorders for which the underlying pathology involves significant oxidative stress. In the future, it will be of interest to further assess the therapeutic benefit of GSH precursor supplementation with Immunocal in additional preclinical animal models of neurodegeneration and ultimately in clinical trials of patients afflicted with neurodegenerative disorders.
Acknowledgments
This study was supported in part by funding from Immunotec Inc. (Quebec, Canada).
Abbreviations
- Aβ:
Amyloid beta
- ALS:
Amyotrophic lateral sclerosis
- BH3:
Bcl-2 homology domain-3
- BSA:
Bovine serum albumin
- BSO:
Buthionine sulfoximine
- CGN:
Cerebellar granule neuron
- DAPI:
4,6-Diamidino-2-phenylindole
- γ-GCL:
γ-Glutamyl-cysteine ligase
- GSH:
Glutathione
- GSH-MEE:
Glutathione monoethylester
- GSS:
Glutathione synthase
- HA14-1:
2-Amino-6-bromo-α-cyano-3-(ethoxycarbonyl)-4H-1-benzopyran-4-acetic acid ethyl ester
- ICAL:
Immunocal
- NAC:
N-Acetyl cysteine
- NMDA:
N-Methyl-D-aspartate
- Nrf2:
Nuclear factor erythroid 2-related factor-2
- ROS:
Reactive oxygen species
- RNS:
Reactive nitrogen species
- SOD1:
Copper-zinc superoxide dismutase
- SNP:
Sodium nitroprusside
- Sxc−:
System xc−
- TDP-43:
TAR DNA binding protein-43.
Disclosure
Significant portions of this work have previously been published as part of one of the coauthor's master's thesis (Erika K. Ross, “Nutraceutical Antioxidants and Their Therapeutic Potential in Neurodegeneration” (2012). Electronic Theses and Dissertations. Paper 563). This work has previously been presented in poster form at the Society for Neuroscience Annual Meeting.
Conflicts of Interest
The authors have received funding from Immunotec Inc. (Quebec, Canada) to support the research on the neuroprotective effects of Immunocal.
Authors' Contributions
Erika K. Ross, Vamsi Daliparthi, Aimee N. Winter, Whitney A. Sumner, Danielle M. Kirchhof, Evan Manning, and Heather M. Wilkins had substantial contributions to the conception and design, acquisition, and analysis or interpretation of data. Aimee N. Winter, Erika K. Ross, and Daniel A. Linseman are involved in drafting the article or revising it critically for important intellectual content. Daniel A. Linseman is responsible for the final approval of the version to be published.
References
- 1.Lin M. T., Beal M. F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 2.Smith E. F., Shaw P. J., De Vos K. J. The role of mitochondria in amyotrophic lateral sclerosis. Neuroscience Letters. 2017 doi: 10.1016/j.neulet.2017.06.052. [DOI] [PubMed] [Google Scholar]
- 3.Cassano T., Pace L., Bedse G., et al. Glutamate and mitochondria: two prominent players in the oxidative stress-induced neurodegeneration. Current Alzheimer Research. 2016;13:185–197. doi: 10.2174/1567205013666151218132725. [DOI] [PubMed] [Google Scholar]
- 4.Jiang T., Sun Q., Chen S. Oxidative stress: a major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Progress in Neurobiology. 2016;147:1–19. doi: 10.1016/j.pneurobio.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Dawson T. M., Dawson V. L. Molecular pathways of neurodegeneration in Parkinson’s disease. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- 6.Zuo L., Motherwell M. S. The impact of reactive oxygen species and genetic mitochondrial mutations in Parkinson’s disease. Gene. 2013;532:18–23. doi: 10.1016/j.gene.2013.07.085. [DOI] [PubMed] [Google Scholar]
- 7.Nunomura A., Perry G., Aliev G., et al. Oxidative damage is the earliest event in Alzheimer disease. Journal of Neuropathology & Experimental Neurology. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 8.Leuner K., Müller W. E., Reichert A. S. From mitochondrial dysfunction to amyloid beta formation: novel insights into the pathogenesis of Alzheimer’s disease. Molecular Neurobiology. 2012;46:186–193. doi: 10.1007/s12035-012-8307-4. [DOI] [PubMed] [Google Scholar]
- 9.Ferri A., Cozzolino M., Crosio C., et al. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:13860–13865. doi: 10.1073/pnas.0605814103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franco R., Cidlowski J. A. Apoptosis and glutathione: beyond an antioxidant. Cell Death and Differentiation. 2009;16:1303–1314. doi: 10.1038/cdd.2009.107. [DOI] [PubMed] [Google Scholar]
- 11.Babu G. N., Kumar A., Chandra R., et al. Oxidant-antioxidant imbalance in the erythrocytes of sporadic amyotrophic lateral sclerosis patients correlates with the progression of disease. Neurochemistry International. 2008;52:1284–1289. doi: 10.1016/j.neuint.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 12.Calabrese V., Sultana R., Scapagnini G., et al. Nitrosative stress, cellular stress response and thiol homeostasis in patients with Alzheimer’s disease. Antioxidants & Redox Signaling. 2006;8:1975–1986. doi: 10.1089/ars.2006.8.1975. [DOI] [PubMed] [Google Scholar]
- 13.Pearce R. K. B., Owen A., Daniel S., Jenner P., Marsden C. D. Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. Journal of Neural Transmission. 1997;104:661–677. doi: 10.1007/BF01291884. [DOI] [PubMed] [Google Scholar]
- 14.Garrido M., Tereshchenko Y., Zhevtsova Z. Glutathione depletion and overproduction both initiate degeneration of nigral dopaminergic neurons. Acta Neuropathologica. 2011;121:475–485. doi: 10.1007/s00401-010-0791-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu M., Srinivas B., Kumar J., Subramanian R., Andersen J. Glutathione depletion resulting in selective mitochondrial complex I inhibition in dopaminergic cells is via an NO-mediated pathway not involving peroxynitrite: implications for Parkinson’s disease. Journal of Neurochemistry. 2005;92:1091–1103. doi: 10.1111/j.1471-4159.2004.02929.x. [DOI] [PubMed] [Google Scholar]
- 16.Muyderman H., Hutson P. G., Matusica D., Rogers M. L., Rush R. A. The human G93A-superoxide dismutase-1 mutation, mitochondrial glutathione and apoptotic cell death. Neurochemical Research. 2009;34:1847–1856. doi: 10.1007/s11064-009-9974-z. [DOI] [PubMed] [Google Scholar]
- 17.Du T., Ciccotosto G. D., Cranston G. A., et al. Neurotoxicity from glutathione depletion is mediated by Cu-dependent p53 activation. Free Radical Biology and Medicine. 2008;44:44–55. doi: 10.1016/j.freeradbiomed.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 18.Iguchi Y., Katsuno M., Takagi S., et al. Oxidative stress induced by glutathione depletion reproduces pathological modifications of TDP-43 linked to TDP-43 proteinopathies. Neurobiology of Disease. 2012;45:862–870. doi: 10.1016/j.nbd.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 19.Jaiswal A. K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radical Biology and Medicine. 2004;36:1199–1207. doi: 10.1016/j.freeradbiomed.2004.02.074. [DOI] [PubMed] [Google Scholar]
- 20.Sun L., Gu L., Wang S., Yuan J., Yang H., Zhu J. N-acetylcysteine protects against apoptosis through modulation of group I metabotropic glutamate receptor activity. PLoS One. 2012;7, article e32503 doi: 10.1371/journal.pone.0032503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andreassen O. A., Dedeoglu A., Klivenyi P., Beal M. F., Bush A. I. N-acetyl-L-cysteine improves survival and preserves motor performance in an animal model of familial amyotrophic lateral sclerosis. Neuroreport. 2000;11:2491–2493. doi: 10.1097/00001756-200008030-00029. [DOI] [PubMed] [Google Scholar]
- 22.Farr S. A., Fai Poon H., Dogrukol-Ak D., et al. The antioxidants α-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. Journal of Neurochemistry. 2003;84:1173–1183. doi: 10.1046/j.1471-4159.2003.01580.x. [DOI] [PubMed] [Google Scholar]
- 23.Zeevalk G. D., Manzino L., Sonsalla P. K., Bernard L. P. Characterization of intracellular elevation of glutathione (GSH) with glutathione monoethyl ester and GSH in brain and neuronal cultures: relevance to Parkinson’s disease. Experimental Neurology. 2007;203:512–520. doi: 10.1016/j.expneurol.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mischley L. K., Leverenz J. B., Lau R. C., et al. A randomized, double-blind phase I/IIa study of intranasal glutathione in Parkinson’s disease. Movement Disorders. 2015;30:1696–1701. doi: 10.1002/mds.26351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen P. C., Vargas M. R., Pani A. K., et al. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: critical role for the astrocyte. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:2933–2938. doi: 10.1073/pnas.0813361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vargas M. R., Johnson D. A., Sirkis D. W., Messing A., Johnson J. A. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. The Journal of Neuroscience. 2008;28:13574–13581. doi: 10.1523/JNEUROSCI.4099-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neymotin A., Calingasan N. Y., Wille E., et al. Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radical Biology and Medicine. 2011;51:88–96. doi: 10.1016/j.freeradbiomed.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanninen K., Heikkinen R., Malm T., et al. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:16505–16510. doi: 10.1073/pnas.0908397106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olsson B., Johansson M., Gabrielsson J., Bolme P. Pharmacokinetics and bioavailability of reduced and oxidized N-acetylcysteine. European Journal of Clinical Pharmacology. 1988;34:77–82. doi: 10.1007/BF01061422. [DOI] [PubMed] [Google Scholar]
- 30.Tsai W. Y., Chang W., Chen C., Lu F. Enhancing effect of patented whey protein isolate (Immunocal) on cytotoxicity of an anti-cancer drug. Nutrition and Cancer. 2000;38:200–208. doi: 10.1207/S15327914NC382_9. [DOI] [PubMed] [Google Scholar]
- 31.Micke P., Beeh K. M., Buhl R. Effects of long-term supplementation with whey proteins on plasma glutathione levels of HIV-infected patients. European Journal of Nutrition. 2002;41:12–18. doi: 10.1007/s003940200001. [DOI] [PubMed] [Google Scholar]
- 32.Grey V., Mohammed S. R., Smountasm A. A., Bahlool R., Lands L. C. Improved glutathione status in young adult patients with cystic fibrosis supplemented with whey protein. Journal of Cystic Fibrosis. 2003;2:195–198. doi: 10.1016/S1569-1993(03)00097-3. [DOI] [PubMed] [Google Scholar]
- 33.Bounous G., Gold P. The biological activity of undenatured dietary whey proteins: role of glutathione. Clinical and Investigative Medicine. 1991;14:296–309. [PubMed] [Google Scholar]
- 34.Linseman D. A., Laessig T., Meintzer M. K., et al. An essential role for Rac/Cdc42 GTPases in cerebellar granule neuron survival. The Journal of Biological Chemistry. 2001;276:39123–39131. doi: 10.1074/jbc.M103959200. [DOI] [PubMed] [Google Scholar]
- 35.Baruchel S., Viau G. In vitro selective modulation of cellular glutathione by a humanized native milk protein isolate in normal cells and rat mammary carcinoma model. Anticancer Research. 1996;16:1095–1100. [PubMed] [Google Scholar]
- 36.Baruchel S., Viau G., Olivier R., Bounous G., Wainberg M. A. Nutraceutical modulation of glutathione with a humanized native milk serum protein isolate, Immunocal®: application in AIDS and cancer. In: Montagnier L., Olivier R., Pasquier C., editors. Oxidative Stress in Cancer, AIDS, and Neurodegenerative Diseases. 1, Chapter 42. New York, NY, USA: Marcel Dekker, Inc.; 1998. pp. 447–462. [Google Scholar]
- 37.Zimmermann A. K., Loucks F. A., Schroeder E. K., Bouchard R. J., Tyler K. L., Linseman D. A. Glutathione binding to the Bcl-2 homology-3 domain groove: a molecular basis for Bcl-2 antioxidant function at mitochondria. The Journal of Biological Chemistry. 2007;282:29296–29304. doi: 10.1074/jbc.M702853200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilkins H. M., Marquardt K., Lash L. H., Linseman D. A. Bcl-2 is a novel interacting partner for the 2-oxyglutarate carrier and a key regulator of mitochondrial glutathione. Free Radical Biology and Medicine. 2012;52:410–419. doi: 10.1016/j.freeradbiomed.2011.10.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drew R., Miners J. O. The effects of buthionine sulphoximine (BSO) on glutathione depletion and xenobiotic biotransformation. Biochemical Pharmacology. 1984;33:2989–2994. doi: 10.1016/0006-2952(84)90598-7. [DOI] [PubMed] [Google Scholar]
- 40.Arciello M., Rotilio G., Rossi L. Copper-dependent toxicity in SH-SY5Y neuroblastoma cells involves mitochondrial damage. Biochemical and Biophysical Research Communications. 2005;327:454–459. doi: 10.1016/j.bbrc.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 41.Yurkova I. L., Arnhold J., Fitzl G., Huster D. Fragmentation of mitochondrial cardiolipin by copper ions in the Atp7b−/− mouse model of Wilson’s disease. Chemistry and Physics of Lipids. 2011;164:393–400. doi: 10.1016/j.chemphyslip.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 42.Fukushima T., Koide M., Ago Y., Baba A., Matsuda T. T-817MA, a novel neurotrophic agent, improves sodium nitroprusside-induced mitochondrial dysfunction in cortical neurons. Neurochemistry International. 2006;48:124–130. doi: 10.1016/j.neuint.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 43.Wei T., Chen C., Hou J., Zhao B., Xin W., Mori A. The antioxidant EPC-K1 attenuates NO-induced mitochondrial dysfunction, lipid peroxidation and apoptosis in cerebellar granule cells. Toxicology. 1999;134:117–126. doi: 10.1016/S0300-483X(99)00030-X. [DOI] [PubMed] [Google Scholar]
- 44.Kumar V., Gill K. D. Aluminum neurotoxicity: neurobehavioural and oxidative aspects. Archives of Toxicology. 2009;83:965–978. doi: 10.1007/s00204-009-0455-6. [DOI] [PubMed] [Google Scholar]
- 45.Niu P. Y., Niu Q., Zhang Q. L., et al. Aluminum impairs rat neural cell mitochondria in vitro. International Journal of Immunopathology and Pharmacology. 2005;18:683–689. doi: 10.1177/039463200501800410. [DOI] [PubMed] [Google Scholar]
- 46.Whittemore E. R., Loo D. T., Watt J. A., Cotman C. W. A detailed analysis of hydrogen peroxide-induced cell death in primary neuronal culture. Neuroscience. 1995;67:921–932. doi: 10.1016/0306-4522(95)00108-U. [DOI] [PubMed] [Google Scholar]
- 47.Eggett C. J., Crosier S., Manning P., et al. Development and characterization of a glutamate-sensitive motor neuron cell line. Journal of Neurochemistry. 2000;74:1895–1902. doi: 10.1046/j.1471-4159.2000.0741895.x. [DOI] [PubMed] [Google Scholar]
- 48.Mao P., Reddy P. H. Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: implications for early intervention and therapeutics. Biochimica et Biophysica Acta. 2011;1812:1359–1370. doi: 10.1016/j.bbadis.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bounous G., Baruchel S. Whey proteins as a food supplement in HIV-seropositive individuals. Clinical and Investigative Medicine. 1993;16:204–209. [PubMed] [Google Scholar]
- 50.Jang J. H., Surh Y. J. Bcl-2 attenuation of oxidative cell death is associated with upregulation of gamma-glutamylcysteine ligase via constitutive NF-kappaB activation. The Journal of Biological Chemistry. 2004;279:38779–38786. doi: 10.1074/jbc.M406371200. [DOI] [PubMed] [Google Scholar]
- 51.Meredith M. J., Cusick C. L., Soltninassab S., Sekhar K. S., Lu S., Freeman M. L. Expression of Bcl-2 increases intracellular glutathione by inhibiting methionine-dependent GSH efflux. Biochemical and Biophysical Research Communications. 1998;248:458–463. doi: 10.1006/bbrc.1998.8998. [DOI] [PubMed] [Google Scholar]
- 52.Hochman A., Stemin H., Gorodin S., et al. Enhanced oxidative stress and altered antioxidants in brains of Bcl-2 deficient mice. Journal of Neurochemistry. 1998;71:741–748. doi: 10.1046/j.1471-4159.1998.71020741.x. [DOI] [PubMed] [Google Scholar]
- 53.Freedman J. H., Ciriolo M., Peisach J. The role of glutathione in copper metabolism and toxicity. The Journal of Biological Chemistry. 1989;264:5596–5605. [PubMed] [Google Scholar]
- 54.Sayre L. M., Perry G., Harris P. L., Liu Y., Schubert K. A., Smith M. A. In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease: a central role for bound transition metals. Journal of Neurochemistry. 2000;74:270–279. doi: 10.1046/j.1471-4159.2000.0740270.x. [DOI] [PubMed] [Google Scholar]
- 55.White A. R., Cappai R. Neurotoxicity from glutathione depletion is dependent on extracellular trace copper. Journal of Neuroscience Research. 2003;71:889–897. doi: 10.1002/jnr.10537. [DOI] [PubMed] [Google Scholar]
- 56.Trumbull K. A., Beckman J. S. A role for copper in the toxicity of zinc-deficient superoxide dismutase to motor neurons in amyotrophic lateral sclerosis. Antioxidants & Redox Signaling. 2009;11:1627–1639. doi: 10.1089/ars.2009.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Di Filippo M., Chisserini D., Tozzi A., Picconi B., Calabresi P. Mitochondria and the link between neuroinflammation and neurodegeneration. Journal of Alzheimer's Disease. 2010;20:S369–S379. doi: 10.3233/JAD-2010-100543. [DOI] [PubMed] [Google Scholar]
- 58.Yuste J. E., Tarragon E., Campuzano C. M., Ros-Bernal F. Implications of glial nitric oxide in neurodegenerative diseases. Frontiers in Cellular Neuroscience. 2015;9:p. 322. doi: 10.3389/fncel.2015.00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dias C., Lourenco C. F., Ferreiro E., Barbosa R. M., Laranjinha J., Ledo A. Age-dependent changes in the glutamate-nitric oxide pathway in the hippocampus of the triple transgenic model of Alzheimer’s disease: implications for neurometabolic regulation. Neurobiology of Aging. 2016;46:84–95. doi: 10.1016/j.neurobiolaging.2016.06.012. [DOI] [PubMed] [Google Scholar]
- 60.Fernández E., Garcia-Moreno J. M., Martin de Pablos A., Chacón J. May the evaluation of nitrosative stress through selective increase of 3-nitrotyrosie proteins other than nitroalbumin and dominant tyrosine-125/136 nitrosylation of serum α-synuclein serve for diagnosis of sporadic Parkinson’s disease? Antioxidants & Redox Signaling. 2013;19:912–918. doi: 10.1089/ars.2013.5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murakami K., Yoshino M. Aluminum decreases the glutathione regeneration by the inhibition of NADP-iscocitrate dehydrogenase in mitochondria. Journal of Cellular Biochemistry. 2004;93:1267–1271. doi: 10.1002/jcb.20261. [DOI] [PubMed] [Google Scholar]
- 62.Shaw C., Petrik M. S. Aluminum hydroxide injections lead to motor deficits and motor neuron degeneration. Journal of Inorganic Biochemistry. 2009;103:1555–1562. doi: 10.1016/j.jinorgbio.2009.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haley R. W. Excess incidence of ALS in young Gulf War veterans. Neurology. 2003;61:750–756. doi: 10.1212/WNL.61.6.750. [DOI] [PubMed] [Google Scholar]
- 64.Cashman N. R., Durham H. D., Blusztain J. K., et al. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Developmental Dynamics. 1992;194:209–221. doi: 10.1002/aja.1001940306. [DOI] [PubMed] [Google Scholar]
- 65.Lewerenz J., Maher P. Chronic glutamate toxicity in neurodegenerative diseases – what is the evidence? Frontiers in Neuroscience. 2015;9:p. 469. doi: 10.3389/fnins.2015.00469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Caspersen C., Wang N., Yao J., et al. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. The FASEB Journal. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 67.Manczak M., Calkins M. J., Reddy P. H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Human Molecular Genetics. 2011;20:2495–2509. doi: 10.1093/hmg/ddr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Walls K. C., Coskun P., Gallegos-Perez J. L., et al. Swedish Alzheimer mutation induces mitochondrial dysfunction mediated by HSP60 mislocalization of amyloid precursor protein (APP) and beta-amyloid. The Journal of Biological Chemistry. 2012;287:30317–30327. doi: 10.1074/jbc.M112.365890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Song W., Tavitian A., Cressatti M., Galindez C., Liberman A., Schipper H. M. Cysteine-rich whey protein isolate (Immunocal®) ameliorates deficits in the GFAP.HMOX1 mouse model of schizophrenia. Free Radical Biology and Medicine. 2017;110:162–175. doi: 10.1016/j.freeradbiomed.2017.05.025. [DOI] [PubMed] [Google Scholar]
- 70.Ross E. K., Winter A. N., Wilkins H. M., et al. A cystine-rich whey supplement (Immunocal®) delays disease onset and prevents spinal cord glutathione depletion in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. Antioxidants. 2014;3:843–865. doi: 10.3390/antiox3040843. [DOI] [PMC free article] [PubMed] [Google Scholar]