

Abstract
Background
Recent findings suggesting that Abelson helper integration site 1 (AHI1) is involved in non-syndromic retinal disease have been debated, as the functional significance of identified missense variants was uncertain. We assessed whether AHI1 variants cause non-syndromic retinitis pigmentosa (RP).
Methods
Exome sequencing was performed in three probands with RP. The effects of the identified missense variants in AHI1 were predicted by three-dimensional structure homology modelling. Ciliary parameters were evaluated in patient’s fibroblasts, and recombinant mutant proteins were expressed in ciliated retinal pigmented epithelium cells.
Results
In the three patients with RP, three sets of compound heterozygous variants were detected in AHI1 (c.2174G>A; p.Trp725* and c.2258A>T; p.Asp753Val, c.660delC; p.Ser221Glnfs*10 and c.2090C>T; p.Pro697Leu, c.2087A>G; p.His696Arg and c.2429C>T; p.Pro810Leu). All four missense variants were present in the conserved WD40 domain of Jouberin, the ciliary protein encoded by AHI1, with variable predicted implications for the domain structure. No significant changes in the percentage of ciliated cells, nor in cilium length or intraflagellar transport were detected. However, expression of mutant recombinant Jouberin in ciliated cells showed a significantly decreased enrichment at the ciliary base.
Conclusions
This report confirms that mutations in AHI1 can underlie autosomal recessive RP. Moreover, it structurally and functionally validates the effect of the RP-associated AHI1 variants on protein function, thus proposing a new genotype–phenotype correlation for AHI1 mutation associated retinal ciliopathies.
Keywords: AHI1, Jouberin, WD40 domain, non-syndromic retinitis pigmentosa, exome sequencing
Introduction
Retinitis pigmentosa (RP) is a highly heterogeneous inherited retinal dystrophy characterised by progressive degeneration of photoreceptor cells primarily affecting the rods in early stages. Typical early signs of RP are night blindness and progressive loss of peripheral vision followed by visual acuity impairment at later stages and eventual blindness. RP is the most common form of inherited retinal dystrophy with a prevalence of 1 in 4000.1 Over 2800 mutations in 81 genes have been implicated RP.2 Several genes associated with syndromic disease are also now implicated in non-syndromic RP (BBS1, USH2A, CLN3).3–5
Abelson helper integration site 1 (AHI1) was one of the first genes to be associated with Joubert syndrome (JBTS),6 7 a ciliopathy characterised by cerebellar vermis dysplasia accompanied by hypotonia in infancy, developmental delay, abnormal eye movements and irregular respiratory pattern in infancy.8 Other features including retinal dystrophy, late onset nephronophthisis and polydactyly are only detected in a subset of patients and are often associated with the causative gene.8 Mutations in AHI1 are the most common genetic cause of JBTS and are often associated with retinal dystrophy and nephronopthisis as additional features in such patients.9 10
The AHI1 gene encodes the Jouberin protein, comprising a small predicted N-terminal coiled-coil domain, a WD40 repeat containing domain and a C-terminal SH3 domain, with multiple SH3-binding sites. This suggests that Jouberin may function as a scaffold protein, for example in protein complex assembly or cell signalling processes.6 Jouberin expression is ubiquitous, but highest in brain and testis and is regulated during development.7 11 At the subcellular level, Jouberin localises to ring-like structures at the transition zone of the primary cilium.12 Depletion of Ahi1 in vitro by siRNA in mouse kidney cell line IMCD3 causes a defect in ciliogenesis.13 Furthermore, Ahi1 knockout mouse models suffer from retinal degeneration caused by damage/defects in Rab8a-associated intracellular trafficking14 and Wnt-dependent cerebellar development defects.15
Although there has been accumulative evidence supportive of an important function of Jouberin in the retina, only a single study has recently suggested AHI1 as a non-syndromic RP candidate gene based on next-generation sequencing data;16 and in the absence of functional validation, the pathogenicity of the variants reported were questioned.17 Here, we report the identification of three patients with non-syndromic RP with compound heterozygous AHI1 mutations. To examine the pathogenicity of the detected AHI1 variants, we assessed their effects via multiple in silico and in vitro techniques.
Materials and methods
Subjects and clinical evaluation
This study was approved by the institutional review boards of the participating centres and adhered to the tenets of the Declaration of Helsinki. All participants provided informed consent.
A Dutch non-consanguineous family (family A) and two English non-consanguineous families (families B and C) each with one affected person with RP were included in this study (figure 1A). The Dutch patient (A-II:1) and English patients (B-II:2 and C-II:1) belong to larger cohorts of patients with visual impairment on whom Whole Exome Sequencing (WES) was performed. DNA samples of 209 RP probands of mainly Dutch ophthalmic centres were available.
Figure 1.

AHI1 variants detected in three patients with non-syndromic retinitis pigmentosa; (A) pedigrees of three families with AHI1 variants. Filled symbols represent affected individuals, whereas open symbols represent unaffected individuals. The father in family A is deceased, depicted by the crossed box. The AHI1 genotype is depicted below each individual. M1: c.(2174G>A; 2488C>T) (p.(Trp725*; Arg830Trp)); M2: c.2258A>T (p.Asp753Val); M3: c.660delC (p.Ser221Glnfs*10); M4: c.2090C>T (p.Pro697Leu); M5: c.2087A>G (p.His696Arg) and M6: c.2429C>T (p.Pro810Leu). (B) Cross-species alignment of various organism spanning the regions containing the Jouberin amino acids p.D753, p.P697, p.H696 and p.P810.
Ophthalmological examinations were carried out on several occasions and the standard clinical examination included best-corrected visual acuity measurement using Snellen visual acuity charts and ophthalmoscopy. Colour vision was analysed using Hardy-Rand-Rittler and Ishihara colour vision test. Retinal fundus imaging was obtained by ultra-widefield confocal scanning laser image (Optos plc, Dunfermline, UK), 55 degree fundus autofluorescence (FAF) imaging (Spectralis, Heidelberg Engineering, Heidelberg, Germany) and Spectralis optical coherence tomography (OCT). A full field and pattern electroretinogram (ERG and PERG) were obtained in patients B-II:2 and C-II:1 using gold foil electrodes and in patient A-II:1 using DTL electrodes according to International Society for Clinical Electrophysiology of Vision (ISCEV) standards.18 19 Visual filed testing was performed using a Humphrey Field Analyser (Carl Zeiss Meditec AG, Jena, Germany) or by Goldmann perimetry.
DNA analysis
Genomic DNA was isolated from leucocytes following standard procedures. Individual A-I:1 was deceased and DNA was extracted from paraffin-stored material.
WES of patient A-II:1 was performed at Beijing Genomics Institute (BGI)-Europe (Copenhagen, Denmark) using Agilent’s SureSelect XT Human All Exon V4 (Agilent Technologies, Santa Clara, California, USA), followed by sequencing on a HiSeq2000 next-generation sequencer (Illumina, San Diego, California, USA). At BGI, sequence reads were aligned to the human reference genome (GrCH37/hg19) using Burrows-Wheeler Aligner software and variant calling using Genome Analysis Toolkit (GATK) software. Variants were annotated using an in-house strategy. WES of patient B-II:2 and C-II:1 was performed at AROS Applied Biotechnology using a solution-phase Agilent SureSelect 38 Mb exome capture (SureSelect Human All Exon Kit; Agilent Technologies, Santa Clara, USA) and the Illumina HiSeq2000 sequencer (Illumina). Reads were aligned to the hg19 human reference sequence (build GRCh37) using Novoalign V.2.08. Calling was performed using GATK software. Variants were annotated using Annotate Variation (ANNOVAR) based on Ensembl gene and transcript definitions.
AHI1 exons were amplified from genomic DNA and Sanger sequenced using primers listed in supplementary tables. Sanger sequencing was used to confirm the detected variants to perform segregation analysis in family members, to exclude any additional variants in coding exons with a coverage below 10× in the WES data and to screen additional patients with RP for variants.
jmedgenet-2016-104200supp002.docx (39.2KB, docx)
Modelling of WD40 domain of Jouberin
The WD40 repeat protein structure predictor (WDSP) Jouberin WD40 domain structure (retrieved on June 2016 from http://wu.scbb.pkusz.edu.cn/wdsp/getResults?id=2328) was used to model the effect of the four ‘non-syndromic’ and seven ‘syndromic’ missense mutations and the retinal modifier on the structure of the WD40 domain.20–22 The protein model was loaded into the Yet Another Scientific Artificial Reality Application (YASARA) software, and images of the wild-type and mutant amino acid were made using the YASARA software.23
cDNA constructs
A synthetic cDNA construct of full-length human AHI1 isoform 1 (Accession number AM393493), gateway adapted with pENTR201 as a backbone vector, was purchased from Thermo Fisher Scientific (Catalogue no. FL1002). Site-directed mutagenesis, using Phusion (R) High-fidelity DNA polymerase (Bioke, the Netherlands) was performed to obtain the reference AHI1 coding sequence (NM001134831.1) and to introduce four missense variants (c.2090C>T, c.2258A>T (current study), c.2156A>G and c.2173T>C (Joubert syndrome variants)). All resulting sequences were validated by Sanger sequencing. All N-terminal monomeric Red Fluorescent Protein (mRFP) fusion expressing constructs were generated using the Gateway cloning system (Life Technologies) and pDest-733 as the Destination expression vector (as previously described24). Primer sequences are available on request. The expression of all full-length mRFP fusion proteins in HEK 293 T cells was validated by immunoblotting supplementary figure S5) using standard procedures, with anti-RFP antibodies (rabbit, ab62341 (Abcam); 1:1000 dilution in phosphate-buffered saline (PBS)) to stain recombinant mRFP, and antialpha tubulin antibodies (mouse, ab7291(Abcam); 1:2000 dilution in PBS) to stain endogenous alpha-tubulin as a loading control.
jmedgenet-2016-104200supp001.pdf (1.5MB, pdf)
Immunocytochemistry hTERT-RPE cells (ATCC) were plated at 25%–30% confluency in 12-well format and induced to form cilia by changing to starvation medium (Dulbecco’s Modified Eagle Medium (DMEM)/F12 with 0.2% Fetal Calf Serum (FCS), 1% Pen/Strep and 1% sodium pyruvate) after 24 hours. Expression plasmids containing sequences encoding either AHI1 wild-type or mutants were transfected using lipofectamine according to manufacturer’s protocol (Thermo Fisher Scientific). Twenty-four hours post transfection, cells were fixed in 2% PFA for 20 min, permeablised in 1% TritonX100/PBS for 5 min and blocked in 2% Bovine Serum Albumin (BSA)/PBS for 30 min, sequentially. Next, cells were incubated with primary antibody against ARL13B (ProteinTech; dilution 1:1000 in 2% BSA/PBS) for 1 hour and then with secondary goat antirabbit-antibody Alexa Fluor 488 (Molecular Probes; dilution 1:500 in blocking buffer). Finally, cells were mounted on slide with vectashield containing DAPI (Brunschwig, The Netherlands). Slides were imaged using Axio Carl Zeiss Z2 with Apotome at ×63 magnification (Zeiss, Sliedrecht, The Netherlands). The experiment was repeated independently three times.
Fibroblast cells from RP patient A-II:1 (CL14-00043) and control individuals (male child CL10-00010, female adult CL09-00025and female adult 0 43 687) were plated at 25%–30% confluency in 12-well format in standard DMEM with 20% FCS and induced to form cilia by changing to starvation medium (DMEM/F12 with 0.2% FCS, 1% Pen/Strep and 1% Sodium Pyruvate) after 24 hours. Seventy-two hours post starvation, cells were fixed in 2% PFA for 20 min, permeablised in 1% TritonX100/PBS for 5 min and blocked in 2% BSA/PBS for 30 min, sequentially. Next, the staining procedure was the same as described above for hTERT-RPE cell with primary antibodies to the following proteins: Acetylated alpha-tubulin (mouse monoclonal, Life Technologies, dilution 1:1000), ARL13B (rabbit polyclonal, ProteinTech, dilution 1:500), RPGRIP1L (guinea pig polyclonal, SNC040, dilution 1:500),25 IFT88 (rabbit polyclonal, ProteinTech, dilution 1:100) and WDR19 (mouse polyclonal, Abnova, dilution 1:100). And these following secondary antibodies were used: antimouse IgG Alexa Fluor 568 and 488 (1:500, Life Technologies), antirabbit IgG Alexa Fluor 568 and 488 (1:500, Molecular Probes) and anti-guinea pig IgG Alexa Fluor 647 (1:500, Molecular Probes). Finally, cells were mounted on slide with vectashield containing 4’,6-Diamidino-2-Phenylindole (DAPI) (Brunschwig, The Netherlands). Slides were imaged using Axio Carl Zeiss Z2 with Apotome at ×63 magnification (Zeiss, Sliedrecht, The Netherlands).
Measurement and statistical analysis of signal intensity and ciliary length
For every AHI1 variant of each experiment, we measured the signal intensity of at least 25 cells. The mRFP signal channel was used to mark and measure the signal of Jouberin in the whole cell area. Both mRFP and green channel (ARL13B) were used to define and measure the signal of Jouberin at the base of the cilium. Corrected total cell fluorescence values were measured by intensity density, subtracted by the fluorescence signal of the background.
Cells with overexpression of different AHI1 proteins were evaluated for cilium formation and cilium length using Fiji Is Just ImageJ (FIJI). The mRFP and green channel for ARL13B were used to define cells with overexpression of AHI1 proteins and examine cilium formation. For cilium length analysis, the green channel (ARL13B staining) was processed using autothreshold. Finally, two plug-ins from FIJI (skeletonise and analyse skeleton, sequentially) were used to measure cilium length.
Pair-wise comparisons between wild-type versus mutants were analysed with unpaired two-tailed t-test using InStat. Significance of pair-wise comparisons indicated as: ns,not significant, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Results reported are from three independent biological replicates.
Results
Detection of AHI1 variants in non-syndromic retinal dystrophy patients
Family A
Patient A-II:1 was diagnosed with RP, prior screening of known recessive and dominant RP-associated variants via microarray (Asper Biotech) was negative and subsequently, WES was performed. The bases in the targeted region of the WES had a mean coverage of 103× and 94% of the bases in the targeted region had a coverage >30×, the WES yielded 1 22 737 variants. Variants in a panel of 299 genes involved in non-syndromic and syndromic visual impairment were examined, yielding 2041 variants, of which 11 were coding or in the splice site region (20 bp of the splice acceptor and donor site) and had a frequency of ≤1% in Single-Nucleotide Polymorphism database (dbSNP) and in an in-house exome database, containing results of 5036 exomes (10 072 alleles) of primarily Dutch individuals. The only plausible disease causing variants were two heterozygous variants in AHI1 c.2174G>A; p.Trp725* and c.2258A>T; p.Asp753Val (online supplementary table S2). This patient had an additional third variant in the AHI1 gene c.2488C>T; p.Arg830Trp; however, due the high frequency of this variant in control populations (3.85% Exome Aggregation Consortium (ExAc) (0.3.1)), it was considered not to be disease causing. Furthermore, segregation analysis determined that the c.2488C>T variant is on the same haplotype as the non-sense variant c. 2174G>A located upstream (figure 1A).
The c.2174G>A; p.Trp725* nonsense variant occurs in exon 16 of 29 and would be predicted to undergo nonsense-mediated decay (NMD), thus representing a null mutation. The missense mutation, c.2258A>T; p.Asp753Val, was previously unreported in JBTS and absent from the ExAC data set and from the 10 072 alleles of in-house exomes of primarily Dutch individuals and alters a highly conserved amino acid within the WD40 repeat region (figure 1B) and was predicted damaging in silico (online supplementary table S3).
Family B
Patient B-II:2 underwent WES to determine the genetic basis of her RP. The bases in the targeted region of the WES had a mean coverage of 90×. WES revealed 22 554 variants that passed standard quality filter, of which 394 had a minor allele frequency (MAF) ≤0.005 in the National Heart Lung Blood Institute (NHLBI) GO Exome Sequencing Project (URL: http://evs.gs.washington.edu/EVS/ release 13 May 2013) and an in-house exome sequencing cohort of over 4000 individuals (UCLex) and were coding or in the splice region (5 bp of the splice acceptor and donor site). Regarding autosomal recessive inheritance, seven genes had ≥2 variants, of which, AHI1 was the most plausible candidate (online supplementary table S2). Segregation analysis confirmed the compound heterozygous genotype (figure 1A).
The frameshift variant c.660delC; p.Ser221Glnfs*10 occurs in exon 7 of 29 and is expected to represent a null allele due to NMD. The c.2090C>T; p.Pro697Leu missense variant was also absent from in-house and ExAC variant databases and alters a highly conserved amino acid within the WD40 repeat region (figure 1B), and was predicted damaging in silico (online supplementary table S3).
Family C
Patient C-II:1 underwent WES to determine the genetic basis of her RP. The bases in the targeted region of the WES had a mean coverage of 38×. WES revealed 23 003 variants that passed a standard quality filter, of which 936 had an MAF ≤0.005 in the NHLBI GO Exome Sequencing Project and an in-house exome sequencing cohort of over 4000 individuals (UCLex), and were coding or in the splice region (5 bp splice acceptor and donor site). Regarding autosomal recessive inheritance, 15 genes had ≥2 variants, of which, AHI1 was the most plausible candidate (online supplementary table S2). Segregation analysis confirmed the compound heterozygous genotype (figure 1A). Both the c.2087A>G; p.His696Arg and c.2429C>T; p.Pro810Leu missense variants were absent from in-house and ExAC variant databases, alter highly conserved amino acids in the WD40 repeat region (figure 1B), and were predicted damaging in silico (online supplementary table S3).
Additional screening of a cohort of 209 patients with RP
Patients in all three of the above families carried at least one AHI1 allele with a missense mutation in the WD40 domain (WD40 repeat 3, 4 or 5). We hypothesised that non-syndromic RP may be associated with mutations in the WD40 repeat region of AHI1, and therefore screened a cohort of 209 RP probands for variants in exon 16–19 of AHI1 (encompassing WD40 repeat 3 until 6) by Sanger sequencing. No plausible pathogenic variants were identified. Three polymorphisms were observed with MAF too high in control datasets to be considered pathogenic alleles (online supplementary table S4).
Clinical evaluations
Invariably, all patients were developmentally normal at last review ages 38 to 55 years. Specifically, none of the patients had symptoms or signs of cerebellar ataxia or hypotonia. No patient had undergone neuroradiological imaging as there was no clinical indication. All three patients had normal renal investigations comprising renal ultrasound and serum urea, creatinine and electrolytes.
Ophthalmic examinations of patient A-II:1, B-II:2 and C-II:1
Clinical data of patients A-II:1, B-II:2 and C-II:1 are summarised in supplementary tables.
Patient A-II:1 was diagnosed with RP at the age of 53 years. In retrospect, she was aware of dark adaptation problems since the age of 20 years, but experienced night blindness only since the age of 47 years. Despite quite pronounced macular changes on ophthalmoscopy and FAF (figure 2A,B), her visual acuity was still 1.0 and 0.8 at recent examination with preserved outer retinal layers on OCT (figure 2C). Goldmann perimetry revealed an altitudinal defect of the upper quadrants with midperipheral absolute scotomas in the lower quadrants. Full-field ERG showed a barely recognisable rod response (mainly in the right eye) with still relatively well preserved cone derived responses in both eyes.
Figure 2.
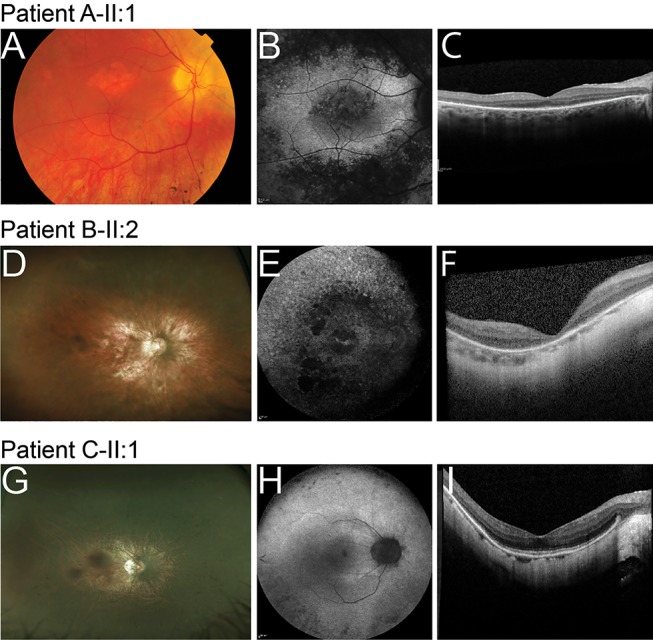
Ophthalmic images of the right eyes of patients with retinitis pigmentosa caused by AHI1 variants. (A, D and G) Fundus photographs; (B, E and H) fundus autofluorescence (FAF) images; (C, F and I) spectral-domain optical coherence tomography images. (A, B and C) images of patient A-II:1; (D, E and F) images of patient B-II:2; (G, H and I) images of patient C-II:1.
Patient B-II:2 developed nyctalopia in her 20s with progressive restriction of visual fields. At the age of 27 years, visual acuity was R 6/36 Snellen, L 6/24 with refractive correction of R −7.75 dioptre sphere (DS) and L −7.50 DS. Vision had deteriorated to hand movements vision in each eye by the age of 33 years. Bilateral cataract surgery was performed and at last review age 39 vision was 1/60 each eye with fields reduced to less than the central 5 degrees. Dilated fundus examination identified pale discs, attenuated vessels and widespread retinal atrophy, without hyperpigmentation in both eyes (figure 2D), and in addition a circular chorioretinal atrophic lesion in the left macula. There was generalised loss of autofluorescence corresponding to atrophic regions, with small islands of central hyperautofluorescence remaining in the macula (figure 2E). OCT demonstrated a degeneration of the outer retina (figure 2F). Electrophysiology at the age of 27 years demonstrated a moderate-to-severe generalised retinal dysfunction involving both rod and cone systems.
Patient C-II:1 developed nyctalopia at the age of 37 years with subjective deterioration over time and the development of restricted peripheral visual fields. At last review at the age of 41 years, VA was R 6/12 and L 6/9 with myopic refractive correction. Colour vision testing demonstrated a mild red-green colour defect (Hardy-Rand-Rittler (HRR) plates). On dilated examination, posterior subcapsular cataracts were present in both eyes. There were myopic fundus changes (tilted discs and peripapillary atrophy) and disc pallor with peripheral intraretinal pigment migration (figure 2G). FAF imaging demonstrated a ring of increased autofluorescence in both maculae, with reduced autofluorescence in the midperipheral retina (figure 2H). OCT imaging demonstrated a preserved central inner segment ellipsoid band (figure 2I). At the age of 38 years, electrophysiology was consistent with a moderate to severe generalised retinal dysfunction of rods slightly more than cones, with the right eye slightly more affected than the left eye.
In silico evaluation of the pathogenicity of the missense variants
To predict the effect of the detected AHI1 variants, we performed three-dimensional structure homology modelling of wild-type and mutant Jouberin. The Jouberin protein contains a WD40 domain, consisting of seven WD40 repeats within its C-terminal half (aa 607–926) and an SH3 domain close to the C terminus (aa 1051–1111). All four missense variants reported herein are located within the WD40 domain (figure 3A). Interestingly, 7 of the 11 missense variants that have been implicated to cause Joubert syndrome are also located in the WD40 domain, indicating the importance of the WD40 domain in the proper function of Jouberin (figure 3A). The only missense variant with a frequency above 1% in the WD40 domain of Jouberin in the ExAc database, the p.Arg830Trp variant, has been described as a retinal modifier in Joubert patient with a non-AHI genetic cause. The predicted secondary structure of the Jouberin WD40 domain was used to model the effect of the four ‘non-syndromic’ and seven ‘syndromic’ missense mutations and the retinal modifier. The p.Thr671Ile, p.His696Arg, p.Asp719Gly, p.Arg723Gln, p.Trp725Arg and p.Ser761Leu most likely disrupt the WD40 structure, because these amino acids changes are at essential positions in the core of their respective WD40 repeat motif (figure 3B and supplementary figure S1). The p.Ile641Asn, p.Pro697Leu, p.Asp753Val, p.Arg830Trp and p.His896Arg are predicted to have a milder effect on the WD40 structure because these amino acid residues are located at the surface of their respective WD40 repeat motif. These changes, however, do have a local effect on the stability of the domain in which they are located (figure 3B and supplementary figure S1). The p.Pro810Leu substitution is predicted to have no clear effect on the WD40 structure, the residue is located at the surface of WD40 repeat 5 and the substitution of the hydrophobic proline with a hydrophobic leucine does not seem to have any effect on the WD40 structure itself (figure 3B). However, the mutated residue might play an important role in the interaction with other proteins.
Figure 3.

Disease-causing missense mutations in Jouberin. (A) Localisation of the missense mutations in Jouberin. Open box depicts coiled coin domain, light grey boxes depict the seven repeats of the WD40 domain and the dark grey box depicts the SH3 domain. Missense variants detected in patients with non-syndromic retinitis pigmentosa are depicted in the upper part, while missense variants detected in JBTS patients are depicted in the bottom part. (B) Predicted effect of the amino acid change of non-syndromic RP (p.His696Arg, p.Pro697Leu, p.Asp753Val and p.Pro810Leu) and JBTS missense variants (p.Asp719Gly and p.Trp725Arg) in a protein homology model of the WD40 domain of Jouberin. In green the wild-type amino acids, in magenta the amino acid changes of a non-syndromic RP-associated missense variants and in cyan the amino acid changes of a JBTS-associated missense variants are depicted. JBTS, Joubert syndrome; RP, retinitis pigmentosa.
RP-associated AHI1 variants do not affect core cilium parameters of fibroblasts
To determine the effect of the identified missense variants on the cilium function of Jouberin, we compared primary cilium formation of patient A-II:1-derived fibroblasts with control fibroblasts. The fibroblasts were cultured in starvation condition to induce ciliogenesis. Approximately 90% of both control and A-II:1 patient-derived fibroblasts were able to form a primary cilium (figure 4A, supplementary figure S2B). The primary cilia length did not differ from controls, that is, approximately 4.88±1.5 µm in patient-derived cells versus 4.83±1.22 µm in controls (figure 4B, supplementary figure S2A,C).
Figure 4.

AHI1 variants do not impair core ciliary parameters in patient’s fibroblasts. Primary cilia from patient with retinitis pigmentosa A-II:1 fibroblasts (CL14-00043) did not differ from control fibroblasts (CL043687, CL09-00025 and CL10-00010) in terms of percentage of ciliated cells (A) and length of the cilia (B). There is also no significant difference in overall distribution of IFT88 (C) and WDR19 (D) along the cilium. The graph is representative for three independent repeated experiments. Representative images of the immunocytochemistry experiments are shown in supplementary figures.
In addition, we evaluated intraflagellar transport (IFT) in the fibroblasts of patient A-II:1 by IFT88 (IFT-B, supplementary figure S2A,B) and WDR19/IFT144 (IFT-A, supplementary figure S2C,D) immunostaining. We observed no significant difference in the overall distribution of IFT88 and WDR19 along the cilium (base, base + tip and base + axoneme) between RP patient-derived and control fibroblasts (figure 4C,D).
RP-associated AHI1 missense mutations decrease its enrichment at the basal body
To evaluate the effect of the detected AHI1 variants on localisation of Jouberin to the ciliary base, we overexpressed mRFP fusion proteins of wild-type and mutant AHI1/Jouberin in immortalised retinal pigmented epithelium cells (hTERT-RPE cells). We selected two missense mutations c.2090C>T. (p.Pro697Leu) and c.2258A>T (p.Asp753Val) detected in patients with non-syndromic RP; and also included two JBTS-associated missense variants c.2156A>G (p.Asp719Gly) and c.2173T>C (p.Trp725Arg), located within the WD40 domain, to evaluate whether there is any different effect on AHI1 localisation between RP-associated variants and JBTS-associated ones.
Wild-type mRFP-AHI1 showed a diffused cytoplasmic localisation and a strong enrichment at the ciliary base of ciliated hTERT-RPE1 cells (figure 5A,B and supplementary figure S3A). Although RP-associated mutant Jouberin was still able to localise to the ciliary base, we observed a significant decrease of this enrichment compared with the wild-type protein (figure 5A,C,D and supplementary figure S3B,C). Interestingly, the localisation of JBTS-associated mutant Jouberin showed a similar difference compared with the wild-type protein, but it was not different from the RP-associated mutant Jouberin (figure 5A,E,F and supplementary figure S3D,E). Results of staining of the ciliary protein ARL13B, used as a marker for cilia in these experiments, were also used to assess any effects of overexpression of the recombinant mutated Jouberin on ciliogenesis. Neither ciliary frequency nor cilium length varied significantly (online supplementary figure S4).
Figure 5.
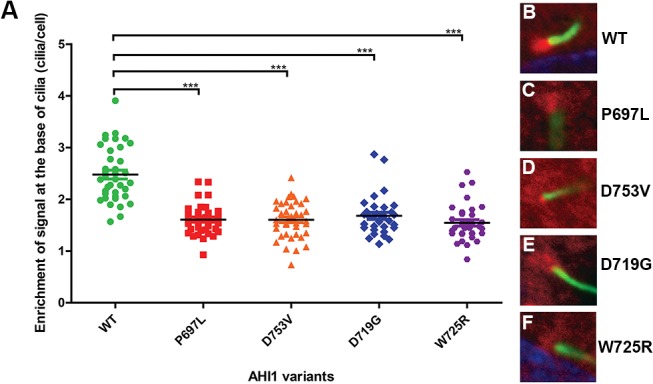
RP-associated and Joubert syndrome-associated missense variants in AHI1 caused a significant decrease in Jouberin enrichment to the basal bodies of cilia in hTERT-RPE1 cells, compared with wild-type Jouberin (A). ***The p value<0.0001 using unpaired t-test. Values of means±SE are provided in supplementary table S6. (B–F) Representative images of the localisation of wild-type (B) and mutant (C–F) Jouberin/AHI1 protein (red) at the ciliary base of hTERT-RPE1 cells by immunofluorescence. ARL13B (green) was used as a marker for the primary cilium. RP, retinitis pigmentosa.
Discussion
It is well established that mutations in AHI1 cause JBTS due to disruption of cilium formation and function in ciliated cells of diverse tissues in the body. In this study, we identified compound heterozygous mutations in AHI1 in three patients with non-syndromic RP. Five of the identified mutations are novel while one (c.2174G>A; p.Trp725*) has been identified in patients with JBTS with two deleterious AHI1 variants.26 The patients had a relatively late onset of RP and the disease progression was mild in all three patients. No other clinical features of JBTS were evident in the patients. Although no neurological imaging was performed, the lack of any of the syndromic clinical characteristics at the advanced age of all three patients makes the presence of a mild form of JBTS in the patients unlikely.
Thirty-nine of 48 reported variants in AHI1 in patients with JBTS are presumed loss of function. The remaining nine missense (of which seven are located within the WD40 domain and three before the WD40 domain) were observed in a homozygous state in typical patients with JBTS, suggesting they have a severe effect on the Jouberin protein localisation and/or function, which has indeed been shown for the p.Val443Asp variant.27
Using homology modelling, we show that of the seven Joubert-associated missense mutations, four have a predicted severe effect and two have a mild effect on the WD40 structure, while of the four non-syndromic retinal dystrophy missense mutations one has a severe, two have a mild and one has no clear effect on the WD40 structure. This indicates that most likely missense mutations detected in patients with Joubert syndrome have a more disruptive effect on the WD40 structure compared with the missense mutations detected in patients with non-syndromic RP.
Two of the patients with RP described here carry a loss-of-function allele and a missense variant affecting a conserved amino acid residue of the WD40 domain, and the third patient carries a predicted severe missense in trans with a milder mutation in the WD40 domain. We hypothesise that patients with Joubert syndrome have two severe AHI1 mutations that completely disrupt Jouberin function, while our patients with non-syndromic RP have one severe and one mild mutation and most likely have some remaining Jouberin activity. The remaining Jouberin activity seems to be sufficient for proper ciliary function in all relevant tissues, except for the retinal photoreceptor cells. A similar tissue-dependent activity-effect model was also suggested for other genes, for instance CLN3, CEP290, HGSNAT and MFSD8 that can be associated with syndromic and non-syndromic retinal diseases.28–31
The hypothesis that the missense variants in the WD40 domain do not completely abolish Jouberin function is further supported by our observation that cultured fibroblasts of patient A-II:1 (c.2174G>A and c.2258A>T) had no disrupted cilium formation, while Tuz et al showed that mutations in individuals with JBTS cause impaired ciliogenesis in patient’s fibroblasts.27 Additionally, we did not observe mislocalisation of either IFT-B (IFT88) or IFT-A (WDR19), a phenotype that was previously detected in other severe ciliopathies.32–35
Moreover, on expression of the recombinant proteins, the RP-associated variants cause a decreased enrichment of Jouberin at the ciliary basal bodies compared with the wild-type protein. This could be due to improper folding of protein leading to destabilisation of mutant Jouberin or due to interference of the protein interaction which is necessary to target Jouberin to the basal body. Intriguingly, JBTS-associated AHI1 mutations within the WD40 domain have a similar distortion of the ability of Jouberin to localise to the basal body. This suggests that phenotypic differences of RP versus JBTS might not be primarily due to mislocalisation of AHI1 mutants, but rather to a more tissue-specific loss of protein function, such as interaction with a particular protein or protein complex in which Jouberin is active, presumably at the described ring-shaped domain at the cilium–centriole interface.12 Alternatively, as the frequent AHI1 variant p.Arg830Trp has been described to be a genetic modifier for retinal degeneration in nephronophthisis, other epistatic interactions outside of the retina could also potentially be important either in exacerbation of the syndromic JBTS phenotype or in protection against it.
Our results suggest that the identified AHI1 variants in patients with RP might not disrupt the essential role of Jouberin in ciliogenesis nor its role in axonemal IFT trafficking, but rather mildly affect IFT balance or docking/undocking at the ciliary base without causing major ciliary defects, which is in line with the comparatively mild RP phenotype. This hypothesis is further supported by the proposed function of Jouberin as scaffold protein at the ciliary transition zone.12 36 37 AHI1 was already implicated to cause non-syndromic RP in a single patient who underwent WES. However, the pathogenicity of the detected AHI1 variants was questioned and further experiments were needed to assess the role of AHI1 in non-syndromic RP. As exome sequencing or screening of gene panels is now being performed on a regular basis, it remains of critical importance to perform an extensive evaluation of the pathogenicity of rare variants. Even more when functional evidence about the association with a specific disease phenotype has remained elusive, which was recently highlighted in Heller et al. 17 We detected three additional patients with compound heterozygous AHI1 mutations and combined with our functional data identified a new genotype–phenotype correlation for AHI1. With the broad use of WES, additional novel AHI1 variants will be detected that proposes a challenge in terms of how to investigate and distinguish the molecular mechanism by which AHI1 mutations cause RP versus JBTS, as well as emphasises the necessity in careful evaluation and validation of the pathogenicity of newly identified AHI1 variants in light of the presented clinical data. This knowledge is critically important to avoid the false interpretation of variants. In family C of this study, the pathogenicity of the detected variants in AHI1 (c.2087A>G; p.His696Arg and c.2429C>T; p.Pro810Leu) were determined using in silico prediction models. To ensure these variants are disease causing, additional functional tests should be performed.
In conclusion, we determined that variants in the AHI1 gene can cause non-syndromic RP next to Joubert syndrome. Interestingly, patients with RP and with pathogenic AHI1 variants have a late onset of disease and mild progression. We hypothesise that the AHI1 variants that cause RP most likely give rise to residual Jouberin function that can overcome all symptoms of Joubert syndrome, except for retinal degeneration.
Acknowledgments
We would like to thank Fight for Sight UK and the National Institute for Health Research Biomedical Research Centre (NIHR BRC) at Moorfields Eye Hospital, London and UCL Institute of Ophthalmology. Furthermore, we would like to thank Hanka Venselaar for het help with the protein modelling.
Footnotes
Contributors: T-MTN performed AHI1 transfection experiments and immunohistochemistry; T-MTN, LH and EdV performed zebrafisch knock-down and analysis of zebrafish sequence; SH, LIvdB and PAvdZ collected clinical cases and performed clinical examinations of patients; MMO and T-MTN performed fibroblast evaluations; SJL performed yeast-2-hybrid analysis; SEB performed TAP analysis; SH, EAB and LH-W performed Sanger sequencing; HY, GA and LH-W analyzed the exome data; LH-W performed modeling of the AHI1 variants in the WD-40 domain; RR, FPMC, EvW, ARW and LH-W designed the study; T-MTN, SH, RR, ARW and LH-W wrote the manuscript.
Funding: This work was supported by research grants from the Rotterdamse Stichting Blindenbelangen (to EV, FPMC, EW and LH-W), Stichting Blindenhulp (to EV, FPMC, EW and LH-W), Stichting tot Verbetering van het Lot der Blinden (to EV, FPMC, EW and LH-W), Stichting voor Ooglijders (to EV, FPMC, EW and LH-W), the European Community’s Seventh Framework Programmes FP7/2009 under grant agreement no: 241955 (SYSCILIA) to RR, by the Netherlands Organization for Scientific Research (NWO Vici-865.12.005) to RR, and by the Foundation Fighting Blindness (C-CMM-0811-0546-RAD02) to RR.
Competing interests: None declared.
Patient consent: The patients of this study already signed internal Radboudumc and Univeristy College of London informed consent forms that were specifically designed to consent for exome sequencing and publishment of results.
Ethics approval: Institutional review board Radboudumc and institutional review board University College London.
Provenance and peer review: Not commissioned; externally peer reviewed.
Correction notice: This paper has been updated since it first published online. Funding information have been added.
References
- 1. Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis 2006;1:40 10.1186/1750-1172-1-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Daiger SP, Sullivan LS, Bowne SJ, Rossiter BJF. Retnet. https://sph.uth.edu/retnet/ (accessed 1 Jan 2016).
- 3. Rivolta C, Sweklo EA, Berson EL, Dryja TP. Missense mutation in the USH2A gene: association with recessive retinitis pigmentosa without hearing loss. Am J Hum Genet 2000;66:1975–8. 10.1086/302926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet 1999;36:437–46. [PMC free article] [PubMed] [Google Scholar]
- 5. Wang F, Wang H, Tuan HF, Nguyen DH, Sun V, Keser V, Bowne SJ, Sullivan LS, Luo H, Zhao L, Wang X, Zaneveld JE, Salvo JS, Siddiqui S, Mao L, Wheaton DK, Birch DG, Branham KE, Heckenlively JR, Wen C, Flagg K, Ferreyra H, Pei J, Khan A, Ren H, Wang K, Lopez I, Qamar R, Zenteno JC, Ayala-Ramirez R, Buentello-Volante B, Fu Q, Simpson DA, Li Y, Sui R, Silvestri G, Daiger SP, Koenekoop RK, Zhang K, Chen R. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype–phenotype correlation and clinical refinements. Hum Genet 2014;133:331–45. 10.1007/s00439-013-1381-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, Gururaj A, Al-Gazali L, Al-Tawari AA, Kayserili H, Sztriha L, Gleeson JG. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet 2004;75:979–87. 10.1086/425985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, Al-Nouri D, Al-Rumayyan A, Topcu M, Gascon G, Bodell A, Shugart YY, Ruvolo M, Walsh CA. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet 2004;36:1008–13. 10.1038/ng1419 [DOI] [PubMed] [Google Scholar]
- 8. Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol 2013;12:894–905. 10.1016/S1474-4422(13)70136-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brancati F, Dallapiccola B, Valente EM, Syndrome J. And related disorders. Orphanet J Rare Dis 2010;5:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Parisi MA, Doherty D, Eckert ML, Shaw DW, Ozyurek H, Aysun S, Giray O, Al Swaid A, Al Shahwan S, Dohayan N, Bakhsh E, Indridason OS, Dobyns WB, Bennett CL, Chance PF, Glass IA. AHI1 mutations cause both retinal dystrophy and renal cystic disease in joubert syndrome. J Med Genet 2006;43:334–9. 10.1136/jmg.2005.036608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Doering JE, Kane K, Hsiao YC, Yao C, Shi B, Slowik AD, Dhagat B, Scott DD, Ault JG, Page-McCaw PS, Ferland RJ. Species differences in the expression of Ahi1, a protein implicated in the neurodevelopmental disorder joubert syndrome, with preferential accumulation to stigmoid bodies. J Comp Neurol 2008;511:238–56. 10.1002/cne.21824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee YL, Santé J, Comerci CJ, Cyge B, Menezes LF, Li FQ, Germino GG, Moerner WE, Takemaru K, Stearns T. Cby1 promotes Ahi1 recruitment to a ring-shaped domain at the centriole-cilium interface and facilitates proper cilium formation and function. Mol Biol Cell 2014;25:2919–33. 10.1091/mbc.E14-02-0735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hsiao YC, Tong ZJ, Westfall JE, Ault JG, Page-McCaw PS, Ferland RJ. Ahi1, whose human ortholog is mutated in Joubert syndrome, is required for Rab8a localization, ciliogenesis and vesicle trafficking. Hum Mol Genet 2009;18:3926–41. 10.1093/hmg/ddp335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Westfall JE, Hoyt C, Liu Q, Hsiao YC, Pierce EA, Page-McCaw PS, Ferland RJ. Retinal degeneration and failure of photoreceptor outer segment formation in mice with targeted deletion of the joubert syndrome gene, Ahi1. J Neurosci 2010;30:8759–68. 10.1523/JNEUROSCI.5229-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lancaster MA, Schroth J, Gleeson JG. Subcellular spatial regulation of canonical wnt signalling at the primary cilium. Nat Cell Biol 2011;13:702–9. 10.1038/ncb2259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang XF, Huang F, Wu KC, Wu J, Chen J, Pang CP, Lu F, Qu J, Jin ZB. Genotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet Med 2015;17:271–8. 10.1038/gim.2014.138 [DOI] [PubMed] [Google Scholar]
- 17. Heller R, Bolz HJ. The challenge of defining pathogenicity: the example of AHI1. Genet Med 2015;17:508. 10.1038/gim.2015.46 [DOI] [PubMed] [Google Scholar]
- 18. Bach M, Brigell MG, Hawlina M, Holder GE, Johnson MA, McCulloch DL, Meigen T, Viswanathan S. ISCEV standard for clinical pattern electroretinography (PERG): 2012 update. Doc Ophthalmol 2013;126:1–7. 10.1007/s10633-012-9353-y [DOI] [PubMed] [Google Scholar]
- 19. McCulloch DL, Marmor MF, Brigell MG, Hamilton R, Holder GE, Tzekov R, Bach M. ISCEV standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol 2015;130:1–12. 10.1007/s10633-014-9473-7 [DOI] [PubMed] [Google Scholar]
- 20. Wu XH, Wang Y, Zhuo Z, Jiang F, Wu YD. Identifying the hotspots on the top faces of WD40-repeat proteins from their primary sequences by β-bulges and DHSW tetrads. PLoS One 2012;7:e43005 10.1371/journal.pone.0043005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Y, Jiang F, Zhuo Z, Wu XH, Wu YD. A method for WD40 repeat detection and secondary structure prediction. PLoS One 2013;8:e65705 10.1371/journal.pone.0065705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang Y, Hu XJ, Zou XD, Wu XH, Ye ZQ, Wu YD. WDSPdb: a database for WD40-repeat proteins. Nucleic Acids Res 2015;43:D339–D344. 10.1093/nar/gku1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krieger E, Vriend G. YASARA View - molecular graphics for all devices - from smartphones to workstations. Bioinformatics 2014;30:2981–2. 10.1093/bioinformatics/btu426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roepman R, Letteboer SJ, Arts HH, van Beersum SE, Lu X, Krieger E, Ferreira PA, Cremers FP. Interaction of nephrocystin-4 and RPGRIP1 is disrupted by nephronophthisis or Leber congenital amaurosis-associated mutations. Proc Natl Acad Sci U S A 2005;102:18520–5. 10.1073/pnas.0505774102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Arts HH, Doherty D, van Beersum SE, Parisi MA, Letteboer SJ, Gorden NT, Peters TA, Märker T, Voesenek K, Kartono A, Ozyurek H, Farin FM, Kroes HY, Wolfrum U, Brunner HG, Cremers FP, Glass IA, Knoers NV, Roepman R. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet 2007;39:882–8. 10.1038/ng2069 [DOI] [PubMed] [Google Scholar]
- 26. Seong MW, Seo SH, Yu YS, Hwang JM, Cho SI, Ra EK, Park H, Lee SJ, Kim JY, Park SS. Diagnostic application of an extensive gene panel for Leber congenital amaurosis with severe genetic heterogeneity. J Mol Diagn 2015;17:100–5. 10.1016/j.jmoldx.2014.09.003 [DOI] [PubMed] [Google Scholar]
- 27. Tuz K, Hsiao YC, Juárez O, Shi B, Harmon EY, Phelps IG, Lennartz MR, Glass IA, Doherty D, Ferland RJ. The Joubert syndrome-associated missense mutation (V443D) in the Abelson-helper integration site 1 (AHI1) protein alters its localization and protein-protein interactions. J Biol Chem 2013;288:13676–94. 10.1074/jbc.M112.420786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kitzmüller C, Haines RL, Codlin S, Cutler DF, Mole SE. A function retained by the common mutant CLN3 protein is responsible for the late onset of juvenile neuronal ceroid lipofuscinosis. Hum Mol Genet 2008;17:303–12. 10.1093/hmg/ddm306 [DOI] [PubMed] [Google Scholar]
- 29. Coppieters F, Lefever S, Leroy BP, De Baere E. CEP290, a gene with many faces: mutation overview and presentation of CEP290base. Hum Mutat 2010;31:1097–108. 10.1002/humu.21337 [DOI] [PubMed] [Google Scholar]
- 30. Haer-Wigman L, Newman H, Leibu R, Bax NM, Baris HN, Rizel L, Banin E, Massarweh A, Roosing S, Lefeber DJ, Zonneveld-Vrieling MN, Isakov O, Shomron N, Sharon D, Den Hollander AI, Hoyng CB, Cremers FP, Ben-Yosef T. Non-syndromic retinitis pigmentosa due to mutations in the mucopolysaccharidosis type IIIC gene, heparan-alpha-glucosaminide N-acetyltransferase (HGSNAT). Hum Mol Genet 2015;24:3742–51. 10.1093/hmg/ddv118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roosing S, van den Born LI, Sangermano R, Banfi S, Koenekoop RK, Zonneveld-Vrieling MN, Klaver CC, van Lith-Verhoeven JJ, Cremers FP, den Hollander AI, Hoyng CB. Mutations in MFSD8, encoding a lysosomal membrane protein, are associated with nonsyndromic autosomal recessive macular dystrophy. Ophthalmology 2015;122:170–9. 10.1016/j.ophtha.2014.07.040 [DOI] [PubMed] [Google Scholar]
- 32. Perrault I, Saunier S, Hanein S, Filhol E, Bizet AA, Collins F, Salih MA, Gerber S, Delphin N, Bigot K, Orssaud C, Silva E, Baudouin V, Oud MM, Shannon N, Le Merrer M, Roche O, Pietrement C, Goumid J, Baumann C, Bole-Feysot C, Nitschke P, Zahrate M, Beales P, Arts HH, Munnich A, Kaplan J, Antignac C, Cormier-Daire V, Rozet JM. Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am J Hum Genet 2012;90:864–70. 10.1016/j.ajhg.2012.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kessler K, Wunderlich I, Uebe S, Falk NS, Gießl A, Brandstätter JH, Popp B, Klinger P, Ekici AB, Sticht H, Dörr HG, Reis A, Roepman R, Seemanová E, Thiel CT. DYNC2LI1 mutations broaden the clinical spectrum of dynein-2 defects. Sci Rep 2015;5:11649. 10.1038/srep11649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Halbritter J, Bizet AA, Schmidts M, Porath JD, Braun DA, Gee HY, McInerney-Leo AM, Krug P, Filhol E, Davis EE, Airik R, Czarnecki PG, Lehman AM, Trnka P, Nitschké P, Bole-Feysot C, Schueler M, Knebelmann B, Burtey S, Szabó AJ, Tory K, Leo PJ, Gardiner B, McKenzie FA, Zankl A, Brown MA, Hartley JL, Maher ER, Li C, Leroux MR, Scambler PJ, Zhan SH, Jones SJ, Kayserili H, Tuysuz B, Moorani KN, Constantinescu A, Krantz ID, Kaplan BS, Shah JV, Hurd TW, Doherty D, Katsanis N, Duncan EL, Otto EA, Beales PL, Mitchison HM, Saunier S, Hildebrandt F. UK10K Consortium. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am J Hum Genet 2013;93:915–25. 10.1016/j.ajhg.2013.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McInerney-Leo AM, Schmidts M, Cortés CR, Leo PJ, Gener B, Courtney AD, Gardiner B, Harris JA, Lu Y, Marshall M, Scambler PJ, Beales PL, Brown MA, Zankl A, Mitchison HM, Duncan EL, Wicking C. UK10K Consortium. Short-rib polydactyly and jeune syndromes are caused by mutations in WDR60. Am J Hum Genet 2013;93:515–23. 10.1016/j.ajhg.2013.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, Baye LM, Wen X, Scales SJ, Kwong M, Huntzicker EG, Sfakianos MK, Sandoval W, Bazan JF, Kulkarni P, Garcia-Gonzalo FR, Seol AD, O'Toole JF, Held S, Reutter HM, Lane WS, Rafiq MA, Noor A, Ansar M, Devi AR, Sheffield VC, Slusarski DC, Vincent JB, Doherty DA, Hildebrandt F, Reiter JF, Jackson PK. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell 2011;145:513–28. 10.1016/j.cell.2011.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, Sandoval W, Peterson AS. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol 2012;14:61–72. 10.1038/ncb2410 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jmedgenet-2016-104200supp002.docx (39.2KB, docx)
jmedgenet-2016-104200supp001.pdf (1.5MB, pdf)