


Abstract
Mitochondrial gene expression was studied using an electrotransformation protocol to introduce foreign DNA into purified wheat mitochondria. Optimal conditions for DNA uptake and transient gene expression were determined. We show here that a DNA plasmid containing either a cognate or a non-cognate gene under the control of a plant mitochondrial promoter is incorporated into the organelle and faithfully recognized by the transcription machinery. Transcripts generated by a plasmid bearing the intron-containing cox II gene were correctly spliced. Moreover, the transcripts were edited at the expected target C residues. The expression and maturation process of the transgene is dependent on the integrity of functional elements such as the promotor or the presence of structural domains necessary for splicing. The mitochondrial transformation described in this report is an important tool to study the multiple steps involved in plant mitochondrial gene expression at conditions closer to those found in vivo.
INTRODUCTION
Plant mitochondrial gene expression is a complex process involving multiple steps such as transcription, cis- and trans-splicing, RNA trimming, RNA editing and translation. A landmark in plant mitochondrial gene expression is RNA editing. All mitochondrial transcripts in land plant mitochondria undergo specific C→U modifications at some residues via a deamination mechanism (1 and references therein). RNA editing is required to fill the gap between the information encoded in plant mitochondrial DNA and the synthesis of functional organellar proteins (2,3). These processes are poorly understood because of the lack of appropriate experimental approaches. Most current studies on mitochondrial gene expression in plant mitochondria are either based on the analysis of intermediate molecules found in vivo or on laborious in vitro approaches. Nevertheless, many interesting insights have been reported by using such approaches as the promoter function and RNA processing (4–9) or the RNA editing mechanism (10–12). However, many problems concerning gene expression, especially RNA splicing and RNA editing, are not yet fully understood. Unlike chloroplasts, the integration of exogenous genes into the mitochondrial genome has not been obtained. In addition, the absence of appropriate selective markers hampers the maintenance of a transgene inside mitochondria.
Transformation of isolated plant mitochondria may be a key approach to study gene expression, thereby enabling the use of site-directed mutagenesis. Previously, Collombet et al. (13) reported the introduction of plasmid DNA into isolated mice mitochondria by electroporation. By using an analogous procedure, To et al. (14) studied the expression of reporter genes in isolated chloroplasts.
Here, we describe that chimeric plasmids containing either a mitochondrial or a non-cognate gene, under the control of a mitochondrial promoter, are transcribed when electroporated into isolated mitochondria. We show that the exogenous transcript undergoes post-transcriptional processing such as RNA splicing and RNA editing. Furthermore, electroporation-mediated gene transfer into mitochondria is a valuable approach to address various questions related to processes involved in mitochondrial gene expression such as transcription, RNA maturation, RNA editing and RNA stability.
MATERIALS AND METHODS
Mitochondria purification
Wheat embryos were obtained from Triticum aestivum var. Fortal seeds as previously described (15). Embryos were sterilized with 0.6% sodium hypochlorite and rapidly washed with sterile-distilled water before use. Seven grams of embryos were set out on a filter paper saturated with sterile water in a Petri dish, and incubated for 18 h at 22°C. Embryos were homogenized with a Polytron (Kinematica CH 6010, Kriens/Luzer, Suize) at 70% full speed for 15 s in 150 ml of solution containing 0.4 M mannitol, 25 mM MOPS pH 7.8, 1 mM EGTA, 8 mM cysteine and 1 mg/ml fatty acid-free bovine serum albumin. The extract was filtered through a 30 µm nylon membrane and mitochondria were purified as described by Leaver et al. (16) by centrifugation on a sucrose gradient. Purified mitochondria were transferred to an Eppendorf tube and collected by centrifugation at 15 000 g for 10 min and washed twice with 0.33 M sucrose. All experiments were performed with freshly purified mitochondria.
Plasmid construction
The sequences of cytochrome oxidase 2 (cox II) and apocytochrome b (cob) from Triticum timopheevi mitochondria used in this study are registered in GenBank under the accession numbers AF336134 and AF337547, respectively. The T.timopheevi cox II gene presents the following differences compared to the endogenous cox II: a GG instead of TA at position –107, an octanucleotide inserted at –90, a G inserted at –67, an A→G transition at 1345, a heptanucleotide insertion at 1939, a G to C change at 2046, a dinucleotide insertion at 2048 and the sequence AAAT replacing TTCG at 2257. The chimeric plasmid pLuc (Fig. 1, top) was constructed by replacing the SV40 promoter regions of pGL3-control plasmid (Promega) vector with 882 bp of T.timopheevi cox II mitochondrial promoter containing the initiation region described by Hanic-Joyce and Gray (4). The plasmid pCox II (Fig. 1, bottom) based on the pBluescribe vector contains 882 bp of the upstream region and 2009 bp of the coding sequences of cox II from T.timopheevi. A 23 bp insert was introduced into the promoter region at position –60 in plasmid pCox II using QuickChange™ Site-Directed Mutagenesis kit (Stratagene). The 23 bp insertion provides a specific sequence to isolate transgene transcripts during RT–PCR analysis using primer 1. pLuc and pCox II plasmids contain 533 bp of the terminator region (nucleotides 1898–2430, AF337547) of T.timopheevi cob gene (17) at the 3′ non-coding region (Fig. 1). In pLuc, the cob terminator sequence replaces the SV40 late poly(A) signal and the SV40 enhancer from vector pGL3. Three deletion plasmids derived from pCox II were constructed: pCoxII-ΔP lacking the cox II promoter region, pCoxII-ΔD6 containing a deletion of nucleotides 2479–2492 forming the stem–loop domain 6 of cox II intron and pCoxII-Δcob deleted from the structured double stem–loop structure in the 3′ untranslated region (terminator region). For PCR analyses of deletion mutants pCoxII-ΔP and pCoxII-Δcob, the primers 1 and 3 were replaced by primers 8 and 9, respectively.
Figure 1.
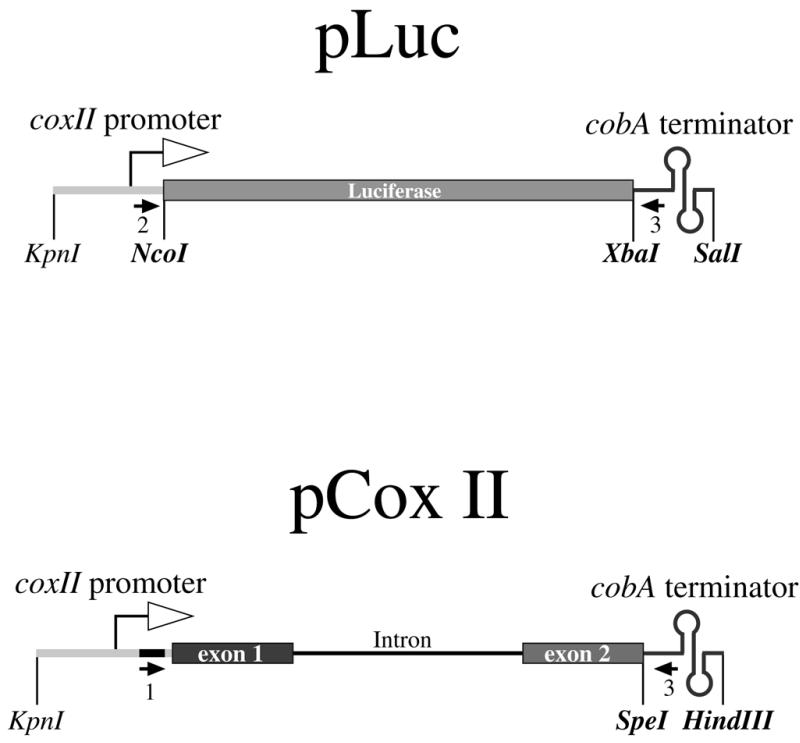
Scheme of the plasmids used. pLuc contains 1656 bp of the Photinus pyralis luciferase ORF obtained from plasmid pGL3 (Promega). pCox II contains a 2 kb fragment from the T.timopheevi cox II ORF formed by two exons interrupted by a 1.2 kb intron. Both constructions are controlled by an 882 bp cox II promoter region from T.timopheevi and 533 bp of a cob 3′ terminator region from T.timopheevi mitochondria (17). Restriction sites indicated in bold letters were artificially generated using appropriate PCR primers. Arrows indicate the position of the primers used in PCR analyses. Open arrows indicate the transcription initiation region as proposed by Hanic-Joyce and Gray (4). The black box in the promoter region of pCox II shows the 23 bp insert used for specific PCR amplification.
PCR primers
Primer 1, GCGGTGCAGTCATACAGATCTGC; primer 2, CAACGCCGGACGTCAAGATCAG; primer 3, TATCCAGATTTGGTACCAAAC; primer 4, CTACCCATGGAAAAGATCATGA; primer 5, AGTAGATCTTCGTTGTTCGGATCTTG; primer 6, GATTGGGTCAACCAGGCCAGA; primer 7, AGGGAGCCATCGAAAGGTGACTGAA; primer 8, ATGATTCTTCGTTCATTATCA; primer 9, AACAGCTATGACCATGATT.
Mutagenesis primer
Only sense primers are indicated (23 bp insert):
AACGCCGGACGTCAAGCGGTGCAGTCATACAGATCTGCGATCAGTCTCCTTTC.
Electroporation
Electrotransfer experiments were carried out with a Bio-Rad Gene Pulser at 4°C in 0.1 cm electrode gap cuvettes (Bio-Rad). The settings were: 25 µF and 400 Ω. Various electric pulses in the range 8–20 kV/cm were tested. One microgram of plasmid purified with the Qiagen™ plasmid midi kit was added to fresh mitochondria (1 mg total protein) resuspended in 50 µl of 0.33 M sucrose. After electroporation, the mitochondrial suspension was withdrawn and the cuvette washed with 50 µl of 0.33 M sucrose. Mitochondria were collected by centrifugation at 15 000 g for 10 min and then resuspended in 250 µl of the expression buffer containing 330 mM mannitol, 90 mM KCl, 10 mM MgCl2, 12 mM tricine pH 7.2, 5 mM KH2PO4, 1.2 mM EGTA, 1 mM GTP, 2 mM DTT, 2 mM ADP, 10 mM sodium succinate and 0.15 mM each CTP and UTP. Mitochondria were incubated at 25°C for different periods of time with constant stirring at 150 t/min. After incubation, the mitochondrial pellet was recovered by centrifugation at 15 000 g for 15 min at 4°C.
DNase I protection assay and DNA purification
After electroporation and centrifugation, the mitochondrial pellet was resuspended in 100 µl buffer [10 mM Tris–HCl pH 7.5, 2 mM magnesium acetate and 0.33 M sucrose, containing 60 U of DNase I (Gibco BRL)] and incubated for 1 h at room temperature. The DNase reaction was stopped by adding 4 µl of 0.5 M EDTA and then heated for 10 min at 65°C. Mitochondria were incubated with 100 µg of Proteinase K (Merck) for 4 h at 37°C. One microliter of 20% SDS was added to achieve mitochondrial lysis and the DNA was extracted with phenol/chloroform (18) and precipitated with 0.1 vol of 3 M sodium acetate pH 5.2, 3 vol of 100% ethanol and 100 ng of carrier yeast tRNA and left overnight at –20°C. After centrifugation, the DNA pellet was resuspended in TE buffer (10 mM Tris–HCl pH 8, 1 mM EDTA).
RNA purification
After electroporation, the mitochondria were recovered by centrifugation at 15 000 g for 10 min at 4°C. The RNA was purified with 200 µl of TRIzol™ Reagent (Gibco BRL) according to the protocol suggested by the supplier. The RNA was resuspended in 20 µl DEPC-treated water.
RT–PCR
One microgram of RNA was treated with 2 U of amplification grade DNase I (Gibco BRL). cDNA synthesis was performed with 200 U of Superscript II RT using 100 ng of random hexamers as proposed by the supplier. The PCR reactions were performed with Advantage™ 2 Polymerase Mix (Clontech) as follows: 95°C for 1 min; 5 cycles at 95°C for 30 s and 68°C for 1 min; 30 cycles at 95°C for 30 s, 58°C for 30 s and 68°C for 30 s; and finally 68°C for 1 min.
DNA sequencing
Sequence analyses were performed directly on the RT–PCR product or, after cloning, on pGEM-T (Promega) vectors, using either the Thermo Sequenase™ radiolabeled terminator cycle sequencing kit (Amersham) or the BigDye™ Terminator Cycle Sequencing Kit (Applied Biosystems).
RESULTS
Electroporation conditions
The plasmid pLuc was used to optimize the mitochondrial transfection conditions. Fresh mitochondria were incubated with the purified plasmid and submitted to different voltage conditions as shown in Figure 2A and B. After electroporation, the mitochondrial suspension was incubated with DNase I and the internalized DNA was purified. The rationale was that the internalization of DNA into mitochondria should protect the plasmid from nuclease digestion, thereby making it available for PCR analysis. The PCR products were analyzed by gel electrophoresis (Fig. 2A). Specific regions of the chimeric plasmid were PCR-amplified using primers 2 and 3. Primer 2 is complementary to the promoter region of cox II and primer 3 is complementary to the 3′ untranslated region of cob. Neither of the primers, which are also complementary to two different mtDNA loci, gave any amplification product with T.aestivum mtDNA used as template (Fig. 2A, lane 2). The foreign DNA was protected from DNase I digestion only after electroporation (Fig. 2A, lanes 8–20). A control experiment without electroporation showed no protection (Fig. 2A, lane 0), indicating that plasmid DNA entry into the organelle was driven by the electric field. The optimal electroporation conditions were found between 10 and 13 kV/cm (Fig. 2B); DNA was not protected when higher voltages were used. The sharp increase in DNA uptake we observed was also described for animal mitochondria (13). An internal control was performed using primers 4 and 5 specific to the maturase domain of mat-r endogenous gene (19,20). At each voltage used, the amount of mat-r PCR product was the same, indicating that our results reflected the efficiency of electroporation.
Figure 2.
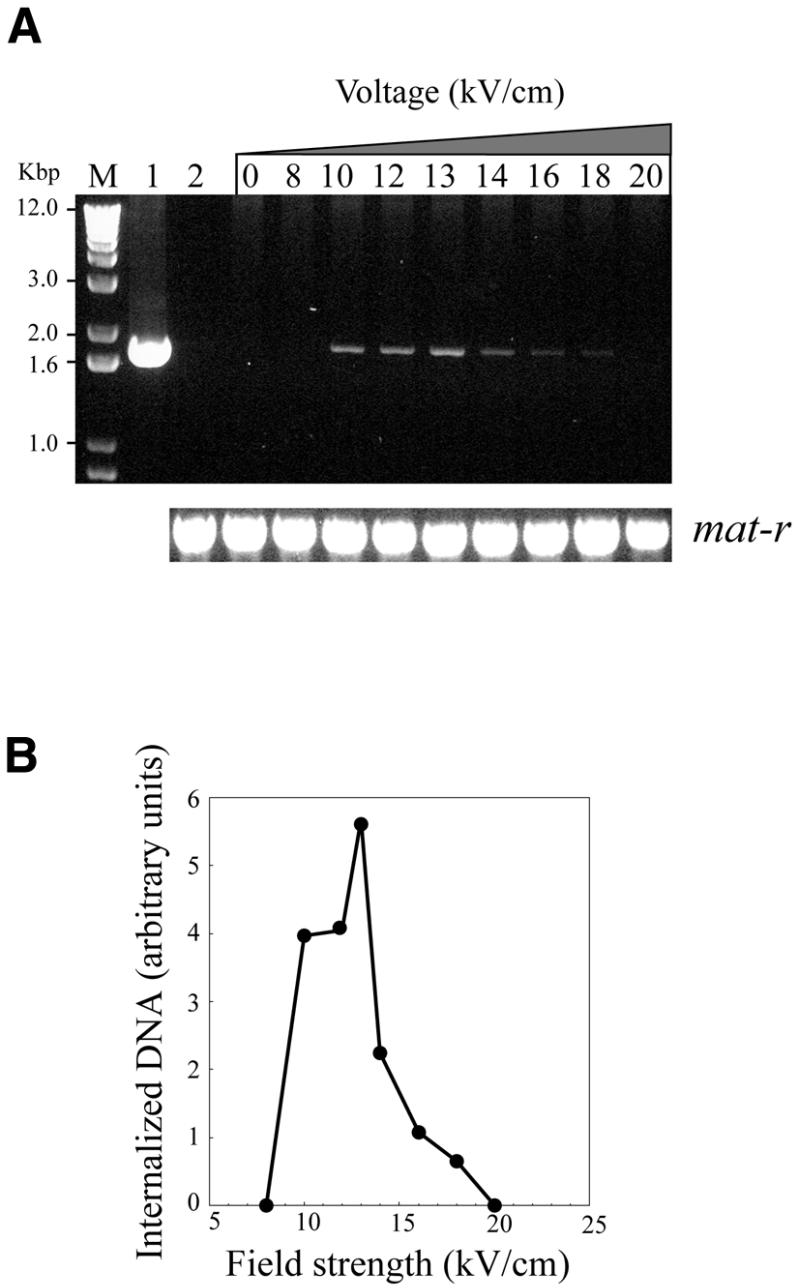
DNase I protection experiments. Purified wheat embryo mitochondria were electroporated in the presence of 1 µg of pLuc plasmid at electric field strengths ranging from 0 to 20 kV/cm. After incubation with DNase I to digest the non-internalized vector, DNA was extracted and the target sequence was revealed by PCR using primers 2 and 3. (A) Agarose gel electrophoresis of PCR products. Lower panel, control PCR experiment with endogenous mtDNA mat-r sequences performed with primers 4 and 5. Lane 1, PCR control reaction with plasmid pLuc; lane 2, untreated wheat mitochondria. (B) The ethidium bromide UV fluorescence signal from the gel electrophoresis experiment in (A) was recorded with a CCD video camera and plotted as arbitrary units using the NIH Image software.
The electrotransferred recombinant plasmid is transcribed
The expression of the luciferase mRNA was analyzed by RT–PCR to verify that entry of the chimeric plasmid into mitochondria resulted in a functional event. For this purpose, total DNA-free RNA was extracted from mitochondria electroporated with pLuc after incubation for 16 h at 25°C in complete ‘expression’ buffer. Synthesis of cDNA using random primers and PCR was carried out with primers 2 and 3. As expected, a 1787 bp fragment was obtained (Fig. 3, lane 2). No amplification product was detected when mitochondria were not submitted to the electric field pulse (Fig. 3, lane 1). Control PCR experiments were carried out on the same cDNA preparation using primers 6 and 7 to amplify the mature form of the endogenous nad1 mRNA (20); the amount of nad1 transcript was identical either with or without an applied electric field pulse. This result indicated that the introduced cox II promoter is recognized by the mitochondrial transcription machinery.
Figure 3.
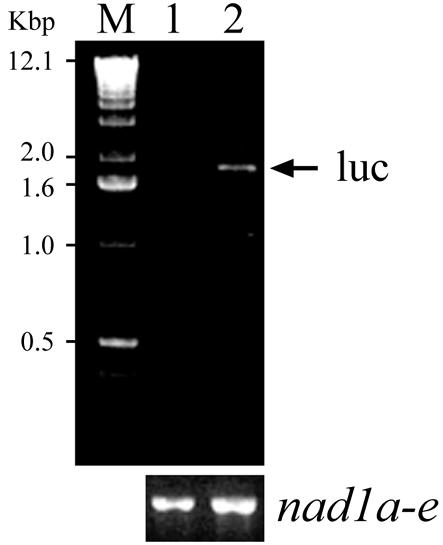
Transcription analysis of electroporated luciferase bearing plasmid. Purified wheat embryo mitochondria were electroporated in the presence of 1 µg of pLuc plasmid. After incubation with DNase I, RNA was extracted and total cDNA was synthesized with random primers. The target sequence was revealed by PCR using primers 2 and 3. Control PCR experiments (lower panel) were performed on the same cDNA with primers 6 and 7 which reveal the mature form of nad1 mRNA. In all cases, PCR experiments gave no amplification products when the reverse transcription step was omitted. Lane 1, without electric pulse; lane 2, electroporation carried out at 13 kV/cm. After the electric pulse, mitochondria were incubated in the expression buffer for 16 h at 25°C (see Materials and Methods). M, DNA size markers.
Incubation conditions of transfected mitochondria
After electroporation, transfected mitochondria were incubated in a complex reaction mixture (for details see Materials and Methods). The first electroporation analyses were done in a complete medium described for in organello protein synthesis assay (16), supplied with UTP and CTP. The requirements necessary for transgene expression were studied and results are summarized in Table 1. As expected, the omission of the amino acid pool had no effect on transcription and further processing of the RNA while lowering the potassium and magnesium ion concentration resulted in a dramatic decrease in the transcription efficiency. Furthermore, in the absence of the four rNTPs precursors transcription was abolished, indicating that the internal nucleotide pool was too low to sustain transcription of the exogenous template. A high concentration (1 mM) of GTP was necessary for expression since transcription was reduced at lower concentrations (0.15 mM) and abolished in the absence of GTP. The addition of ADP and succinate was necessary for expression, but exogenous ATP did not replace the metabolically generated precursor. The latter observation is important because it indicates unambiguously that during the assay wheat mitochondria are intact and functional.
Table 1. Effect of incubation conditions on transcription of pCox II by electrotransformed mitochondria.
| Complete (+ 1µg
DNA) |
++++ |
| Complete (+ 10 µg DNA) | +++ |
| –Amino acids | ++++ |
| 7.5 mM KCl; 5 mM MgCl2 | + |
| –Succinate, –ADP | – |
| –Succinate, –ADP, +ATP (0.15 mM) | – |
| 0.15mM GTP | ++ |
| –GTP | – |
| –rNTP | – |
Purified wheat embryo mitochondria were electroporated in the presence of pCox II plasmid. The RNA was extracted and total cDNA was synthesized with random primers. The target cox II transcript was revealed by PCR using primers 1 and 3. The complete reaction mixture contained 1 µg DNA, 330 mM mannitol, 90 mM KCl, 10 mM MgCl2, 12 mM tricine pH 7.2, 5 mM KH2PO4, 1.2 mM EGTA, 1 mM GTP, 2 mM DTT, 2 mM ADP, 10 mM sodium succinate, 0.15 mM each of CTP and UTP and 25 µM amino acid mixture. Relative transcription efficiency was determined by ethidium bromide UV fluorescence after gel electrophoresis. The data were normalized using the signal obtained with the endogenous nad1 transcripts (primers 6 and 7) as internal standard.
The transgenic cox II mitochondrial transcript is correctly spliced
To study the processing of the mRNA generated by the electroporated construct, we used the plasmid pCox II carrying the complete coding sequence of the cox II gene which is formed by two exons linked by a 1226 bp intron. Experiments were carried out with primers 1 and 3, which are unable to amplify the endogenous cox II mitochondrial transcript. Moreover, the presence of nucleotide variants and the chimeric cox II/cob configuration of the transgene distinguished it from the endogenous cox II (see Materials and Methods). RT–PCR analysis of electroporated mitochondria gave two bands: 2133 bp corresponding to the precursor cox II transcript and 907 bp generated by the processed cox II mRNA (Fig. 4A, lane 3). No transcripts were found when mitochondria were incubated with the plasmid in the absence of an electric field pulse (Fig. 4A, lane 1). As expected, deletion of the promoter region (pCoxII-ΔP) abolished transgene expression (Fig. 4A, lane 2), whereas deletion of cob 3′ untranslated region (pCoxII-Δcob) did not affect the expression of the transgene (Fig. 4A, lane 4). Sequence analysis of the processed fragment clearly showed that the exon 1–exon 2 junction corresponded to that observed in vivo, indicating that the foreign cox II transcript underwent a faithful splicing process (Fig. 4B).
Figure 4.
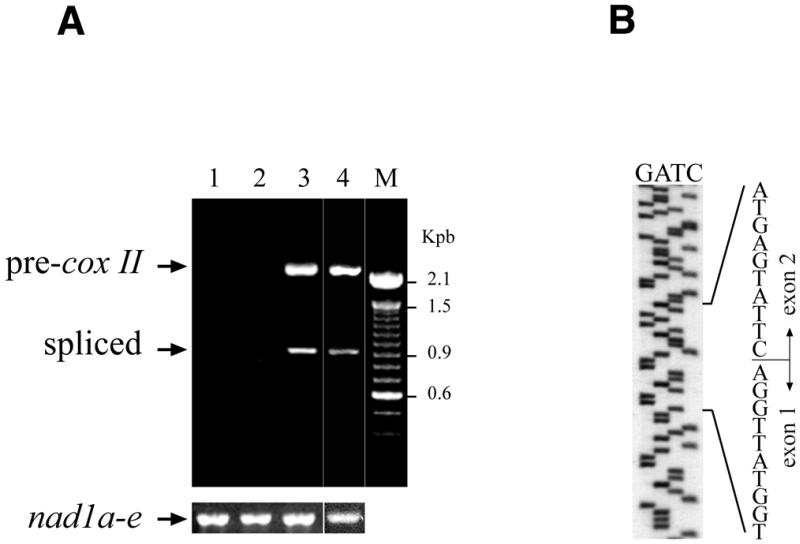
Transcription and maturation of mRNA from electroporated pCox II. Purified wheat embryo mitochondria were electroporated in the presence of 1 µg of pCox II as described in Figure 3. The target cox II sequence was revealed by PCR using primers 1 and 3. (A) Lane 1, without electric pulse; lane 2, electroporation performed with the promoter-free plasmid pCoxII-ΔP; lane 3, electroporation with pCox II; lane 4, electroporation of plasmid pCoxII-Δcob. Lower panel, control RT–PCR of nad1 endogenous transcripts using primers 6 and 7. (B) Sequence gel of the mature foreign cox II transcript showing the exon 1–exon 2 junction.
Splicing is impaired in the absence of intron domain 6
The deletion of the domain VI (Fig. 5A) characteristic of group II introns (21,22) in construct pCoxII-ΔD6 dramatically inhibited mRNA splicing (Fig. 5B, lane 2) compared to the wild-type construct (Fig. 5B, lane 1). This resulted in the accumulation of precursor mRNA.
Figure 5.
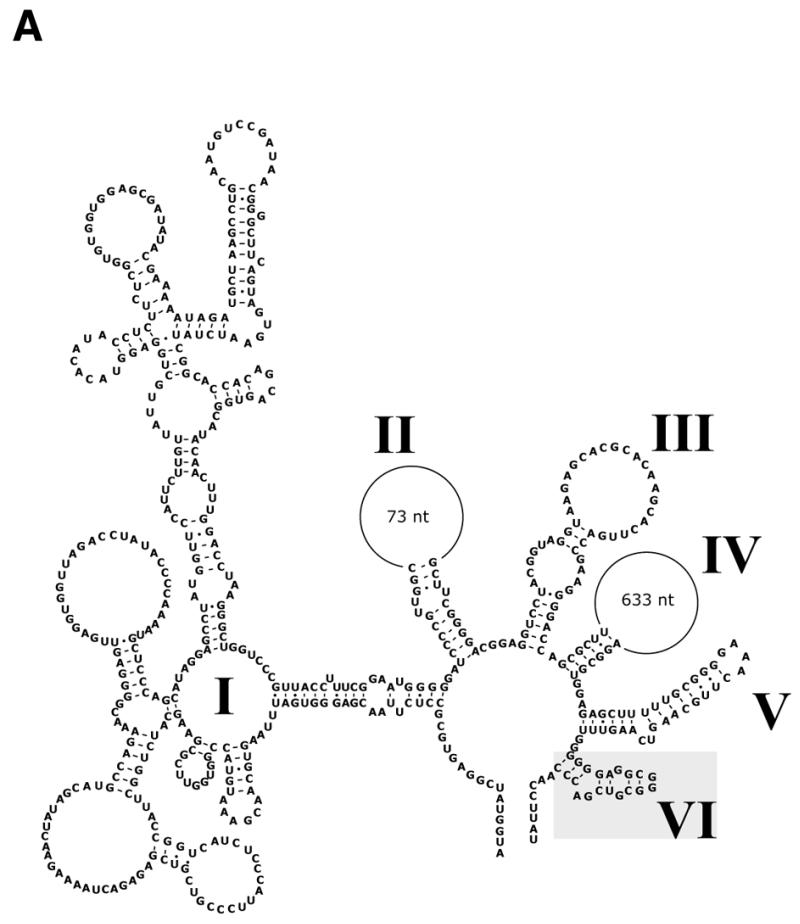

Splicing of cox II mRNA deleted in intron domain VI. Incubation conditions were as described in Figure 3. (A) Putative secondary structure of cox II intron shown according to the group II intron model proposed by Michel et al. (21). The conserved intron domains I–VI are indicated by roman numerals; deleted domain VI is indicated. (B) Lane 1, electroporation of mitochondria with pCox II; lane 2, electroporation performed with plasmid pCoxII-ΔD6. Control PCR experiments on endogenous nad1 transcripts (lower panel) were performed on the same cDNA with primers 6 and 7. M, DNA molecular weight markers.
Time course of cox II transgene splicing
The kinetics of pCox II expression were studied after electroporation. The cox II precursor transcript rapidly accumulated, reaching a plateau at 6 h of incubation and remaining stable until 18 h, followed by a slow decrease (Fig. 6A and B). The spliced form increased steadily over the same period of time, indicating that the cox II transgene was efficiently transcribed and processed. For further analysis, the electroporated mitochondria were incubated for 16 h in expression buffer. Control RT–PCR experiments on the endogenous nad1 transcripts were performed with primers 6 and 7 (Fig. 6A, lower panel). No significant changes in the endogenous transcript were observed throughout the incubation period.
Figure 6.
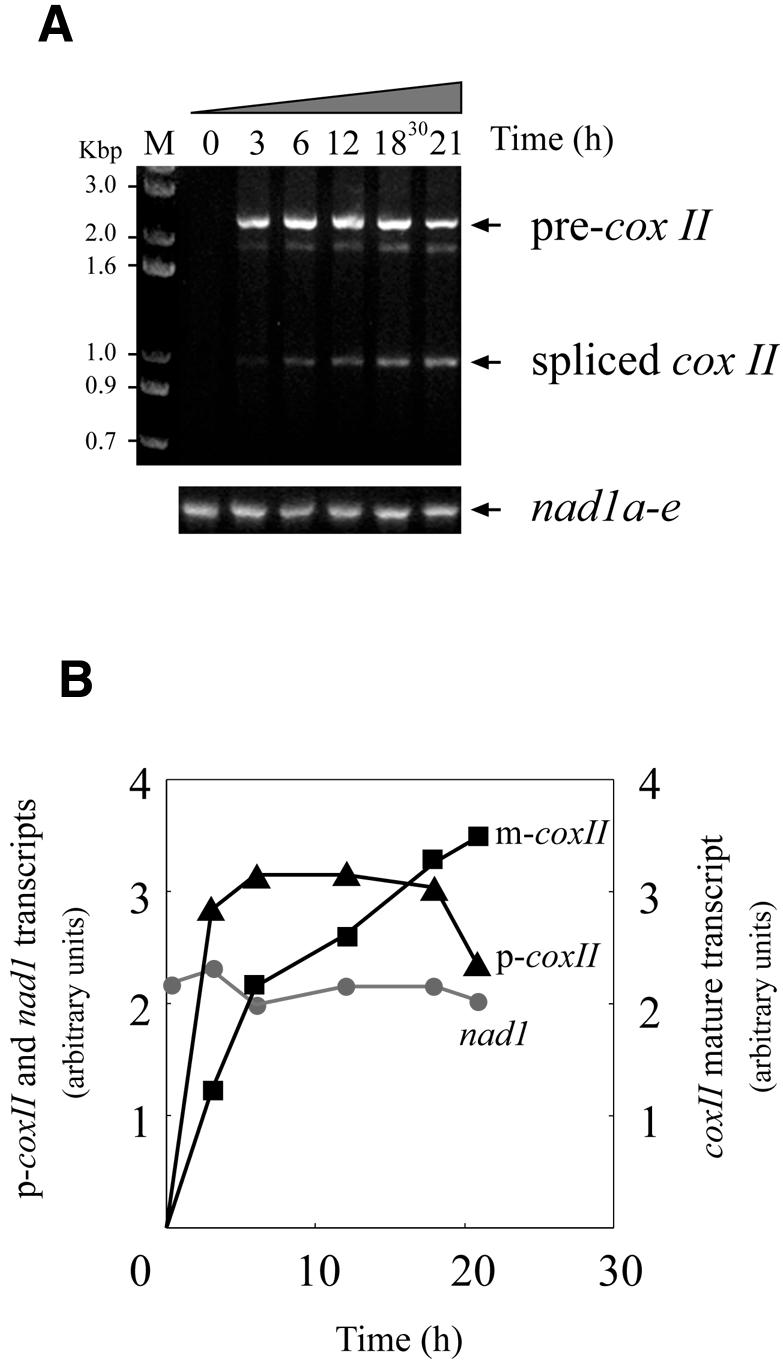
Time course of transcription and maturation of the exogenous cox II. Purified wheat embryo mitochondria were treated as described in Figure 3. (A) Mitochondria electroporated in the presence of pCox II were incubated for different periods of time. After incubation with DNase I, the RNA was extracted and total cDNA was synthesized with random primers. The target cox II sequence was revealed by PCR using primers 1 and 3. A control PCR using endogenous transcripts (lower panel) was carried out with primers 6 and 7 (see Fig. 3). (B) Kinetics of accumulation of the precursor (p-cox II) and mature (m-cox II) mRNAs from the introduced coxII gene. Circles represent the endogenous mature nad1 transcripts. The ethidium bromide fluorescence signal obtained in (A) was scanned and plotted using arbitrary units (see Fig. 2).
The exogenous transcripts are edited
To analyze the editing process, the RT–PCR products from the precursor and mature cox II transcripts were cloned and sequenced. As shown in Figure 7, the transcripts from the introduced cox II gene were edited at 16 out of 18 naturally occurring editing sites. Two C residues in exon 2 (sites 482 and 638) were found unedited in the clones analyzed (Fig. 7B) When comparing the editing status of the precursor with the mature exogenous cox II transcripts, we found that all the spliced transcripts analyzed were strongly edited, while precursor molecules showed partial editing.
Figure 7.
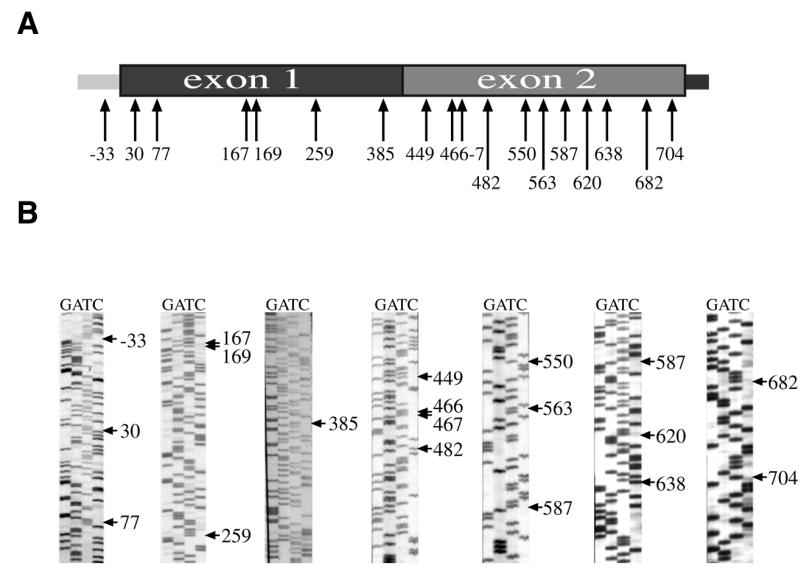
Editing sites in transgenic cox II transcripts. (A) The edited C residues found in the exogenous cox II transcripts after electroporation are indicated by arrows. The figures refer to the residue number with the A residue of the initiation codon as the first base. (B) The sequences concerning all the editing sites are shown.
DISCUSSION
The flow of information from DNA to proteins is exquisitely regulated to ensure an optimal degree of fidelity in the transmission of the encoded message. Several other steps have been described in addition to transcription and translation. Thus, precursor mRNAs require a processing step involving cleavage and ligation (splicing) to give a mature messenger. More recently a new process was described called RNA editing, which implies very specific changes of the mRNA sequence (1). In higher plant mitochondria, RNA editing is linked to C→U modifications, and a lesser extent to U→C changes, leading to amino acid changes as well as to the emergence of new initiation or stop codons (23,24). Multiprotein complexes are involved in the transcription, splicing and RNA editing steps. The specificity of all these processes is given by the recognition of particular sequences which are known to play a major role in transcription and splicing, while very little is known about sequence recognition and the enzymatic machinery in plant mitochondrial RNA editing. The difficulty in transforming plant organelles is a major difficulty in the study of RNA synthesis and maturation in this compartment. The goal of the present work was to optimize the entry of a given gene into wheat mitochondria to obtain a more detailed picture of the molecular and biochemical mechanisms occurring during transcription, splicing and RNA editing. A major finding in this work is that a transgene can be efficiently transcribed when incorporated into mitochondria by electroporation, and that these transcripts are faithfully processed and edited. Previous reports have described DNA delivery into isolated animal mitochondria as an approach towards the study of gene corrections in mitochondrial disorders (13). These authors described the protection of DNA after electroporation and the integrity of the organelle after the electric pulse. Using a similar approach, we have developed a new procedure of exogenous DNA delivery into isolated higher plant mitochondria.
The entry into mitochondria of a chimeric plasmid pLuc of 5.9 kb was followed by DNase I protection assay. A possible explanation for the intactness of the internalized DNA was that protection against DNase I digestion reflected the formation of membrane vesicles wrapping DNA during the electroporation process. In this eventuality, a concomitant loss of endogenous DNA should be observed. Under the optimal conditions we used, the amount of endogenous DNA was not affected by the electric pulse as shown by PCR amplification of endogenous mat-r gene. This indicates that it is essentially the entry of the exogenous DNA into mitochondria that is observed. Moreover, the fact that the chimeric gene harboring the luciferase gene resulted in an efficient transcription when driven by the wheat mitochondria cox II promoter strongly suggests that DNA is incorporated into the mitochondrial matrix, thus becoming accessible to the transcription machinery. The presence of a cob terminator sequence in our constructions was chosen for two reasons: first, to confer a higher stability to the transcript and secondly, to provide a specific foreign sequence to select the chimeric insert during RT–PCR analysis. The T.timopheevi 3′ cob sequence used in this study is postulated to form a double stem–loop structure to protect the transcripts from degradation (17,25). When this structure was omitted, no significant differences were found in the steady state amounts of chimeric transcripts (data not shown). More detailed studies are required to understand the role of these structures in plant mitochondrial transcripts. The cob 3′ sequence was kept in the construction used here, thereby allowing the specific PCR and RT–PCR amplification of the transgene and their transcripts.
To study gene expression, wheat mitochondria carrying the transgene were incubated for different periods of time. The incubation system required potassium and magnesium ions and all four ribonucleotides with GTP at a higher concentration than the other rNTPs, probably reflecting the additional functions of GTP in gene expression. Interestingly, the ATP regenerating system formed by succinate and ADP could not be substituted by the addition of exogenous ATP. This observation is important because it may be correlated with the integrity and the functionality of mitochondria after the stressful electric pulse event. As in the case of chloroplasts (14), gene transcription of plasmid constructs occurs in wheat organelles only after electroporation since no expression was observed by incubating the vector with mitochondria in the absence of an electric field pulse. The expression of the 6.6 kb pCox II plasmid was very efficient; the steady state was reached after 2–3 h incubation and remained stable up to 18 h. This is probably due to both the depletion of the nucleotide precursor pool and the gradual functional impairment of the organelle. During the 18 h of incubation, mature cox II mRNA levels increased steadily. In the case of some reporter genes like β-glucuronidase and chloramphenicol acetyltransferase, gene expression in transformed chloroplasts for incubation periods longer than 24 h has been described (14).
The fidelity of transgene expression was confirmed by the analysis of the sequence at the junction of exons 1 and 2. Indeed, the nucleotide sequence at this point was identical to that found in vivo (26). Under different conditions, our group and several others have been unable to produce in vitro self-splicing of group II introns from plant mitochondria (unpublished results). Our next step was to investigate the maturation of mitochondrial transcripts after electroporation. This approach should allow the detailed molecular study of the splicing process by site-directed mutagenesis. In a first attempt, we deleted the stem–loop structure corresponding to domain VI, which has been described as necessary for splicing (22). Mitochondria electroporated with the deleted construction gave rise to a precursor transcript unable to undergo splicing. A similar result was described in an in vivo chloroplast group II intron splicing model (27). Some plant mitochondrial transcripts are submitted either to cis-splicing, as in the case of the cox II gene used here, or to more complex situations as found in nad1 (28), nad5 (29) or nad2 (30), where transcript maturation combines cis- and trans-splicing events. We are currently studying the structural determinants involved in the splicing of wheat cox II transcripts.
RNA editing is a crucial step in plant mitochondria gene expression. Cox II mRNA presents 18 cytidine residues, susceptible to editing, scattered on both exons (26). We found that cox II transcribed in wheat mitochondria from the electroporated gene were edited at the expected C residues. This suggests that the editing machinery faithfully recognized de novo generated transgenic transcripts after electroporation. This is a further argument in favor of the functionality of isolated mitochondria following the electric field pulse. Interestingly, precursor RNA molecules were less edited than the spliced mRNA. In particular, residues 482 and 638 were found unedited in most of the clones analyzed; the reason for this situation is not clear and is being studied. The presence of partially edited molecules among the mature transcripts argues against the hypothesis that RNA editing takes place before splicing in the case of most of the editing sites in cox II mRNA. As in the case of transcription and splicing described above, electroporation of exogenous DNA may become an important tool to study the RNA editing process. We are currently focusing our attention on understanding how the editing machinery can recognize the cis-elements surrounding the C residues involved in plant mitochondria RNA editing.
The present results show that wheat mitochondria electroporation should be extremely useful in future studies on plant organelle gene expression. Thus, access to mitochondrial transcription, RNA processing and RNA editing can be attained in one experiment. Moreover, all these events can be followed in a situation closer to that found under in vivo conditions. As described here for the mitochondrial cox II gene, the introduced plasmid is recognized by the mitochondrial machinery and transcribed, processed and edited, as are the endogenous mitochondrial genes. This strategy should help to answer many questions concerning mitochondrial gene expression, a process that still remains poorly understood because of the difficulty of generating transgenic plants with a recombinant chondriome.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Simon Litvak and Dr Dominique Bégu for helpful discussions and critical reading of the manuscript, and Ms Evelyne Sargos and Ms Beata Matusiak for technical assistance. This research was supported by the French Ministère de l’Enseignement Supérieur et de la Recherche, the Université Victor Segalen Bordeaux 2, French Ministère de l’Agriculture et de la Pêche, the Pôle Génie Biologique et Medical Aquitaine.
References
- 1.Brennicke A., Marchfelder,A. and Binder,S. (1999) RNA editing. FEMS Microbiol. Rev., 23, 297–316. [DOI] [PubMed] [Google Scholar]
- 2.Bégu D., Graves,P.V., Domec,C., Arselin,G., Litvak,S. and Araya,A. (1990) RNA editing of wheat mitochondrial ATP synthase subunit 9: direct protein and cDNA sequencing. Plant Cell, 2, 1283–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hernould M., Suharsono,S., Litvak,S., Araya,A. and Mouras,A. (1993) Male-sterility induction in transgenic tobacco plants with an unedited atp9 mitochondrial gene from wheat. Proc. Natl Acad. Sci. USA, 90, 2370–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanic-Joyce P.J. and Gray,M.W. (1991) Accurate transcription of a plant mitochondrial gene in vitro. Mol. Cell. Biol., 11, 2035–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mulligan R.M., Leon,P. and Walbot,V. (1991) Transcriptional and posttranscriptional regulation of maize mitochondrial gene expression. Mol. Cell. Biol., 11, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rapp W.D. and Stern,D.B. (1992) A conserved 11 nucleotide sequence contains an essential promoter element of the maize mitochondrial atp1 gene. EMBO J., 11, 1065–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rapp W.D., Lupold,D.S., Mack,S. and Stern,D.B. (1993) Architecture of the maize mitochondrial atp1 promoter as determined by linker-scanning and point mutagenesis. Mol. Cell. Biol., 13, 7232–7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Binder S., Hatzack,F. and Brennicke,A. (1995) A novel pea mitochondrial in vitro transcription system recognizes homologous and heterologous mRNA and tRNA promoters. J. Biol. Chem., 270, 22182–22189. [DOI] [PubMed] [Google Scholar]
- 9.Lupold D.S., Caoile,A.G. and Stern,D.B. (1999) Genomic context influences the activity of maize mitochondrial cox2 promoters. Proc. Natl Acad. Sci. USA, 96, 11670–11675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Araya A., Domec,C., Begu,D. and Litvak,S. (1992) An in vitro system for the editing of ATP synthase subunit 9 mRNA using wheat mitochondrial extracts. Proc. Natl Acad. Sci. USA, 89, 1040–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blanc V., Litvak,S. and Araya,A. (1995) RNA editing in wheat mitochondria proceeds by a deamination mechanism. FEBS Lett., 373, 56–60. [DOI] [PubMed] [Google Scholar]
- 12.Yu W. and Schuster,W. (1995) Evidence for a site-specific cytidine deamination reaction involved in C to U RNA editing of plant mitochondria. J. Biol. Chem., 270, 18227–18233. [DOI] [PubMed] [Google Scholar]
- 13.Collombet J.M., Wheeler,V.C., Vogel,F. and Coutelle,C. (1997) Introduction of plasmid DNA into isolated mitochondria by electroporation. A novel approach toward gene correction for mitochondrial disorders. J. Biol. Chem., 272, 5342–5347. [DOI] [PubMed] [Google Scholar]
- 14.To K.Y., Cheng,M.C., Chen,L.F. and Chen,S.C. (1996) Introduction and expression of foreign DNA in isolated spinach chloroplasts by electroporation. Plant J., 10, 737–743. [DOI] [PubMed] [Google Scholar]
- 15.Johnston F.B. and Stern,H. (1957) Mass isolation of viable wheat embryos. Nature, 179, 160–161. [DOI] [PubMed] [Google Scholar]
- 16.Leaver C.J., Hack,E. and Forde,B.G. (1983) Protein synthesis by isolated plant mitochondria. Methods Enzymol., 97, 476–484. [DOI] [PubMed] [Google Scholar]
- 17.Saalaoui E., Litvak,S. and Araya,A. (1990) The apocytochrome b from an alloplasmic line of wheat (T.aestivum, cytoplasm-T.timopheevi) exists in two differently expressed forms. Plant Sci., 66, 237–246. [Google Scholar]
- 18.Sambrook J.E., Fritsch,F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 19.Bégu D., Mercado,A., Farré,J.C., Moenne,A., Holuigue,L., Araya,A. and Jordana,X. (1998) Editing status of mat-r transcripts in mitochondria from two plant species: C-to-U changes occur in putative functional RT and maturase domains. Curr. Genet., 33, 420–428. [DOI] [PubMed] [Google Scholar]
- 20.Farré J.C. and Araya,A. (1999) The mat-r open reading frame is transcribed from a non-canonical promoter and contains an internal promoter to co-transcribe exons nad1e and nad5III in wheat mitochondria. Plant Mol. Biol., 40, 959–967. [DOI] [PubMed] [Google Scholar]
- 21.Michel F., Umesono,K. and Ozeki,H. (1989) Comparative and functional anatomy of group II catalytic introns—a review. Gene, 82, 5–30. [DOI] [PubMed] [Google Scholar]
- 22.Michel F. and Ferat,J.L. (1995) Structure and activities of group II introns. Annu. Rev. Biochem., 64, 435–461. [DOI] [PubMed] [Google Scholar]
- 23.Araya A., Bégu,D. and Litvak,S. (1994) RNA editing in plants. Physiol. Plant., 91, 543–550. [Google Scholar]
- 24.Maier R.M., Zeltz,P., Kössel,H., Bonnard,G., Gualberto,J.M. and Grienenberger,J.M. (1996) RNA editing in plant mitochondria and chloroplasts. Plant Mol. Biol., 32, 343–365. [DOI] [PubMed] [Google Scholar]
- 25.Kaleikau E.K., Andre,C.P. and Walbot,V. (1992) Structure and expression of the rice mitochondrial apocytochrome b gene (cob-1) and pseudogene (cob-2). Curr. Genet., 22, 463–470. [DOI] [PubMed] [Google Scholar]
- 26.Covello P.S. and Gray,M.W. (1990) Differences in editing at homologous sites in messenger RNAs from angiosperm mitochondria. Nucleic Acids Res., 18, 5189–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hollander V. and Kuck,U. (1999) Group II intron splicing in chloroplasts: identification of mutations determining intron stability and fate of exon RNA. Nucleic Acids Res., 27, 2345–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chapdelaine Y. and Bonen,L. (1991) The wheat mitochondrial gene for subunit I of the NADH dehydrogenase complex: a trans-splicing model for this gene-in-pieces. Cell, 65, 465–472. [DOI] [PubMed] [Google Scholar]
- 29.Pereira de Souza A., Jubier,M.F., Delcher,E., Lancelin,D. and Lejeune,B. (1991) A trans-splicing model for the expression of the tripartite nad5 gene in wheat and maize mitochondria. Plant Cell, 3, 1363–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morawala-Patell V., Gualberto,J.M., Lamattina,L., Grienenberger,J.M. and Bonnard,G. (1998) Cis- and trans-splicing and RNA editing are required for the expression of nad2 in wheat mitochondria. Mol. Gen. Genet., 258, 503–511. [DOI] [PubMed] [Google Scholar]