

Abstract
Background.
Prodrug-activator gene therapy with Toca 511, a tumor-selective retroviral replicating vector (RRV) encoding yeast cytosine deaminase, is being evaluated in recurrent high-grade glioma patients. Nonlytic retroviral infection leads to permanent integration of RRV into the cancer cell genome, converting infected cancer cell and progeny into stable vector producer cells, enabling ongoing transduction and viral persistence within tumors. Cytosine deaminase in infected tumor cells converts the antifungal prodrug 5-fluorocytosine into the anticancer drug 5-fluorouracil, mediating local tumor destruction without significant systemic adverse effects.
Methods.
Here we investigated mechanisms underlying the therapeutic efficacy of this approach in orthotopic brain tumor models, employing both human glioma xenografts in immunodeficient hosts and syngeneic murine gliomas in immunocompetent hosts.
Results.
In both models, a single injection of replicating vector followed by prodrug administration achieved long-term survival benefit. In the immunodeficient model, tumors recurred repeatedly, but bioluminescence imaging of tumors enabled tailored scheduling of multicycle prodrug administration, continued control of disease burden, and long-term survival. In the immunocompetent model, complete loss of tumor signal was observed after only 1–2 cycles of prodrug, followed by long-term survival without recurrence for >300 days despite discontinuation of prodrug. Long-term survivors rejected challenge with uninfected glioma cells, indicating immunological responses against native tumor antigens, and immune cell depletion showed a critical role for CD4+ T cells.
Conclusion.
These results support dual mechanisms of action contributing to the efficacy of RRV-mediated prodrug-activator gene therapy: long-term tumor control by prodrug conversion-mediated cytoreduction, and induction of antitumor immunity.
Keywords: antitumor immunity, prodrug activator gene therapy, retroviral replicating vector, Toca 511
Importance of the study
Prodrug-activator gene therapy with an RRV, Toca 511, has shown promising evidence of clinical benefit, radiographic responses, and increased survival in multicenter first-in-human phase I dose-escalation trials for recurrent high-grade glioma, and a phase IIB/III registrational trial is now under way. This study improves our understanding of mechanisms of action underlying the therapeutic efficacy of this approach, by demonstrating through real-time optical imaging that persistent infection by Toca 511 achieves long-term disease control through repeated cycles of prodrug-activated tumor mass reduction after a single virus injection in immunodeficient human glioma models. Furthermore, despite the low intrinsic immunogenicity of RRV, we definitively demonstrate for the first time that long-term cellular antitumor immunity develops after Toca 511–mediated prodrug-activator gene therapy in immunocompetent syngeneic glioma models. Together with its companion paper (Mitchell et al), this study provides an important conceptual foundation for developing further immunotherapeutic strategies with RRV.
Unique among replicating viruses being developed as anticancer agents, retroviral replicating vectors (RRVs) maintain viral persistence in tumors through the combined characteristics of nonlytic replication, stable integration into the cancer cell genome, and reduced viral immunogenicity.1–3 RRVs lack signals for active uptake into nuclei in quiescent cells and integrate into the host genome only when the nuclear membrane dissolves during mitosis4; they are highly restricted in normal tissues by innate and adaptive immune mechanisms, which are generally inactive in tumors.5,6 In pre-established intracranial glioma models, tumor-selective viral replication enables RRVs to achieve up to >98% transduction throughout entire tumor masses within a few weeks after injection at multiplicities of infection (MOI; ie, virus to glioma cell ratios) as low as 0.01.7–12 In contrast, tumor transduction levels were ≤1% after injecting the same dose of conventional nonreplicating retroviral vectors.7,13
While not intrinsically cytolytic, RRVs can be engineered with a prodrug-activator gene such as yeast cytosine deaminase (yCD), which generates 5-fluorouracil (5-FU) directly within cancer cells from its precursor 5-fluorocytosine (5-FC), an antifungal agent approved for clinical use and commonly used to treat CNS fungus infections.7,8 For clinical use, we developed a new RRV construct, Toca 511 (vocimagene amiretrorepvec), containing genome-stabilizing modifications in the virus backbone and a codon-optimized/temperature-stabilized yCD transgene.14 Toca 511 and Toca FC (extended release formulation of 5-FC) have been evaluated in first-in-human clinical trials of RRV-mediated prodrug-activator gene therapy in patients with recurrent high-grade glioma, with the vector administered via different routes of administration (clinicaltrials.gov: NCT01156584, NCT01470794, NCT01985256). To date, the treatment has been well tolerated and shows promising signs of clinical benefit, radiological responses, and increased survival compared with historical controls,15–17 and an international phase IIB/III randomized trial in recurrent high-grade glioma is now under way (NCT02414165).
In the present study, we further investigated mechanisms underlying the therapeutic efficacy of Toca 511 followed by 5-FC (Toca 511+5-FC) in intracranial xenograft models of human glioma and syngeneic models of murine glioma. Previous studies in immunodeficient models had indicated that long-term tumor control could be achieved but was dependent upon continued administration of prodrug using a cyclic dosing schedule.7,8 In contrast, prodrug could be discontinued after a few cycles without further recurrence in immunocompetent syngeneic models.9 However, the equivalence of these models in terms of virus replication kinetics and transduction efficiency had not been established, and the specific role of the immune system in contributing to therapeutic efficacy has not been evaluated to date. As noted, RRV itself is not highly immunogenic, and in fact is capable of suppressing interferon signaling.3 Hence, to date, it has not been clear whether RRV could activate antitumor immune responses.
Our present results now definitively demonstrate that the therapeutic efficacy of direct tumor cell killing after Toca 511+5-FC treatment, as demonstrated in the immunodeficient tumor model, is significantly augmented by induction of anticancer immunity in the immunocompetent tumor model. In the accompanying manuscript (Mitchell et al), we further investigate the mechanisms by which antitumor immune responses are generated. Taken together, our results indicate that repeated cycles of intratumoral prodrug conversion by RRV not only achieves long-term disease control but, through local “bystander effects” on immunosuppressive tumor stroma, also permits development of durable cellular immune responses directed against endogenous tumor antigens, contributing to apparent complete eradication of residual disease.
Materials and Methods
Please see the Supplementary material for additional details.
Cell Culture
The human glioma cell line U-87MG (American Type Culture Collection) and the mouse glioma cell line Tu-2449 were cultured as previously described.8–11,18
Viral Vector Plasmids
Plasmids encoding RRV (pAC3-GFP, pAC3-yCD2 [Toca 511]; Fig. 1A) and lentiviral vector pRRL-sin-cPPT-hCMV-FLuc2 have been described previously.14,19
Fig. 1.

Vector design, replication, and functionality. (A) Design of RRV AC3-GFP and Toca 511. Proviral versions of these vectors are shown; U3/R/U5, domains of viral long terminal repeat; gag/pol/ampho env, viral coding sequences. (B) Replication kinetics of Toca 511 in glioma cell lines. U-87 and Tu-2449 (4 × 105) cells were infected with Toca 511 (5 × 103 TU / 5 µL, MOI 0.005) on day 0. Infected cells were analyzed for integrated viral copy number by genomic qPCR every third day after virus infection. (C) MTS assay to assess 5-FC drug cytotoxicity. U-87 and Tu-2449 were fully transduced with AC3-GFP or Toca 511 before plating. Cell viability was measured by MTS dye conversion after exposure to 5-FC at the indicated concentrations, and normalized to nontreated cells. Note: “No vector” almost completely coincides with “AC3-GFP” for both cell lines.
Replication Kinetics of RRV In Vitro
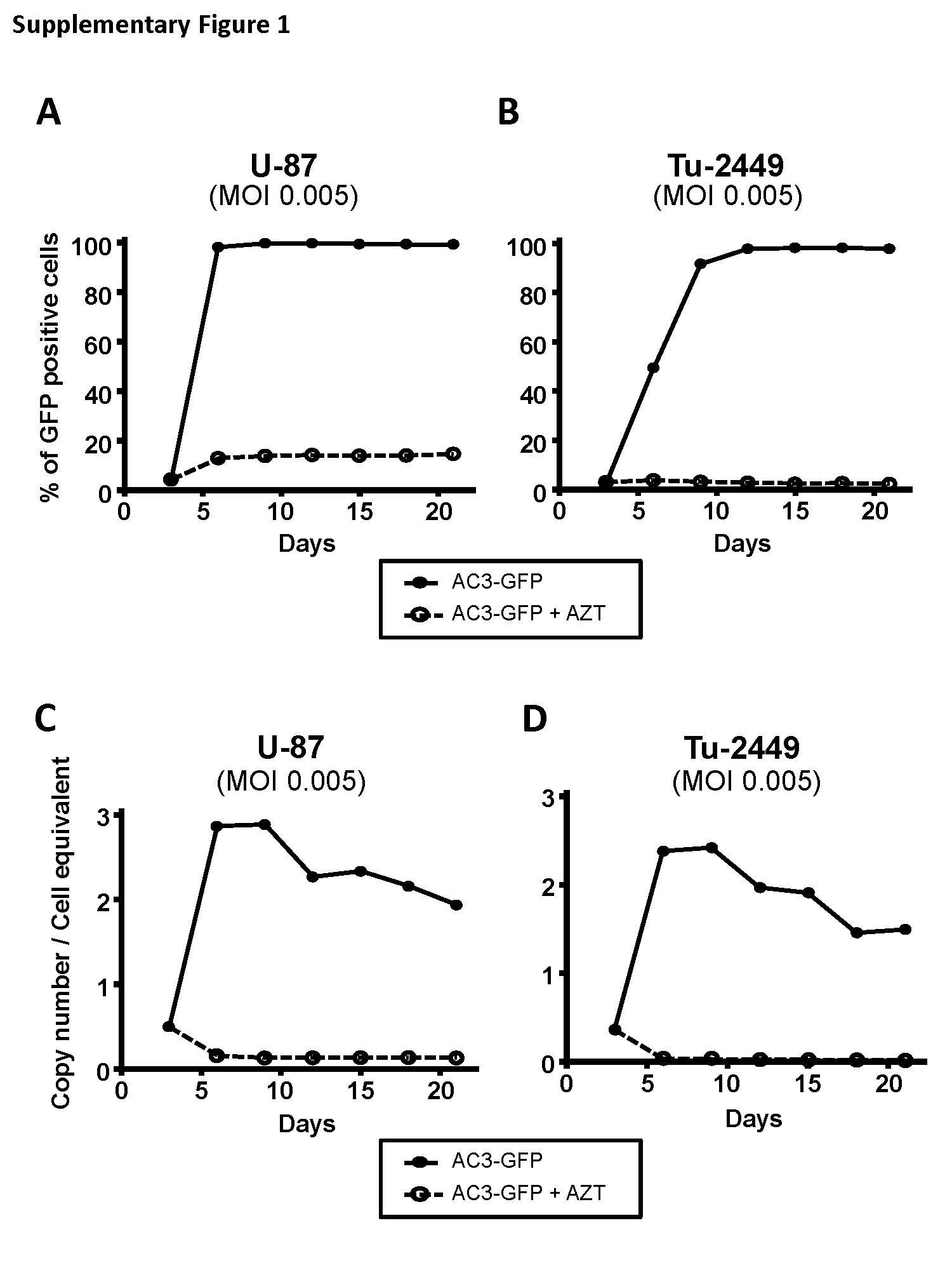
AC3-GFP or Toca 511 was used for infection of U-87 or Tu-2449 cells at MOI of 0.005 with Polybrene (Sigma). At serial time points, viral copy number was determined by genomic quantitative (q)PCR. As nonreplicating controls, 50 µmol 3ʹ-azide-3ʹ-deoxythymidine (AZT) was added to the culture medium to prevent virus spread.
Quantitative Real-Time PCR Analysis
Quantitative analysis of RRV copy number in the infected cells was performed as described previously19 (see also the Supplementary material).
Cytotoxicity Assay In Vitro
To assess drug cytotoxicity, cell viability was determined using 3-(4-5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy phenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay (Promega) in triplicate 96-well plates cultured with various concentrations of 5-FC. Cytotoxicity was determined by calculation of the absorbance of viable cells as measured against wells without 5-FC on day 9.
Prodrug-Activator Gene Therapy in Intracranial Tumor Models
All animal protocols and experiments were approved by the UCLA or Explora institutional animal care and use committees. Six- to 8-week-old female nude mice (Hsd:Athymic Nude-Foxn1nu; Envigo) or B6C3F1/J mice (Jackson Laboratory) were bred and maintained under specific pathogen-free conditions. To establish intracranial gliomas, U-87 (1 × 105) or Tu-2449 (1 × 104) cells were stereotactically injected into the right frontal lobe in mice, as described previously.8 In U-87 tumor models, Toca 511 (low dose: 3.4 × 104 transducing units [TU] or high dose: 2 × 106 TU) was given by stereotactic injection on day 5, and daily intraperitoneal (i.p.) administration of 5-FC (500 mg/kg once daily or twice daily) or phosphate buffered saline (PBS) was started on day 18 for 7 consecutive days. This 7-day cycle was repeated at intervals of every 1–2 weeks. In Tu-2449 tumor model studies conducted at UCLA, Toca 511 (2 × 106 TU) was given by stereotactic injection on day 4, and daily i.p. administration of 5-FC or PBS was started on day 10 for 7 consecutive days. This cycle was repeated at intervals of every 10 days. In independent Tu-2449 studies conducted at Tocagen, Toca 511 was administered as above at 3 × 106 TU followed by 4 cycles of 5-FC administered twice a day for 4 days every 2 weeks, or at a lower dose (1.6 × 104 TU), and continuous 5-FC administered once a day for 14 or 21 consecutive days.
Optical Imaging Analysis
Bioluminescent signals were analyzed 5 min after administration of D-luciferin (126 mg/kg, i.p.) in anesthetized mice, with a 1 min acquisition time on a charge-coupled camera system (IVIS Lumina II, Caliper Life Sciences) with Living Image software.
Tumor Rechallenge and Immunological Assays
Tumor rechallenge was performed on all cured mice from intracranial Tu-2449 tumor long-term survival studies, by subcutaneous inoculation with uninfected Tu-2449SC cells on day 0 (= days 105, 182, and 303 after initial tumor establishment, respectively) at low (1 × 105) or high (2 × 106) cell doses, and tumor growth was measured twice a week.
For enzyme-linked immunosorbent spot (ELISPOT) assay, purified splenocytes were stimulated with anti-CD3 and recombinant human interleukin 2 (rhIL-2) (100 U/mL) and plated in the Mouse IFN-γ [interferon gamma] ELISPOT Kit (1 × 104 cells/well; BD Bioscience) and the number of IFN-γ–producing cells counted with an ELISPOT plate reader (CTL Laboratories) at the Immuno/BioSpot and Cytometrics Core, UCLA.
Lymphocyte subset depletions were performed by i.p. injection of 50 µg anti-CD4 (GK1.5) or anti-CD8 (2.43), or 25 µg anti-NK1.1 (PK136) antibodies (Bio X Cell) on days −1, 1, 3, and then twice a week, in previously cured animals. Depletion was verified by immunophenotyping splenocytes from one animal from each group on days −1 and 8 before and after rechallenge.
xCELLigence Analysis
Cytotoxicity of stimulated versus unstimulated splenocytes, from previously cured mice or naïve B6C3F1/J mice, respectively, against Tu-2449SC cells was determined by xCELLigence analysis (ACEA Biosciences) (see also the Supplementary material). For an immune boost, previously cured mice were subcutaneously rechallenged with Tu-2449SC cells on day 0. On day 8, harvested splenocytes from immune-boosted cured mice were cultured without rhIL-2 overnight, then plated in triplicate with target cells on E-Plate 96 at several effector-to-target ratios. Up to 400 sweeps were run at 2-min intervals until data collection 24 h later.
Statistical Analysis
Statistical analysis was performed using Student’s t-test and 1-way or 2-way ANOVA. Survival data were analyzed according to the Kaplan–Meier method. P-values of <.05 were considered statistically significant in all analyses, performed with GraphPad Prism 5 software.
Results
RRVs Mediate Efficient Delivery and Functional Expression of Yeast Cytosine Deaminase Transgene in Glioma Cells
In preliminary studies, AC3-GFP, an RRV expressing the green fluorescent protein (GFP) reporter gene (Fig. 1A), was used to evaluate the equivalence of RRV replication and gene transfer efficiency in vitro in both human U-87 and murine Tu-2449 glioma cells. In both cell lines, RRV transduction levels increased from 0.5% to >90% within 5–10 days in vitro, and stably maintained 1–2 integrated vector copies per cell (Supplementary Fig. S1).
We employed Toca 511 (AC3-yCD2),14 a vector designed for clinical use which expresses an improved yCD2 prodrug-activator gene (Fig. 1A). To evaluate possible differences in replication kinetics between AC3-GFP and Toca 511, we also analyzed the integrated vector copy number by genomic qPCR analysis of cells initially infected with Toca 511 at an MOI of 0.005, using primers targeting the amphotropic envelope gene, at serial time points after infection in the presence or absence of AZT in vitro. In both U-87 and Tu-2449 glioma cells, the time course of changes in Toca 511 vector copy numbers was similar to that of AC3-GFP, showing an initial rapid increase followed by gradual decrease to a plateau level (Fig. 1B). In U-87 human glioma cells, the vector copy number increased rapidly to reach >10 copies/cell by day 6, followed by gradual decrease to plateau at ~2 copies/cell within 15 days. In Tu-2449 murine glioma cells, the viral copy number increased rapidly to reach >4 copies/cell within 9 days, followed by a gradual decrease to plateau at ~3 copies/cell within 15 days.
As the CD enzyme converts the well-tolerated prodrug 5-FC into the antimetabolite 5-FU intracellularly within infected cancer cells, in vitro cytotoxicity was examined after exposure of fully transduced glioma cells to 5-FC at various concentrations (Fig. 1C). Control U-87 cells transduced with AC3-GFP or no vector showed no loss of viability at concentrations of 5-FC up to 10 mM 5-FC treatment. In contrast, the viability of Toca 511 transduced cells was reduced to below 40% even after exposure to only 0.1 mM 5-FC, and complete cell killing was observed after 9 days of exposure to 10 mM 5-FC. Similar results were obtained in Tu-2449 cells transduced with the Toca 511 vector. Control Tu-2449 cells showed no loss of viability up to concentrations of 1 mM, whereas the viability of Toca 511 transduced cells was reduced over 70% even after exposure to only 0.01 mM of 5-FC, and 95% cell killing was achieved at 1 mM 5-FC.
Toca 511 and Cyclic 5-FC Achieve Long-Term Survival in Intracranial Glioma Models
We previously showed in the intracranial U-87 xenograft model in athymic mice that a single dose of a preclinical prototype RRV expressing the CD prodrug-activator gene followed by continuous administration of 5-FC for over 2 weeks resulted in doubling of median survival time, but animals all eventually succumbed to tumor recurrence.8 In contrast, multiple cycles of 5-FC could achieve prolonged survival, which we have now confirmed using the clinical vector Toca 511. After establishment of intracranial U-87 gliomas, 2 × 106 TU of Toca 511 was given by stereotactic injection at the same coordinates, and multicycle daily 5-FC prodrug treatment was commenced on day 18 for 7 consecutive days at intervals of 1–2 weeks. As expected, none of the mice in the control groups, either untreated or treated with vector only without prodrug, survived longer than 38 days. In contrast, the group treated with Toca 511+5-FC showed significant survival benefit (P < .0001) and 87.5% survival for over 120 days (Fig. 2A).
Fig. 2.

Survival benefit of RRV gene therapy in intracranial glioma models. (A) U-87 model. Toca 511+5-FC showed significantly increased survival compared with control groups (87.5% survival for >120 days; P < .0001). Shaded areas: daily 5-FC cycles. (B–D) Tu-2449 models. (B) Low dose: 1.6 × 104 TU. Daily 5-FC was commenced continuously on day 10 for either 14 or 21 consecutive days (Toca 511+5-FC × 14 or 21). Toca 511+5-FC showed significantly increased survival compared with Toca 511+PBS (40% survival for >240 days; P < .005). Hatched area: daily 5-FC × 14 days, shaded area: daily 5-FC × 21 days. (C) High dose: 3 × 106 TU. Twice-daily 5-FC was commenced on day 10 for 4 consecutive days at 2-week intervals (Toca 511+5-FC). Toca 511+5-FC showed significantly increased survival compared with Toca 511+PBS (82% survival for >160 days; P < .0001). Gray areas: twice-daily 5-FC cycles. (D) High dose: 2 × 106 TU. Daily 5-FC was commenced on day 10 for 7 consecutive days at 10-day intervals (Toca 511+5-FC). Toca 511+5-FC showed significantly increased survival compared with control groups (100% survival for >300 days; P < .0001). Gray areas: daily 5-FC cycles.
We then evaluated the survival of Toca 511+5-FC in intracranial Tu-2449 syngeneic models in immunocompetent B6C3F1/J mice. Tu-2449, derived from a spontaneous tumor in GFAP-v-src-transgenic mice, has morphological features and growth characteristics reminiscent of human gliomas.20,21 In previous studies, we found that Toca 511+5-FC could achieve eradication of intracranial Tu-2449 tumors in syngeneic hosts,9 but not in immunodeficient hosts.11
We hypothesized that local intratumoral production of 5-FU avoids drug-induced myelosuppression as seen with systemic 5-FU treatment, thereby permitting induction of antitumor immune responses. However, while the 5-FC prodrug itself is well tolerated in humans and only extended high-level exposure can result in clinically relevant myelotoxicity, it is also possible that prolonged production of 5-FU from the tumor might be counterproductive.
Therefore we investigated different dosing regimens for efficacy and induction of antitumor immunity. First, we examined Toca 511 at a low dose (104 TU), followed by continuous daily 5-FC for 14 or 21 days. This is equivalent to the dose used in previous U-87 studies with the preclinical vector followed by continuous 5-FC, and as there is a dose response effect for Toca 511 in the Tu-2449 model, this lower vector dose enables us to see either improvement or detriment compared with our previous 5-FC dosing regimens. Results in Fig. 2B show that these continuous dosing regimens give similar outcomes: significantly improved survival compared with controls (P < .005) and 40% survival for >240 days after a continuous 14- or 21-day single course of 5-FC. It should be noted that long-term survival is significantly improved compared with that seen after the equivalent dose of virus followed by continuous 5-FC in the U-87 model,8 again suggesting the critical role of an intact immune system in achieving long-term survival, and indicating that the 5-FC dosing regimen or Toca 511 dose can impact induction of antitumor immunity.
We next investigated a shorter and more intense cyclic dosing regimen with high dose Toca 511 (3 × 106 TU) followed by 5-FC for twice-daily 4-day cycles, spaced 10 days apart. This regimen also results in significantly improved survival compared with controls (P < .0001) and 82% survival for >160 days without further prodrug treatment after 4 cycles (Fig. 2C). Notably, previously published results from a 4-day on/10-day off dosing regimen using a lower dose of virus also showed long-term survival benefit, but in a lower percentage of animals,9 again suggesting that the initial effectiveness of prodrug-activator gene therapy may impact subsequent development of antitumor immunity.
Finally, we evaluated the survival of prodrug-activator gene therapy in the Tu-2449 model using the same cyclic 5-FC dosing regimen employed in the U-87 studies described above, that is, 2 × 106 TU Toca 511 injected into pre-established intracranial tumors followed by daily 5-FC prodrug treatment for 7 days at 1- to 2-week intervals. As expected, both control groups (no vector and Toca 511 followed by saline vehicle instead of 5-FC) showed similar results and did not survive beyond 28 days. However, the group treated with Toca 511 plus 5-FC for 7-day cycles showed significantly longer survival than control groups (P < .0001), achieving 100% survival for over 360 days even without further prodrug treatment after the third cycle (Fig. 2D).
In Vivo Bioluminescence Imaging of Tumor Response to Toca 511 and Multicycle 5-FC
To enable real-time assessment of the therapeutic effect of Toca 511 followed by multicycle 5-FC in individual animals, U-87 and Tu-2449 cells used in the optimized cyclic 5-FC studies above were pre-transduced with a replication-defective lentivirus vector expressing firefly luciferase. Stable expression of luciferase in these cells was confirmed by optical imaging in vitro (Fig. 3A), which showed a quantitative correlation between cell number and luminescent signal intensity (Supplementary Fig. S2). After intratumoral injection of Toca 511 into established U-87 intracranial gliomas, each mouse was analyzed by optical imaging weekly. In both tumor models, control mice that received saline instead of 5-FC showed tumor progression, as evidenced by increasing bioluminescence signal intensities. However, animals treated with Toca 511 followed by 5-FC exhibited stable disease or decreased tumor burden, and overall, average tumor growth was significantly inhibited compared with control groups after the initial cycle of 5-FC treatment in both U-87 (P < .0001) and Tu-2449 (P < .0001) tumor models (Fig. 3BC, Supplementary Fig. S3).
Fig. 3.

Bioluminescence imaging of tumor responses in vivo. Luciferase-marked U-87 and Tu-2449 gliomas were monitored by bioluminescence imaging in vivo. (A) Representative examples comparing imaging signals and hematoxylin and eosin–stained brain sections from the same animal. (B and C, upper panels) Imaging results from representative animals bearing U-87 (B) or Tu-2449 (C) intracranial tumors at different time points (Fig. 2A and D). (B and C, lower panels) Signal intensities (photons/sec/mouse) from each animal at different time points. (D) qPCR analysis to detect luciferase sequences from residual tumor cells in brain after Toca 511+5-FC in Tu-2449 model on day 361 (Fig. 2D). Detection limit was determined based on background signal from naïve Tu-2449 cells. Copy number/cell equivalent = luciferase copy number/1 × 105 cell genomes based on mouse β-actin copy number.
Subsequently, in the U-87 human glioma xenograft tumor model, the Toca 511+5-FC treated mice repeatedly showed reemergence of increased luminescence signal intensities during intervals between 5-FC treatment cycles, indicating tumor recurrence upon cessation of prodrug treatment after each cycle (Fig. 3B). Increased tumor burden as indicated by luminescence imaging was associated with characteristic behavioral changes, including hunched posture and reduced feeding and grooming activity, while reduction of tumor burden with each prodrug cycle restored normal activity without obvious ill effects. Thus, with repeated cycles of prodrug treatment, long-term tumor control was possible without incurring any adverse effects.
Consistent with imaging results, qPCR for both human genomic and RRV sequences showed residual signals in brain tissues in this immunodeficient model, supporting the existence of a reservoir of stably infected glioma cells enabling continued therapeutic benefit with prodrug administration upon tumor recurrence for at least 125 days until study termination (Supplementary Fig. S4).
In contrast, in the immunocompetent Tu-2449 model, bioluminescent signals of all treated tumors decreased rapidly after the initial cycle of 5-FC prodrug, stayed below detection threshold after the second cycle, and never recurred even without further prodrug treatment after the third cycle (Fig. 3C). At the molecular level, firefly luciferase and RRV sequences were both undetectable by qPCR analysis of brain tissues from long-term surviving animals even 280 days after the last prodrug injection (ie, at study termination on day 361 after initial tumor establishment) (Fig. 3D, Supplementary Fig. S4).
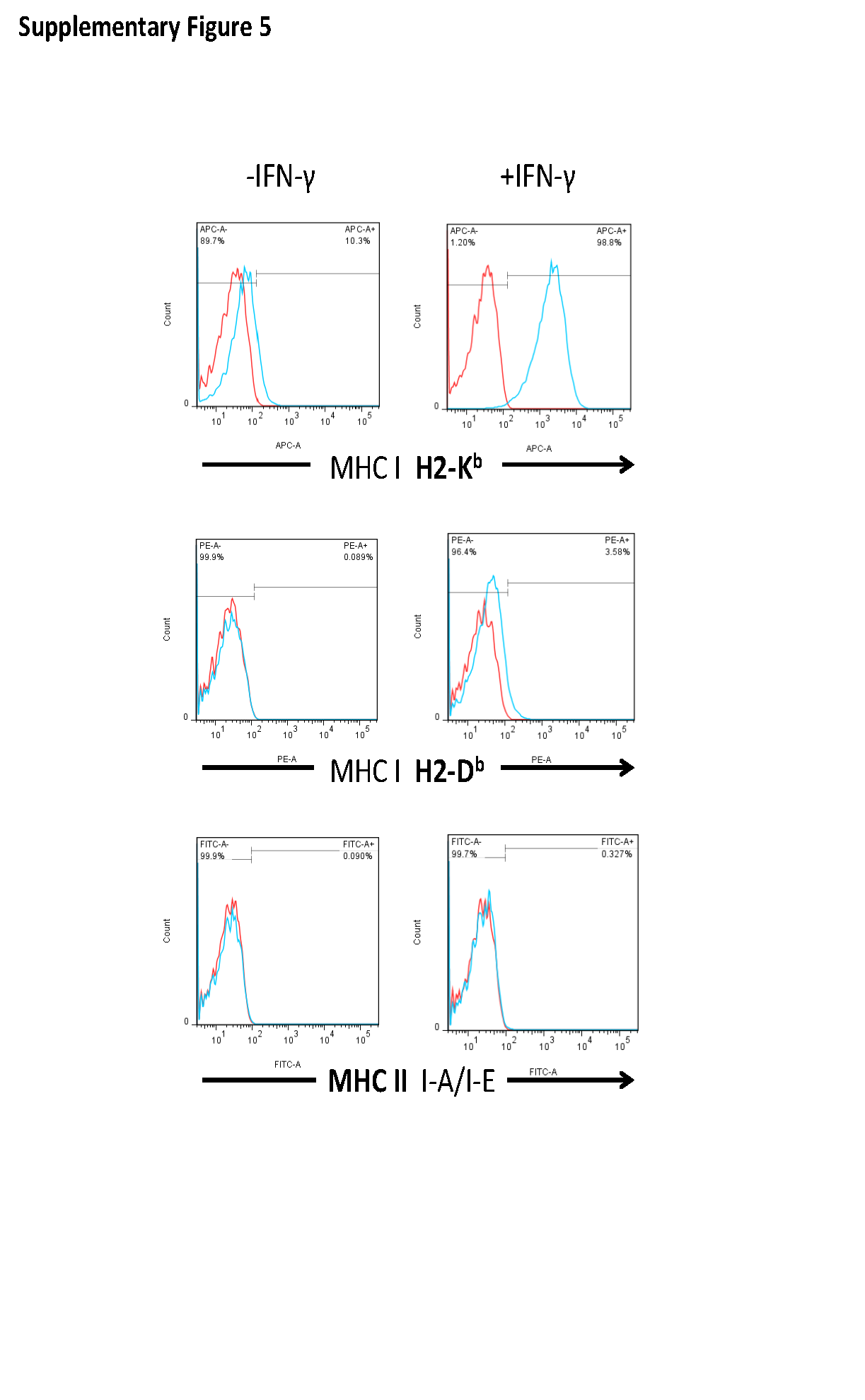
Immunological Memory Induced in Long-Term Survivors After Toca 511+5-FC Treatment
Major histocompatibility complex (MHC) antigens are not highly expressed on Tu-2449 cells at baseline, but MHC class I expression is upregulated by IFN-γ (Supplementary Fig. S5). Accordingly, tumor engraftment is robust in naïve syngeneic hosts, but pre-immunization leads to rejection,21 indicating that antitumor immunity can be elicited in this model. To evaluate whether antitumor immunity had developed in immunocompetent hosts with Tu-2449 intracranial gliomas after administration of different dosing regimens, we challenged surviving animals subcutaneously with a low dose (1 × 105) of uninfected Tu-2449SC cells (previously passaged as subcutaneous tumors) at earlier time points post-establishment of the original intracranial tumors (Fig. 4A). Some mice that had shown long-term survival with low-dose Toca 511/continuous 5-FC (Fig. 2B) completely controlled growth of subcutaneous tumors (4/8 treated vs 0/4 control) after rechallenge on day 105, although the mean tumor volume difference did not reach significance (Fig. 4B). However, mice that had been cured of brain tumor after receiving high-dose Toca 511/cyclic 5-FC (Fig. 2C) showed significant inhibition of subcutaneous tumor growth after rechallenge on day 182, compared with naïve animals (P < .005, Fig. 4B). To further evaluate the durability of antitumor immunity, we challenged cured mice after high dose Toca 511/cyclic 5-FC with a higher inoculum (2 × 106) of uninfected Tu-2449SC cells (ie, uninfected with either RRV or lentiviral luciferase marking vector) on day 303 post-original tumor implantation (Fig. 4A). Tumors successfully engrafted in all control animals, which had not previously been exposed to Tu-2449 tumors and were naïve to any prior treatment. In contrast, in the previously cured mice, although subcutaneous tumors were observed initially on day 7 after challenge, these tumors all spontaneously disappeared by day 14 without recurrence (Fig. 4C).
Fig. 4.

Tumor rechallenge studies. (A) Timeline of tumor rechallenge. Long-term survivors from intracranial Tu-2449 tumor studies (Fig. 2B–D) were subcutaneously inoculated with uninfected parental Tu-2449SC cells on day 0 (= days 105, 182, and 303 after initial tumor establishment, respectively) at low (1 × 105) or high (2 × 106) cell doses. As controls, Tu-2449SC cells were implanted into naïve B6C3F1/J mice. (B) Tu-2449SC tumor growth after low dose challenge (1 × 105 cells) in previously cured mice at early time points (day 105 or day 182, respectively) after continuous 5-FC or 4-day cyclic 5-FC regimens (Fig. 2B, C). Open circles: naïve controls. Closed triangles: cured mice from Fig. 2B. Closed circles: cured mice from Fig. 2C. Tumor growth was significantly inhibited in mice cured of gliomas with high-dose Toca 511 and cyclic 5-FC, compared with naïve controls (P = .0004). (C) Tu-2449SC cells (2 × 106 cells) implanted in naïve versus previously cured mice (from Fig. 2D) at a late time point (day 303) after 7-day cyclic 5-FC regimen. Tumors spontaneously regressed in cured mice (P = .0008). Open circles: naïve controls. Closed circles: cured mice.
Cellular immunity was evaluated in splenocyte populations obtained from these previously cured mice by measuring the number of IFN-γ producing cells with ELISPOT assay. Target cell–stimulated IFN-γ responses of splenocytes derived from cured mice were significantly higher than splenocytes obtained from naïve control mice (P = .0005; Fig. 5A). Furthermore, mixed lymphocyte–target cell reactions using splenocytes from previously cured animals, monitored by xCELLigence electrical impedance analysis, showed progressive killing of Tu-2449 target cells over time in an effector:target ratio–dependent manner, compared with splenocytes from naïve mice (Fig. 5B, C), and were similar to those obtained after stimulating splenocytes from naïve animals with INF-γ treated Tu-2449 cells (Supplementary Fig. S6). Taken together, these results indicate that RRV-mediated prodrug-activator gene therapy stimulated endogenous antitumor responses that could be reactivated >300 days later.
Fig. 5.

Cured animals show T cell–mediated antitumor responses. (A) ELISPOT assay quantitating the number of IFN-γ–producing cells per 104 splenocytes from cured mice in Fig. 2D versus naïve controls. (B, C) xCELLigence real-time cell impedance analysis showed effector cell dose-dependent antitumor activity of splenocytes from previously cured, immune-boosted mice against Tu-2449SC target cells at the effector:target (E:T) ratios shown (10:1, 30:1, 50:1). Results are shown as (B) real-time target cell growth curves, and (C) cell growth inhibition index values at the terminal 24-hour post-incubation time point, normalized to baseline values from wells plated with target cells only.
Specific lymphocyte subsets that contribute to tumor eradication upon tumor rechallenge were assessed by antibody-mediated depletion of CD4+, CD8+, and natural killer (NK) cells. Depletion was verified by flow cytometry of splenocytes isolated from a randomly selected animal from each depletion group, and tumor establishment versus rejection was compared between lymphocyte subset–depleted groups and nondepleted controls. Notably, tumors were still rejected in all previously cured animals subjected to NK or CD8+ T-cell depletion, as in nondepleted controls. In contrast, CD4+ T-cell or combination CD4+/CD8+ T-cell depletion prevented tumor rejection in previously cured animals (Fig. 6).
Fig. 6.

Lymphocyte depletion prevents tumor rejection in previously cured animals. Cured mice from Fig. 2D were injected with anti-CD4 and/or anti-CD8, or anti-NK antibodies for immunocyte depletion prior to subcutaneous challenge with parental Tu-2449SC cells. Tumor measurements were performed twice a week.
Discussion
The present studies underscore the advantages of RRV therapy, which integrates a prodrug-activator transgene into the cancer cell genome nonlytically, thereby converting infected cancer cells permanently into virus-producing cells. Previous studies relied solely on survival and endpoint immunohistochemical analyses to evaluate the efficacy of multicycle prodrug treatment in intracranial brain tumor models.8–11 This represents the first study to monitor RRV-mediated prodrug-activator gene therapy in intracranial glioma models using bioluminescence optical imaging, which allowed more detailed assessments of tumor response to each cycle of treatment by sequentially visualizing and quantifying tumor burden in the same individual over time, and by qPCR analysis for sensitive detection of residual disease.
In the U-87 immunodeficient intracranial tumor model, the group treated with Toca 511+5-FC showed a significant reduction of tumor burden compared with control groups after the initial cycle of 5-FC. However, bioluminescence imaging revealed the ongoing presence of residual disease, resulting in periodic tumor recurrence, and variable responses to repeated 5-FC were observed in individual animals (ie, tumor responses were either attenuated, lost, or recovered).
Theoretically, response to prodrug treatment depends on the number of RRV-transduced tumor cells undergoing division at the time of prodrug administration. Although transduction efficiencies were relatively uniform among infected tumors at the initial cycle of 5-FC treatment, variation in the number of residual infected cells at later time points may have caused variations in response to prodrug treatment. Nonetheless, relatively consistent control of tumor burden could be achieved over long durations with continued prodrug cycles, although not completely eliminated.
Viral replication kinetics and overall transduction levels were confirmed to be relatively equivalent between U-87 and Tu-2449 models. However, with optimal vector dose and prodrug scheduling in the Tu-2449 model, intracranial tumor signals became undetectable, without recurrence even upon cessation of prodrug, and tumor eradication was confirmed by qPCR analysis for residual tumor cells in brain tissues from long-term survivors. An obvious difference between these models is the presence of a functioning cellular immune system in the immunocompetent syngeneic host. As noted, previous studies showed that Toca 511+5-FC was not curative in intracranial Tu-2449 models established in immunodeficient mice.11 Furthermore, baseline levels of MHC expression are low in Tu-2449 cells in the absence of IFN-γ (Supplementary Fig. S7), and intact RRVs cause inhibition of type I interferon responses,3 so it was not clear whether antitumor immunity would develop after RRV-mediated gene therapy in this model.
In fact, our results indicate that the strong therapeutic efficacy with apparent tumor clearance observed in the immunocompetent model was achieved not only by cytoreduction, but also by the development of antitumor immune responses. Once denatured, RRV particles show immunostimulatory activity3 and hence may act as an adjuvant. Uninfected parental tumor cells were used to rechallenge long-term survivors after Toca 511+5-FC, so the rejection responses observed were not directed against viral or luciferase reporter epitopes, but presumably against endogenous tumor antigens released by Toca 511+5-FC–induced cell killing.
ELISPOT and xCELLigence assays confirmed the presence of cellular immunity against the parental tumor cells. As this was observed in animals that had survived for over 300 days without further prodrug treatment beyond the third cycle, these responses may be mediated by memory T cells. Notably, lymphocyte subset–depletion studies demonstrated that CD4+ T cells were required for tumor rejection, while NK and CD8+ cell depletion did not abrogate rejection. Recent data indicate the importance of CD4+ T effector cells in antitumor and antiviral immunity, and CD4+ cells may be even more efficient as cytolytic effectors than CD8+ cells, even with MHC class II–negative cancer cell targets.22 It has also recently been reported that cytolytic CD4+ and CD8+ T cells can cooperatively attack retrovirus-infected cells,23 as also suggested by the data from our studies suggesting that depletion of both CD4+ and CD8+ subsets together was more effective in inhibiting tumor rejection than depletion of CD4+ cells alone. While Tu-2449 cells do not appear to express class II MHC (Supplementary Fig. S5), it has been reported that CD4+ T cells may exert class II–unrestricted cytolytic effects via Fas signaling.24 An alternative possibility is that class II–negative tumor cells are killed by CD4+ Th1 cell-dependent IFN-γ stimulated M1-like macrophages.25 Of note, detailed studies of intratumoral cell populations show reduction of myeloid-derived suppressor cells and initial reduction followed by increased percentage of CD4+ cells overall, but with reduced Th2 and Th17 subsets, after Toca 511+5-FC treatment in Tu-2449 subcutaneous tumor models (Mitchell et al, accompanying manuscript).
In previous studies, a dose-response effect was noted, with fewer animals showing long-term survival with lower vector doses,9 suggesting a threshold for effective RRV infection. However, in the present studies employing high-dose vector, therapeutic results were robust with respect to 5-FC dosing schedules, as either daily 7-day cycles or twice-a-day 4-day cycles showed similar survival and tumor rejection. Furthermore, while longer-term continuous 5-FC dosing yielded some long-term survivors, cyclic 5-FC dosing schedules resulting in intermittent release of tumor antigens may be more effective at inducing long-term immune memory, analogous to prime-boost vaccination.
In summary, one major advantage of the use of a replicative integrating vector compared with other virotherapies is the ability to trigger repeated cycles of cytoreduction by systemic administration of a well-tolerated prodrug. A second major advantage as demonstrated here is the ability to induce antitumor immune responses after Toca 511+5-FC mediated tumor ablation. Importantly, prodrug conversion is intracellular and metabolic inactivation of 5-FU is rapid, so that cytotoxicity is confined locally to the tumor. Adverse systemic effects characteristic of conventional chemotherapy, such as myelosuppression, are avoided and the immune system remains intact. Antitumor immunity can be further enabled by local generation of 5-FU leading to ablation of myeloid-derived suppressor cells selectively within the tumor26 (Mitchell et al, accompanying manuscript).
As noted, Toca 511+Toca FC is now being evaluated in phase II/III trials for recurrent high-grade glioma, and we are investigating whether anticancer immunity is similarly induced and contributes to clinical activity. Given recent interest in cancer immunotherapy and immune checkpoint blockade, the pro-immunogenic effects of RRV-mediated prodrug conversion for tumor cell killing and tumor antigen release and for bystander killing of immunosuppressive tumor stromal cells may prove widely useful.
Supplementary Material
Supplementary material is available at Neuro-Oncology online.
Funding
This work was supported in part by U01NS059821 from the National Institute of Neurological Diseases and Stroke (to N.K.). ABC2 Foundation, the National Brain Tumor Society, the American Brain Tumor Association, the Musella Foundation, and Voices Against Brain Cancer provided financial support to Tocagen.
Conflict of interest statement. T.T.H., L.A.M., D.O., J.M.R., H.E.G. and D.J.J. are employees and/or shareholders of Tocagen Inc.; N.K. owns stock and receives consulting fees from Tocagen Inc.
Companion Paper. Please note that this manuscript is one of a pair, with its companion paper: Mitchell et al, “Toca 511 gene transfer and treatment with the prodrug, 5-fluorocytosine, promotes durable antitumor immunity in a mouse glioma model.”
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We dedicate this paper to the memory of our dear colleague, Dr Carol Kruse. We are grateful to Dr Christopher Logg for technical advice, and to Dr Carlos Ibanez and the UCLA Vector Core for vector production.
References
- 1. Dalba C, Klatzmann D, Logg CR, Kasahara N. Beyond oncolytic virotherapy: replication-competent retrovirus vectors for selective and stable transduction of tumors. Curr Gene Ther. 2005;5(6):655–667. [DOI] [PubMed] [Google Scholar]
- 2. Logg CR, Robbins JM, Jolly DJ, Gruber HE, Kasahara N. Retroviral replicating vectors in cancer. Methods Enzymol. 2012;507:199–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lin AH, Burrascano C, Pettersson PL, Ibañez CE, Gruber HE, Jolly DJ. Blockade of type I interferon (IFN) production by retroviral replicating vectors and reduced tumor cell responses to IFN likely contribute to tumor selectivity. J Virol. 2014;88(17):10066–10077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Miller DG, Adam MA, Miller AD. Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol Cell Biol. 1990;10(8):4239–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Butt AQ, Mills KH. Immunosuppressive networks and checkpoints controlling antitumor immunity and their blockade in the development of cancer immunotherapeutics and vaccines. Oncogene. 2014;33(38):4623–4631. [DOI] [PubMed] [Google Scholar]
- 6. Naik S, Russell SJ. Engineering oncolytic viruses to exploit tumor specific defects in innate immune signaling pathways. Expert Opin Biol Ther. 2009;9(9):1163–1176. [DOI] [PubMed] [Google Scholar]
- 7. Wang WJ, Tai CK, Kasahara N, Chen TC. Highly efficient and tumor-restricted gene transfer to malignant gliomas by replication-competent retroviral vectors. Hum Gene Ther. 2003;14(2):117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tai CK, Wang WJ, Chen TC, Kasahara N. Single-shot, multicycle suicide gene therapy by replication-competent retrovirus vectors achieves long-term survival benefit in experimental glioma. Mol Ther. 2005;12(5):842–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ostertag D, Amundson KK, Lopez Espinoza F, et al. Brain tumor eradication and prolonged survival from intratumoral conversion of 5-fluorocytosine to 5-fluorouracil using a nonlytic retroviral replicating vector. Neuro Oncol. 2012;14(2):145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang TT, Hlavaty J, Ostertag D, et al. Toca 511 gene transfer and 5-fluorocytosine in combination with temozolomide demonstrates synergistic therapeutic efficacy in a temozolomide-sensitive glioblastoma model. Cancer Gene Ther. 2013;20(10):544–551. [DOI] [PubMed] [Google Scholar]
- 11. Huang TT, Parab S, Burnett R, et al. Intravenous administration of retroviral replicating vector, Toca 511, demonstrates therapeutic efficacy in orthotopic immune-competent mouse glioma model. Hum Gene Ther. 2015;26(2):82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hlavaty J, Jandl G, Liszt M, et al. Comparative evaluation of preclinical in vivo models for the assessment of replicating retroviral vectors for the treatment of glioblastoma. J Neurooncol. 2011;102(1):59–69. [DOI] [PubMed] [Google Scholar]
- 13. Solly SK, Trajcevski S, Frisén C, et al. Replicative retroviral vectors for cancer gene therapy. Cancer Gene Ther. 2003;10(1):30–39. [DOI] [PubMed] [Google Scholar]
- 14. Perez OD, Logg CR, Hiraoka K, et al. Design and selection of Toca 511 for clinical use: modified retroviral replicating vector with improved stability and gene expression. Mol Ther. 2012;20(9):1689–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cloughesy TF, Landolfi J, Hogan DJ, et al. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci Transl Med. 2016;8(341):341ra375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aghi M, Vogelbaum MA, Kesari S, et al. AT-02. Intratumoral delivery of the retroviral replicating vector (RRV) Toca 511 in subjects with recurrent high grade glioma: interim report of a phase I study (NCT 01156584). Neuro Oncol. 2014;16(suppl 5):v8. [Google Scholar]
- 17. Vogelbaum MA, Kesari S, Kalkanis SN, et al. SURG-09: results of a dose escalation trial of Toca 511 with Toca Fc in recurrent HGG Undergoing repeat resection. Neuro Oncol. 2015;17(suppl 5):v216. [Google Scholar]
- 18. Tai CK, Wang W, Lai YH, et al. Enhanced efficiency of prodrug activation therapy by tumor-selective replicating retrovirus vectors armed with the Escherichia coli purine nucleoside phosphorylase gene. Cancer Gene Ther. 2010;17(9):614–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hiraoka K, Kimura T, Logg CR, et al. Therapeutic efficacy of replication-competent retrovirus vector-mediated suicide gene therapy in a multifocal colorectal cancer metastasis model. Cancer Res. 2007;67(11):5345–5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pohl U, Wick W, Weissenberger J, et al. Characterization of Tu-2449, a glioma cell line derived from a spontaneous tumor in GFAP-v-src-transgenic mice: comparison with established murine glioma cell lines. Int J Oncol. 1999;15(4):829–834. [DOI] [PubMed] [Google Scholar]
- 21. Smilowitz HM, Weissenberger J, Weis J, Brown JD, O’Neill RJ, Laissue JA. Orthotopic transplantation of v-src-expressing glioma cell lines into immunocompetent mice: establishment of a new transplantable in vivo model for malignant glioma. J Neurosurg. 2007;106(4):652–659. [DOI] [PubMed] [Google Scholar]
- 22. Perez-Diez A, Joncker NT, Choi K, et al. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood. 2007;109(12):5346–5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Johnson S, Eller M, Teigler JE, et al. Cooperativity of HIV-specific cytolytic CD4 T cells and CD8 T cells in control of HIV viremia. J Virol. 2015;89(15):7494–7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stalder T, Hahn S, Erb P. Fas antigen is the major target molecule for CD4+ T cell-mediated cytotoxicity. J Immunol. 1994;152(3):1127–1133. [PubMed] [Google Scholar]
- 25. Haabeth OA, Tveita AA, Fauskanger M, et al. How do CD4(+) T cells detect and eliminate tumor cells that either lack or express MHC class II molecules? Front Immunol. 2014;5:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vincent J, Mignot G, Chalmin F, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70(8):3052–3061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.