

ABSTRACT
CsrA is a global regulatory RNA binding protein that has important roles in regulating carbon metabolism, motility, biofilm formation, and numerous other cellular processes. IraD functions as an antiadapter protein that inhibits RssB-mediated degradation of RpoS, the general stress response and stationary-phase sigma factor of Escherichia coli. Here we identified a novel mechanism in which CsrA represses iraD translation via translational coupling. Expression studies with quantitative reverse transcriptase PCR, Western blotting, and lacZ fusions demonstrated that CsrA represses iraD expression. Gel mobility shift, footprint, and toeprint studies identified four CsrA binding sites in the iraD leader transcript, all of which are far upstream of the iraD ribosome binding site. Computational modeling and RNA structure mapping identified an RNA structure that sequesters the iraD Shine-Dalgarno (SD) sequence. Three open reading frames (ORFs), all of which are translated, were identified in the iraD leader region. Two of these ORFs do not affect iraD expression. However, the translation initiation region of the third ORF contains three of the CsrA binding sites, one of which overlaps its SD sequence. Furthermore, the ORF stop codon overlaps the iraD start codon, a sequence arrangement indicative of translational coupling. In vivo expression and in vitro translation studies with wild-type and mutant reporter fusions demonstrated that bound CsrA directly represses translation initiation of this ORF. We further established that CsrA-dependent repression of iraD translation occurs entirely via translational coupling with this ORF, leading to accelerated iraD mRNA decay.
KEYWORDS: CsrA, IraD, RpoS, translational coupling, translational control
IMPORTANCE
CsrA posttranscriptionally represses gene expression associated with stationary-phase bacterial growth, often in opposition to the transcriptional effects of the stationary-phase sigma factor RpoS. We show that CsrA employs a novel regulatory mechanism to repress translation of iraD, which encodes an antiadapter protein that protects RpoS against proteolysis. CsrA binds to four sites in the iraD leader transcript but does not directly occlude ribosome binding to the iraD SD sequence. Instead, CsrA represses translation of a short open reading frame encoded upstream of iraD, causing repression of iraD translation via translational coupling. This finding offers a novel mechanism of gene regulation by the global regulator CsrA, and since RpoS can activate csrA transcription, this also highlights a new negative-feedback loop within the complex Csr and RpoS circuitry.
INTRODUCTION
Bacteria sense and respond to environmental signals via global regulatory networks, which coordinate sweeping changes in gene expression. The Csr system, also called Rsm in some organisms, is one such network that globally controls gene expression posttranscriptionally. Depending on the particular species, the Csr/Rsm system regulates a variety of cellular processes, including carbon metabolism, motility, biofilm formation, quorum sensing, and virulence (1). CsrA, the central component of the Escherichia coli Csr system, is a homodimeric RNA binding protein containing two identical RNA binding surfaces. These two surfaces can simultaneously bind to two sites within a target transcript (2, 3). GGA is a critical motif in CsrA binding sites and is often present in the loop of short RNA hairpins (4, 5). CsrB and CsrC (CsrB/C) are two small RNAs (sRNAs) that function by antagonizing CsrA activity; each sRNA contains several CsrA binding sites and is capable of sequestering multiple CsrA dimers (4, 6). Transcription of csrB and csrC is activated by the BarA-UvrY two-component signal transduction system in response to short-chain-carboxylic-acid-containing metabolites (7, 8) and by ppGpp (9, 10). A membrane-bound GGDEF-EAL domain protein, CsrD, is required for initiation of CsrB/C decay by RNase E cleavage (11, 12). Glucose activates CsrB/C decay by binding of CsrD to the unphosphorylated form of EIIAGlc, which predominates during glucose uptake by the phosphotransferase system (PTS) (13). Thus, the availability of preferred carbon favors depressed CsrB/C levels and increased CsrA availability, while the combination of depletion of preferred carbon and accumulation of end products causes CsrB/C accumulation and CsrA sequestration. In addition, transcription of csrA is driven by five promoters, two of which utilize RpoS (σS) (14), the general stress response and stationary-phase sigma factor of E. coli. CsrA can also repress its own translation by binding to four sites in the leader RNA, one of which overlaps its Shine-Dalgarno (SD) sequence, thereby directly competing with ribosome binding (14). Thus, the level of available CsrA is tightly controlled in the cell. Negative feedback within the Csr circuitry is mediated via indirect CsrA-dependent activation of CsrB/C synthesis (15) and repression of csrD (11) and has been found to improve response times and signaling dynamics in the Csr system (16).
A global study employing transcriptome sequencing (RNA-seq) previously identified several hundred mRNAs bound by CsrA (9). In two cases, it was shown that bound CsrA alters the structure surrounding the SD sequence, leading to repression or activation of expression by affecting translation efficiency (17, 18). However, the most common CsrA-mediated regulatory mechanism involves CsrA binding to multiple GGA motif-containing sites, one of which overlaps the cognate SD sequence, such that bound CsrA represses translation initiation by directly occluding the ribosome binding site (1). Although in some instances CsrA binds with high affinity to the site overlapping the SD sequence, in other cases one RNA binding surface of the CsrA dimer binds to a high-affinity site, thereby bridging the other surface to a lower affinity site overlapping the SD sequence (3, 19). In addition, CsrA-mediated translational repression often leads to destabilization of the mRNA because the mRNA is then accessible to RNases (1, 19).
It is well documented that gene expression can be controlled by translational coupling, a process in which translation of a downstream cistron is at least partially dependent on translation of the cistron immediately upstream (20, 21). In some coupling mechanisms, translation of the upstream cistron disrupts an SD-sequestering hairpin that would otherwise block translation of the downstream cistron, while other coupling mechanisms involve overlapping stop and start codons of the two cistrons.
IraD functions as an antiadapter protein that prevents RssB adapter-mediated degradation of RpoS by the ClpXP protease (22). Induction of IraD expression occurs in response to DNA damage and during the transition to stationary phase via ppGpp-dependent promoters P1 and P2. Transcription from P1 occurs with RNA polymerase (RNAP) containing σ70, whereas transcription from P2 occurs with RNAP containing either σ70 or σS (Fig. 1) (23, 24). In this study, we found that CsrA regulates iraD expression by binding to iraD leader RNA. The iraD leader forms a strong SD-sequestering structure. In addition, an open reading frame (ORF) was identified whose stop codon overlaps the iraD start codon. We demonstrate that CsrA represses translation initiation of the ORF, leading to translational repression of iraD, exclusively by preventing translational coupling with the ORF. This finding expands the known interactions between two global regulators of stationary-phase gene expression and stress responses, CsrA and RpoS, and reveals a new feedback loop of the Csr circuitry.
FIG 1 .

Nucleotide sequence of the iraD promoter and leader region. −35 and −10 elements of promoters P1 and P2 and the corresponding transcription start sites (+1) are in bold. The ORF23, ORF47, and ORF27 translation start and stop codons are in bold type and marked with lines. The iraD Shine-Dalgarno (SD) sequence and start codon are also marked. Authenticated CsrA binding sites BS1 to BS4 are marked with a line, with critical GGA motifs in red. A fifth GGA motif (indicated by three asterisks [***]) overlaps the iraD SD sequence, but we determined in this study that CsrA does not bind to this region of the transcript. Numbering is with respect to the start of iraD translation.
RESULTS
CsrA represses iraD expression.
Visual inspection of the iraD leader region led to the identification of five potential CsrA binding sites, each containing a critical GGA motif (Fig. 1). We first examined expression of P1-P2-iraD'-'lacZ and P2-iraD'-'lacZ translational fusions in wild-type (WT) and CsrA-deficient (csrA::kan) strains. This csrA::kan allele contains a transposon insertion near the C-terminal coding sequence of csrA, resulting in a mutant CsrA protein that retains about 12% of the binding activity observed in the WT protein (25). Expression of both fusions in the WT background was low during exponential growth and then increased as cells entered stationary phase (Fig. 2A and B). Notably, in the csrA::kan mutant background, the levels of expression of the P1-P2-iraD'-'lacZ and P2-iraD'-'lacZ translational fusions were elevated about 7- and 3-fold, respectively, indicating that CsrA represses iraD expression. We also examined expression of a leader fusion in which the iraD P1-P2 promoter region was replaced with the lacUV5 promoter (PlacUV5) (Fig. 2A and C). Since CsrA does not affect transcription from PlacUV5 (9), an effect of the csrA::kan mutation is inferred to reflect CsrA-dependent posttranscriptional effects of the iraD leader RNA. As was seen with the P2-iraD'-'lacZ translational fusion, expression of the PlacUV5-iraD'-'lacZ leader fusion was ~3-fold higher in the csrA::kan strain (Fig. 2C). When similar experiments were performed with P1-iraD-lacZ and P2-iraD-lacZ transcriptional fusions in which the iraD leader region was absent, expression levels were similar in WT and csrA::kan strains, confirming that CsrA-dependent regulation of iraD expression is independent of the iraD promoters (Fig. 2A and D). We also performed quantitative reverse transcriptase PCR (qRT-PCR) experiments and found that iraD mRNA levels were 5-fold higher in the csrA::kan strain (Fig. 3A). In addition, a Western blot demonstrated that IraD protein levels increased ~5-fold in the csrA::kan strain during exponential phase (Fig. 3B). However, IraD reached the same levels in WT and csrA::kan strains in overnight cultures, suggesting that the physiological role of CsrA is to prevent premature accumulation of IraD. Taken together, these data suggest that CsrA directly represses iraD expression posttranscriptionally.
FIG 2 .

CsrA represses iraD expression. (A) Schematic representation of the fusions used in this analysis. Relative positions of the iraD promoters (P1 and P2), PlacUV5, and the start codons (ATG) driving translation of each fusion are shown. The region that includes the iraD promoter and leader is depicted with a thin black line, while the iraD and lacZ coding sequences are depicted with thick black and red lines, respectively. Dashed lines indicate that the corresponding sequence is absent. (B to D) β-Galactosidase activities (in Miller units) ± standard errors were determined throughout growth. Experiments were performed at least three times. Representative growth curves are shown with dashed lines (Klett). (B) Expression of P1-P2-iraD'-'lacZ and P2-iraD'-'lacZ translational fusions in wild-type (WT) and csrA::kan mutant strains. (C) Expression of the PlacUV5-iraD'-'lacZ leader fusion in WT and csrA::kan mutant strains. (D) Expression of P1-P2-iraD-lacZ and P2-iraD-lacZ transcriptional fusions in WT and csrA::kan mutant strains.
FIG 3 .
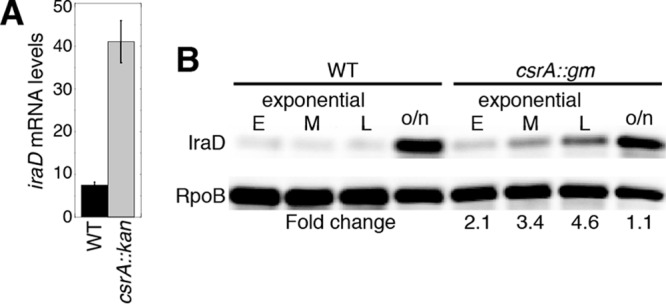
CsrA represses iraD mRNA and IraD protein levels. (A) qRT-PCR analysis of iraD mRNA levels in WT and csrA::kan mutant strains at mid-exponential phase. Values represent averages of results from 3 experiments ± standard deviations. (B) Western blot analysis of IraD levels in WT and csrA::gm strains grown to early (E), mid (M), and late (L) exponential phase. o/n, overnight cultures. Fold change values (csrA::gm/WT) are shown. Values are normalized to RpoB levels. Results are representative of two independent experiments.
CsrA binds specifically to iraD leader RNA.
Quantitative gel mobility shift assays were performed to determine whether CsrA bound to an iraD transcript extending from the P2 transcription start site into the early iraD coding sequence. Although this transcript contained five potential CsrA binding sites (Fig. 1), only one shifted complex was observed. A distinct band was observed between 2 and 128 nM CsrA, indicating that CsrA formed a tight complex with this transcript with an apparent Kd (dissociation constant) value of 15 nM (Fig. 4A), which is similar to the values seen with other known mRNA targets (1, 3, 14, 19). The specificity of CsrA-iraD RNA interaction was investigated by performing competition experiments with specific (iraD) and nonspecific (phoB) unlabeled RNA competitors. Unlabeled iraD was an effective competitor, whereas phoB was not (Fig. 4B). We next tested binding to a transcript in which the GGA motifs of BS2, BS3, and BS4 were all changed to CCA. These mutations eliminated CsrA binding (Fig. 4C). We conclude that CsrA binds to iraD with high affinity and specificity and that BS2, BS3, and/or BS4 function as CsrA binding sites.
FIG 4 .
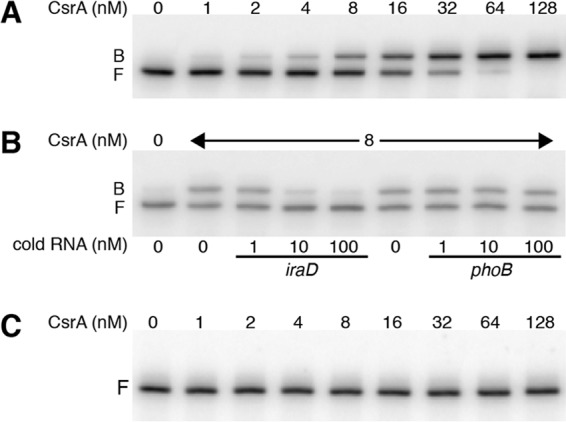
Gel mobility shift analysis of CsrA-iraD leader RNA interaction. (A to C) Labeled iraD RNA (0.1 nM) was incubated with the concentration of CsrA shown at the top of each lane. Positions of bound (B) and free (F) RNA are marked. Experiments were performed at least twice, and representative gels are shown. (A) CsrA forms a single high-affinity complex with WT iraD RNA. (B) RNA competition experiment demonstrating binding specificity. Labeled iraD RNA was incubated with the indicated concentration of unlabeled specific (iraD) or nonspecific (phoB) competitor RNA. (C) CsrA binding to iraD RNA is eliminated when the GGA motifs in BS2, BS3, and BS4 are changed to CCA.
CsrA-iraD RNA footprint experiments using RNase T1 as a single-strand G-specific probe were performed to identify the authentic CsrA binding sites in the iraD leader transcript. Bound CsrA resulted in strong protection of RNase T1-mediated cleavage of the G residues between −112 and −103 (BS2) and between −90 to −84 (BS3) and in weak protection at −78 and −77 (BS4) (Fig. 5A). Notably, bound CsrA did not protect the GGA sequence within the iraD SD sequence. We also performed CsrA toeprint experiments as an alternative method to observe positions of bound CsrA. A strong CsrA-dependent toeprint band was observed just downstream from BS2, whereas toeprints were not observed downstream from the other binding sites, suggesting that CsrA has the highest affinity for BS2 (Fig. 5B).
FIG 5 .

CsrA-iraD RNA footprint and toeprint analyses. (A) CsrA-iraD RNA footprint. Labeled iraD RNA was treated with RNase T1 in the presence of the CsrA concentration shown at the top of the lane. Partial alkaline hydrolysis (OH) and RNase T1 digestion (T1) ladders, as well as a control lane without RNase T1 treatment (C), are marked. Positions of BS2, BS3, BS4, the iraD start codon (Met), and the Shine-Dalgarno (SD) sequence are shown. Residues that were protected by bound CsrA from RNase T1 cleavage are marked (–). Numbering is with respect to the start of iraD translation. (B) CsrA-iraD RNA toeprint. The concentration of CsrA used is shown at the top of the lane. Positions of BS2, BS3, BS4, and the CsrA toeprint (carat) are marked. Lanes corresponding to results of sequencing to reveal A, C, G, and U residues are labeled.
Since the transcript in this analysis began at the P2 transcription start site, we were unable to obtain information for BS1 because the GGA motif was too close to the 5′ end of the transcript. Thus, footprint experiments were performed with a transcript whose 5′ end was extended 17 nucleotides (nt) upstream of the P2 transcription start site, which would mimic a transcript originating from P1. In this case, we observed weak protection of BS1 and BS4, strong protection of BS2 and BS3, and no protection of the GGA motif overlapping the iraD SD sequence (see Fig. S1 in the supplemental material). We also performed a footprint experiment with RNA containing a GGA-to-CGA mutation in BS2. In addition to complete loss of protection of BS2, this mutation caused a small reduction in the level of protection of BS3 (Fig. S1). We conclude that CsrA is capable of binding to four sites in the iraD leader RNA, with tight binding to BS2 and BS3.
CsrA-iraD RNA footprint analysis. Wild-type (WT) or BS2 mutant iraD RNA was treated with RNase T1 in the presence of the CsrA concentration shown at the top of the lane. Partial alkaline hydrolysis (OH) and RNase T1 digestion (T1) ladders, as well as a control lane without RNase T1 treatment (c), are shown. Positions of BS1, BS2, BS3, BS4, the iraD start codon (Met), and the Shine-Dalgarno (SD) sequence are shown. Residues that are protected by bound CsrA from RNase T1 cleavage are marked (–). Numbering is with respect to the start of iraD translation. The position of the BS2 GGA-to-CGA mutation is marked with a carat. Positions in which CsrA-mediated protection was lost in the mutant transcript are marked (*). Sequencing lanes revealing A, C, G, and U residues are labeled. Download FIG S1, TIF file, 3.4 MB (3.5MB, tif) .
Copyright © 2017 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
BS2 is critical for CsrA-mediated repression of iraD.
Since our gel shift assay indicated that mutating BS2, BS3, and BS4 eliminated CsrA binding, we tested the effect of single nucleotide substitutions in BS1 (GGA to GCA), BS2 (GGA to CGA), BS3 (GGA to GCA), or BS4 (GGA to GCA) in our reporter assay. The BS1 mutation resulted in elevated expression of the P1-P2-iraD'-'lacZ translational fusion but not the P2-iraD'-'lacZ translational fusion (Fig. 6), indicating that BS1 functions as a CsrA binding site only in transcripts that originate from P1, presumably because the GGA motif is too close to the 5′ end of transcripts originating from P2; there are only 3 nt upstream of this GGA motif in transcripts that initiate from P2 (Fig. 1). This interpretation is consistent with the systematic evolution of ligands by exponential enrichment (SELEX)-derived CsrA binding site consensus sequence containing 6 nt upstream and 3 nt downstream from the GGA motif (5). The mutation in BS2 resulted in a dramatic increase in expression of both the P1-P2-iraD'-'lacZ and P2-iraD'-'lacZ translational fusions throughout all stages of growth (Fig. 6A). In contrast, the BS3 and BS4 mutations resulted in only a modest increase in expression of the P1-P2-iraD'-'lacZ fusion, particularly in stationary phase (Fig. 6B). We also noted a modest decrease in expression of the P2-iraD'-'lacZ translational fusion containing the BS3 mutation in the overnight culture (Fig. 3B). These results for BS3 were surprising in the context of the footprint data showing that CsrA bound to both BS2 and BS3 with high affinity (Fig. 5A). This discrepancy is addressed below.
FIG 6 .

Effects of CsrA binding site mutations (BS1, BS2, BS3, and BS4) on expression of P1-P2-iraD'-'lacZ and P2-iraD'-'lacZ translational fusions. (A and B) β-Galactosidase activities (Miller units) ± standard deviations were determined in exponential-phase (exp), stationary-phase (stat), and overnight (o/n) cultures. Experiments were performed at least three times. (A) Wild-type (WT) and mutant fusions containing the indicated changes in BS1, BS2, BS3, and BS4. (B) WT and mutant fusions containing the indicated changes in BS1, BS3, and BS4 to emphasize small differences caused by these mutations.
Since BS2 was critical for repression of iraD expression, we examined the effect of this binding site in more detail. First, we compared the levels of expression of the WT and BS2 mutant P1-P2-iraD'-'lacZ translational fusions in WT and csrA::kan strains. While the BS2 mutation resulted in a more dramatic increase in expression than the csrA::kan mutation, the BS2 csrA::kan mutant combination exhibited the highest level of expression (Fig. 7A). These results indicate that despite complete loss of binding to BS2 as determined by footprinting, CsrA is still capable of repressing expression of the fusion by binding to one or more of the remaining sites. Furthermore, the finding that iraD expression was not fully derepressed by the csrA::kan mutation is consistent with the mutant CsrA protein encoded by the csrA::kan allele retaining ~12% of its WT RNA binding activity (25).
FIG 7 .

Effects of csrA::kan and BS2 mutations on iraD expression. (A) β-Galactosidase activities (Miller units) ± standard deviations of P1-P2-iraD'-'lacZ translational fusions were determined throughout growth. Experiments were performed at least three times. A representative growth curve is shown with a dashed line (Klett). Symbols: black, WT fusion WT csrA strain; orange, WT fusion csrA::kan strain; blue, BS2 mutant fusion WT csrA strain; gray, BS2 mutant fusion csrA::kan strain. (B) β-Galactosidase activities ± standard deviations of P1-P2-iraD'-'lacZ and P2-iraD'-'lacZ translational fusions in the presence of the indicated CsrA concentration were determined usinjg a PURExpress system. Experiments were performed at least three times. Values for samples without CsrA were set to 100. Symbols: black, WT P1-P2-iraD'-'lacZ fusion; orange, BS2 mutant P1-P2-iraD'-'lacZ fusion; blue, WT P2-iraD'-'lacZ fusion; gray, BS2 mutant P2-iraD'-'lacZ fusion; green, control pnp'-'lacZ translational fusion that is not repressed by CsrA (19). (C) Cultures were grown to mid-exponential phase prior to the addition of rifampin. Samples were harvested at the indicated times and then analyzed by primer extension for iraD'-'lacZ mRNA levels. mRNA half-lives (T1/2) are shown at the bottom of the gel. This experiment was performed twice.
We next used an in vitro coupled transcription-translation PURExpress system to determine whether CsrA represses iraD translation. Four different plasmids carrying iraD'-'lacZ translational fusions were used in this analysis, all of which were driven by identical T7 RNAP promoters. Two plasmids gave rise to transcripts that initiated at the P1 transcription start site, while the other two plasmids gave rise to transcripts that initiated at the P2 transcription start site. In each case, one of the plasmids contained the WT sequence and the other contained the BS2 mutation. Addition of CsrA in increasing concentrations resulted in 80% to 90% repression of the WT fusions, whereas only 50% repression was observed for the BS2 mutant fusions (Fig. 7B). A pnp'-'lacZ translational fusion that we had previously shown was not repressed by CsrA (19) served as a negative control. These results indicate that CsrA represses translation of iraD despite the fact that the 3′ end of the closest binding site (BS4) is 58 nt upstream from the iraD SD sequence (Fig. 1).
Since translational repression often leads to destabilization of mRNA, we determined the iraD mRNA half-life of the WT and BS2 mutant P1-P2-iraD'-'lacZ translational fusions in WT and csrA::kan strains. Since we were unable to detect the transcript of the WT fusion in the WT (csrA+) genetic background by Northern analysis, we performed primer extension experiments using a primer that hybridized to lacZ just downstream of the fusion junction. We found that the csrA::kan and BS2 mutations increased the iraD mRNA half-life (Fig. 7C), suggesting that CsrA-mediated translational repression reduces iraD mRNA stability.
Two ORFs that do not affect iraD expression are expressed in the iraD leader.
We identified sequences capable of encoding ORFs of 23 and 47 amino acids (aa) in the 280-nt stretch that lies between the P1 and P2 transcription start sites (Fig. 1, ORF23 and ORF47). To test whether either of these ORFs was expressed, we generated P1-ORF23'-'lacZ and P1-ORF47'-'lacZ translational fusions. Both of these fusions were expressed, with ORF23 expression being higher (Fig. S2A). To test whether expression of ORF23 or ORF47 affected expression of iraD, we changed each of the start codons to stop codons independently in the context of the P1-P2-iraD'-'lacZ translational fusion. Since expression of the mutant fusions was comparable to that seen with the WT fusion, we conclude that expression of these ORFs did not affect iraD expression (Fig. S2B). Thus, we did not explore these ORFs further.
Expression of ORF23 and ORF47 does not affect iraD expression. (A and B) β-Galactosidase activity data (Miller units) ± standard deviations were determined throughout growth. Experiments were performed at least three times. Representative growth curves are shown with dashed lines (Klett). (A) Expression of P1-ORF23′-′lacZ and P1-ORF47′-′lacZ translational fusions. (B) Expression of wild-type (WT) or mutant P1-P2-iraD′-′lacZ translational fusions. Stop indicates that the fusion contains a stop codon soon after translation initiation of ORF23 or ORF47. Download FIG S2, TIF file, 3.6 MB (3.7MB, tif) .
Copyright © 2017 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
CsrA-mediated translational repression of a third iraD leader ORF regulates iraD expression via translational coupling.
CsrA has been shown to repress translation of several genes (1), although in all cases at least one CsrA binding site is located in a position in which bound CsrA could directly block 30S ribosome binding or promote formation of an SD-sequestering hairpin. However, in the case of iraD, the 3′ end of the closest binding site is 58 nt upstream of the iraD SD sequence, a position that is too remote for CsrA to directly occlude ribosome binding. Using a combination of RNA structure predictions with Mfold data (26) and RNA structure mapping information (Fig. 5A, 0 μM CsrA), we identified an RNA secondary structure that sequesters the iraD ribosome binding site, including its SD sequence (Fig. 8A). We also identified a 27-aa ORF with an appropriately spaced SD sequence. Importantly, the ORF27 stop codon overlaps the iraD start codon, a sequence arrangement typical of translational coupling (Fig. 8A). Moreover, since BS3 overlaps the ORF27 SD sequence, translational coupling would provide an explanation for the unexpected result in which the GGA-to-GCA mutation in BS3 had little to no effect on iraD expression despite BS3 being a high-affinity site (Fig. 5A; Fig. 6; Fig. S1). Although we had predicted that this mutation would cause a substantial increase in expression of the iraD'-'lacZ translational fusions due to reduced CsrA binding, this was not observed. However, this mutation also disrupted the ORF27 SD sequence, which in turn would reduce ORF27 translation and therefore translational coupling with iraD, ultimately leading to reduced iraD expression.
FIG 8 .

CsrA represses translation of ORF27 and iraD via translation coupling. (A) Translational coupling model. CsrA-mediated repression of ORF27 translation leads to repression of iraD translation via translational coupling. Critical GGA motifs of CsrA binding sites BS1 to BS4 (red) and the iraD start codon are also marked. The ORF27 start and stop codons are boxed in yellow. The ORF27 and iraD SD sequences are marked. (B) β-Galactosidase activities ± standard deviations of WT and stop codon mutant ORF27'-'lacZ and iraD'-'lacZ translational fusions determined in vitro with PURExpress. "stop" indicates an ORF27 start-to-stop codon mutation. Experiments were performed at least three times. Values are reported as (1,000)(optical density at 420 nm [OD420]) in a 12.5-min reaction. Symbols: solid blue, wild-type (WT) ORF27'-'lacZ fusion; striped blue, stop codon mutant ORF27'-'lacZ fusion; solid black, WT iraD'-'lacZ fusion; striped black, stop codon mutant iraD'-'lacZ fusion. (C) β-Galactosidase activities (Miller units) ± standard deviations of P1-P2-ORF27'-'lacZ and P1-P2-iraD'-'lacZ translational fusions determined in WT (+) and csrA::kan mutant (–) strains during exponential-phase and stationary-phase growth. Experiments were performed at least three times. Symbols: blue, WT ORF27'-'lacZ fusion; black, WT iraD'-'lacZ fusion; red, iraD'-'lacZ fusion with an ORF27 stop codon mutation.
We performed experiments to test a model in which CsrA directly represses translation of ORF27, leading to repression of iraD translation via translational coupling. We first employed an in vitro PURExpress system to determine whether ORF27 was expressed. A plasmid containing an ORF27'-'lacZ translational fusion in which a T7 RNAP promoter gave rise to transcripts initiating from the P1 transcription start site was used for this analysis. High-level expression was observed from this fusion, whereas expression was greatly reduced when the ORF27 start codon was replaced with a UAG stop codon (Fig. 8B). The effect of the ORF27 stop codon mutation was also examined in the context of an iraD'-'lacZ translational fusion in which T7 RNAP initiated transcription from the P1 transcription start site. As with the ORF27'-'lacZ translational fusion, expression of the iraD'-'lacZ translational fusion was reduced by the ORF27 stop codon mutation (Fig. 8B). From these data, we conclude that ORF27 is expressed and that loss of ORF27 expression reduces expression of iraD.
We next determined the effect of CsrA on expression of a P1-P2-ORF27'-'lacZ translational fusion in vivo and found that expression was much higher in the csrA::kan strain, indicating that bound CsrA represses ORF27 translation (Fig. 8C). We also examined the effect of CsrA and the ORF27 stop codon mutation on expression of the P1-P2-iraD'-'lacZ translational fusion. As was observed previously (Fig. 2B), expression of the WT fusion was higher in the csrA::kan mutant background, especially in stationary phase (Fig. 8C). We also found that, relative to expression of the WT fusion, introduction of the ORF27 stop codon reduced expression of the P1-P2-iraD'-'lacZ translational fusion, particularly in the csrA::kan mutant background during stationary phase. The level of expression that remained in the ORF27 stop codon fusion reflects the level of iraD translation that occurs in the absence of translational coupling. Taken together, these data indicate that CsrA represses translation initiation of ORF27, leading to repression of iraD translation due to the loss of translational coupling. Moreover, the finding that loss of coupling by the ORF27 stop codon mutant had only a minor effect on expression in the WT (csrA+) background indicates that CsrA tightly represses ORF27 synthesis.
DISCUSSION
It is well established that RNA binding proteins and RNA structural features present in the 5′ leader region of mRNA can regulate translation initiation (20, 21). In some cases, the bound protein directly competes with ribosome binding (1, 27), while in others the protein promotes formation of an RNA structure that sequesters the SD sequence (17, 27). Since translating ribosomes can protect the mRNA from ribonucleases, translational repression often leads to decreased mRNA stability. In E. coli, CsrA represses translation initiation of many genes, typically by binding two or more sites in target transcripts, where one site overlaps the cognate SD sequence and/or initially translated region. In these instances, bound CsrA represses translation by directly occluding 30S ribosome binding (1, 3, 14, 19). In this study, we found that CsrA represses translation of iraD, leading to decreased stability of the transcript and reduced IraD protein levels (Fig. 2, 3, and 7). Although CsrA is capable of binding to four sites in the iraD leader RNA, tight binding to BS2 is critical for regulation (Fig. 4 to 6; see also Fig. S1 in the supplemental material). Our data also indicate that CsrA binding to BS1 contributes to regulation of transcripts derived from P1 (Fig. 6). Even though CsrA binds tightly to BS3 (Fig. 5), a mutation in BS3 had little effect on iraD expression (Fig. 6). The finding that CsrA represses translation of ORF27 provides an explanation for this discrepancy; the BS3 mutation simultaneously disrupts CsrA binding and inhibits ribosome binding by disrupting the ORF27 SD sequence (Fig. 1 and 8). Thus, the opposing consequences of this mutation essentially cancel each other out.
Translational coupling within a polycistronic transcript is a process in which translation of a downstream cistron depends at least partially on translation of a cistron immediately upstream. In some coupling mechanisms, translation of the upstream gene disrupts an SD-sequestering hairpin for the downstream cistron, thereby freeing the SD sequence for translation initiation by another ribosome. In other examples, coupling involves overlapping stop and start codons of the two cistrons, such that following termination of the upstream cistron, the same ribosome can reinitiate translation of the downstream cistron (20). Of particular interest, translation of iraD may be controlled by both of these coupling mechanisms (Fig. 8). Translation of ORF27 would disrupt the iraD SD-sequestering hairpin such that a different ribosome could initiate translation of iraD. Moreover, since the ORF27 stop codon overlaps the iraD start codon, the same ribosome could initiate translation of iraD once ORF27 synthesis is complete. In this context, CsrA-dependent repression of iraD translation is mediated entirely through its ability to directly repress ORF27 translation, which constitutes a novel CsrA-mediated regulatory mechanism.
RpoS, the general stress response and stationary-phase sigma factor, is controlled by a variety of mechanisms, including regulated proteolysis. IraD functions as an antiadapter protein that binds to the adapter protein RssB, thereby preventing RssB from targeting RpoS for degradation by the ClpXP protease (22). Thus, CsrA-dependent repression of iraD translation would lead to increased proteolysis of RpoS (Fig. 9). Furthermore, ppGpp induces iraD expression, causing RpoS levels to increase during the stringent response and at the transition to stationary phase (24). Because ppGpp also activates transcription of CsrB/C, the sRNA antagonists of CsrA (9, 10), our finding that CsrA represses iraD translation offers a new pathway for increasing IraD and RpoS levels in response to ppGpp, by decreasing CsrA availability. In addition to the connection of CsrA to the general stress response via IraD, RpoS is responsible for activating transcription of csrA, leading to increased csrA expression during the transition from exponential to stationary phase (14). Thus, iraD repression by CsrA apparently creates a negative-feedback loop affecting CsrA and RpoS expression. CsrA activity is also subject to negative feedback through its own effects on csrB/C transcription and CsrB/C RNA stability, leading to improved signaling response times in the Csr system (11, 15, 16). CsrA, RpoS, and ppGpp play complex roles in coordinating the expression of genes responsible for bacterial lifestyle decisions, e.g., between exponential growth and stationary-phase growth (28–30). The finely tuned interactions between these factors are evidence of a highly evolved regulatory network that is only beginning to be understood.
FIG 9 .
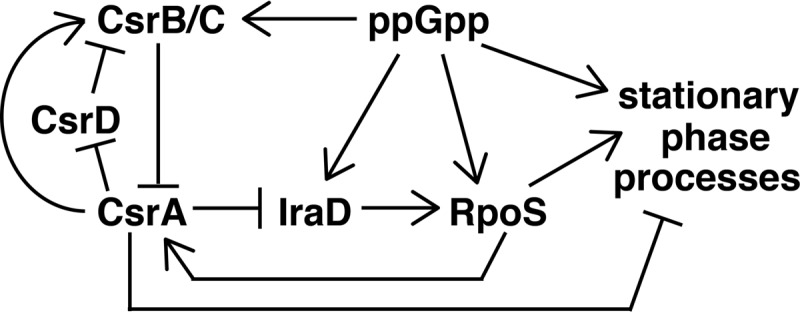
Regulatory circuitry of the Csr, stringent response, and general stress response systems. ppGpp activates transcription of csrB, csrC, iraD, and rpoS. CsrA represses IraD synthesis via coupling with ORF27. IraD stabilizes RpoS by inhibiting RssB-mediated degradation of RpoS. RpoS activates transcription of genes involved in stationary-phase processes. RpoS activates transcription of csrA, while CsrA represses stationary-phase processes. CsrB/C sRNAs bind to and sequester CsrA from its mRNA targets, while CsrA indirectly activates csrB/C expression. CsrD targets CsrB/C for degradation by RNase E, and CsrA indirectly represses csrD expression. See text for additional details.
MATERIALS AND METHODS
Bacterial strains and plasmids.
All bacterial strains used in this study are listed in Table 1. E. coli strain S17-1 λ pir+ (31) was used for plasmid construction performed by the conditional-replication, integration, and modular (CRIM) method (32). All numbering throughout the manuscript is with respect to the iraD translation initiation codon (Fig. 1). Plasmids pLFT, pLFX, and pUV5 (9) were used to generate translational, transcriptional, and leader fusions, respectively. Plasmid pLM12 contains a P1-P2-iraD'-'lacZ translation fusion (nt −498 to +20 relative to the iraD start codon cloned into the PstI and BamHI sites of pLFT). Plasmid pLM13 contains a P2-iraD'-'lacZ translation fusion (nt −234 to +20 cloned into the PstI and BamHI sites of pLFT). Plasmid pLM16 contains a P1-iraD-lacZ transcriptional fusion (nt −547 to −413 cloned into the PstI and EcoRI sites of pLFX). Plasmid pLM17 contains a P2-iraD-lacZ transcriptional fusion (nt −254 to −133 cloned into the PstI and EcoRI sites of pLFX). Plasmid pLM15 contains a PlacUV5-iraD'-'lacZ leader fusion (nt −138 to +20 cloned into the EcoRI and BamHI sites of pUV5) such that the iraD P1 and P2 promoter region was replaced with the lacUV5 promoter. Plasmid pLM30 contains a P1-ORF23'-'lacZ translational fusion (nt −498 to −376 cloned into the PstI and BamHI sites of pLFT). Plasmid pLM31 contains a P1-ORF47'-'lacZ translational fusion (nt −498 to −323 cloned into the PstI and BamHI sites of pLFT). Plasmid pHP25 contains a P1-P2-ORF27'-'lacZ translational fusion (nt −498 to −58 cloned into the PstI and BamHI sites of pLFT). Mutations in the CsrA binding sites (BS1, BS2, BS3, and BS4) in the P1-P2-iraD'-'lacZ and P2-iraD'-'lacZ translational fusions, or mutations of start codons to stop codons in P1-ORF23'-'lacZ, P1-ORF47'-'lacZ, and P1-P2-ORF27'-'lacZ translational fusions, were introduced using the QuikChange protocol (Agilent Technologies). WT and mutant fusions were integrated into the chromosomal λ att site of strain CF7789 as previously described (32). P1-mediated transduction was used to introduce the csrA::kan allele from TRMG1655 (7) into strains containing integrated WT and mutant fusions.
TABLE 1 .
E. coli strains used in this study
| Strain | Descriptiona | Source or reference |
|---|---|---|
| CF7789 | ΔlacI-lacZ (MluI) | M. Cashel |
| MG1655 | Prototrophic | M. Cashel |
| PLB2296 | CF7789 P1-P2-iraD'-'lacZ Apr | This study |
| PLB2297 | CF7789 csrA::kan P1-P2-iraD'-'lacZ Apr Kmr | This study |
| PLB2298 | CF7789 P2-iraD'-'lacZ Apr | This study |
| PLB2299 | CF7789 csrA::kan P2-iraD'-'lacZ Apr Kmr | This study |
| PLB2484 | CF7789 P2-iraD'-'lacZ (CsrA BS2, GGA to CGA) Apr | This study |
| PLB2485 | CF7789 P2-iraD'-'lacZ (CsrA BS3, GGA to GCA) Apr | This study |
| PLB2492 | CF7789 P1-P2-iraD'-'lacZ (CsrA BS2, GGA to CGA) Apr | This study |
| PLB2493 | CF7789 P1-P2-iraD'-'lacZ (CsrA BS1, GGA to GCA) Apr | This study |
| PLB2495 | CF7789 P2-iraD'-'lacZ (CsrA BS1, GGA to GCA) Apr | This study |
| PLB2497 | CF7789 P1-P2-iraD'-'lacZ (CsrA BS3, GGA to GCA) Apr | This study |
| PLB2498 | CF7789 P1-P2-iraD'-'lacZ (CsrA BS4, GGA to GCA) Apr | This study |
| PLB2499 | CF7789 P2-iraD'-'lacZ (CsrA BS4, GGA to GCA) Apr | This study |
| PLB2605 | CF7789 PlacUV5-iraD'-'lacZ Apr | This study |
| PLB2606 | CF7789 csrA::kan PlacUV5-iraD'-'lacZ Apr Kmr | This study |
| PLB2609 | CF7789 P1-iraD-lacZ Apr | This study |
| PLB2610 | CF7789 csrA::kan P1-iraD-lacZ Apr Kmr | This study |
| PLB2611 | CF7789 P2-iraD-lacZ Apr | This study |
| PLB2612 | CF7789 csrA::kan P2-iraD-lacZ Apr Kmr | This study |
| PLB2638 | CF7789 P1-ORF23′-'lacZ Apr | This study |
| PLB2639 | CF7789 P1-ORF47′-'lacZ Apr | This study |
| PLB2649 | CF7789 P1-iraD'-'lacZ (ORF47 start to stop, ATG to TAA) Apr | This study |
| PLB2650 | CF7789 P1-iraD'-'lacZ (ORF23 start to stop, ATG to TAA) Apr | This study |
| PLB2651 | CF7789 csrA::kan P1-P2-iraD'-'lacZ (CsrA BS2, GGA to CGA) Apr Kmr | This study |
| PLB2668 | CF7789 P1-P2-iraD'-'lacZ (ORF27 start to stop, ATG to TAG) Apr | This study |
| PLB2671 | CF7789 P1-P2-ORF27′-'lacZ Apr | This study |
| PLB2672 | CF7789 csrA::kan P1-P2-ORF27′-'lacZ Apr Kmr | This study |
| PLB2674 | CF7789 csrA::kan P1-P2-iraD'-'lacZ (ORF27 start to stop, ATG to TAG) Apr Kmr | This study |
| S17-1 λpir | recA thi pro hsdR-M + RP4: 2-tc::mu km::tn7 λpir+ | 31 |
| TRCF7789 | CF7789/csrA::kan Kmr | 7 |
| TRMG1655 | MG1655/csrA::kan Kmr | 7 |
| FLAG-iraD | MG1655/3xFLAG-iraD Kmr | This study |
| FLAG-iraD csrA | MG1655/3xFLAG-iraD Kmr csrA::gm Gmr | This study |
All fusions were integrated into the λ att site via the CRIM system (32). The iraD sequences in each fusion are indicated relative to the iraD translation initiation codon. P1-P2-iraD'-'lacZ translational fusions contain nt −498 to +20. P2-iraD'-'lacZ translation fusions contain nt −234 to +20. The P1-iraD-lacZ transcriptional fusion contains nt −547 to −413. The P2-iraD-lacZ transcriptional fusion contains nt −253 to −134. The PlacUV5-iraD'-'lacZ leader fusion contains nt −138 to +20. ORF23'-'lacZ translational fusions contain nt −498 to −376. ORF47'-'lacZ translational fusions contain nt −498 to −323. P1-P2-ORF27'-'lacZ translational fusions contain nt −498 to −58. Apr, ampicillin resistance Gmr, gentamicin resistance; Kmr, kanamycin resistance.
The C-terminal 3×FLAG-tagged iraD strain was constructed using the λ Red recombinase method as previously described (32). P1-mediated transduction was then used to introduce the csrA::gm allele into this strain. The csrA::gm allele was derived from the TRMG1655 csrA::kan strain, where the kanamycin marker was replaced with a gentamicin marker (12).
β-Galactosidase assay.
Bacterial cultures containing lacZ fusions were grown at 37°C in Luria-Bertani (LB) broth supplemented with 100 μg/ml ampicillin and 50 μg/ml kanamycin for csrA::kan strains. Cells were harvested at various times throughout growth. β-Galactosidase activity was measured as described previously (19). At least three independent experiments were performed for each strain.
Quantitative reverse transcriptase PCR (qRT-PCR).
qRT-PCR was carried out as previously described (33). Bacterial cultures grown to mid-exponential phase in LB at 37°C were added directly to RNAprotect Bacteria Reagent (Qiagen) according to the instructions of the manufacturer to stabilize RNA. Total RNA was purified with an RNeasy minikit (Qiagen), and DNA was removed with a Turbo DNA-free kit (Ambion). An IScript One-Step RT-PCR kit with SYBR green (Bio-Rad) was used to measure RNA or DNA control levels according to the manufacturer’s instructions. Reaction mixtures (20 µl) contained 100 ng of RNA (or an appropriate concentration of DNA standard), 1× SYBR green RT-PCR reaction mix, and a 300 nM concentration of each primer. The thermal cycling was done with an iCycler thermocycler (Bio-Rad) with 10 min reverse transcription at 50°C for 2 min at 95°C and then 45 cycles of PCR at 95°C for denaturation for 10 s and at 60°C for annealing, extension, and detection for 20 s. Melting curves were used to verify the specificity of the PCR product after the qRT-PCR reaction. ICycler iQ software (Bio-Rad) was used to determine RNA abundance relative to a standard curve of PCR products and 16S rRNA levels.
Western blotting.
Western blotting followed previously published protocols (10). Bacterial cultures were grown in LB at 37°C, and samples were collected throughout growth. Cell pellets were resuspended in Laemmli buffer and lysed with boiling and sonication. Samples were separated by SDS-PAGE on 15% bis-Tris gels. Proteins were transferred to 0.2-µm-pore-size polyvinylidene difluoride membranes with electroblotting performed using a Mini Trans Blot system (Bio-Rad). 3×FLAG-tagged IraD was detected with anti-FLAG M2 monoclonal antibody (Sigma), and RpoB was detected with anti-RpoB monoclonal antibody (Neoclone). Horseradish peroxidase-linked ECL anti-mouse IgG antibody (GE Healthcare) and SuperSignal West Femto chemiluminescent substrate (Pierce) were used to detect signal. Images were collected using a ChemiDoc XRS+ system (Bio-Rad) and quantified using Quantity One software (Bio-Rad).
Gel mobility shift assay.
Quantitative gel mobility shift assays followed a published procedure (14). His-tagged CsrA (CsrA-H6) was purified as described previously (34). WT and mutant RNAs started at the transcription initiation site from the iraD P2 promoter and were synthesized using an RNAMaxx kit (Agilent Technologies) and PCR-generated DNA templates. Gel-purified RNA was dephosphorylated and then 5′ end labeled using T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (7,000 Ci/mmol). Labeled RNAs were renatured by heating for 1 min at 85°C followed by slow cooling to room temperature. Binding reaction mixtures (10 µl) contained 0.1 nM labeled RNA, 10 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 100 mM KCl, 32.5 ng of yeast RNA, 7.5% glycerol, 20 mM dithiothreitol, 4 U of RNase inhibitor (Promega), 0.1 mg/ml xylene cyanol, and various concentrations of purified CsrA-H6. Previously frozen CsrA was thawed and activated by incubation for 15 min at 37°C. Reaction mixtures were incubated for 30 min at 37°C to allow CsrA-RNA complex formation and then fractionated through 15% polyacrylamide gels. Free and bound RNA species were visualized with a Typhoon 9410 phosphorimager (GE Healthcare). CsrA-RNA interactions were quantified as described previously (14).
Footprint assay.
CsrA-RNA footprint assays followed a published procedure (19). Labeled WT RNA beginning at the P2 transcription start site described for the gel mobility shift analysis was used. Additional WT and CsrA BS2 mutant RNAs started 17 nt upstream of the P2 transcription start site. Reaction mixtures were identical to those in the gel shift assay except that RNase inhibitor was omitted, the concentration of labeled RNA was increased to 10 nM, and 200 μg/ml acetylated bovine serum albumin (BSA) was added. After a 30-min incubation at 37°C to allow CsrA-RNA complex formation, RNase T1 (0.12 U) was added and incubation was continued for 15 min at 37°C. Reactions were stopped by adding 10 µl of Stop buffer (95% formamide, 0.025% SDS, 20 mM EDTA, 0.025% bromophenol blue, and 0.025% xylene cyanol) and cooling on ice. RNAs were fractionated through the use of standard 6% (vol/vol) polyacrylamide-8 M urea sequencing gels. Cleaved patterns were examined using a phosphorimager.
Toeprint assay.
CsrA-RNA toeprint assays followed a published procedure (14). The iraD RNA sequence used for toeprinting extends from the iraD P2 transcription start site through +20 relative to the iraD translation initiation codon and includes a 3′ extension derived from plasmid vector sequence. Gel-purified iraD RNA (150 nM) was hybridized in Tris-EDTA (TE) buffer to a 5′-end-labeled DNA oligonucleotide (150 nM) complementary to the vector-derived 3′ extension by heating for 3 min at 85°C followed by slow cooling to room temperature. Toeprint reaction mixtures (10 µl) contained 2 µl of the hybridization mixture (30 nM final concentration), 1.5 µM CsrA-H6, a 375 µM concentration of each deoxynucleoside triphosphate (dNTP), 10 mM dithiothreitol (DTT), and avian myeloblastosis virus (AMV) reverse transcriptase buffer. Previously frozen CsrA was thawed and activated by incubation for 15 min at 37°C. Mixtures were incubated for 30 min at 37°C to allow CsrA-RNA complex formation. AMV reverse transcriptase (0.3 U) was then added, and incubation was continued for 15 min at 37°C. Reactions were terminated by the addition of 6 µl of gel loading buffer. Samples were fractionated through the use of standard 6% (vol/vol) polyacrylamide–8 M urea sequencing gels. Toeprint patterns were visualized with a phosphorimager.
Coupled transcription-translation assay.
In vitro coupled transcription-translation assays using PURExpress (New England Biolabs) followed a published procedure (19). Plasmid pT7-P1iraD'-'lacZ contains a T7 promoter driving transcription of the translational fusion from the natural iraD P1 transcription start site (Fig. 1). Plasmid pT7-P1(BS2)iraD'-'lacZ is identical to pT7-P1iraD'-'lacZ except that it contains a GGA-to-CGA mutation in BS2. Similarly, plasmids pT7-P2iraD'-'lacZ and pT7-P2(BS2)iraD'-'lacZ contain a T7 promoter that drives transcription from the natural iraD P2 transcription start site. These four plasmids were used as templates for coupled transcription-translation reactions using the PURExpress in vitro protein synthesis kit according to the manufacturer’s instructions. Reaction mixtures contained a 20 nM concentration of plasmid DNA template and various concentrations of purified CsrA-His6 with 10 U of RNase inhibitor (Promega). The mixtures were incubated for 2 h at 37°C, and β-galactosidase activity was determined according to the manufacturer’s instructions.
mRNA half-life analysis.
Cultures were grown at 37°C in LB broth to exponential phase prior to the addition of rifampin (200 µg/ml) to prevent transcription initiation. After 1 min following rifampin addition, 0.8-ml aliquots were removed at various times and total cellular RNA was prepared by the hot phenol extraction method. To measure the iraD mRNA half-life, total cellular RNA (30 µg) was annealed to 2 pmol of an iraD-specific 5′ end-labeled primer in a 25-µl reaction mixture. Reaction mixtures were renatured by heating for 1 min at 80°C and slowly cooling to 25°C. Primer extension procedures were performed according to the manufacturer’s instructions (Superscript III; Life Technologies, Inc.) for 2 h at 50°C. The resulting products were fractionized through the use of 8% (vol/vol) polyacrylamide-8 M urea sequencing gels. Quantitative analysis was performed using a phosphorimager.
ACKNOWLEDGMENTS
We thank Susan Gottesman for pointing out the significance of ORF27, whose stop codon overlaps the iraD start codon.
This work was funded by NIH grant R01 GM059969 to T.R. and P.B. and NSF grant DGE-1315138 to A.H.P.
Footnotes
Citation Park H, McGibbon LC, Potts AH, Yakhnin H, Romeo T, Babitzke P. 2017. Translational repression of the RpoS antiadapter IraD by CsrA is mediated via translational coupling to a short upstream open reading frame. mBio 8:e01355-17. https://doi.org/10.1128/mBio.01355-17.
REFERENCES
- 1.Vakulskas CA, Potts AH, Babitzke P, Ahmer BM, Romeo T. 2015. Regulation of bacterial virulence by Csr (Rsm) systems. Microbiol Mol Biol Rev 79:193–224. doi: 10.1128/MMBR.00052-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schubert M, Lapouge K, Duss O, Oberstrass FC, Jelesarov I, Haas D, Allain FH-t. 2007. Molecular basis of messenger RNA recognition by the specific bacterial repressing clamp RsmA/CsrA. Nat Struct Mol Biol 14:807–813. doi: 10.1038/nsmb1285. [DOI] [PubMed] [Google Scholar]
- 3.Mercante J, Edwards AN, Dubey AK, Babitzke P, Romeo T. 2009. Molecular geometry of CsrA (RsmA) binding to RNA and its implications for regulated expression. J Mol Biol 392:511–528. doi: 10.1016/j.jmb.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu MY, Gui G, Wei B, Preston JF III, Oakford L, Yüksel U, Giedroc DP, Romeo T. 1997. The RNA molecule CsrB binds to the global regulatory protein CsrA and antagonizes its activity in Escherichia coli. J Biol Chem 272:17502–17510. doi: 10.1074/jbc.272.28.17502. [DOI] [PubMed] [Google Scholar]
- 5.Dubey AK, Baker CS, Romeo T, Babitzke P. 2005. RNA sequence and secondary structure participate in high-affinity CsrA-RNA interaction. RNA 11:1579–1587. doi: 10.1261/rna.2990205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weilbacher T, Suzuki K, Dubey AK, Wang X, Gudapaty S, Morozov I, Baker CS, Georgellis D, Babitzke P, Romeo T. 2003. A novel sRNA component of the carbon storage regulatory system of Escherichia coli. Mol Microbiol 48:657–670. doi: 10.1046/j.1365-2958.2003.03459.x. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki K, Wang X, Weilbacher T, Pernestig AK, Melefors O, Georgellis D, Babitzke P, Romeo T. 2002. Regulatory circuitry of the CsrA/CsrB and BarA/UvrY systems of Escherichia coli. J Bacteriol 184:5130–5140. doi: 10.1128/JB.184.18.5130-5140.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chavez RG, Alvarez AF, Romeo T, Georgellis D. 2010. The physiological stimulus for the BarA sensor kinase. J Bacteriol 192:2009–2012. doi: 10.1128/JB.01685-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edwards AN, Patterson-Fortin LM, Vakulskas CA, Mercante JW, Potrykus K, Vinella D, Camacho MI, Fields JA, Thompson SA, Georgellis D, Cashel M, Babitzke P, Romeo T. 2011. Circuitry linking the Csr and stringent response global regulatory systems. Mol Microbiol 80:1561–1580. doi: 10.1111/j.1365-2958.2011.07663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zere TR, Vakulskas CA, Leng Y, Pannuri A, Potts AH, Dias R, Tang D, Kolaczkowski B, Georgellis D, Ahmer BM, Romeo T. 2015. Genomic targets and features of BarA-UvrY (-SirA) signal transduction systems. PLoS One 10:e0145035. doi: 10.1371/journal.pone.0145035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suzuki K, Babitzke P, Kushner SR, Romeo T. 2006. Identification of a novel regulatory protein (CsrD) that targets the global regulatory RNAs CsrB and CsrC for degradation by RNase E. Genes Dev 20:2605–2617. doi: 10.1101/gad.1461606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vakulskas CA, Leng Y, Abe H, Amaki T, Okayama A, Babitzke P, Suzuki K, Romeo T. 2016. Antagonistic control of the turnover pathway for the global regulatory sRNA CsrB by the CsrA and CsrD proteins. Nucleic Acids Res 44:7896–7910. doi: 10.1093/nar/gkw484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leng Y, Vakulskas CA, Zere TR, Pickering BS, Watnick PI, Babitzke P, Romeo T. 2016. Regulation of CsrB/C sRNA decay by EIIA(Glc) of the phosphoenolpyruvate: carbohydrate phosphotransferase system. Mol Microbiol 99:627–639. doi: 10.1111/mmi.13259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yakhnin H, Yakhnin AV, Baker CS, Sineva E, Berezin I, Romeo T, Babitzke P. 2011. Complex regulation of the global regulatory gene csrA: CsrA-mediated translational repression, transcription from five promoters by Eσ70 and EσS, and indirect transcriptional activation by CsrA. Mol Microbiol 81:689–704. doi: 10.1111/j.1365-2958.2011.07723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Camacho MI, Alvarez AF, Chavez RG, Romeo T, Merino E, Georgellis D. 2015. Effects of the global regulator CsrA on the BarA/UvrY two-component signaling system. J Bacteriol 197:983–991. doi: 10.1128/JB.02325-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adamson DN, Lim HN. 2013. Rapid and robust signaling in the CsrA cascade via RNA-protein interactions and feedback regulation. Proc Natl Acad Sci U S A 110:13120–13125. doi: 10.1073/pnas.1308476110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Irie Y, Starkey M, Edwards AN, Wozniak DJ, Romeo T, Parsek MR. 2010. Pseudomonas aeruginosa biofilm matrix polysaccharide Psl is regulated transcriptionally by RpoS and post-transcriptionally by RsmA. Mol Microbiol 78:158–172. doi: 10.1111/j.1365-2958.2010.07320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ren B, Shen H, Lu ZJ, Liu H, Xu Y. 2014. The phzA2-G2 transcript exhibits direct RsmA-mediated activation in Pseudomonas aeruginosa M18. PLoS One 9:e89653. doi: 10.1371/journal.pone.0089653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park H, Yakhnin H, Connolly M, Romeo T, Babitzke P. 2015. CsrA participates in a PNPase autoregulatory mechanism by selectively repressing translation of pnp transcripts that have been previously processed by RNase III and PNPase. J Bacteriol 197:3751–3759. doi: 10.1128/JB.00721-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laursen BS, Sørensen HP, Mortensen KK, Sperling-Petersen HU. 2005. Initiation of protein synthesis in bacteria. Microbiol Mol Biol Rev 69:101–123. doi: 10.1128/MMBR.69.1.101-123.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Babitzke P, Baker CS, Romeo T. 2009. Regulation of translation initiation by RNA binding proteins. Annu Rev Microbiol 63:27–44. doi: 10.1146/annurev.micro.091208.073514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Battesti A, Majdalani N, Gottesman S. 2011. The RpoS-mediated general stress response in Escherichia coli. Annu Rev Microbiol 65:189–213. doi: 10.1146/annurev-micro-090110-102946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merrikh H, Ferrazzoli AE, Bougdour A, Olivier-Mason A, Lovett ST. 2009. A DNA damage response in Escherichia coli involving the alternative sigma factor, RpoS. Proc Natl Acad Sci U S A 106:611–616. doi: 10.1073/pnas.0803665106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merrikh H, Ferrazzoli AE, Lovett ST. 2009. Growth phase and (p)ppGpp control of IraD, a regulator of RpoS stability, in Escherichia coli. J Bacteriol 191:7436–7446. doi: 10.1128/JB.00412-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yakhnin AV, Baker CS, Vakulskas CA, Yakhnin H, Berezin I, Romeo T, Babitzke P. 2013. CsrA activates flhDC expression by protecting flhDC mRNA from RNase E-mediated cleavage. Mol Microbiol 87:851–866. doi: 10.1111/mmi.12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gollnick P, Babitzke P, Antson A, Yanofsky C. 2005. Complexity in regulation of tryptophan biosynthesis in Bacillus subtilis. Annu Rev Genet 39:47–68. doi: 10.1146/annurev.genet.39.073003.093745. [DOI] [PubMed] [Google Scholar]
- 28.Hengge R. 2011. Stationary-phase gene regulation in Escherichia coli. Ecosal Plus 4. doi: 10.1128/ecosalplus.5.6.3. [DOI] [PubMed] [Google Scholar]
- 29.Romeo T, Vakulskas CA, Babitzke P. 2013. Post-transcriptional regulation on a global scale: form and function of Csr/Rsm systems. Environ Microbiol 15:313–324. doi: 10.1111/j.1462-2920.2012.02794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hauryliuk V, Atkinson GC, Murakami KS, Tenson T, Gerdes K. 2015. Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat Rev Microbiol 13:298–309. doi: 10.1038/nrmicro3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Lorenzo V, Herrero M, Jakubzik U, Timmis KN. 1990. Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in gram-negative eubacteria. J Bacteriol 172:6568–6572. doi: 10.1128/jb.172.11.6568-6572.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haldimann A, Wanner BL. 2001. Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J Bacteriol 183:6384–6393. doi: 10.1128/JB.183.21.6384-6393.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vakulskas CA, Pannuri A, Cortés-Selva D, Zere TR, Ahmer BM, Babitzke P, Romeo T. 2014. Global effects of the DEAD box RNA helicase DeaD (CsdA) on gene expression over a broad range of temperatures. Mol Microbiol 92:945–958. doi: 10.1111/mmi.12606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mercante J, Suzuki K, Cheng X, Babitzke P, Romeo T. 2006. Comprehensive alanine-scanning mutagenesis of Escherichia coli CsrA defines two subdomains of critical functional importance. J Biol Chem 281:31832–31842. doi: 10.1074/jbc.M606057200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CsrA-iraD RNA footprint analysis. Wild-type (WT) or BS2 mutant iraD RNA was treated with RNase T1 in the presence of the CsrA concentration shown at the top of the lane. Partial alkaline hydrolysis (OH) and RNase T1 digestion (T1) ladders, as well as a control lane without RNase T1 treatment (c), are shown. Positions of BS1, BS2, BS3, BS4, the iraD start codon (Met), and the Shine-Dalgarno (SD) sequence are shown. Residues that are protected by bound CsrA from RNase T1 cleavage are marked (–). Numbering is with respect to the start of iraD translation. The position of the BS2 GGA-to-CGA mutation is marked with a carat. Positions in which CsrA-mediated protection was lost in the mutant transcript are marked (*). Sequencing lanes revealing A, C, G, and U residues are labeled. Download FIG S1, TIF file, 3.4 MB (3.5MB, tif) .
Copyright © 2017 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Expression of ORF23 and ORF47 does not affect iraD expression. (A and B) β-Galactosidase activity data (Miller units) ± standard deviations were determined throughout growth. Experiments were performed at least three times. Representative growth curves are shown with dashed lines (Klett). (A) Expression of P1-ORF23′-′lacZ and P1-ORF47′-′lacZ translational fusions. (B) Expression of wild-type (WT) or mutant P1-P2-iraD′-′lacZ translational fusions. Stop indicates that the fusion contains a stop codon soon after translation initiation of ORF23 or ORF47. Download FIG S2, TIF file, 3.6 MB (3.7MB, tif) .
Copyright © 2017 Park et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.