

Abstract
Inflammatory bowel disease (IBD) is a multifactorial disease including both genetic and environmental factors. We compared the diversity of intestinal microbesamong a cohort of IBD patients to study the microbial ecological effects on IBD. Fecal samples from patients were sequenced with next generation sequence technology at 16S rDNA region. With statistical tools, microbial community was investigated at different level. The gut microbial diversity of Crohn’s disease (CD) patients and colonic polyp (CP) patients significantly different from each other. However, the character of ulcerative colitis (UC) patients has of both CD and CP features. The microbial community from IBD patients can be very different (CD patient) or somewhat similar (UC patients) to non-IBD patients. Microbial diversity can be an important etiological factor for IBD clinical phenotype.
Electronic supplementary material
The online version of this article (doi:10.1007/s12088-017-0652-6) contains supplementary material, which is available to authorized users.
Keywords: Microbial diversity, 16S rDNA, Inflammatory bowel disease (IBD), Statistical analysis
Introduction
Inflammatory bowel disease (IBD) is a chronic inflammation of digestive disorder which in general, includes two major types: ulcerative colitis (UC) and Crohn’s disease (CD). They share several pathological and clinical symptoms, such as severe diarrhea, pain, fatigue and weight loss, at the same time, they also show clearly distinct characters. UC is confined to the innermost lining of large intestine and rectum and causes long-lasting inflammation. However, CD leads to inflammation of the lining of digestive tract, this inflammation can occur throughout the large intestine, small intestine or both, and often spreads deep into affected tissues [1].
More than five million people are suffering from IBD, and at the same time the newly diagnosed cases of IBD have been increasing gradually worldwide, especially in western world, such as North America, Europe, Australia and New Zealand [2–4]. A large number of reports have shown that many agents or mechanisms may contribute to IBD, but by no evidence, single factor can result in the disease. In general, causes of IBD can be sorted into two types: genetic (intrinsic) factors and non-genetic (external) factors. In UC, phenotypic concordance in monozygotic twins is less than 20%. However, in CD the rate is over 50%, and the relative risk of developing CD is much higher than the regular population [5]. Among these genetic factors, accumulating data from animal model demonstrated that nucleotide oligomerization domain 2 (NOD2) [6, 7], autophagy genes [8–11], components of the type 17 helper T cell (Th17) pathway, and multiple genes along the interleukin-23 signaling pathway have been involved in IBD [12–15]. Compared with genetic factors, non-genetic factors, such as changes in diet, antibiotic use, and intestinal colonization, have probably contributed to the increased prevalence of IBD [14, 15]. Increasing publications have proved that the intestinal microbiota is likely the most important environmental effect on IBD as the target of the inflammatory response [16] which is observed in humans and mouse models [17].
In mammalian gastrointestinal tract all three domains of life (Archaea, Bacteria and Eukarya) can be found [18], and there are more than 1014 gastrointestinal microorganisms in human [19, 20]. The gastrointestinal microbiota plays a key role in the regulation of the intestinal immune system [21], and the dysbiosis of intestinal microbiota, either in quantity or their ratio, may result in gut diseases, like IBD. Aminosalicylates and corticosteroids are drugs most common used for IBD treatment. However, they do not have long-term clinical healing and strong side effects have also been observed [22].
To overcome these shortcomings, several new generation of biopharmaceuticals such as monoclonal antibodies infliximab, adalimumab have been developed for IBD treatment. They are demonstrated to be more selective therapeutic drugs, particularly in some given cases. However, an increased risk of malignancies, such as non-Hodgkin’s lymphoma and nonmelanoma skin cancers have been observed [23]. These limitations highlight the therapeutic gaps in IBD treatment, one of the most important reasons for the difficulty to develop effective drugs is the complex causes of IBD, and among these reasons, the diversity of gut microbiota changes in IBD patients is the key one. To overcome this obstacle, scientists have been pureeing great efforts on identifying gastrointestinal microbiota variations between IBD patients and healthy people and between different types of IBD. Thanks to developing of next-generation sequencing technologies, currently a large number of microorganisms in human gut have been identified by 16S rDNA sequence analysis and metagenomics [23, 24]. Study revealed that Bacteroidetes and the Firmicutes constitute over 90% of the known phylogenetic categories of the distal gut microbiota [25].
Currently scientists have tried to determine whether specific variations can be identified in the intestinal microbiota in IBD. Results from 16S rRNA sequencing showed a visible difference between the intestinal microbiota in IBD patients compared to healthy control [26]. Patients with CD and those with UC have reduced diversity of members of the mucosa-associated phyla Firmicutes and Bacteroidetes [27, 28]. Whether these changes leads to IBD or whether they have same effects on people from different races is largely unknown.
To address these challenges, we analyzed 22 independent gut microbiome samples, which are from 6 CD patients, 9 UC patients and 7 colonic polyp (CP) patients., using Illumina PE250 sequencing and bioinformatics analysis. According to our results, the structure of microbial communities from IBD patients has a clear shift compared with CP patients, such as the relative composition of Collinsella, Dorea, Faecalibacterium obviously decreased in the gut of IBD patients, and this may imply the protection from IBD initiation or progression by this set of intestinal microbiota. Moreover, we also found a marked structure variation between CD and CP patients as well as between UC and CP patients. Our data also showed that Erysipelatoclostridium, Gemella, Granulicatella, Mogibacterium, Rothia, and Streptococcus increased dramatically in CD patients, while Lachnoclostridium and Tyzzerella-4 selectively rose in UC patients. Among these findings, the possible association between decrease of Faecalibacterium and CD is also supported by previous report [29], however many gut microbiota changes in IBD in our study were observed first time. Our study further revealed not only the complexity of causes of IBD but also the diversity between its subtypes, and shed new light on IBD prevention and new drug development.
Method
Patients
From January 2015 to December 2015, Patients, including 6 CD and 9 UC, were selected from those undergoing routine colonoscopic assessment of IBD at Department of Digestive Diseases, Huashan Hospital, Fudan University, Shanghai, China. IBD was diagnosed on the basis of combined gross and microscopic features. As controls, asymptomatic individuals undergoing colonoscopy were diagnosed as colon polyps. The CDAI of CD patients and Mayo score of UC patients were got. Tissue samples at lesion sites were collected from patients with assistant of endoscope with informed consent, and controls at normal sites.
DNA Preparation
Total DNA of samples were extracted and by and checked with 1% agarose electrophoresis. Then sample DNAs were normalized at 5 ng/μl in 10 mM Tris pH8.5 and amplified with GeneAmp® 9700 thermocycler (ABI, U.S.) using TransStart Fastpfu DNA Polymerase (TransGen, Beijing). Target 16srDNA regions were amplified by polymerase chain reaction (PCR). Illumina adapter overhang nucleotide sequences are added to the gene‐specific sequences. The full length primer sequences targeting this region are:
16S Amplicon PCR Forward Primer = 5′TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG
16S Amplicon PCR Reverse Primer = 5′GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC
PCR program is as following: 95 °C for 3 min; 25 cycles of: 95 °C for 30 s, 55 °C for 30 s, 72 °C for 30 s then 72 °C for 5 min and hold at 4 °C.
Each sample was repeated 3 times individually. After amplification, all amplicons from three repeats were pooled together, separated by 2% agarose and purified with AxyPrepDNA gel extraction kit (AXYGEN, Hangzhou, China). Purified PCR products were quantified with QuantiFluor™-ST system (Promega) and mixed according to manufacturer’s instruction to generate library and then loaded to Illumina PE250 sequencer (illumine, SD, USA) for sequencing.
Data Analysis
To optimize sequences, all PE reads were aligned and filtered to remove bad reads. The final sequences were clustered as Operational Taxonomic Units (OTU) based on similarity using Usearch software (version 7.1 http://drive5.com/uparse/) with thread hold line of 97% homology. The whole procedures are roughly as following:
A dereplication procedure was applied to extract non-repeat sequences from optimized sequences to remove redundant calculation. All singletons (unique reads among all reads) were also discarded to remove sequencer errors. All non-repeat reads (single non-repeat reads and chimeras were excluded) with ≥97% homology were clustered to generate reprehensive OTUs. Each optimized sequence was then mapped to reprehensive OTUs and sequences that have over 97% homology were selected to generate OTU table.
For taxonomic analysis, Bayesian Algorithm based RDP classifier was applied to reprehensive OTUs. Principal co-ordinates analysis (PCoA) was also used to classify OTUs.
Hierarchical clustering based on unweighted pair group method with arithmetic mean (UPGMA) was used to generate likelihood tree, Qiime was used to calculate distance of matrix, where the distance is defined by Bray Curtis
where: SA,i = the number of sequences in sample A included in the ith OTU; SB,i = the number of sequences in sample B included in the ith OTU.
The structure tree was finally drawn by R.
Nonmetric multidimensional scaling (NMDS) was used to analyze differences among samples. Community data at genus level were calculate by count absolute number of each sequence read and were shown as bar plot and heatmap. Box-whisker Plot was used to show the differences between samples at genus level. All reference databases, platform and soft wares are as following:
The 16S rDNA database of bacteria and archaea (If not specified, Silva database will be used as default):
Silva (Release 119 http://www.arb-silva.de); RDP (Release 11.1 http://rdp.cme.msu.edu/); Greengene (Release 13.5 http://greengenes.secondgenome.com/); ITS fungi:Unite [7] (Release 6.0 http://unite.ut.ee/index.php) Functional Gene data: GeneBank (Release 7.3 http://fungene.cme.msu.edu/:) Qiime (http://qiime.org/scripts/assign_taxonomy.html), RDP Classifier [9] (version 2.2 http://sourceforge.net/projects/rdp-classifier/), Confidence threshold 0.7.
Results
Characteristics of the Subjects
Patients are not only from Shanghai area but also other place, which makes our results representative. Twenty-two mucosal biopsies were collected from patients, including 6 patients with active CD, 9 patients with active UC and 7 biopsies from non-IBD controls. There was no age difference between CD and UC cases but, due to the indication for colonoscopy, the average age of the non-IBD control patients was higher. The median ages were 42.2 (34–60) years for the CD group, 52.8(17–82) years for the UC group and 60.1 (29–70) years for the controls. The characteristics of patients were shown as in Table 1. The IBD patients (including CD and UC) covered both males and females and the ages were from young to old (Table 2).
Table 1.
Characteristics of patients and biopsy tissue at time of sampling
| Diagnosis | No. | Age | Sex | Biopsy site | CDAI | Mayo score |
|---|---|---|---|---|---|---|
| CD | 1 | 34 | F | Ileocecal valve | 167 | |
| CD | 2 | 59 | M | Ileocecal valve | 210 | |
| CD | 3 | 35 | M | Ileocecal valve | 231 | |
| CD | 4 | 60 | M | Transverse colon | 190 | |
| CD | 5 | 42 | M | Transverse colon | 195 | |
| CD | 6 | 53 | F | Transverse colon | 208 | |
| UC | 1 | 45 | F | Rectum | 5 | |
| UC | 2 | 49 | F | Descending colon | 5 | |
| UC | 3 | 38 | M | Ascending colon | 3 | |
| UC | 4 | 67 | M | Rectum | 3 | |
| UC | 5 | 59 | M | Sigmoid colon | 7 | |
| UC | 6 | 60 | F | Sigmoid colon | 8 | |
| UC | 7 | 17 | M | Ascending colon | 6 | |
| UC | 8 | 82 | M | Cecum | 4 | |
| UC | 9 | 58 | M | Rectum | 4 | |
| CP | 1 | 70 | M | Transverse colon | ||
| CP | 2 | 29 | M | Rectum | ||
| CP | 3 | 67 | M | Transverse colon | ||
| CP | 4 | 65 | M | Sigmoid colon | ||
| CP | 5 | 68 | M | Transverse colon | ||
| CP | 6 | 61 | M | Transverse colon | ||
| CP | 7 | 61 | F | Descending colon |
CD Crohn’s disease, UC ulcerative colitis, CP colonic polyp
Table 2.
Characteristics of subjects
| Group | Sample label | Gender | Age |
|---|---|---|---|
| CD | 37-1 | F | 34 |
| 52-1 | M | 59 | |
| 55-1 | M | 35 | |
| 60-1 | M | 60 | |
| 74-1 | M | 42 | |
| 75-1 | F | 53 | |
| UC | 30-1 | F | 45 |
| 48-1 | F | 49 | |
| 49-1 | M | 38 | |
| 51-1 | M | 67 | |
| 56-1 | M | 59 | |
| 63-1 | F | 60 | |
| 64-1 | M | 17 | |
| 68-1 | M | 82 | |
| 77-1 | M | 58 | |
| CP | 57-1 | M | 70 |
| 58-1 | M | 29 | |
| 72-1 | M | 67 | |
| 76-1 | M | 65 | |
| 78-1 | M | 68 | |
| 80-1 | M | 61 | |
| 81-1 | F | 61 |
CD Crohn’s disease, UC ulcerative colitis, CP colonic polyp
Metagenome Analysis
We collected samples from lesion sites of patients and analyzed metagenomes from all 6 CD, 9 UC patients and 7 CP patients with 16S rDNA method. To reveal the composition and diversity of gut microbiota from IBD patients and CP patients, we used OTU to do the taxonomic analysis, which was based on sequence homology. The OTU tables were shown as in supplemental data. Since OTU were multi-dimensional data so we use weighted PCoA to characterize OTUs. The PCoA cluster showed that CD group was significantly separated from CP group (Fig. 1, red and blue), however, UC group was close to both CD and CP (Fig. 1, green). These results strongly indicate variation of microbiota in these diseases.
Fig. 1.
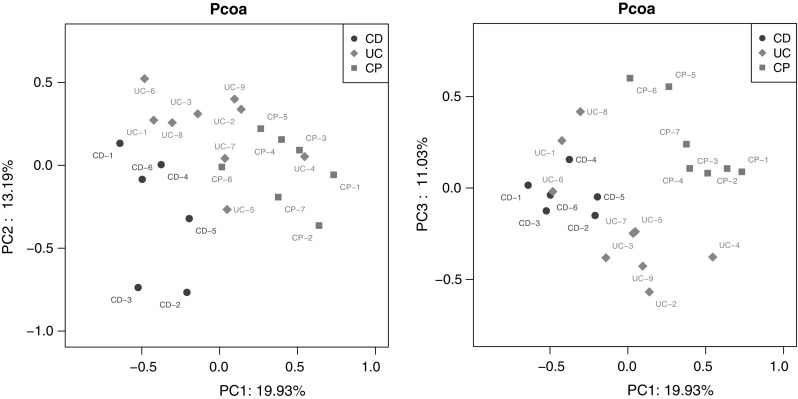
Principal co-ordinates analysis (PCoA) of OTUs. PCoA based on total OTUs level information, cluster analysis of OTUs profile according to Bray Curtis distance (the average linkage). a PC1 and PC2 were used to plot PCoA results. b PC1 and PC3 were used to plot PCoA results
Microbiota Diversity Between Groups
We took advantage of dendritic structure to describe and compare the similarity and diversity between samples. Hierarchical clustering was characterized based on spatial scales on beta diversity and then visible dendritic structure was constructed by unweighted pair group method with arithmetic mean (UPGMA). From multiple samples similarity tree analysis, we can draw a conclusion that gut microbiota communities show more similarity inner group than between any of them (Fig. 2a). The differences in bacterial community structure among groups were also reflected in NMDS based on spatial scales of beta diversity. According the data, microbiota community distribution is clearly separated into three parts which are correlated with sample groups (Fig. 2b). Similar conclusion can be drawn based on PCoA (Fig. 1). What is more, difference between CD and CP is more significant than between UC and CP (Fig. 2a, b). These results further illuminated the fact that the inappropriate changes of gut microbiota probably contribute to IBD, and the changes between UC and CD patient are clearly different.
Fig. 2.
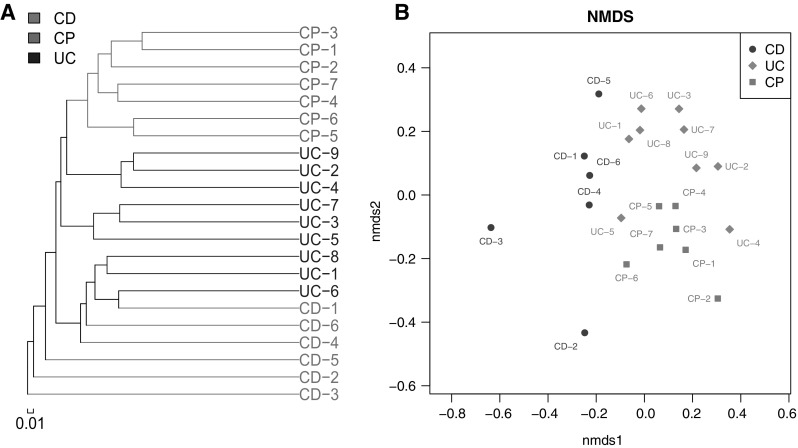
Multivariate analysis based on OTUs levels. a Hierarchical clustering based on unweighted pair group method with arithmetic mean (UPGMA) was used to generate likelihood tree. b Nonmetric multidimensional scaling (NMDS) analysis of samples
Microbiota Composition
To further reveal the difference of microbiota composition, we quantified the relative abundance of the microorganism among groups (Fig. 3a). Ratio of Faecalibacterium decreases obviously in CD patients compared with CP controls, however this is not observed in UC patients. At the same time, Streptococcus is significantly increased in CD patients compared with CP patients, and similarly this is detected in UC patients. The reduction of Faecalibacterium in CD is consistent with previous report [29], and these data imply that Faecalibacterium is probably a probiotic which may help to prevent CD initiation or progression. Nevertheless, Lachnoclostridium is notably increased only in UC patient samples, while Subdoligranulum declines accordingly, which indicates that these gut microbiotas may be related with UC formation. All samples in each of three groups were compared using a heat map of 18 different microbiotas as well (Fig. 3b). This data further consolidated the conclusion drawn from Fig. 3a.
Fig. 3.
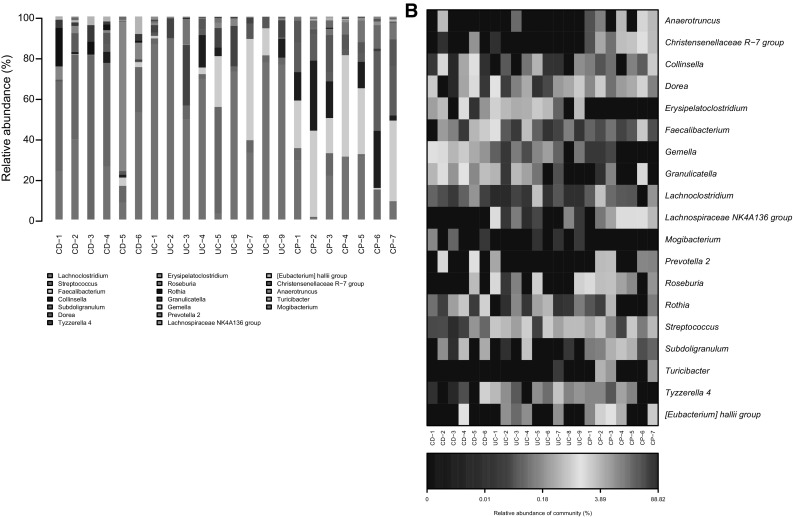
The community distribution among samples. a Compositions of microbiota at genus within UC, CD or CP. b Compositions of microbiota at genus by heat map analysis
Classification of Changed Communities
The most important purpose of this study is to find the specific or definite IBD related gut microbiota. We further classified the 18 gut microbiotas which were dramatically changed (Fig. 3b) into 3 groups: decreased in IBD (including both CD and UC) patients, specifically increased in CD patients or specifically increased in UC patients. IBD is a dysregulated mucosal immune response to antigens from the commensal microbiota in a genetically susceptible host, and there are probably some common shifts of gut microbiota communities. Studies showed that abundance of several types of microbiota were increased while some were decreased in general IBD patients [30–32]. In the first group, there are 10 different types of microbial community, including Collinsella, Dorea, Faecalibacterium, Hallii-group, Lachnospiraceae, Prevatella-2, R-7-group, Roseburia, Subdoligranulum and Turicibacter (Fig. 4a). Our results not only broadly replenished previous reports [30–32] which showed that Faecalibacterium and Lachnospiraceae were decreased in IBD patients, but also further revealed that IBD related bacteria may be different when put into different genetic and/or environmental background. Meanwhile, we can easily conclude that these bacteria may be probiotics which offer protection from IBD initiation or progression.
Fig. 4.

Classification of gut bacteria according to its distinct distribution among UC, CD and CP using Barplot. a Bacteria that are decreased both in CD and UC compared to CP group. b Bacteria that are increased only in CD group. c Bacteria that are increased only in UC group
Based on great difference of the clinical symptoms and occurring position of CD and UC, it is logical to say that probably different gut microbiota communities are responsible to CD and UC development. Researchers demonstrated less diversity in patients with CD compared to healthy control, however, they failed to describe which exact kinds of bacteria are directly related with CD [30, 33]. There are 6 members in the second group, including Erysipelatoclostridium, Gemella, Granulicatella, Mogibacterium, Rothia, Streptococcus (Fig. 4b). These increased microbiotas may, at least partially, explain the environmental effect on IBD. Although a great number of publications described the impact of gut microbiota on IBD, few of them were involved in UC. There are 2 communities in the third group, Lachnoclostridium and Tyzzerella-4 (Fig. 4c), and this observation perhaps gives new sights into understanding the pathological mechanism of UC. Overall, these findings may point out a novel direction into developing new drugs or creating new strategies, such as specific gut flora transplantation on IBD treating.
Discussion
Inflammatory bowel disease (IBD) includes two major phenotypes Crohn’s disease (CD) and Ulcerative colitis (UC), and is developing into a globally prevailing disease. Though the precise aetiology still remains unclear, it obviously involves a complex interplay of microbiological, immunological and genetic elements. Innate immune cell can be activated by bacterial components such as lipopolysaccharides (LPS) and flagellin etc. and cause inflammatory reaction. So microbiota diversity may play key roles in inflammatory bowel disease. More and more evidence has been uncovering that interaction between the host’s immune system and the commensal microbiota plays critical role in the pathogenesis of UC and CD. Using 16S ribosomal RNA gene-based single strand confirmation polymorphism analysis, several studies have reported that 50% and 30% reduced mucosa-associated colonic microbiota diversity were associated with active CD and UC respectively [24].
With similar technique, the present study provides a detailed experimental comparison of microbial community in patients of three different types of intestinal disease. Within our determined samples, according statistical analysis CD patients showed clear segregation from CP patients (Figs. 1, 2). This indicates a significant different underlying mechanism between these two diseases. However, the UC group, although as another typical IBD, the statistical characters of that showed some inter between features of both CD and CP patients (Figs. 1, 2). That may suggest different therapeutical strategies should be used for UC patients from CD.
There are some common shifts of gut microbiota communities in IBD patients and we identified 10 such groups as Streptococcus, Collinsella, Dorea, Hallii-group etc. These common shifts in microbiota communities may reflect the similarity between CD and UC patients. Nevertheless, there is indeed clear difference of community abundances between these two IBD phenotypes. Particularly Faecalibacterium was obviously observed decrease in CD patients, however, this is not true in UC patients. On the other hand, Lachnoclostridium is notably increased only in UC patient samples and Subdoligranulum also declines, but these changes were not observed in CD samples. Since the reduction of Faecalibacterium in CD has been reported previously (1), these data may confirm that Faecalibacterium is probably a probiotic which may prevent CD initiation or progression. Meanwhile, the unique changes of Lachnoclostridium and Subdoligranulum in UC patients suggested these gut microbiotas may be associated with UC formation.
In conclusion, microbial communities from three different intestinal diseases showed obvious differences. Particularly CD patients were obviously segregated from CP patients. However, samples from UC patients were also different from CD patients and showed some similarity to CP samples. The mechanism of the varieties still needs to be elucidated. However, using high-throughput NGS sequencing method, our work provided a detailed comparison of three prevalent intestinal diseases on the microbial community. The results may shed a light on the personalised therapeutical strategies for IBD in future.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by Grants from the Shanghai Municipal Commission of Health and Family Planning (No. 20124011), the Shanghai Municipal Commission of Health and Family Planning (No. 20164Y0153).
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s12088-017-0652-6) contains supplementary material, which is available to authorized users.
References
- 1.Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol. 2003;3:521–533. doi: 10.1038/nri1132. [DOI] [PubMed] [Google Scholar]
- 2.Harlan WR, Meyer A, Fisher J. Inflammatory bowel disease: epidemiology, evaluation, treatment, and health maintenance. N C Med J. 2016;77:198–201. doi: 10.18043/ncm.77.3.198. [DOI] [PubMed] [Google Scholar]
- 3.Burisch J, Munkholm P. The epidemiology of inflammatory bowel disease. Scand J Gastroenterol. 2015;50:942–951. doi: 10.3109/00365521.2015.1014407. [DOI] [PubMed] [Google Scholar]
- 4.Xiao B, Merlin D. Oral colon-specific therapeutic approaches toward treatment of inflammatory bowel disease. Expert Opin Drug Deliv. 2012;9:1393–1407. doi: 10.1517/17425247.2012.730517. [DOI] [PubMed] [Google Scholar]
- 5.Halme L, Paavola-Sakki P, Turunen U, Lappalainen M, Farkkila M, Kontula K. Family and twin studies in inflammatory bowel disease. World J Gastroenterol. 2006;12:3668–3672. doi: 10.3748/wjg.v12.i23.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Limbergen J, Russell RK, Nimmo ER, Satsangi J. The genetics of inflammatory bowel disease. Am J Gastroenterol. 2007;102:2820–2831. doi: 10.1111/j.1572-0241.2007.01527.x. [DOI] [PubMed] [Google Scholar]
- 7.Cho JH, Weaver CT. The genetics of inflammatory bowel disease. Gastroenterology. 2007;133:1327–1339. doi: 10.1053/j.gastro.2007.08.032. [DOI] [PubMed] [Google Scholar]
- 8.Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 9.Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, Roberts RG, Nimmo ER, Cummings FR, Soars D, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P, Zody MC, Hall JL, Brant SR, Cho JH, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat Genet. 2008;40:1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wellcome Trust Case Control C Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Franke A, Balschun T, Karlsen TH, Sventoraityte J, Nikolaus S, Mayr G, Domingues FS, Albrecht M, Nothnagel M, Ellinghaus D, et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat Genet. 2008;40:1319–1323. doi: 10.1038/ng.221. [DOI] [PubMed] [Google Scholar]
- 14.Silverberg MS, Cho JH, Rioux JD, McGovern DP, Wu J, Annese V, Achkar JP, Goyette P, Scott R, Xu W, et al. Ulcerative colitis-risk loci on chromosomes 1p36 and 12q15 found by genome-wide association study. Nat Genet. 2009;41:216–220. doi: 10.1038/ng.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernando MM, Stevens CR, Walsh EC, De Jager PL, Goyette P, Plenge RM, Vyse TJ, Rioux JD. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4:e1000024. doi: 10.1371/journal.pgen.1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 18.Donaldson GP, Lee SM, Mazmanian SK. Gut biogeography of the bacterial microbiota. Nat Rev Microbiol. 2016;14:20–32. doi: 10.1038/nrmicro3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 22.Yadav V, Varum F, Bravo R, Furrer E, Bojic D, Basit AW. Inflammatory bowel disease: exploring gut pathophysiology for novel therapeutic targets. Transl Res. 2016;176:38–68. doi: 10.1016/j.trsl.2016.04.009. [DOI] [PubMed] [Google Scholar]
- 23.Human Microbiome Project C A framework for human microbiome research. Nature. 2012;486:215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Human Microbiome Project C Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eckburg PB, Relman DA. The role of microbes in Crohn’s disease. Clin Infect Dis. 2007;44(2):256–262. doi: 10.1086/510385. [DOI] [PubMed] [Google Scholar]
- 28.Xenoulis PG, Palculict B, Allenspach K, Steiner JM, Van House AM, Suchodolski JS. Molecular-phylogenetic characterization of microbial communities imbalances in the small intestine of dogs with inflammatory bowel disease. FEMS Microbiol Ecol. 2008;66:579–589. doi: 10.1111/j.1574-6941.2008.00556.x. [DOI] [PubMed] [Google Scholar]
- 29.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148:1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spor A, Koren O, Ley R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol. 2011;9:279–290. doi: 10.1038/nrmicro2540. [DOI] [PubMed] [Google Scholar]
- 32.Perry S, de la Luz Sanchez M, Yang S, Haggerty TD, Hurst P, Perez-Perez G, Parsonnet J. Gastroenteritis and transmission of Helicobacter pylori infection in households. Emerg Infect Dis. 2006;12:1701–1708. doi: 10.3201/eid1211.060086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dicksved J, Halfvarson J, Rosenquist M, Jarnerot G, Tysk C, Apajalahti J, Engstrand L, Jansson JK. Molecular analysis of the gut microbiota of identical twins with Crohn’s disease. ISME J. 2008;2:716–727. doi: 10.1038/ismej.2008.37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.