

Abstract
Advanced melanoma has long been treated with chemotherapy using cytotoxic agents like dacarbazine (DTIC), but overall survival rates with these drugs have been generally low. Recently, immunoregulatory monoclonal antibodies and molecularly targeted therapy with a BRAF inhibitor and/or a MEK inhibitor, have been used to treat malignant melanoma and have improved the survival rate of patients with advanced melanoma. However, high prices of these drugs are problematic. In this study, we evaluated the oncolytic efficacy of HF10, an attenuated, replication-competent HSV, with DTIC in immunocompetent mice model of malignant melanoma. For in vitro studies, cytotoxicity assays were conducted in clone M3 mouse melanoma cells. For the in vivo studies, subcutaneous melanoma models were prepared in DBA/2 mice with clone M3 cells, and then HF10 was intratumorally inoculated with/without intraperitoneal DTIC injection. The efficacy of the therapies was evaluated by survival, growth of subcutaneous tumor, and histopathological and immunological analyses. Both HF10 infection and DTIC treatment showed cytotoxic effects in melanoma cells, but combination treatment with HF10 and DTIC showed a rapid and strong cytotoxic effect compared with monotherapy. In the subcutaneous melanoma model, intratumoral HF10 inoculation significantly inhibited tumor growth. HF10 also inhibited the growth of non-inoculated contralateral tumors when it was injected into the ipsilateral tumors of mice. In histologic and immunohistochemical analysis, tumor lysis and inflammatory cell infiltration were observed after intratumoral HF10 inoculation. When mice were treated with HF10 and DTIC, the combination therapy induced a robust systemic anti-tumor immune response and prolonged survival. IFN-γ secretion from splenocytes of the HF10-DTIC combination therapy group showed more IFN-γ secretion than did the other groups. These data showed the efficacy of HF10 and DTIC combination therapy in a mouse melanoma model.
Keywords: Melanoma, oncolytic virotherapy, HF10, dacarbazine, interferon-γ
Introduction
Malignant melanoma is a neoplasm derives from melanocytes and is the most aggressive and life-threatening skin cancer. The incidence of cutaneous malignant melanoma has been increasing in the world. In the United States, there were an estimated 76,690 new patients with melanoma in 2013 and approximately 9480 melanoma-related deaths [1]. The incidence of melanoma in Japan was estimated to be 1500-2500 new cases per year [2]. In spite of this increase, survival rates for melanoma have also improved substantially due to improvements in early diagnosis using dermoscopy and computerized imaging [3]. However, the prognosis of advanced melanoma patients, is still poor. In a survey of the approximate 5-year overall survival rate for Japanese patients with melanoma, patients at early stages had more than 80% survival rate; however, patients at advanced stages had an unfavorable outcome, with 5-year survival rates of less than 40% [2].
Advanced melanoma was treated with chemotherapy using cytotoxic drugs. Dacarbazine (DTIC) was administrated as a first line therapy against metastatic melanoma until 2011, but overall survival rates with DTIC were generally poor; the objective response rate for 1390 patients receiving DTIC monotherapy ranged between 5.3% and 28% (average, 15.3%) [4]. Recently, immunoregulatory monoclonal antibodies such as ipilimumab and nivolumab, or molecularly targeted therapy with a BRAF inhibitor or an MEK inhibitor, have been used for patients with malignant melanoma and have improved the survival rate of patients with advanced melanoma [5]. However, high prices of these drugs are problematic.
We have been studying oncolytic viral therapy with a highly attenuated, replication-competent herpes simplex virus (HSV)-1, HF10. Previously, we reported that HF10 therapy exhibited striking anti-tumor efficacy of peritoneally disseminated internal malignancies of immunocompetent mice models, including malignant melanoma [6-9]. Preclinical studies using HF10 to treat recurrent head and neck squamous cell carcinoma and breast cancer were sufficiently promising [10-12]. Currently, phase I/II trial of HF10 in patients with solid cutaneous tumors, including melanomas, has been completed (NCT01017185) in the United States. Moreover, in 2015, the U.S. Food and Drug Administration (FDA) approved a first oncolytic HSV named talimogene laherparepvec (T-VEC) for the treatment of advanced inoperable malignant melanoma [13].
In this study, we studied the oncolytic efficacy of HF10 combined with DTIC against malignant melanoma in vitro and in vivo. We determined that combination therapy with HF10 and DTIC was more effective in infecting and lysing mouse melanoma cells in vitro and in reducing the growth of local and distant tumor in immunocompetent mice models of malignant melanoma.
Materials and methods
Cell lines
Cloudman S91 clone M3 mouse melanoma cells (clone M3 cells) were obtained from the Cell Resource Center for Biochemical Research Institute of Development, Aging and Cancer of Tohoku University (Sendai, Japan). African green monkey kidney cells (Vero cells) were obtained from the Riken Cell Bank (Tsukuba, Japan). The clone M3 cells were maintained in RPMI 1640 medium (Sigma-Aldrich Corporation, St. Louis, MO) containing 10% fetal bovine serum. Vero cells were grown in Eagle’s minimal essential medium (Nissui Pharmaceutical Co., LTD., Tokyo, Japan) containing 10% calf serum.
Virus
HF10 is a nonselected clone derived from our laboratory as previously described [14]. Briefly, the HF10 genome is characterized by a 3.9-kb deletion downstream of the unique long region and a 2.3-kb deletion and extensive gene rearrangements upstream of the unique long region. Sequence analysis revealed that HF10 lacks the expression of functional UL43, UL49.5, UL55, and UL56 and the latency-associated transcript. HF10 was propagated in Vero cells at a multiplicity of infection (MOI) of 0.03 at 37°C. Infected cultures were harvested when most of cells exhibited cytopathic effects. After three times of freezing and thawing and eliminating cell debris by centrifugation, the supernatant was stored in aliquots at -80°C. Viral titers were determined by plaque assays using Vero cells and were expressed as plaque-forming units (pfu)/mL.
Cytotoxicity assay
For single treatment, clone M3 cells were prepared in 96-well plates (10,000 cells/well), then infected with HF10 at various MOIs from 0.003 to 3 pfu/cell or treated with DTIC at various concentrations from 0.2 to 10 mmol/L. MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assays were performed at 24 h after each treatment, using CellTiter 96 AQueous One Solution Reagent (Promega, Fitchburg, WI) according to the manufacturer’s instructions. Cytotoxicity was expressed as a decrease in cell viability with each treatment relative to the mock-infected cells.
For combination treatment, cells were pre-treated with or without DTIC (0.8 mmol/L) for 24 h. Cells were infected with HF10 (MOI 0.3) or mock for 1 h, then incubated in the media with or without DTIC (0.8 mmol/L). The results of cytotoxicity were examined with MTS assay at 24 and 48 h after infection. All assays were performed in triplicate.
Animal studies
All animal studies were performed in accordance with guidelines issued by the Animal Center at Nagoya University School of Medicine. Six-week-old female DBA/2 Cr mice were obtained from Japan SLC (Hamamatsu, Japan).
Clone M3 cells were harvested from subconfluent monolayer cultures by a 1-min treatment with 0.25% trypsin and 0.02% EDTA. Then the cells were washed in supplemented medium and resuspended in sterilized phosphate-buffered saline (PBS) for injection. Tumor cells (1×106) were inoculated subcutaneously into the bilateral flanks of the mice. Four days after inoculation, when tumors grew 3-4 mm in diameter, animals were randomly divided into six treatment groups: mock (group 1), HF10 (group 2), DTIC1 (group 3), DTIC2 (group 4), HF10+DTIC1 (group 5), and HF10+DTIC2 (group 6). First, 50 mg/kg (1 mL per mouse) DTIC or the same volume of citric acid was injected intraperitoneally once or twice. Then, 1.0×107 pfu/100 µL HF10 or the same volume of PBS was intratumorally inoculated into the right side 3 times every 2 days. Before HF10 injection, the virus was thawed and diluted PBS to adjust the inoculation dose.
Histology and immunohistochemistry
At the indicated times after HF10 treatment, the mice were sacrificed and fixed with formalin in PBS. Then the specimens were used for hematoxylin and eosin staining and immunostaining for HSV-1 antigens, CD4, CD8 and CD49c. For immunohistochemistry, rabbit anti-HSV type 1 antibody (DAKO A/S, Glostrup, Denmark), rat anti-mouse CD4 polyclonal antibody (Angio-Proteomie, Boston, MA) rat anti-mouse CD8 polyclonal antibody (Bioss Antibodies, Woburn, MA), rabbit anti-mouse CD49b polyclonal antibody (GeneTex, Inc. Irvine, CA) were used. CD8-positive area were evaluated according to the following criteria: -, < 1%; +, 1-10%; ++, > 10%.
Analysis of the supernatant from splenic cells co-cultured with clone M3 cells
On Day 14, three mice each from groups, mock, HF10, DTIC2, and HF10+DTIC2 were anesthetized by intraperitoneal administration of chloral hydrate. Then spleen cells were collected, and red cells were lysed using Pharm Lyse lysing solution (BD Biosciences Pharmingen, San Diego, CA). After the splenocytes were washed twice with RPMI 1640 medium (Sigma-Aldrich), they were co-cultured with clone M3 cells at a ratio of 1:20 for 24 h with mouse monoclonal anti-CD4 and/or anti-CD8 antibodies (R&D Systems, Minneapolis, MN). According to the manufacturer’s suggestions, these antibodies were added at a sufficient dose to deplete CD4 and/or CD8 T cells. IFN-γ secretion in the supernatant was measured using a Quantikine mouse IFN-γ ELISA kit (R&D Systems). TNF-α, IFN-α, and IFN-ß were also measured with Quantikine mouse TNF-α ELISA kit (R&D Systems), VeriKine Human Interferon Alpha ELISA Kit, (PBL Assay Science, Piscataway, NJ) and VeriKine Human Interferon Beta ELISA Kit (PBL Assay Science).
Statistical analysis
The survival data of mice were analyzed using the Kaplan-Meier method. Differences of tumor volumes between the treated and control groups and secretion of IFN-γ were analyzed by ANOVA followed by Tukey’s test. P-values less than 0.05 were considered statistically significant.
Results
HF10 and DTIC treatment induced cell death in melanoma cell lines in vitro
First, we checked the cytotoxic activity of HF10 and DTIC in clone M3 cells in vitro. Clone M3 cells were infected with HF10 at various MOIs from 0.003 to 3. At 24 h after infection, little cytotoxicity was observed at an MOI of 0.003, but cell viability declined as the increase of MOI. The calculated IC50 was an MOI of 0.2 (Figure 1A). Then, cytotoxic activity of DTIC was examined. Clone M3 cells were treated with DTIC at various molar concentrations from 0.2 to 10 mmol/L for 24 h. Cell viability decreased as the molar concentration increased. The calculated IC50 was 0.8 mmol/L (Figure 1B).
Figure 1.
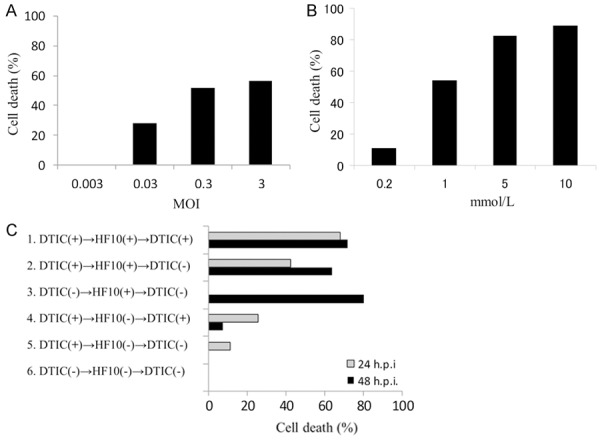
Oncolytic effect in vitro. A: Cytotoxic effect of HF10. Clone M3 cells were infected with HF10 at an MOI of 0.003-3. The viability of the cells was determined at 24 h post-infection, Cell viability is represented as the percent of the uninfected control. B: Cytotoxic effect of DTIC. Clone M3 cells were treated with DTIC at 0.2-10 mmol/L. The viability of the cells was determined at 24 h post treatment. C: Combination effect of HF10 and DTIC. Clone M3 cells were treated with or without DTIC (0.8 mmol/L) for 24 h. Each cells were mock-infected or infected with HF10 (MOI 0.3) for 1 h, and incubated with/without DTIC.
Next, we examined the effect of combination treatment with HF10 and DTIC. Clone M3 cells were treated with or without HF10 (MOI 0.3) and DTIC (0.8 mmol/L). The results of the six different treatment groups are shown in Figure 1C. HF10-DTIC combination treatment groups (groups 1 and 2) showed tumor cell death at 24 h after infection. The HF10 treatment group (group 3) also showed robust cytotoxicity only at 48 h after infection. DTIC-treated groups (group 4 and 5) showed a reduced cytotoxic effect compared with the virus-treated groups. These results suggested that combination treatment with HF10 and DTIC showed a rapid and strong cytotoxic effect compared with monotherapy.
HF10 virotherapy with DTIC induced persistent systemic anti-tumor effects and prolonged survival in melanoma mice
Next, we observed the effects of HF10-DTIC combination therapy in a bilateral subcutaneous melanoma model. Mice were inoculated subcutaneously with 1×106 clone M3 cells in their bilateral flanks. When subcutaneous tumors grew approximately 3-4 mm in maximal diameter (day 0), animals were randomly divided into six treatment groups: mock (group 1), HF10 (group 2), DTIC1 (group 3), DTIC2 (group 4), HF10+DTIC1 (group 5), and HF10+DTIC2 (group 6). First, 50 mg/kg DTIC or the same volume of citric acid was injected intraperitoneally once or twice on day 4 and 5. Then, 100 µL of HF10 (1.0×107 pfu) or PBS was inoculated into the right side of the tumor three times every 3 days (day 7, 10, 13). Because DTIC is an alkylating agent and inhibits DNA replication, virus inoculation was started two days after injection of DTIC. The treatment protocol is summarized in Figure 2A.
Figure 2.
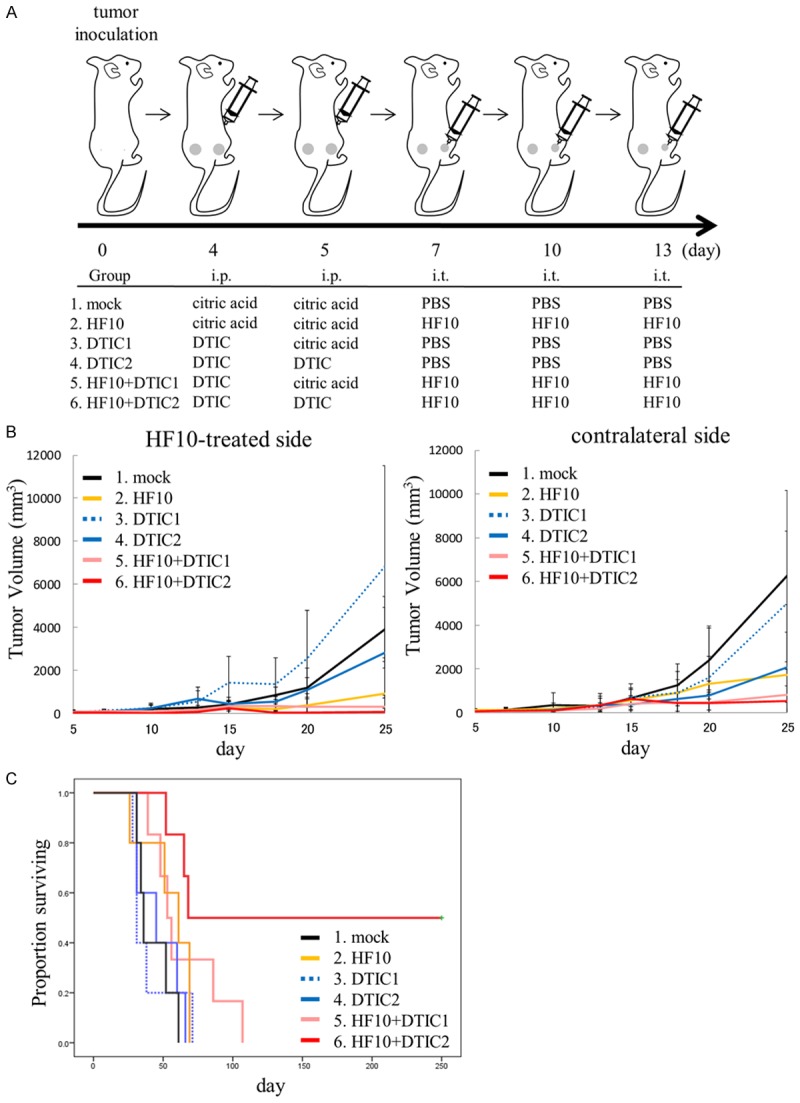
A: Treatment procedure of the bilateral subcutaneous melanoma model. DBA/2 mice were subcutaneously inoculated with 1×106 clone M3 cells in their bilateral flanks. When tumors reached approximately 3-4 mm in maximal diameter (day 0), mice were randomly divided into six treatment groups: mock (group 1), HF10 (group 2), DTIC1 and 2 (groups 3 and 4, respectively) and HF10+DTIC1 and 2 (groups 5 and 6, respectively). i.p.; intraperitoneal injection, i.t.; intratumoral injection. B: Tumor growth inhibition by HF10 local injection and/or DTIC intraperitoneal injection. Subcutaneous tumor were treated with HF10 and/or DTIC. Volume of the treated tumor (right flank) or the untreated tumor (left flank) was measured for the indicated days after treatment. C: Survival analysis of mice after treatment. HF10-DTIC combination therapy showed a significant prolonged survival effect compared to the control group (P = 0.004).
Regardless of DTIC pre-injection, intratumoral inoculation with HF10 (groups 2, 5 and 6) showed a significant inhibition in tumor growth in the right flank compared with mock-inoculated controls (group 1). In the DTIC-monotherapy groups (groups 3 and 4), the effect of tumor growth inhibition was limited (Figure 2B). Anti-tumor effects on the non-inoculated, left flank tumor were also observed in the HF10-inoculated group. After day 15, tumor regrowth was observed in the HF10-monotherapy group (group 2) but not in the combination therapy groups (groups 5 and 6) (Figure 2B). Survival analysis of the mice showed that HF10+DTIC2 (group 6) exhibited a significant prolonged survival effect compared to the control (group 1, P = 0.004) (Figure 2C). In group 6, complete tumor disappearance in both sides was observed in three of the six mice, with a survival time of over a year after treatment without tumor recurrence, showing complete rejection of the tumor. These results indicated that the treatment of mice melanoma with HF10-DTIC combination therapy induced a robust systemic anti-tumor immune response and prolonged survival.
HF10 virotherapy with DTIC induced tumor necrosis and lymphoid cell infiltration
Histopathology and immunohistochemical staining were performed on the tumors of treated and mock-treated mice. With hematoxylin and eosin staining, no histological changes in tumors of either side were observed in control and DTIC-treated mice (Figure 3). In the HF10-DTIC treated group, robust tumor necrosis was observed from day 14 in HF10-injected tumors. Tumor necrosis was also observed in contralateral tumors on day 14 and the necrotic area was expanded at day 16. In the HF10-monotherapy group, necrosis of the tumor was also detected from day 14 in HF10-injected tumors. On the other hand, tumor necrosis in contralateral tumors was not observed until day 16. By immunohistochemical staining, HSV antigen staining cells were only observed in tumors of the HF10-treated side at day 14 (Figure 4). Infiltrating CD8-positive cells were detected in the HF10-inoculated tumor in the HF10-monotherapy and HF10-DTIC treated groups; in contrast, very few CD8-positive cells were observed in the contralateral tumor at day 14 (Figure 5A and 5B). At day 16, CD8-positive cells were sill observed in the HF10-inoculated tumor; however, in the contralateral tumor, more CD8 positive cells were observed in the HF10-DTIC treated groups. In the mock- and DTIC-treated group, very few CD8-positive cells were observed. There were no significant differences in the infiltration of CD4-positive cells in the HF10 and HF10-DTIC treated tumors (data not shown). Next, we investigated CD49b-positive cells to detect infiltrating NK cells after HF10 injection. A fraction of CD49b-positive cells were observed in the both tumor in the HF10-DTIC treated group, and in the HF10-inoculated tumor in the HF10-monotherapy group (Figure 5C). However, no CD49b-positive cells were observed in control and DTIC-treated groups.
Figure 3.
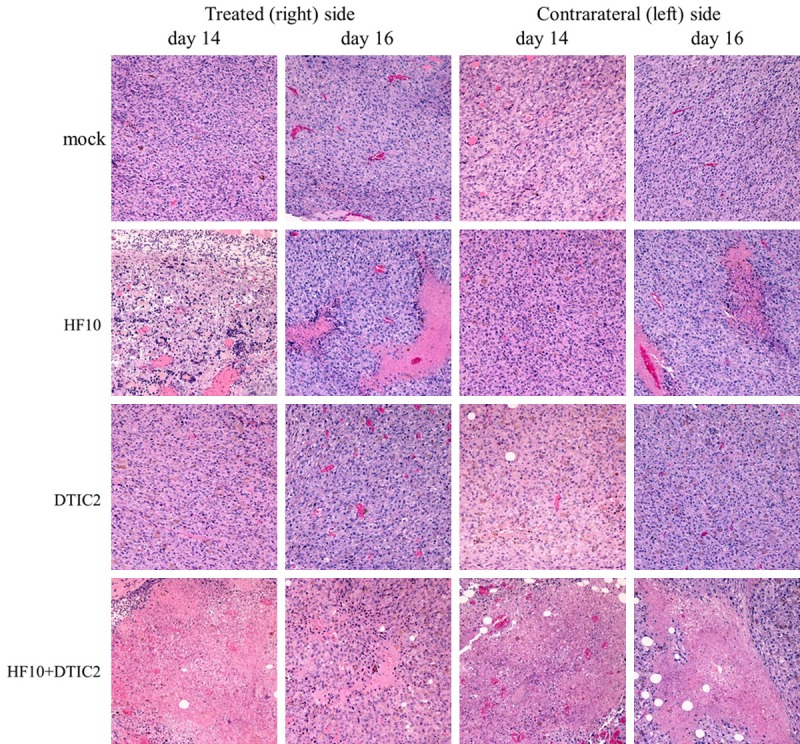
Histological analysis after treatment with hematoxylin eosin staining. No histological changes in tumors of either side were observed in control and DTIC-treated mice. In the HF10-DTIC treated group, tumor necrosis was observed on both sides on day 16, and expanded on day 16. In the HF10-monotherapy group, necrosis of the tumor was detected from day 14 in HF10-injected tumors. However, tumor necrosis in contralateral tumors was not observed until day 16. Original magnification, ×100.
Figure 4.
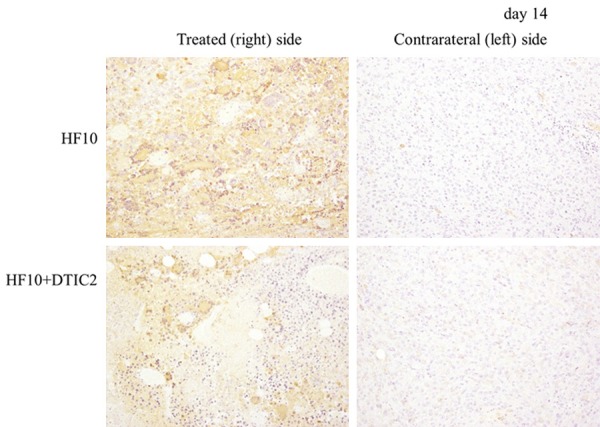
Immunohistochemical analysis of the HSV-1 antigen at day 14. HSV antigen-positive cells were only detected in the subcutaneous tumors of the HF10-treated side. Original magnification, ×100.
Figure 5.
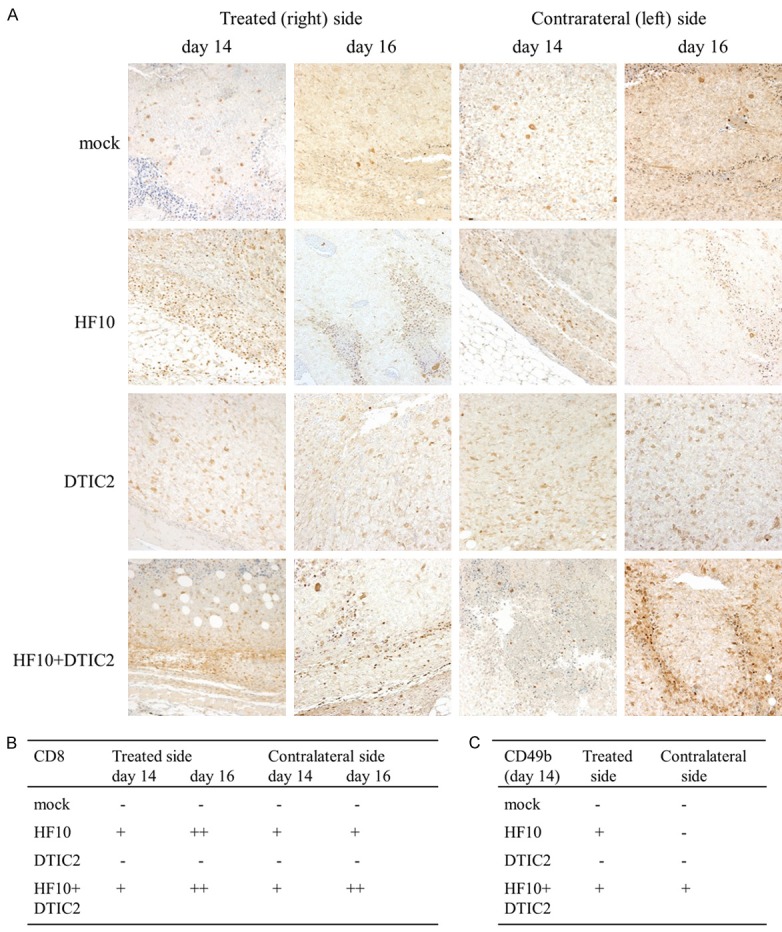
Immunohistochemical analysis of the CD8 antigen after treatment at days 14 and 16. A: CD8-positive cells were detected in the HF10-inoculated tumor of the HF10-monotherapy and HF10-DTIC treated groups, and fewer CD8-positive cells were observed in the contralateral tumor at day 14. At day 16, CD8-positive cells were sill observed in the HF10-inoculated tumor, and more CD8 positive cells were observed only in the HF10-DTIC treated groups in the contralateral tumor. B: CD8-positive area were evaluated according to the following criteria: -, < 1%; +, 1-10%; ++, > 10%. C: CD49b-positive cells were counted and evaluated according to the following criteria: -, 0 cells; +, 1-10 cells; ++, > 10 cells.
Splenic cells from HF10 virotherapy with DTIC induced IFN-γ by co-culture with melanoma cells
To determine the induction of tumor-specific cytotoxic T lymphocyte response in each treatment, effector cells were generated from splenocytes in vitro. On 14 days after treatment, splenocytes were collected from three mice of each group. These splenocytes were co-cultured with clone M3 cells at a ratio of 1:20 for 24 h and secreted IFN-γ in the supernatant was measured. In the HF10-monotherapy group, IFN-γ secretion from splenocytes was observed. In the HF10-monotherapy and HF10-DTIC combination therapy group, the amount of IFN-γ secretion was significantly higher than that of other groups (Figure 6A). This secretion was inhibited by the addition of an anti-CD4 antibody but not an anti-CD8 antibody (Figure 6B). These results suggested that the activation of CD4-positive T cells might be strongly induced in HF10 virotherapy with DTIC. TNF-α, INF-α, and INF-ß were also measured, but none of these were elevated (data not shown).
Figure 6.
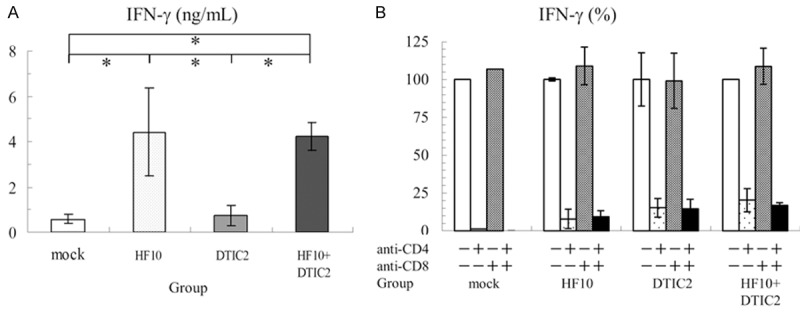
Analysis of the supernatant from spleen cells co-cultured with clone M3 cells. A: Increased IFN-γ in the supernatant of the co-culture. Splenocytes from three mice from each group were co-cultured with clone M3 cells for 24 h IFN-γ secretion in the supernatant was measured. In the HF10 and HF10-DTIC2 groups, the amount of IFN-γ secretion was significantly higher than that of the other groups (*; P < 0.05). B: Inhibition of IFN-γ with anti-CD4 and anti-CD8 antibodies. Splenocytes and clone M3 cells were co-cultured with mouse monoclonal anti-CD4 and/or anti-CD8 antibodies. Increased IFN-γ secretion in the supernatant was suppressed by adding the anti-CD4 antibody (*; P < 0.05).
Discussion
Genetically altered, replication-competent viruses have been developed for cancer therapies as well as for use as gene delivery vectors or vaccine vectors [15]. The use of these highly attenuated viruses that selectively replicate in and kill tumor cells is called “oncolytic virotherapy”. Oncolytic virotherapies using HSV, adenovirus and vaccinia virus have been engineered and studied widely in the last decade [16]. To date, T-VEC is the only oncolytic virus presently approved by the FDA that approved for the treatment of advanced inoperable melanoma [17]. This virus has been engineered to selectively replicate within tumor cells and to express granulocyte-macrophage colony-stimulating factor (GM-CSF). In an open-label phase III trial, intratumoral treatment with T-VEC showed significantly higher durable response rate, overall response rate and median overall survival were in treated patients than those treated with GM-CSF [18].
Oncolytic virotherapy against malignant melanoma induces anti-tumor effects through not only a direct oncolytic effect on viral injected lesions but also induction of systemic anti-tumor immunity. In phase II study of T-VEC, directly injection with the virus induced complete resolution of tumor in 46.1%. Moreover, in uninjected non-visceral lesions, 41.1% decreased in size by ≥ 30%, and the majority of which (30.1%) completely resolved. And of 32 visceral lesions, 12.5% decreased in size by ≥ 30%, and 9.4% completely resolved [19]. In this study, combination therapy with HF10 and DTIC induced more persistent systemic anti-tumor effects and prolonged survival in melanoma mice compared with HF10 or DTIC monotherapy. The reason for this effectiveness might be explained by various pharmacological actions of DTIC in both direct killing of tumor cells and the induction of anti-tumor immunity.
In cell viability assays, combination treatment with HF10 and DTIC induced rapid cell death. DTIC is a cell cycle non-specific anti-neoplastic agent that functions as an alkylating agent after activation in the liver [4]. To enhance the efficacy of oncolytic virotherapy, several combination therapies with GM-CSF, chemotherapy or radiation therapy have been studied [20,21]. In addition, the combination therapy using oncolytic adenoviruses with the alkylating agent temozolomide (TMZ) enhanced the anti-tumor response against melanoma and glioblastoma significantly [22]. This enhanced anti-tumor response was thought to be the induction of autophagy by TMZ. The efficacy of combination therapy with TMZ and the oncolytic HSV G47Δ has also been reported to have a synergistic effect in killing glioblastoma stem cells through enhancement of G47Δ-mediated DNA damage responses [23]. Taken together, our data may support combination therapy with an alkylating agent and oncolytic HSV.
It is well known that oncolytic virotherapy can induce anti-tumor effects through not only a direct oncolytic effect in injected lesions but also the induction of systemic anti-tumor immunity [24]. In the immune responses, dendritic cells (DCs) activate and expand CD8-positive cytotoxic T cells (CTLs) and CD4-positive T cells. CTLs recognize and destroy tumor cells, and CD4-positive T cells enhance the capacity of DCs to induce CTLs by the interaction between CD40 on DCs and CD40 ligand on activated CD4-positive T cells. In addition, CD4-positive T cells provide help for the maintenance and expansion of CTLs by secreting cytokines, such as IFN-γ [25]. In the current study, HF10 injection induced systemic anti-tumor immunity in the spleen. CD4-neutralizing antibody strongly suppressed the anti-tumoral immunity, which was in consistent with our previous study [21]. We speculate that HF10 induced systemic anti-tumoral immunity by CD4-positive T cells, which stimulated tumor-infiltrating CD8-positive T cells.
In a previous study, DTIC triggered the up-regulation of NKG2D ligands in melanoma cells, leading to activation of NK cells and secretion of IFN-γ in mice and humans [26]. NK cell-derived IFN-γ subsequently induced up-regulation of major histocompatibility complex (MHC) class I molecules in tumor cells, resulting these cells sensitive to CTLs. This result demonstrates that upon treatment with DTIC, the tumor participates in the initiation of an immune response. In this study, HF10 in combination following twice DTIC injection strongly reduced subcutaneous tumor on both side, resulting in complete rejection of tumors. Although only a fraction of NK cells were detected in the tumor, we speculate that these immunomodulating effects of DTIC enhanced HF10 oncolytic therapy by increased recognition of tumor antigens, secretion of IFN-γ, infiltration of CD8+ T cells and cell lysis of untreated tumor sides.
In conclusion, our data indicate the efficacy of HF10 and DTIC combination therapy. Though immunoregulatory monoclonal antibodies or molecularly targeted agents are the first-line therapy for advanced melanoma, our combination therapy would be a complementary therapy for advanced melanoma, especially in patients with severe side effects or resistance to immunoregulatory monoclonal antibodies or molecularly targeted therapy.
Acknowledgements
We thank Ms. T. Kunogi and Mr. S. Tanaka for their support. This work was supported by Grant-in-Aids for Scientific Research from the Ministry of Education, Culture, Sports, Sciences, and Technology of Japan (24501351 and 16K07166 to F.G.).
Disclosure of conflict of interest
None.
Abbreviations
- DTIC
dacarbazine
- FDA
the U.S. Food and Drug Administration
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- HSV
herpes simplex virus
- IFN
interferon
- MOI
a multiplicity of infection
- TMZ
temozolomide
- TNF
tumor necrosis factor
- T-VEC
talimogene laherparepvec
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Ishihara K, Saida T, Otsuka F, Yamazaki N Prognosis and Statistical Investigation Committee of the Japanese Skin Cancer Society. Statistical profiles of malignant melanoma and other skin cancers in Japan: 2007 update. Int J Clin Oncol. 2008;13:33–41. doi: 10.1007/s10147-007-0751-1. [DOI] [PubMed] [Google Scholar]
- 3.Thompson JF, Scolyer RA, Kefford RF. Cutaneous melanoma. Lancet. 2005;365:687–701. doi: 10.1016/S0140-6736(05)17951-3. [DOI] [PubMed] [Google Scholar]
- 4.Lui P, Cashin R, Machado M, Hemels M, Corey-Lisle PK, Einarson TR. Treatments for metastatic melanoma: synthesis of evidence from randomized trials. Cancer Treat Rev. 2007;33:665–680. doi: 10.1016/j.ctrv.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 5.Tang T, Eldabaje R, Yang L. Current status of biological therapies for the treatment of metastatic melanoma. Anticancer Res. 2016;36:3229–3241. [PubMed] [Google Scholar]
- 6.Kimata H, Takakuwa H, Goshima F, Teshigahara O, Nakao A, Kurata T, Sata T, Nishiyama Y. Effective treatment of disseminated peritoneal colon cancer with new replicationcompetent herpes simplex viruses. Hepatogastroenterology. 2003;50:961–966. [PubMed] [Google Scholar]
- 7.Kohno S, Luo C, Goshima F, Nishiyama Y, Sata T, Ono Y. Herpes simplex virus type 1 mutant HF10 oncolytic viral therapy for bladder cancer. Urology. 2005;66:1116–1121. doi: 10.1016/j.urology.2005.05.041. [DOI] [PubMed] [Google Scholar]
- 8.Teshigahara O, Goshima F, Takao K, Kohno S, Kimata H, Nakao A, Nishiyama Y. Oncolytic viral therapy for breast cancer with herpes simplex virus type 1 mutant HF 10. J Surg Oncol. 2004;85:42–47. doi: 10.1002/jso.20005. [DOI] [PubMed] [Google Scholar]
- 9.Watanabe D, Goshima F, Mori I, Tamada Y, Matsumoto Y, Nishiyama Y. Oncolytic virotherapy for malignant melanoma with herpes simplex virus type 1 mutant HF10. J Dermatol Sci. 2008;50:185–196. doi: 10.1016/j.jdermsci.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Fujimoto Y, Mizuno T, Sugiura S, Goshima F, Kohno S, Nakashima T, Nishiyama Y. Intratumoral injection of herpes simplex virus HF10 in recurrent head and neck squamous cell carcinoma. Acta Otolaryngol. 2006;126:1115–1117. doi: 10.1080/00016480600702100. [DOI] [PubMed] [Google Scholar]
- 11.Kimata H, Imai T, Kikumori T, Teshigahara O, Nagasaka T, Goshima F, Nishiyama Y, Nakao A. Pilot study of oncolytic viral therapy using mutant herpes simplex virus (HF10) against recurrent metastatic breast cancer. Ann Surg Oncol. 2006;13:1078–1084. doi: 10.1245/ASO.2006.08.035. [DOI] [PubMed] [Google Scholar]
- 12.Nakao A, Kimata H, Imai T, Kikumori T, Teshigahara O, Nagasaka T, Goshima F, Nishiyama Y. Intratumoral injection of herpes simplex virus HF10 in recurrent breast cancer. Ann Oncol. 2004;15:988–989. doi: 10.1093/annonc/mdh225. [DOI] [PubMed] [Google Scholar]
- 13.Pol J, Kroemer G, Galluzzi L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology. 2016;5:e1115641. doi: 10.1080/2162402X.2015.1115641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ushijima Y, Luo C, Goshima F, Yamauchi Y, Kimura H, Nishiyama Y. Determination and analysis of the DNA sequence of highly attenuated herpes simplex virus type 1 mutant HF10, a potential oncolytic virus. Microbes Infect. 2007;9:142–149. doi: 10.1016/j.micinf.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 15.Watanabe D. Medical application of herpes simplex virus. J Dermatol Sci. 2010;57:75–82. doi: 10.1016/j.jdermsci.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 16.Burke J, Nieva J, Borad MJ, Breitbach CJ. Oncolytic viruses: perspectives on clinical development. Curr Opin Virol. 2015;13:55–60. doi: 10.1016/j.coviro.2015.03.020. [DOI] [PubMed] [Google Scholar]
- 17.Appleton ES, Turnbull S, Ralph C, West E, Scott K, Harrington K, Pandha H, Melcher A. Talimogene laherparepvec in the treatment of melanoma. Expert Opin Biol Ther. 2015;15:1517–1530. doi: 10.1517/14712598.2015.1084280. [DOI] [PubMed] [Google Scholar]
- 18.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, Milhem M, Cranmer L, Curti B, Lewis K, Ross M, Guthrie T, Linette GP, Daniels GA, Harrington K, Middleton MR, Miller WH Jr, Zager JS, Ye Y, Yao B, Li A, Doleman S, VanderWalde A, Gansert J, Coffin RS. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 19.Kaufman HL, Amatruda T, Reid T, Gonzalez R, Glaspy J, Whitman E, Harrington K, Nemunaitis J, Zloza A, Wolf M, Senzer NN. Systemic versus local responses in melanoma patients treated with talimogene laherparepvec from a multi-institutional phase II study. J Immunother Cancer. 2016;4:12. doi: 10.1186/s40425-016-0116-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esaki S, Goshima F, Kimura H, Murakami S, Nishiyama Y. Enhanced antitumoral activity of oncolytic herpes simplex virus with gemcitabine using colorectal tumor models. Int J Cancer. 2013;132:1592–1601. doi: 10.1002/ijc.27823. [DOI] [PubMed] [Google Scholar]
- 21.Goshima F, Esaki S, Luo C, Kamakura M, Kimura H, Nishiyama Y. Oncolytic viral therapy with a combination of HF10, a herpes simplex virus type 1 variant and granulocyte-macrophage colony-stimulating factor for murine ovarian cancer. Int J Cancer. 2014;134:2865–2877. doi: 10.1002/ijc.28631. [DOI] [PubMed] [Google Scholar]
- 22.Liikanen I, Ahtiainen L, Hirvinen ML, Bramante S, Cerullo V, Nokisalmi P, Hemminki O, Diaconu I, Pesonen S, Koski A, Kangasniemi L, Pesonen SK, Oksanen M, Laasonen L, Partanen K, Joensuu T, Zhao F, Kanerva A, Hemminki A. Oncolytic adenovirus with temozolomide induces autophagy and antitumor immune responses in cancer patients. Mol Ther. 2013;21:1212–1223. doi: 10.1038/mt.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanai R, Rabkin SD, Yip S, Sgubin D, Zaupa CM, Hirose Y, Louis DN, Wakimoto H, Martuza RL. Oncolytic virus-mediated manipulation of DNA damage responses: synergy with chemotherapy in killing glioblastoma stem cells. J Natl Cancer Inst. 2012;104:42–55. doi: 10.1093/jnci/djr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dharmadhikari N, Mehnert JM, Kaufman HL. Oncolytic virus immunotherapy for melanoma. Curr Treat Options Oncol. 2015;16:326. doi: 10.1007/s11864-014-0326-0. [DOI] [PubMed] [Google Scholar]
- 25.Jahnisch H, Fussel S, Kiessling A, Wehner R, Zastrow S, Bachmann M, Rieber EP, Wirth MP, Schmitz M. Dendritic cell-based immunotherapy for prostate cancer. Clin Dev Immunol. 2010;2010:517493. doi: 10.1155/2010/517493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hervieu A, Rebe C, Vegran F, Chalmin F, Bruchard M, Vabres P, Apetoh L, Ghiringhelli F, Mignot G. Dacarbazine-mediated upregulation of NKG2D ligands on tumor cells activates NK and CD8 T cells and restrains melanoma growth. J Invest Dermatol. 2013;133:499–508. doi: 10.1038/jid.2012.273. [DOI] [PubMed] [Google Scholar]