

Abstract
Imatinib resistance has become a major obstacle for the treatment of chronic myeloid leukemia (CML). The present study aimed to investigate the effects of the long non-coding RNA, SNHG5 on imatinib resistance in CML and explored the underlying mechanisms. The quantitative real-time PCR results showed that SNHG5 and ABCC2 expressions were up-regulated in the isolated peripheral blood cells of the CML patients when compared with healthy controls, and SNHG5 expression levels was positively correlated with ABCC2 in CML patients. In vitro studies showed that the expressions of SNHG5 and ABCC2 were up-regulated in imatinib resistant cells (K562-R) when compared to K562 cells. Bioinformatics analysis showed the interaction between SNHG5 and miR-205-5p, which was further confirmed by luciferase reporter assay and RNA immune-precipitation in K562 cells. Overexpression of SNHG5 suppressed the expression of miR-205-5p and the expression of SNHG5 was negatively correlated with the miR-205-5p expression in CML patients. In addition, ABCC2 was predicted as a downstream target of miR-205-5p, which was further confirmed by the luciferase reporter assay in K562-R cells, and overexpression of miR-205-5p suppressed the expression of ABCC2 in K562-R cells. In vitro functional assay showed that overexpression of SNHG5 in K562 cells increased imatinib resistance and knock-down of SNHG5 reduced the imatinib resistance in K562-R cells. Further experiments showed that SNHG5 promotes imatinib resistance through regulating ABCC2. Taken together, SNHG5 promotes imatinib resistance in CML via acting as a competing endogenous RNA against miR-205-5p.
Keywords: Imatinib resistance, chronic myeloid leukemia, K562, SNHG5, MiR-205-5p, ABCC2
Introduction
Chronic myeloid leukemia (CML) is a type of cancer that starts in certain blood-forming cells of the bone marrow, and studies have indicated CML as a myeloproliferative disorder resulting from abnormal proliferation of BCR-ABL1-positive bone marrow stem cells [1]. The annual incidence of CML is about 1-2 per 100,000 population worldwide, accounting for 15% of newly diagnosed adult leukemias [2]. The first-line treatment for CML is imatinib, a competitive inhibitor of tyrosine kinase, can bind to BCR-ABL protein and suppress the subsequent signal transduction. As a result, imatinib markedly reduced the proliferation of leukemic cells and greatly improved the 5-year survival rate from 34.2% to 80-90% [3]. However, some of the CML patients developed imatinib resistance, which has been suggested to be associated with copy number mutation of BCR-ABL gene [4], aberrant expression of drug transporters [5], or epigenetic alterations [6]. Unfortunately, the exact underlying mechanisms involved imatinib resistance is largely unknown.
ATP-binding cassette (ABC) transporters are crucial for chemoresistance [5,7]. As imatinib is a substrate for ABC transporters, the members of the ABC family may involve in the imatinib resistance in CML. ABCC2 is one of the family member and also called multidrug resistance protein 2 (MRP2), which has been shown to involve in chemoresistance in various types of cancers [8]. Recently, growing evidence showed that noncoding RNAs take part in the regulation of ABCC2 expression. For example, microRNAs (miRNAs) such as let-7c, miR-27a and miR-490-3p regulated ABCC2 expression in a post-transcriptional manner [7,9,10]. Another kind of noncoding RNA, long noncoding RNAs (lncRNAs) were also found to play critical roles in regulation of gene expression. LncRNAs are transcripts longer than 200 nucleotides, which can not translate proteins [11]. In view of tumor biology, lncRNAs were found to participate in the pathological process of tumorigenesis and the development of drug resistance [12]. One of the well-studied lncRNAs is small nucleolar RNA host gene 5 (SNHG5). SNHG5, also called U50HG, is 524 bp in length, and SNGH5 has six exons and encodes for the snoRNAs U50 and U50’. SNHG5 locates at the chromosomal translocation breakpoint involved in B-cell lymphoma [13]. Aberrant expression of SNHG5 has been reported in various human cancers including colorectal cancer, malignant melanoma, gastric cancer and so on [14-16]. As far as we know, the functional role of SNHG5 in CML, particularly imatinib resistance, is largely unknown.
In the present study, the expression pattern of SNHG5 and ABCC2 in CML patients and cell lines were examined, and the increase expression of SNHG5 in CML patients and cell lines was positively correlated with an increase in ABCC2 expression. Furthermore, the underlying mechanisms involved in SNHG5-regulated imatinib resistance in CML was explored.
Materials and methods
CML patients’ recruitment
Total twenty healthy volunteers (10 males and 10 females, age from 35 to 67), and total forty patients with CML (17 females and 23 males, age from 40 to 72), were recruited in the present study. The CML patients were all under the treatment of imatinib (400 mg/day) for at least two years. The study protocol was approved by the Ethics Committee of Hanzhong Central Hospital, and informed written consent was signed by all the healthy volunteers and patients participating in this study.
Cell culture
The CML cell line K562 was obtained from ATCC (Manassas, USA), and cultured in RPMI 1640, containing 10% fetal bovine serum (FBS, Gibco, Thermo Scientific, Waltham, USA) in a humidified incubator with 5% CO2 at 37°C. Imatinib-resistant K562 cells (K562-R) were established as follows: In brief, K562 cells were first maintained in complete medium containing 0.05 μM imatinib, and the concentration of imatinib was increased progressively until a final concentration of 2.5 μM was reached. Subsequently, K562-R cells were cultured in the presence of 2.5 μM of imatinib to maintain the drug-resistant phenotype.
Vector construction, miRNAs, small interference RNAs (siRNAs) and cell transfection
The SNHG5 overexpression vector, pcDNA3.1-SNHG5 and the relative control, pcDNA3.1 were purchased from Genepharma Company (Shanghai, China); the pCMV6 overexpression vector, pCMV6-ABCC2 and the relative control, pCMV6 were obtained from Shanghai BlueGene Biotech Co., Ltd (Shanghai, China); miR-205-5p mimics and miR-205-5p inhibitors, and the respective controls; the siRNAs for SNHG5 and ABCC2, and their respective controls, were all purchased from RiboBio (Guangzhou, China). The transfection with vector constructs, miRNAs or siRNAs were performed by using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer’s instructions.
RNA isolation and quantitative real-time PCR (qRT-PCR)
Tissues and cells were collected and total RNAs from peripheral blood samples of CML patients and K562/K562-R cells were extracted using TRIzol reagent (Takara, Dalian, China), according to the manufacturer’s instructions. Briefly, cDNA was synthesized by by M-MLV reverse transcriptase (Invitrogen) from extracted RNAs. Real-time PCR was performed in triplicates with SYBR Green Real-Time PCR Master Mixes (Takara) on an ABI 7900 PCR system (Applied Biosystems, Bedford, USA). GAPDH was used as an internal control for lncRNAs and mRNA, U6 was used as internal control for miRNA. The relative expression of respective genes was calculated by using comparative Ct method.
MTT assay
The cell viability of K562/K562-R was measured by the MTT assay. Briefly, cells were seeded in 96-well plate (5000 cells/well) and cultured overnight. After imatinib treatment and/or transfection for 24 h, 10 μl of MTT solution (5 mg/ml) was added to each well and kept in the dark at 37°C for 4 h. Then the supernatant was removed, 200 μl of DMSO was added to dissolve the precipitated formazan. Cell viability was determined by measuring the absorbance at 570 nm on a micro-plate reader.
Luciferase reporter assay
For the interaction between SNHG5 and miR-205-5p, wide type SNHG5 and the mutant SNHG5 were cloned into pmiRGLO reporter vector, respectively. The pmirGLO-SNHG5 or pmirGLO-SNHG5-MUT was co-transfected with miR-205-5p mimics or miRNA mimics control. For the interaction between miR-205-5p and ABCC2, wide type 3’UTR of ABCC2 and the mutant 3’UTR of ABCC2 were cloned into pmiRGLO reporter vector, respectively. The pmirGLO-ABCC2 or pmirGLO-ABCC2-MUT was co-transfected with miR-205-5p mimics or miRNA mimics control. Forty-eight hrs post-transfection, Dual Luciferase Assay (Promega, Madison, USA) was used to determine the luciferase reporter activities according to the manufacturer’s instructions.
RNA immunoprecipitation (RIP)
The RIP assay was performed as follows. K562 cells were co-transfected with pLV-MS2, pLV-SNHG5-MS2, or pLV-SNHG5 (mutant)-MS2 and pMS2-GFP (Addgene, Cambridge, USA). Forty-eight hrs later, Magna RIPTM RNA-Binding Protein Immunoprecipitation Kit (Millipore, Billerica, USA) was used to perform the RIP assay. Cells were lysed and magnetic beads were pre-incubated with anti-GFP antibody or anti-rabbit IgG for 1 h. Then the lysates were immune-precipitated with beads at 4°C overnight. RNA was purified, reverse transcribed and detected by qRT-PCR.
Western blotting
Total proteins were extracted from cells using lysis buffer containing protease inhibitor and protein concentrations were determined by BCA protein assay kit. Total 30 μg protein was separated by SDS-PAGE and electro-transferred to PVDF membranes. The membranes were blocked with 5% skimmed milk for 1 h at room temperature and incubated with anti-ABCC2 antibody (Santa Cruz Biotechnology, Dallas, USA) at 4°C overnight. After washing 3 times with PBST, the membrane was incubated with HRP-conjugated secondary antibodies (Santa Cruz) at room temperature for 2 h. GAPDH (Santa Cruz) was used as an internal control. The expression of protein was visualized by the enhanced chemiluminescence system (Roche, Basel, Switzerland).
Data analysis
All the data are presented as mean ± SD. All in vitro experiments were repeated at least for 3 times. All the data were analyzed by GraphPad Prism Version 6.0 software. Student’s t-test was used for the comparison between two groups and one-way ANOVA was used for the comparison for more than two groups. The association between SNHG5 and ABCC2, and association between SNHG5 and miR-205-5p were analyzed by Pearson correlation test. P<0.05 were considered statistically significant.
Results
Expression pattern of SNHG5 and ABCC2 in healthy donors and CML patients
Firstly, qRT-PCR was performed to examine the expression of lncRNA SNHG5 and ABCC2 in healthy donors and CML patients. The results demonstrated the expression levels of SNHG5 and ABCC2 from peripheral blood cells of CML patients were significantly higher than that from healthy donors (Figure 1A and 1B). In addition, the SNHG5 expression level was positively correlated with the ABCC2 expression level in CML patients (Figure 1C). According to the median mRNA expression of ABCC2, the total 40 CML patients were divided into two groups i.e. high expression group and low expression group (Figure 1D). The expression level of SNHG5 was also compared in these two groups. Expectedly, the expression of SNHG5 was significantly higher in high ABCC2 expression group than that in low ABCC2 expression group (Figure 1E).
Figure 1.
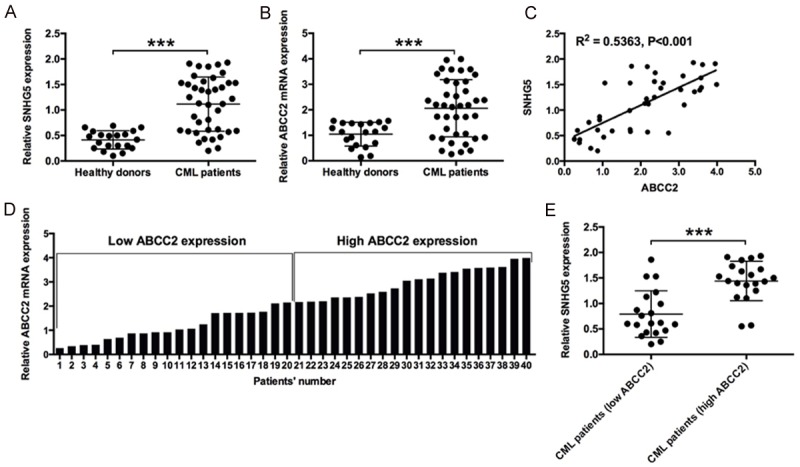
Expressions of SNHG5 and ABCC2 in healthy donors and CML patients. A: Relative expression of SNHG5 in peripheral blood cells of CML patients (n = 40) and healthy subjects (n = 20). B: Relative expression of ABCC2 mRNA in peripheral blood cells of CML patients (n = 40) and Healthy subjects (n = 20). C: The correlation between SNHG5 expression level and ABCC2 expression level in the peripheral blood cells of CML patients. D: The definition of high ABCC2 expression (n = 20) and low ABCC2 expression (n = 20) in the peripheral blood cells of CML patients based on the median values. E: The relative expression of SNHG5 in peripheral blood cells of CML patients with high ABCC2 expression (n = 20) and CML patients with low ABCC2 expression (n = 20). The SNHG5 levels and ABCC2 mRNA levels were determined by qRT-PCR. Significant differences between groups were shown as ***P<0.001.
Expression pattern of SNHG5 and ABCC2 in K562 and K562-R cells
The expression levels of SNHG5 and ABCC2 were further examined in K562 and K562-R cells, and SNHG5 and ABCC2 were up-regulated in K562-R cells when compared to K562 cells. We also measured the protein expression level of ABCC2 in the cells and K562-R cells showed higher expression level of ABCC2 protein when compared to K562 cells (Figure 2A-C). Next, we explored the impact of overexpression of SNHG5 on K562 cells as well as the knock-down effect of SNHG5 on K562-R cells. Twenty-four hrs after transfection, the mRNA and protein expression levels of ABCC2 were detected in K562 and K562-R cells. SNHG5 overexpression dramatically increase the mRNA and protein expression levels of ABCC2 level in K562 cells (Figure 2D-F); while knock-down of SNHG5 significantly suppressed the mRNA and protein expression levels of ABCC2, compared with respective control (Figure 2G-I).
Figure 2.
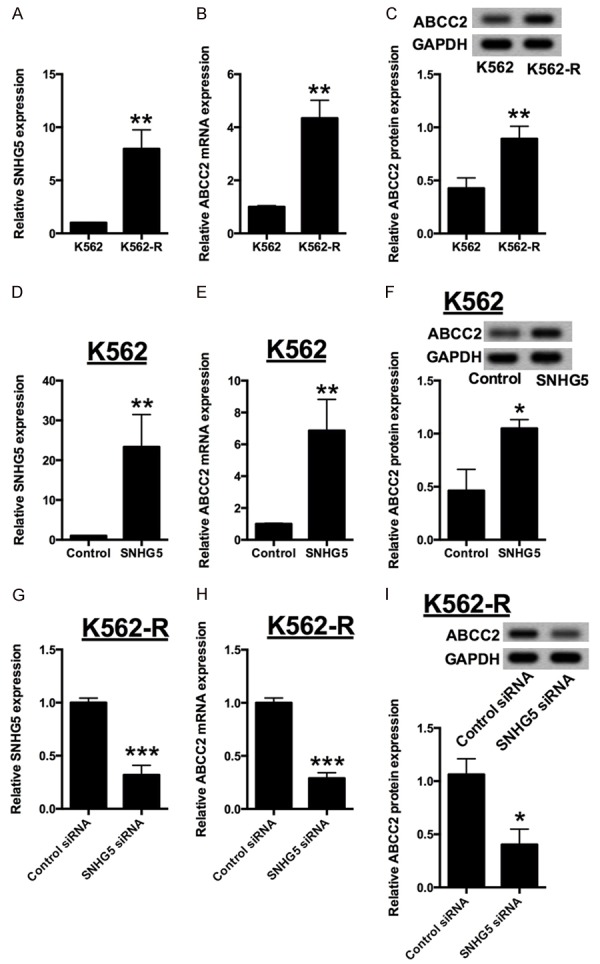
Expressions of SNHG5 and ABCC2 in K562 and K562-R cells. (A) Relative expression of SNHG5 in K562 cells and K562-R cells. Relative expression of (B) ABCC2 mRNA and (C) ABCC2 protein in K562 cells and K562-R cells. (D) Relative expression of SNHG5 in K562 cells transfected with pcDNA3.1 or pcDNA3.1-SNHG5. Relative expression of (E) ABCC2 mRNA and (F) ABCC2 protein in K562 cells transfected with pcDNA3.1 or pcDNA3.1-SNHG5. (G) Relative expression of SNHG5 in K562-R cells transfected with siRNA control or SNHG5 siRNA. Relative expression of (H) ABCC2 mRNA and (I) ABCC2 protein in K562-R cells transfected with siRNA control or SNHG5 siRNA. The SNHG5 levels and ABCC2 mRNA levels were determined by qRT-PCR and ABCC2 protein levels were determined by western blot assay. Significant differences between groups were shown as *P<0.05, **P<0.01.
SNHG5 directly interacts with miR-205-5p
Recent studies suggested lncRNAs can directly bind to miRNAs, therefore preventing the subsequently binding of miRNAs to target mRNAs [17]. We searched an online database starBase (http://starbase.sysu.edu.cn) to look for complementary miRNAs [18,19], which can bind to SNHG5. MiR-205-5p was found to be one of potential targets. Furthermore, two luciferase reporters containing wild type SNHG5 or the mutant SNHG5 (mutations at predicted miR-205-5p binding sites) were constructed. We found that K562 cells transfected with miR-205-5p mimics dramatically inhibited the activities of the wild type SNHG5 reporter, but not that of the mutant one (Figure 3A-C). To validate the direct interaction between SNHG5 and miR-205-5p, RIP assay was used to pull down endogenous miRNAs associated with SNHG5, followed by qRT-PCR analysis. The amount of SNHG5 pull down for miR-205-5p was significantly higher than that of the empty vector (pLV-MS2), negative control IgG, and mutant one (Figure 3D). Next, we explored the correlation between SNHG5 and miR-205-5p in K562 cells and CML patients. After overexpression of SNHG5 in K562 cells, the expression level of miR-205-5p was suppressed dramatically (Figure 3E). A further correlation analysis disclosed that SNHG5 and miR-205-5p were negatively correlated in peripheral blood cells from CML patients (Figure 3F).
Figure 3.
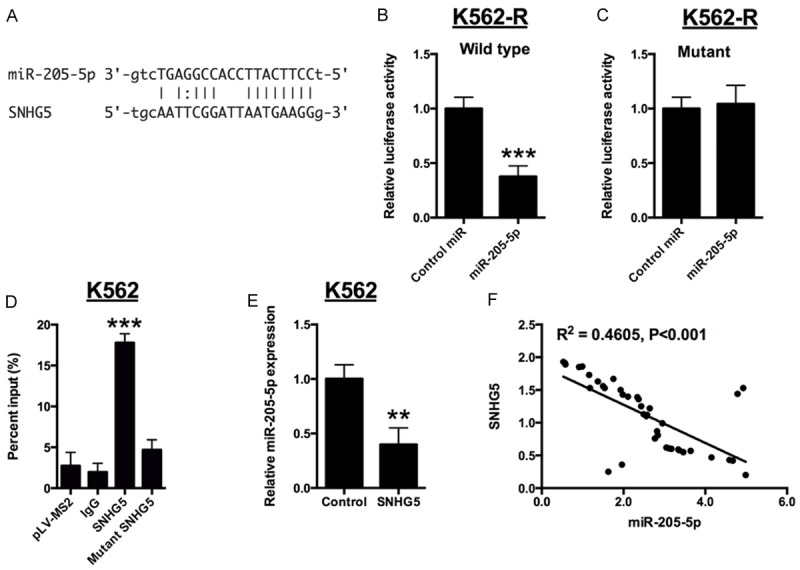
SNHG5 directly interacts with miR-205-5p. A: Bioinformatics analysis showed the prediction for miR-205-5p binding sites on SNHG5. B and C: Luciferase activity in K562-R cells co-transfected with miR-205-5p mimics or mimics control and luciferase reporters containing wild type SNHG5 or mutant SNHG5. D: MS2-RIP, followed by miRNA qRT-CPR to detect endogenous miR-205-5p associated with SNHG5 in K562 cells. E: Relative expression of miR-205-5p in K562 cells transfected with pcDNA3.1 or pcDNA3.1-SNHG5. F: Correlation between SNHG5 expression and miR-205-5p expression in the peripheral blood cells of CML patients. Significant differences between groups were shown as **P<0.01, ***P<0.001.
ABCC2 was a direct target of miR-205-5p
The downstream targets of miR-205-5p were predicted by bioinformatics screening and ABCC2 was found to be one of the predicted targets. Subsequently, the luciferase reporter assay performed. The ABCC2 3’UTR sequence (WT) or the mutant one (MUT) was cloned into a luciferase reporter vector and luciferase reporter assay was performed in K562-R cells (Figure 4A). A significant decrease in luciferase activity was observed in cells transfected with miR-205-5p and wild type 3’UTR of ABCC2 in K562-R cells (Figure 4B). However, the luciferase activity was unaffected when co-transfection with the mutant 3’TUR of ABCC2 and miR-205-5p mimics in K562 cells (Figure 4C). Furthermore, the changes of ABCC2 expression after miR-205-5p overexpression were examined in K562-R cells. We found that the overexpression of miR-205-5p resulted in dramatically reduction of ABCC2 mRNA and protein levels when compared to control miRNA (Figure 4D and 4E).
Figure 4.
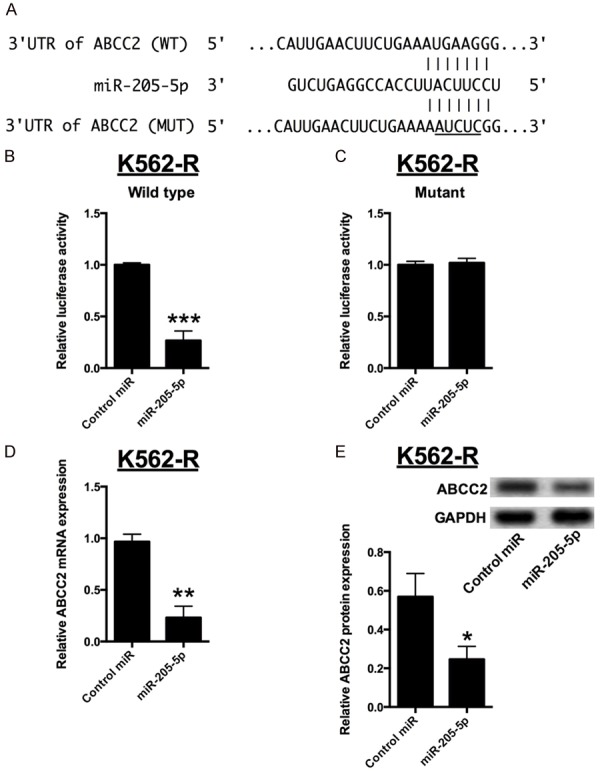
ABCC2 was a direct target of miR-205-5p. A: Bioinformatics analysis showed the prediction for miR-205-5p bindings sites on the 3’TUR of ABCC2. B and C: Luciferase activity in K562-R cells co-transfected with miR-205-5p mimics or mimics control and luciferase reporters containing wild type 3’UTR of ABCC2 or mutant 3’UTR of ABCC2. D and E: Relative expression of ABCC2 mRNA and protein in K562-R cells transfected with miR-205-5p mimics or mimics control. Significant differences between groups were shown as *P<0.05, **P<0.01, ***P<0.001.
Altered expression of SNHG5 affects the chemo-sensitivity of K562/K562-R cells
The measurement of drug sensitivity showed that K562-R cells were much more resistant to imatinib than K562 cells, as the IC50 of imatinib was about ~2 μM in the K562 cells while it reached ~10 μM in the K562-R cells (Figure 5A and 5B). After overexpression of SNHG5, the K562 cells were less resistant to imatinib compared with control, and the IC50 of imatinib in the K562 cells transfected with SNHG5 construct was increased to ~8 μM (Figure 5C and 5D). In contrast, the K562-R cells transfecting with SNHG5 siRNA were more resistant to imatinib when to control siRNA group, and the IC50 of imatinib was decreased from ~8.5 μM to 2.5 μM (Figure 5E and 5F).
Figure 5.
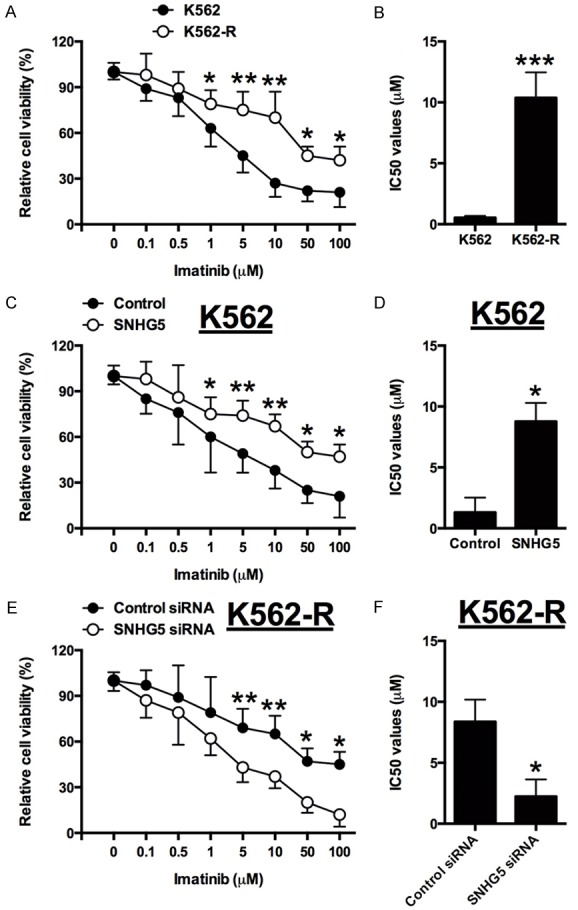
Altered expression of SNHG5 affects the chemo-sensitivity of K562/K562-R cells. A and B: The cell viability and IC50 values of imatinib in K562 and K562-R cells received imatinib treatment. C and D: The cell viability and IC50 values of imatinib in pcDNA3.1 or pcDNA3.1-SNHG5 transfected K562 cells received imatinib treatment. E and F: The cell viability and IC50 values of imatinib in siRNA control or SNHG5 siRNA transfected K562-R cells received imatinib treatment. Significant differences between groups were shown as *P<0.05, **P<0.01, ***P<0.001.
SNHG5 promotes imatinib resistance through regulating ABCC2
K562 cells with SNHG5 overexpression had higher resistance against imatinib, showing about four-fold increase of IC50 and upregulation of ABCC2 mRNA and protein, compared with control. While overexpressing miR-205-5p or silencing ABCC2 partially abolished these effects of SNHG5 overexpression K562 cells (Figure 6A, 6C and 6E). In contrast, K562-R cells with SNHG5 knockdown had reduced imatinib resistance and down-regulation of ABCC2 mRNA and protein, compared to control siRNA group; while knock-down of miR-205-5p or overexpression of ABCC2 could partially rescue the suppressive effects of induced by SNHG5 knock-down (Figure 6B, 6D and 6F). Taken together, these data indicated that SNHG5 promotes resistance against imatinib in CML cells partially through regulating ABCC2 expression.
Figure 6.
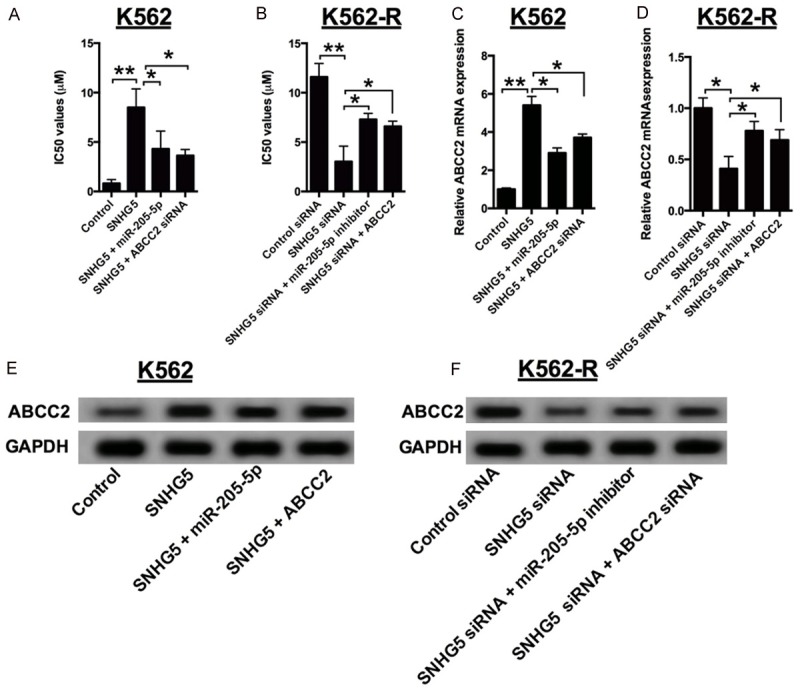
SNHG5 promotes imatinib resistance through regulating ABCC2. A: The IC50 values of imatinib in SNHG5-overexpressed K562 cells transfected with miR-205-5p mimics or ABCC2 siRNA. B: The IC50 values of imatinib in SNHG5 knock-down K562-R cells transfected with miR-205-5p inhibitor or pCMV6-ABCC2. C: The mRNA expression levels of ABCC2 in SNHG5 overexpressed K562 cells transfected with miR-205-5p mimics or ABCC2 siRNA. D: The mRNA expression levels of ABCC2 in SNHG5 knock-down K562-R cells transfected with miR-205-5p inhibitor or pCMV6-ABCC2. E: The protein levels of ABCC2 in SNHG5 overexpressed K562 cells transfected with miR-205-5p mimics or ABCC2 siRNA. F: The protein expression levels of ABCC2 in SNHG5 knock-down K562-R cells transfected with miR-205-5p inhibitor or pCMV6-ABCC2. Significant differences between groups were shown as *P<0.05, **P<0.01, ***P<0.001.
Discussion
Although imatinib is effective in the treatment of CML, drug resistance cannot be avoided in some patients, particularly for those CML patients at advanced stage, and imatinib resistance greatly limits the clinical use of imatinib. Recently, more and more investigators have focused on investigating the roles lncRNAs, which have been well-known for the roles in oncogenic and tumor suppressor pathways. Moreover, some lncRNAs have been reported to involve in the development of imatinib resistance in CML chemotherapy. For example, MEG3 could suppress imatinib resistance by reducing the expression of MRP1, MDR1, and ABCG2. HOTAIR regulates CML cell drug resistance in a PI3K/Akt-dependent way [20,21]. In the present study, we found that the increase of SNHG5 in CML patients was associated with an increase in ABCC2 expression. SNHG5 and ABCC2 were also up-regulated in the K562-R cells, which indicated that abnormal expression of SNHG5 and ABCC2 may contribute to imatinib resistance in CML. Therefore, uncovering the mechanisms underneath will be helpful for us to have a better understanding of imatinib resistance in CML.
In the further study, overexpression of SNGH5 in K562 cells significantly increased ABCC2 mRNA and protein expression; while knock-down of SNHG5 drastically decreased the ABCC2 expression in K562-R cells. Recent studies have demonstrated that the lncRNAs can act as competing endogenous RNA (ceRNA), which serves as molecular sponges for a miRNA through the miRNA binding sites, therefore preventing the subsequently binding of miRNAs to target mRNAs. Bioinformatics analysis showed that one of the binding sites of miR-205-5p span the transcript of SNHG5. Previously, miR-205-5p was shown to regulate cancer cell proliferation and invasion. In breast cancer, miR-205-5p could modulate epithelial to mesenchymal transition [22]. In another study, overexpression of miR-205-5p could re-sensitize gemcitabine-resistant pancreatic cancer cells and reduce the cell proliferation as well as tumor growth [23]. Therefore, in the present study, SNHG5 may mediate imatinib resistance by interacting with miR-205-5p. Indeed, through luciferase reporter assay and RNA immunoprecipitation assay, we found that SNHG5 directly binds to miR-205-5p and their expressions were negatively correlated in CML patients.
The downstream targets of miR-205-5p were predicted by bioinformatics screening and ABCC2 was found to be one of the predicted targets. Overexpression of miR-205-5p resulted in dramatically reduction of ABCC2 mRNA and protein, and SNHG5 regulates ABCC2 expression through binding miR-205-5p, which suppressed the ABCC2 mRNA degradation. In the in vitro functional study, we showed that SNHG5 promoted imatinib resistance of K562 cells, while silence of ABCC2 or overexpression of miR-205-5p partially abolished this effect. In the imatinib-resistant cells, SNHG5 knock-down showed reduced imatinib resistance, and inhibition of miR-205-5p or overexpression of ABCC2 partially rescue the suppression induced by knock-down of SNHG5. Consistently, previous study has demonstrated that SNHG5 is playing functional roles in oxaliplatin resistance by counteracting STAU1-mediated mRNA destabilization in the colorectal cancer cells [15,24]. Moreover, except for ABC transporter pathway, imatinib resistance in CML includes BCR-ABL1 kinase-dependent as well as BCR-ABL1 kinase-independent pathways, such as PI3K, MAPK, or JAK/STAT [25-28]. Taken together, SNHG5 may promote drug resistance by different mechanisms that would be the focus of our future studies.
In conclusion, our results suggested that SNHG5 promotes imatinib resistance in CML via acting as a competing endogenous RNA against miR-205-5p. These findings enlightened us that lncRNAs may be a promising target for CML treatment, especially imatinib resistance cases. However, the mechanism of imatinib resistance in CML may be multifactorial and very complex and the precise molecular mechanism underlying the involvement of SNHG5 in CML require further investigation.
Disclosure of conflict of interest
None.
References
- 1.Goldman JM, Melo JV. Chronic myeloid leukemia--advances in biology and new approaches to treatment. N Engl J Med. 2003;349:1451–1464. doi: 10.1056/NEJMra020777. [DOI] [PubMed] [Google Scholar]
- 2.O’Brien S, Berman E, Borghaei H, Deangelo DJ, Devetten MP, Devine S, Erba HP, Gotlib J, Jagasia M, Moore JO, Mughal T, Pinilla-Ibarz J, Radich JP, Shah Md NP, Shami PJ, Smith BD, Snyder DS, Tallman MS, Talpaz M, Wetzler M. NCCN clinical practice guidelines in oncology: chronic myelogenous leukemia. J Natl Compr Canc Netw. 2009;7:984–1023. doi: 10.6004/jnccn.2009.0065. [DOI] [PubMed] [Google Scholar]
- 3.Wei G, Rafiyath S, Liu D. First-line treatment for chronic myeloid leukemia: dasatinib, nilotinib, or imatinib. J Hematol Oncol. 2010;3:47. doi: 10.1186/1756-8722-3-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hochhaus A, Kreil S, Corbin A, La Rosee P, Lahaye T, Berger U, Cross NC, Linkesch W, Druker BJ, Hehlmann R, Gambacorti-Passerini C, Corneo G, D’Incalci M. Roots of clinical resistance to STI-571 cancer therapy. Science. 2001;293:2163. [PubMed] [Google Scholar]
- 5.Takahashi N, Miura M. Therapeutic drug monitoring of imatinib for chronic myeloid leukemia patients in the chronic phase. Pharmacology. 2011;87:241–248. doi: 10.1159/000324900. [DOI] [PubMed] [Google Scholar]
- 6.Milosevic JD, Kralovics R. Genetic and epigenetic alterations of myeloproliferative disorders. Int J Hematol. 2013;97:183–197. doi: 10.1007/s12185-012-1235-2. [DOI] [PubMed] [Google Scholar]
- 7.Tian J, Xu YY, Li L, Hao Q. MiR-490-3p sensitizes ovarian cancer cells to cisplatin by directly targeting ABCC2. Am J Transl Res. 2017;9:1127–1138. [PMC free article] [PubMed] [Google Scholar]
- 8.Choi JR, Kim JO, Kang DR, Shin JY, Zhang XH, Oh JE, Park JY, Kim KA, Kang JH. Genetic variations of drug transporters can influence on drug response in patients treated with docetaxel chemotherapy. Cancer Res Treat. 2015;47:509–517. doi: 10.4143/crt.2014.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhan M, Qu Q, Wang G, Zhou H. Let-7c sensitizes acquired cisplatin-resistant A549 cells by targeting ABCC2 and Bcl-XL. Pharmazie. 2013;68:955–961. [PubMed] [Google Scholar]
- 10.Zhu H, Wu H, Liu X, Evans BR, Medina DJ, Liu CG, Yang JM. Role of MicroRNA miR-27a and miR-451 in the regulation of MDR1/Pglycoprotein expression in human cancer cells. Biochem Pharmacol. 2008;76:582–588. doi: 10.1016/j.bcp.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao P, Wei GH. Genomic insight into the role of lncRNA in cancer susceptibility. Int J Mol Sci. 2017:18. doi: 10.3390/ijms18061239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng J, Wang Y, Lei J, Lei W, Xiong JP. Insights into the involvement of noncoding RNAs in 5-fluorouracil drug resistance. Tumour Biol. 2017;39:1010428317697553. doi: 10.1177/1010428317697553. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura Y, Takahashi N, Kakegawa E, Yoshida K, Ito Y, Kayano H, Niitsu N, Jinnai I, Bessho M. The GAS5 (growth arrest-specific transcript 5) gene fuses to BCL6 as a result of t(1;3)(q25;q27) in a patient with B-cell lymphoma. Cancer Genet Cytogenet. 2008;182:144–149. doi: 10.1016/j.cancergencyto.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 14.Ichigozaki Y, Fukushima S, Jinnin M, Miyashita A, Nakahara S, Tokuzumi A, Yamashita J, Kajihara I, Aoi J, Masuguchi S, Zhongzhi W, Ihn H. Serum long non-coding RNA, snoRNA host gene 5 level as a new tumor marker of malignant melanoma. Exp Dermatol. 2016;25:67–69. doi: 10.1111/exd.12868. [DOI] [PubMed] [Google Scholar]
- 15.Damas ND, Marcatti M, Come C, Christensen LL, Nielsen MM, Baumgartner R, Gylling HM, Maglieri G, Rundsten CF, Seemann SE, Rapin N, Thezenas S, Vang S, Orntoft T, Andersen CL, Pedersen JS, Lund AH. SNHG5 promotes colorectal cancer cell survival by counteracting STAU1-mediated mRNA destabilization. Nat Commun. 2016;7:13875. doi: 10.1038/ncomms13875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao L, Han T, Li Y, Sun J, Zhang S, Liu Y, Shan B, Zheng D, Shi J. The lncRNA SNHG5/miR-32 axis regulates gastric cancer cell proliferation and migration by targeting KLF4. FASEB J. 2017;31:893–903. doi: 10.1096/fj.201600994R. [DOI] [PubMed] [Google Scholar]
- 17.Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li JH, Liu S, Zhou H, Qu LH, Yang JH. star-Base v2.0: decoding miRNA-ceRNA, miRNAncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–97. doi: 10.1093/nar/gkt1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang JH, Li JH, Shao P, Zhou H, Chen YQ, Qu LH. starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 2011;39:D202–209. doi: 10.1093/nar/gkq1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou X, Yuan P, Liu Q, Liu Z. LncRNA MEG3 regulates imatinib resistance in chronic myeloid leukemia via suppressing MicroRNA-21. Biomol Ther (Seoul) 2017 doi: 10.4062/biomolther.2016.162. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang H, Li Q, Tang S, Li M, Feng A, Qin L, Liu Z, Wang X. The role of long noncoding RNA HOTAIR in the acquired multidrug resistance to imatinib in chronic myeloid leukemia cells. Hematology. 2017;22:208–216. doi: 10.1080/10245332.2016.1258152. [DOI] [PubMed] [Google Scholar]
- 22.Stankevicins L, Barat A, Dessen P, Vassetzky Y, de Moura Gallo CV. The microRNA-205-5p is correlated to metastatic potential of 21T series: A breast cancer progression model. PLoS One. 2017;12:e0173756. doi: 10.1371/journal.pone.0173756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaudhary AK, Mondal G, Kumar V, Kattel K, Mahato RI. Chemosensitization and inhibition of pancreatic cancer stem cell proliferation by overexpression of microRNA-205. Cancer Lett. 2017;402:1–8. doi: 10.1016/j.canlet.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heery R, Finn SP, Cuffe S, Gray SG. Long non-coding RNAs: key regulators of epithelialmesenchymal transition, tumour drug resistance and cancer stem cells. Cancers (Basel) 2017:9. doi: 10.3390/cancers9040038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding J, Romani J, Zaborski M, MacLeod RA, Nagel S, Drexler HG, Quentmeier H. Inhibition of PI3K/mTOR overcomes nilotinib resistance in BCR-ABL1 positive leukemia cells through translational down-regulation of MDM2. PLoS One. 2013;8:e83510. doi: 10.1371/journal.pone.0083510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quentmeier H, Eberth S, Romani J, Zaborski M, Drexler HG. BCR-ABL1-independent PI3-Kinase activation causing imatinib-resistance. J Hematol Oncol. 2011;4:6. doi: 10.1186/1756-8722-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hakansson P, Nilsson B, Andersson A, Lassen C, Gullberg U, Fioretos T. Gene expression analysis of BCR/ABL1-dependent transcriptional response reveals enrichment for genes involved in negative feedback regulation. Genes Chromosomes Cancer. 2008;47:267–275. doi: 10.1002/gcc.20528. [DOI] [PubMed] [Google Scholar]
- 28.Gotlib J. JAK inhibition in the myeloproliferative neoplasms: lessons learned from the bench and bedside. Hematology Am Soc Hematol Educ Program. 2013;2013:529–537. doi: 10.1182/asheducation-2013.1.529. [DOI] [PubMed] [Google Scholar]