

Abstract
Clinical management of malignant mesothelioma (MM) is very challenging due to marked resistance of this tumor to chemotherapy. Various mechanisms lead to a less than ideal drug concentration inside of MM cells, diminishing cytotoxicity. Consequently, single cytotoxic drugs achieve very modest response rates in MM patients, and combination regimens using standard and novel therapies have achieved only limited improvement in overall survival. Here, we demonstrate that MYC has either proliferative or pro-survival effects in MM cells during normal or stressed conditions, respectively. A MYC inhibitor 10058-F4 reduced MM cell proliferation via down regulation of cyclin D. Under serum starvation conditions, MM cells became quiescent, and the addition of MYC inhibitors triggered apoptosis in the resting MM cells. We also found that high concentrations of the PAK inhibitor PF3758309 killed MM cells, but the drug had only cytostatic effects at lower concentrations. These quiescent cells underwent apoptosis upon pharmacological inhibition of MYC. A novel MYC inhibitor KJ-Pyr-9 and a newer PAK inhibitor, FRAX597, also demonstrated marked cytotoxic cooperativity. Collectively, these findings demonstrate that targeting of MYC can sensitize MM cells and provide rationale for inhibition of MYC and PAK as a novel combinatory regimen for the treatment of this otherwise therapy-resistant, clinically incurable malignancy.
Keywords: Mesothelioma, MYC, PAK, cytostasis, apoptosis
Introduction
Malignant mesothelioma (MM) is a treatment-resistant malignancy that is usually associated with exposure to asbestos. This disease is rapidly progressive, with a median overall survival of only 9 to 17 months [1]. Genetic studies of MM have revealed frequent deletions of CDKN2A, which encodes the tumor suppressors p16INK4A and p14ARF, simultaneously resulting in the inactivation of the two gatekeeper tumor suppressor pathways, i.e., the Rb and p53 pathways, respectively [2]. NF2 is one of the most frequently inactivated genes in MM [3,4]. Re-expression of NF2/Merlin in MM cells inhibits cell cycle progression through the down regulation of cyclin D1 [5]. The p21-activated kinase (PAK) has been shown to phosphorylate and inactivate NF2 [6], and we have reported that PAK activation is a common feature in MM cells [7]. PAK is also an upstream regulator of ERK, NFkB, Catenin and Aurora pathways [8,9]. Moreover, ERK2 is essential for the proliferation of MM cells [10]. PAK has also been shown to be activated in many MMs, and we previously reported that PAK inhibitor IPA-3 can induce MM cell death [7].
Pleural MM is a virtually incurable disease because of its marked resistance to conventional chemotherapies. The current preferred first-line treatment is the combination of cisplatin and pemetrexed. Still, nearly all patients fail this treatment [11]. Moreover, targeted reagents such as the EGFR inhibitor gefitinib or the PDGFR inhibitor imatinib mesylate have shown no clinical efficacy. Also, the combination of cisplatin and gemcitabine with the VEGF monoclonal antibody bevacizumab resulted in no significant improvement in patient response rate or survival [1]. Therefore, innovative and effective approaches to eradicate MM cells are urgently needed.
MYC has been found to be frequently amplified in many malignancies such as lung, colorectal, breast and prostate carcinomas [12]. Additionally, chromosome translocation-mediated up regulation of MYC is a causal mechanism for the development of various lymphoid malignancies [13]. Tissue-specific ectopic expression of MYC in mice is also known to induce T- and B-cell neoplasms as well as tumors of the breast, liver, skin, pancreas and bone [14]. MYC up regulation causes cell cycle progression through the up regulation of cyclins and the repression of p15 and p21 [15]. Moreover, MYC promotes genomic instability and angiogenesis. In addition to genes involved in cell proliferation, MYC target genes play a role in cell survival, DNA dynamics, RNA modification, energy metabolism and macromolecular synthesis [16,17]. Mechanistically, MYC binds to the promoters of its target genes and promotes elongation by regulating transcription pause release [18]. Also, because MYC controls multiple components of ribosome biogenesis, it can indirectly regulate gene expression at the translational level [19]. These critical functions underlie MYC-induced tumorigenesis [14]. Furthermore, it is now believed that low but persistent levels of MYC are responsible for the start of cellular transformation, because low, as opposed to high, levels of MYC enable cells to evade ARF-p53 surveillance and thereby circumvent apoptosis [20]. Thus, MYC is essential for both initiation and maintenance of malignancy.
MYC inhibition by a dominant-negative MYC, dubbed Omomyc, was shown to suppress Kras-induced lung cancer in a mouse model [21]. Furthermore, a small-molecule MYC inhibitor, 10058-F4, can induce cell cycle arrest and/or apoptosis in human acute myeloid leukemia and lymphoma [22,23]. This type of inhibitor binds MYC and stabilizes the monomer over the highly ordered MYC-MAX heterodimer, thus abolishing MYC’s function [24,25]. One mechanism of the inhibitor’s efficacy is through the down regulation of RNA polymerase II levels at the promoter of MYC target genes [18]. This inhibitor also showed anti-cancer effects in gastric and prostate tumors [26,27]. It also greatly facilitates Akt inhibitor-induced cell death in T-cell lymphomas [28]. Recently, a novel MYC inhibitor KJ-Pyr-9 was discovered to be able to suppress MYC function and inhibit cancer cell growth via a similar mechanism [29].
In contrast to the extensive reports on the oncogenic role of MYC in other cancers, the role of MYC in the pathogenesis of MM is not well established. Immunohistochemistry studies have revealed that MYC is more frequently expressed in MM tissues than in normal mesothelium [30]. Recently, FUSE-binding protein-interacting repressor (FIR), a c-myc transcriptional repressor, has been shown to induce apoptosis in MM cells [31]. Other than these reports, the role of MYC in MM, and whether it can be targeted efficaciously by using small molecular inhibitors, is largely unknown. Here, we report that MYC is frequently up regulated in MM cells, and MYC inhibitors induce apoptosis in MM cells upon serum depletion or PAK inhibition-induced cell cycle arrest.
Materials and methods
Cell culture
MM cells used in this investigation were as previously described [32]. Cells were maintained in RPMI1640 medium supplemented with 10% FBS containing 100 μg/mL penicillin and streptomycin and 2 mM L-glutamine.
Reagents
Antibodies against cyclin D2, D3, GAPDH, β-actin, and horseradish peroxidase (HRP)-linked secondary antibodies were purchased from Santa Cruz Biotechnology. Antibodies against MYC, cleaved caspase-3, PARP, PAK1,2,3, p-AKT/AKT, and p-ERK/ERK were from Cell Signaling. p-PAK1,2,3 antibody and MYC inhibitors 10058-F4 and 10074-G5 were from Sigma-Aldrich. KJ-Pyr-9 was obtained from Millipore. PAK inhibitor PF3758309, FRAX597 and IPA-3, BRD inhibitor JQ1, as well as MEK inhibitors U0126 and PD0325901 were obtained from Selleckchem.
Cell viability assay
Cell viability was measured by MTS assay (Promega). In brief, cells were seeded at 1×104 cells/well in 96-well plates. OD values at 595 nm was evaluated 1 to 4 h after incubation with 10% MTS using a 96-well microplate reader.
Western blot analysis
Total cellular protein was extracted by using 1× cell lysis buffer (Cell Signaling) supplemented with 2 mM PMSF. Cell debris was removed by spinning at 15,000× g for 15 min at 4°C. Protein concentrations were determined using Bradford reagent. Proteins (50 µg/well) were loaded into the Tris-Glycine buffered SDS-PAGE gel (Invitrogen). Separated protein was then transferred onto PVDF membranes (Millipore). Membranes were blocked with 5% non-fat milk in Tris-buffered saline with tween (TBST) for 1 h and then incubated with primary antibodies at 4°C overnight. After washing 3x with TBST, membranes were incubated with secondary antibody at RT for 1 h, and further washed three times. Film was exposed and developed for HRP signals.
Flow cytometry analysis
Flow cytometry (LSRII, BD Biosciences) was used to analyze cell death markers. An apoptosis detection kit containing AnnexinV-FITC and Propidium Iodide (PI) were purchased from BD Pharmingen. For cell cycle analysis, cells were fixed with 70% ice-cold ethanol, washed twice with PBS, and stained with 50 µg/mL PI. Data were analyzed by using FlowJo.
Results
Inhibition of MYC suppresses cell proliferation
Twenty-three human MM-derived cell lines were used to determine the expression of MYC protein, along with three non-transformed mesothelial cell lines (MeT-5A, 7086 and LP9). Immunoblotting revealed that 18 of 23 (78%) MM cell lines showed up regulation of MYC expression in MYC1 (CUG-initiated), MYC2 (AUG-initiated), or both isoforms (Figure 1A). For further investigations, we chose six cell lines with either medium (Meso1, Meso25 and Meso41) or high (MSTO211H, NCI2052 and NCI2452) levels of MYC expression to determine if suppression of MYC by small molecule inhibitors affects MM cell viability. Mitochondrial activity-based MTS assay showed that MM cells have reduced viability or proliferation when treated with the MYC inhibitor 10058-F4 at 40 µM for 30 h (Figure 1B). Immunoblotting demonstrated that 10058-F4 reduced cyclin D2 and D3 levels and induced p21Waf1/Cip1 expression after 16-24 h of treatment. Interestingly, cyclin D2 exhibited up regulation at an early time point (4 h) in some cell lines, suggesting a transient, non-transcriptional effect of the inhibitor. Activity of the survival factor AKT kinase was not altered following MYC inhibition (Figure 1C). No PARP cleavage was detected, indicating that apoptotic caspase-3 was not engaged (not shown). Therefore, the reduced cell MTS activity was interpreted to be due to a cytostatic effect of 10058-F4 (Figure 1C). We next performed cell cycle analysis on cells treated with 10058-F4. Inhibition of MYC inhibition resulted in a significantly increased proportion of cells arrested at G1 phase (Figure 1D). MM cells cultured without serum (mitogen depletion) similarly underwent cell cycle arrest.
Figure 1.
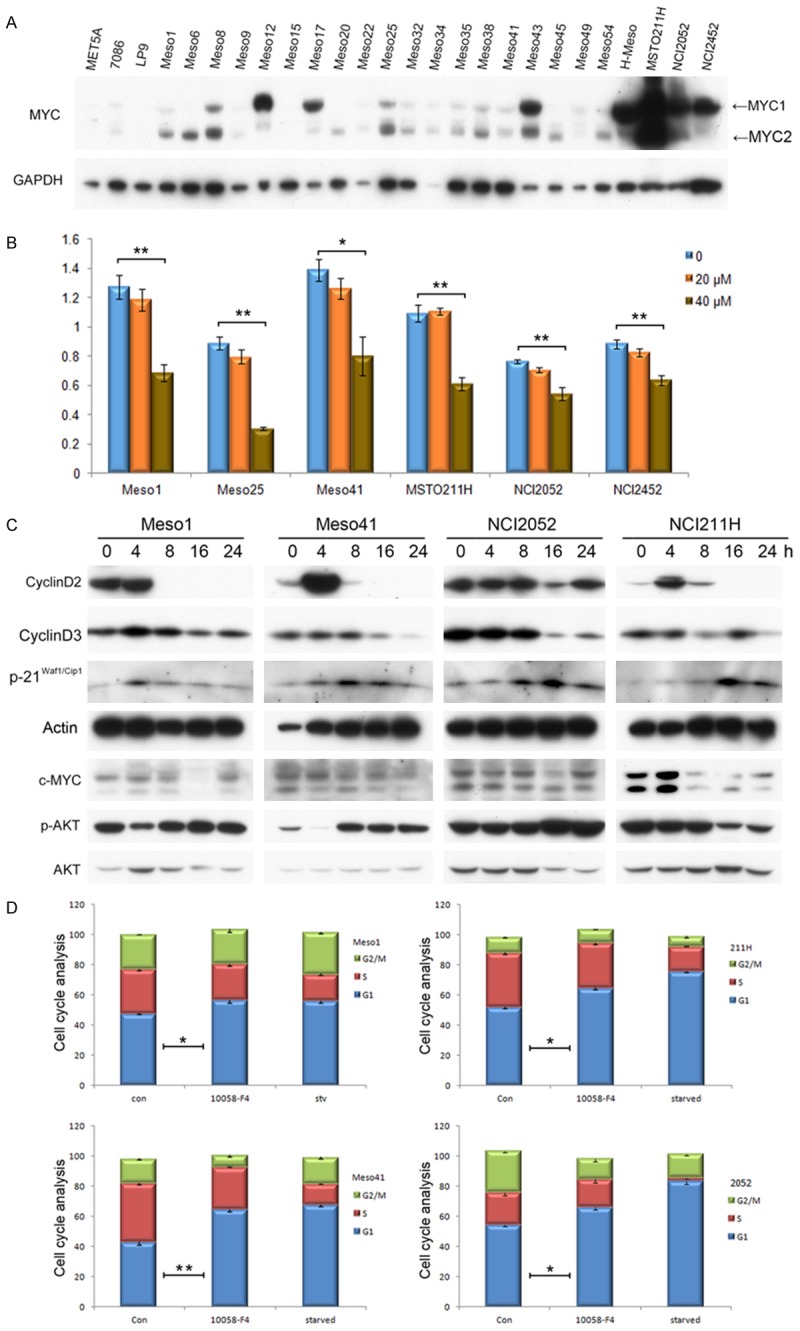
MYC is up regulated in MM cells, and inhibition of MYC suppresses MM cell proliferation. A. MYC is over expressed in 78% MM cell lines as shown by immunoblotting (18 of 23). Note: MeT-5A, 7086 and LP9 are non-malignant mesothelial cell lines. MYC1 (CUG-initiated), MYC2 (AUG-initiated). B. MTS assay depicting viability of MM cells treated with MYC inhibitor 10058-F4 at the indicated concentrations for 30 h. C. Immunoblotting demonstrating that 10058-F4 reduces cyclin D2 and D3 levels and induces p21Waf1/Cip1 expression. No caspase-3 activity was found. D. Cell cycle analysis of MM cells (Meso1, Meso41, 211H, 2052) following 10058-F4 treatment or serum starvation.
MYC blockade induces cell death of serum-starved MM cells
Evasion of growth suppression is one of the cancer hallmarks [33]. When incubated with serum-free medium, MM cells did not show cell death. However, when we treated the cells with 10058-F4 at 40 mM for 20 h either in serum-free or normal media, we found that 10058-F4 dramatically diminishes the viability of cells cultured under serum-free versus serum-containing medium, implying a cytotoxic effect of MYC inhibition under such conditions (Figure 2A). Indeed, the serum-starved MM cells with 10058-F4 demonstrated microscopic morphology of cell death (Figure 2B). Collectively, these findings indicate that MYC has a pro-survival role in MM cells during mitogen depletion. To assess the type of cell death caused by 10058-F4 in serum-starved MM cells, treated cells were stained with AnnexinV-FITC and PI. Apoptotic cells lose membrane integrity, such that the inner side of the membrane can be bound by AnnexinV. FACS analysis showed that serum-starved, 10058-treated MM cells had significantly more cells in early apoptosis (AnnexinV+) and late apoptosis (AnnexinV+PI+) when compared to control cells (either serum starvation alone or 10058-F4 alone), indicating that the type of cell death caused by 10058-F4 is mainly apoptotic (Figure 2C, 2D). In parallel, cells were fixed and stained with PI to visualize the apoptotic sub-G1 population by FACS. This analysis revealed that 10058-F4 significantly increased DNA condensation in serum-starved MM cells (Figure 2E, 2F). Mechanistically, caspase-3 was cleaved and activated (Figure 2G, 2H, Figure S1A-C). Use of a second MYC inhibitor, 10074-G5, produced virtually identical results, including a reduction in cyclin D levels under normal serum conditions and induction of apoptosis when serum was depleted (Figure S2A). The BRD4 inhibitor JQ1 has also been shown to suppress MYC transcription [34]. Similarly, we found the JQ1 can also inhibit MM cell viability under normal culture conditions in the majority of cell lines, and such inhibition was enhanced by serum-depletion in most cell lines (Figure S2B). Notably, however, JQ1 was found to promote the proliferation of MTSO211H. This suggests that in addition to inhibiting MYC, JQ1 inhibits other BRD4-associated super-enhancers, which may include negative regulators of the cell cycle. Thus, the overall effect of JQ1 appears to depend on a balance of the super-enhancer related expression of oncogenic factors as well as tumor suppressors.
Figure 2.
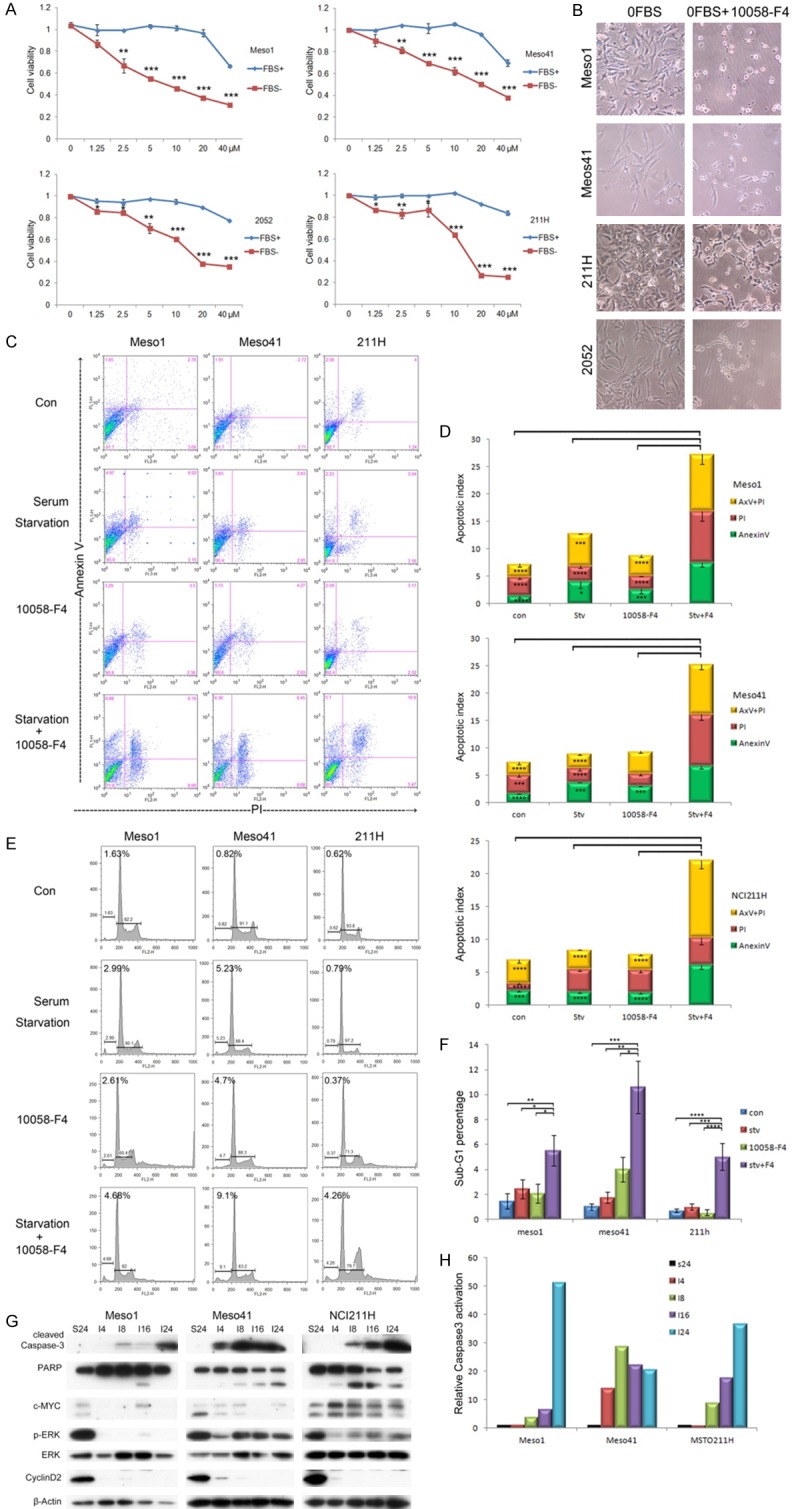
MYC inhibition triggers apoptosis of MM cells when medium is depleted of serum. A. MTS assay to monitor MM cell viability following treated with MYC inhibitor 10058-F4 at 40 mM for 20 h in serum free or normal medium. MTS activities were normalized to each control. B. Microscopic depiction of increased cell death in serum-starved MM cells treated with 10058-F4. C. Membrane integrity was monitored by using AnnexinV staining and analyzed by FACS. Representative set of experimental findings is shown (AnnexinV: Early Apoptosis; PI: Necrosis; AnnexinV/PI: Late Apoptosis). D. MM cells in early apoptosis, late apoptosis, and necrosis following treatment with 10058-F4 plus serum starvation are significantly higher than those in control cells or cells treated with 10058-F4 alone. E. Cells were fixed, stained with PI, and cell cycle analysis was performed by FACS to visualize apoptotic sub-G1 population. F. 10058-F4 plus serum starvation significantly increases the proportion of sub-G1 cells compared with that of control treatment. G. Western blot analysis indicating activation of caspase-3. (S24: starved for 24 h; I4: Inhibitor for 4 h). H. Signal intensity as assessed using fluorchem SP (Alpha Innotech). *P<0.05, **P<0.01, ***P<0.005, ****<0.001.
PAK inhibitor PF3758309 inhibits the viability of MM cells in a dose-dependent manner
We now report that another PAK inhibitor, PF3758309, has increased efficacy compared to IPA-3. MM cells were treated with PF3758309 or IPA-3 at the indicated concentrations for 20 h, and MTS assay demonstrated that treatment with PF3758309 caused decreased cell viability as compared to IPA-3 (Figure 3A, Figure S3A). Low concentrations of PF3758309 induces cell cycle arrest (Figure 3B, 3C, Figure S3B) and reduction of Cyclin A (Figure S3C). Higher concentrations of the drug triggers loss of morphological integrity (Figure S3D). The latter effect was due to apoptosis, as it was characterized by sub-G1 DNA condensation (Figure 3D, 3E, Figure S3E) and loss of membrane polarity (Figure 3F, 3G). Moreover, caspase-3 was found to be activated by higher doses of PF3758309 (Figure S3E). MYC protein levels were unchanged at low concentrations of PF3758309, but were diminished at higher drug concentrations (Figure S3E).
Figure 3.
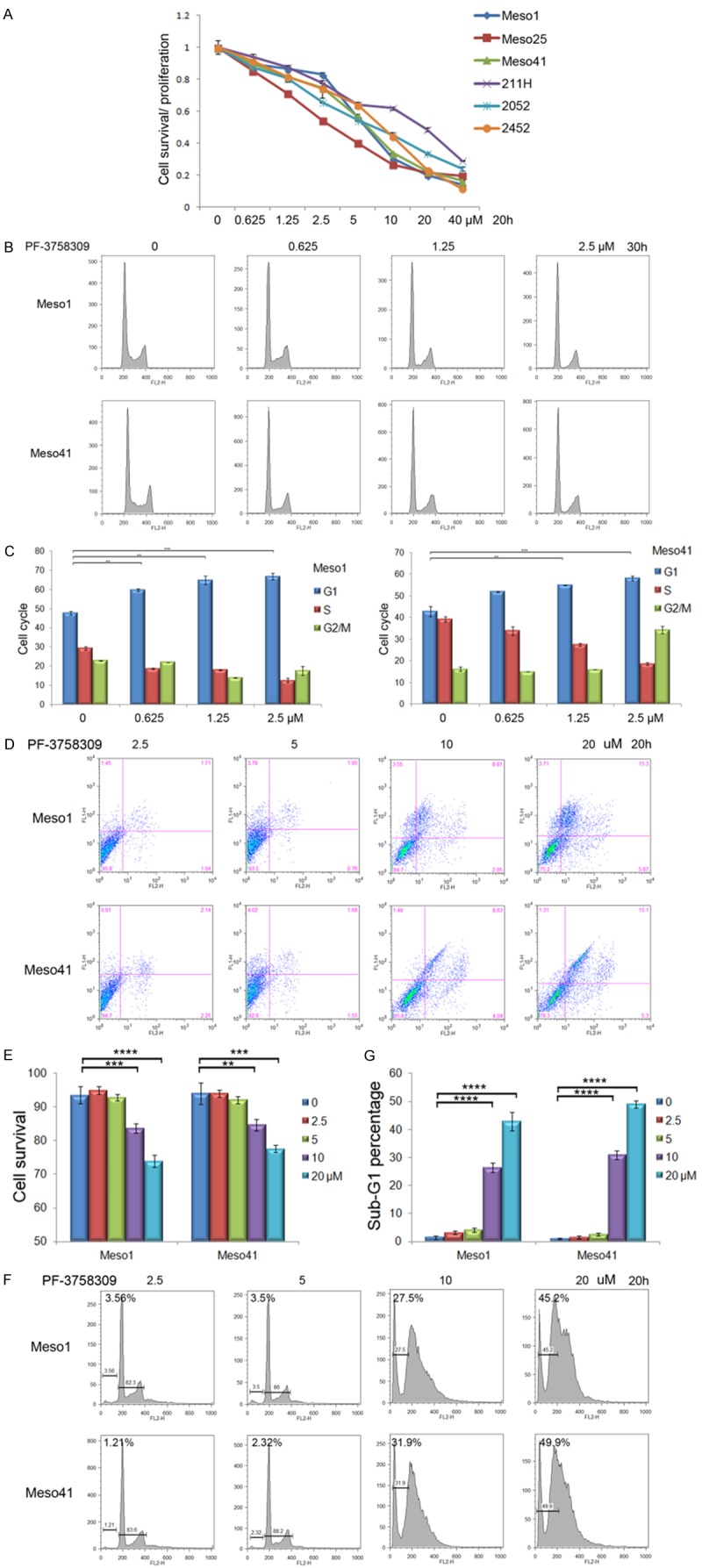
PAK inhibitor PF3758309 induces cell cycle arrest or apoptosis in a dose-dependent manner. A. MM cells were treated with PF3758309 at indicated concentration for 30 h, and MTS assay was performed to evaluate cell viability. B, C. MM cells were treated with PF3758309 at indicated concentration for 24 h. Flow cytometry analysis revealed that low concentrations of PF-3758309 induce cell cycle arrest. D, E. MM cells treated with PF3758309 at indicated concentration for 20 h revealing that higher concentrations of PF3758309 trigger apoptosis, as characterized by loss of membrane integrity and sub-G1 DNA condensation (F, G).
MYC inhibition switches PF3758309-induced cytostasis to cell death
MM cells were treated the 40 µM of 10058-F4 and 2.5 µM of PF3758309 either alone or in combination for 30 h. We found that the combination of 10058-F4 and PF3758309 significantly reduces cell proliferation/viability (Figure 4A). Such combinatorial treatment augmented the sub-G1 apoptotic population (Figure 4B, 4C) and loss of cell membrane integrity (Figure 4D, 4E). Western blotting revealed robust caspase-3 cleavage and activation after treatment for 20 h (Figure 4F, 4G). IPA-3 also showed cooperativity with 10058-F4 (Figure S4A). Interestingly, JQ1 worked well with PF3758309 to diminish cell viability (Figure S4B).
Figure 4.
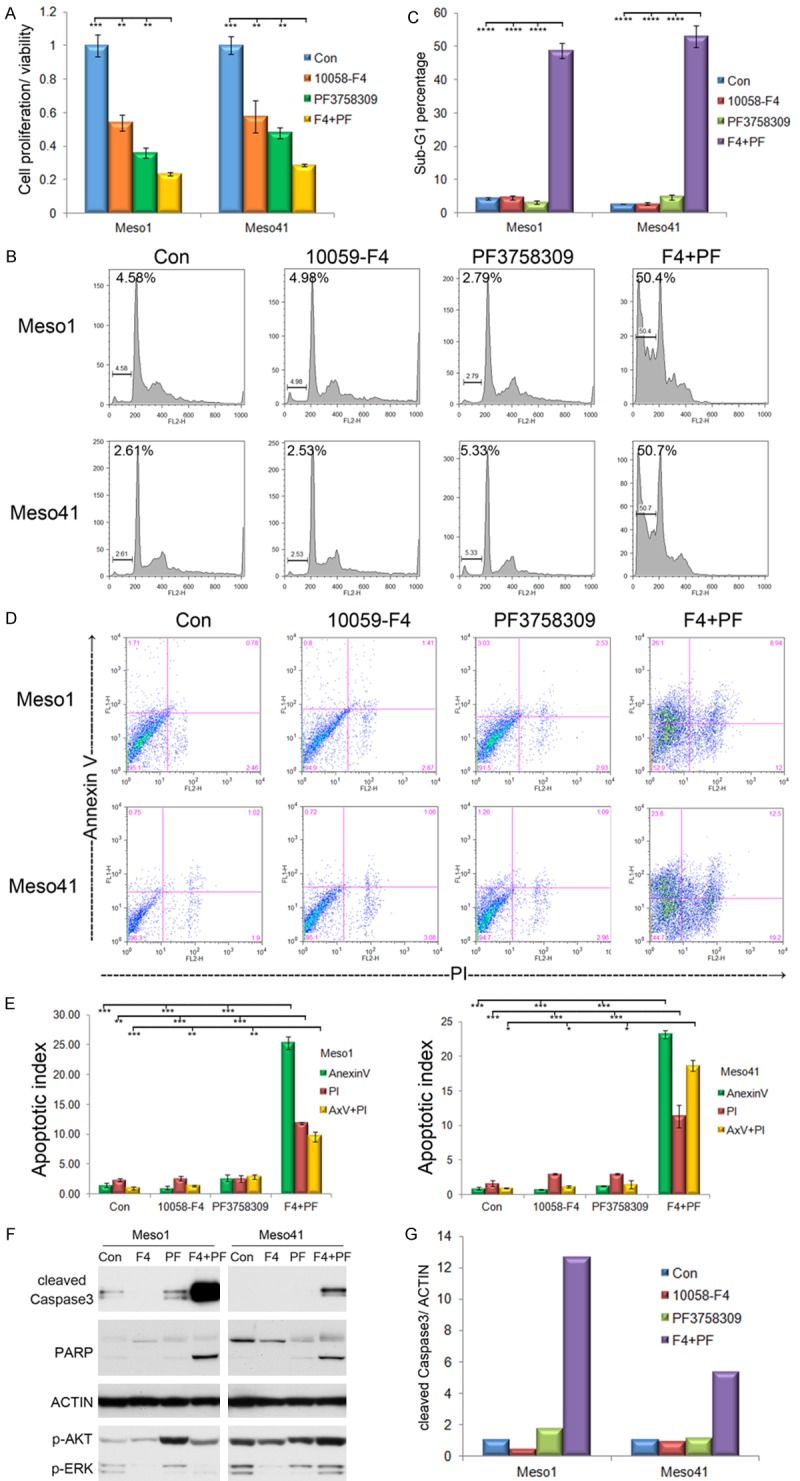
MYC inhibitor switches PF3758309-induced cytostasis to cell death. A. MST assay of viability of cells treated with 40 µM of 10058-F4 and 2.5 µM of PF3758309, either alone or in combination for 30 h. B, C. After treatment for 20 h, sub-G1 population was visualized by FACS. D, E. Loss of membrane integrity was analyzed by using AnnexinV/PI staining. F, G. Caspase-3 cleavage and activation were analyzed by western blotting and densitometry.
The novel MYC inhibitor KJ-Pyr-9 sensitizes cells to cytoxicity induced by a new PAK inhibitor, FRAX597
Since 10058-F4 has been reported to have adverse pharmacological dynamics [35], we decided to test the effects of a newly developed MYC inhibitor, KJ-Pyr-9, with a newer PAK inhibitor, FRAX597. MM cells were treated with KJ-Pyr-9 at the indicated concentrations, either alone or in combination with 2 µM of FRAX597, with or without serum. We found that both KJ-Pyr-9 and FRAX597 had dose-dependent cytotoxic effects and that such effects were amplified when these agents were used in combination (Figure 5A, Figure S5A). Interestingly, like 10058-F4, KJ-Pyr-9 alone had more profound cytotoxic effects under serum starvation conditions. The level of cleaved caspase-3, used as a biochemical marker of apoptosis, supported such conclusions (Figure 5B, Figure S5B).
Figure 5.

A novel MYC inhibitor KJ-Pyr-9 cooperates with FRAX597 in abolishing MM cell viability. A. The PAK inhibitor FRAX597 triggers cell death in a dose-dependent manner, and KJ-Pyr-9 markedly augments FRAX597’s toxicity. Moreover, KJ-Pyr-9 alone induces greater levels of apoptosis in the absence than in the presence of serum in all three MM cell lines tested. B. Induction of caspase-3 with FRAX597 (2 µM) is enhanced by co-treatment with KJ-Pyr-9 (12.5 µM), as shown by Immunoblotting. All treatments in panels A and B were performed for 30 h.
Discussion
MYC is a key pro-survival factor in many human cancers, but its role in the development and maintenance of MM is not well understood. Our studies demonstrate that MYC is frequently up regulated in tumor cells derived from MM patients, and that MYC has either proliferative or pro-survival effects in MM cells during normal or stressed conditions, respectively. MYC inhibition reduced MM cell proliferation via down regulation of cyclin D.
The unusually strong resistance of MM to chemotherapy makes clinical management of this disease extremely challenging. Our studies demonstrate that MYC has pro-survival effects in MM cells during stressed conditions such as upon serum withdrawal or PAK inhibition-induced cell cycle arrest. In our proof-of-principle experiments, MM cells became quiescent upon serum withdrawal from medium, and inhibition of MYC activity by 10058-F4 triggered apoptosis in the resting MM cells. Our study also indicates that MYC inhibition achieved by inhibitors that block MYC/MAX interaction is more efficacious than that of BRD inhibitors such as JQ1, as the latter broadly suppresses BRD-associated gene transcription.
Conventional treatment of MM patients with cisplatin and pemetrexed has a poor response rate when used alone or in combination. Recently, PAK has emerged as a promising drug target. PAK activation is frequently observed in human MM cells in vitro as well as in a mouse model of MM [7]. The six mammalian Paks are categorized into group I (PAK1-3) and group II (PAK4-6), which have overlapping as well as distinct functions. Although PF3758309 has the lowest IC50 with PAK4, it can inhibit both group I and group II PAKs [36]. PF3758309 blocks the growth of multiple human tumor types [37]. We chose PF3758309 over IPA-3, as the former has an IC50 to PAKs of about 1000 times lower than that of IPA-3 [37]. We discovered that PF3758309 exerted either cytostatic or cytotoxic effects in a dose-dependent manner. Low levels of PF3758309 induced cell cycle arrest in MM cells, whereas higher concentrations of this drug efficiently triggered apoptosis. Multidrug resistance is an innate feature of MM, which might reduce the toxicity of PF3758309 to a cytostatic effect. To circumvent this effect, we used MYC inhibitors, which were able to transition this PF3758309-induced quiescent state to apoptosis. Our further studies with KJ-Pyr-9 and FRAX597, novel inhibitors to MYC and PAK, respectively, revealed that such collaborating anti-MM viability is not due to off-target effect from a particular inhibitor.
Collectively, these findings demonstrate that targeting MYC is a feasible strategy to re-sensitize MM cells to mitogen-depletion or PAK-inhibition induced cell death. By targeting MYC in MM patients, it may be possible to overcome drug resistance-induced cytostasis, thus providing rationale for inhibition of MYC and PAK as a novel combinatory regiment for the treatment of this otherwise therapy-resistant, incurable cancer.
Acknowledgements
The authors thank the Flow Cytometry Core Facility at Fox Chase Cancer Center for assistance. This work was supported by NCI grants CA148805 and CA06927, an appropriation from the Commonwealth of Pennsylvania, and a gift from the Local #14 Mesothelioma Fund of the International Association of Heat and Frost Insulators and Allied Workers.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Tsao AS, Wistuba I, Roth JA, Kindler HL. Malignant pleural mesothelioma. J. Clin. Oncol. 2009;27:2081–90. doi: 10.1200/JCO.2008.19.8523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng JQ, Jhanwar SC, Klein WM, Bell DW, Lee WC, Altomare DA, Nobori T, Olopade OI, Buckler AJ, Testa JR. p16 alterations and deletion mapping of 9p21-p22 in malignant mesothelioma. Cancer Res. 1994;54:5547–51. [PubMed] [Google Scholar]
- 3.Bianchi AB, Mitsunaga SI, Cheng JQ, Klein WM, Jhanwar SC, Seizinger B, Kley N, Klein-Szanto AJ, Testa JR. High frequency of inactivating mutations in the neurofibromatosis type 2 gene (NF2) in primary malignant mesotheliomas. Proc Natl Acad Sci U S A. 1995;92:10854–8. doi: 10.1073/pnas.92.24.10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng JQ, Lee WC, Klein MA, Cheng GZ, Jhanwar SC, Testa JR. Frequent mutations of NF2 and allelic loss from chromosome band 22q12 in malignant mesothelioma: evidence for a two-hit mechanism of NF2 inactivation. Genes Chromosomes Cancer. 1999;24:238–42. [PubMed] [Google Scholar]
- 5.Xiao GH, Gallagher R, Shetler J, Skele K, Altomare DA, Pestell RG, Jhanwar S, Testa JR. The NF2 tumor suppressor gene product, merlin, inhibits cell proliferation and cell cycle progression by repressing cyclin D1 expression. Mol Cell Biol. 2005;25:2384–94. doi: 10.1128/MCB.25.6.2384-2394.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiao GH, Beeser A, Chernoff J, Testa JR. p21-activated kinase links Rac/Cdc42 signaling to merlin. J Biol Chem. 2002;277:883–6. doi: 10.1074/jbc.C100553200. [DOI] [PubMed] [Google Scholar]
- 7.Menges CW, Sementino E, Talarchek J, Xu J, Chernoff J, Peterson JR, Testa JR. Group I p21-activated kinases (PAKs) promote tumor cell proliferation and survival through the AKT1 and Raf-MAPK pathways. Mol Cancer Res. 2012;10:1178–88. doi: 10.1158/1541-7786.MCR-12-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Molli PR, Li DQ, Murray BW, Rayala SK, Kumar R. PAK signaling in oncogenesis. Oncogene. 2009;28:2545–55. doi: 10.1038/onc.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Radu M, Lyle K, Hoeflich KP, Villamar-Cruz O, Koeppen H, Chernoff J. p21-activated kinase 2 regulates endothelial development and function through the Bmk1/Erk5 pathway. Mol Cell Biol. 2015;35:3990–4005. doi: 10.1128/MCB.00630-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shukla A, Hillegass JM, MacPherson MB, Beuschel SL, Vacek PM, Butnor KJ, Pass HI, Carbone M, Testa JR, Heintz NH, Mossman BT. ERK2 is essential for the growth of human epithelioid malignant mesotheliomas. Int J Cancer. 2011;129:1075–86. doi: 10.1002/ijc.25763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ceresoli GL, Zucali PA, Gianoncelli L, Lorenzi E, Santoro A. Second-line treatment for malignant pleural mesothelioma. Cancer Treat Rev. 2010;36:24–32. doi: 10.1016/j.ctrv.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Santarius T, Shipley J, Brewer D, Stratton MR, Cooper CS. A census of amplified and overexpressed human cancer genes. Nat Rev Cancer. 2010;10:59–64. doi: 10.1038/nrc2771. [DOI] [PubMed] [Google Scholar]
- 13.Chen J, Odenike O, Rowley JD. Leukaemogenesis: more than mutant genes. Nat Rev Cancer. 2010;10:23–36. doi: 10.1038/nrc2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8:976–90. doi: 10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- 15.Gartel AL, Shchors K. Mechanisms of c-mycmediated transcriptional repression of growth arrest genes. Exp Cell Res. 2003;283:17–21. doi: 10.1016/s0014-4827(02)00020-4. [DOI] [PubMed] [Google Scholar]
- 16.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frye M, Watt FM. The RNA methyltransferase Misu (NSun2) mediates Myc-induced proliferation and is upregulated in tumors. Curr Biol. 2006;16:971–81. doi: 10.1016/j.cub.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 18.Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–45. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer. 2010;10:301–9. doi: 10.1038/nrc2819. [DOI] [PubMed] [Google Scholar]
- 20.Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, Brown-Swigart L, Johnson L, Evan GI. Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell. 2008;14:447–57. doi: 10.1016/j.ccr.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, Evan GI. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455:679–83. doi: 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang MJ, Cheng YC, Liu CR, Lin S, Liu HE. A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp Hematol. 2006;34:1480–9. doi: 10.1016/j.exphem.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 23.Sampson VB, Rong NH, Han J, Yang Q, Aris V, Soteropoulos P, Petrelli NJ, Dunn SP, Krueger LJ. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 2007;67:9762–70. doi: 10.1158/0008-5472.CAN-07-2462. [DOI] [PubMed] [Google Scholar]
- 24.Yin X, Giap C, Lazo JS, Prochownik EV. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene. 2003;22:6151–9. doi: 10.1038/sj.onc.1206641. [DOI] [PubMed] [Google Scholar]
- 25.Hammoudeh DI, Follis AV, Prochownik EV, Metallo SJ. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J Am Chem Soc. 2009;131:7390–401. doi: 10.1021/ja900616b. [DOI] [PubMed] [Google Scholar]
- 26.Kim J, Roh M, Abdulkadir SA. Pim1 promotes human prostate cancer cell tumorigenicity and c-MYC transcriptional activity. BMC Cancer. 2010;10:248. doi: 10.1186/1471-2407-10-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khanna A, Bockelman C, Hemmes A, Junttila MR, Wiksten JP, Lundin M, Junnila S, Murphy DJ, Evan GI, Haglund C, Westermarck J, Ristimäki A. MYC-dependent regulation and prognostic role of CIP2A in gastric cancer. J Natl Cancer Inst. 2009;101:793–805. doi: 10.1093/jnci/djp103. [DOI] [PubMed] [Google Scholar]
- 28.Tan Y, Sementino E, Pei J, Kadariya Y, Ito TK, Testa JR. Co-targeting of Akt and Myc inhibits viability of lymphoma cells from Lck-Dlx5 mice. Cancer Biol Ther. 2015;16:580–8. doi: 10.1080/15384047.2015.1018495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hart JR, Garner AL, Yu J, Ito Y, Sun M, Ueno L, Rhee JK, Baksh MM, Stefan E, Hartl M, Bister K, Vogt PK, Janda KD. Inhibitor of MYC identified in a Krohnke pyridine library. Proc Natl Acad Sci U S A. 2014;111:12556–61. doi: 10.1073/pnas.1319488111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramael M, Van den Bossche J, Buysse C, Deblier I, Segers K, Van Marck E. Immunoreactivity for c-fos and c-myc protein with the monoclonal antibodies 14E10 and 6E10 in malignant mesothelioma and non-neoplastic mesothelium of the pleura. Histol Histopathol. 1995;10:639–43. [PubMed] [Google Scholar]
- 31.Kitamura A, Matsushita K, Takiguchi Y, Shimada H, Tada Y, Yamanaka M, Hiroshima K, Tagawa M, Tomonaga T, Matsubara H, Inoue M, Hasegawa M, Sato Y, Levens D, Tatsumi K, Nomura F. Synergistic effect of non-transmissible Sendai virus vector encoding the c-myc suppressor FUSE-binding protein-interacting repressor plus cisplatin in the treatment of malignant pleural mesothelioma. Cancer Sci. 2011;102:1366–73. doi: 10.1111/j.1349-7006.2011.01931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheung M, Pei J, Pei Y, Jhanwar SC, Pass HI, Testa JR. The promyelocytic leukemia zinc-finger gene, PLZF, is frequently downregulated in malignant mesothelioma cells and contributes to cell survival. Oncogene. 2010;29:1633–40. doi: 10.1038/onc.2009.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 34.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo J, Parise RA, Joseph E, Egorin MJ, Lazo JS, Prochownik EV, Eiseman JL. Efficacy, pharmacokinetics, tisssue distribution, and metabolism of the Myc-Max disruptor, 10058-F4 [Z,E] -5-[4-ethylbenzylidine] -2-thioxothiazolidin-4-one, in mice. Cancer Chemother Pharmacol. 2009;63:615–25. doi: 10.1007/s00280-008-0774-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Radu M, Semenova G, Kosoff R, Chernoff J. PAK signalling during the development and progression of cancer. Nat Rev Cancer. 2014;14:13–25. doi: 10.1038/nrc3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murray BW, Guo C, Piraino J, Westwick JK, Zhang C, Lamerdin J, Dagostino E, Knighton D, Loi CM, Zager M, Kraynov E, Popoff I, Christensen JG, Martinez R, Kephart SE, Marakovits J, Karlicek S, Bergqvist S, Smeal T. Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth. Proc Natl Acad Sci U S A. 2010;107:9446–51. doi: 10.1073/pnas.0911863107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.