

Abstract
Salmonella enterica subsp enterica serovar Typhimurium (S. Typhimurium) is a serovar with broad host range. To determine the genomic diversity of S. Typhimurium, we sequenced 39 isolates (37 Australian and 2 UK isolates) representing 14 Repeats Groups (RGs) determined primarily by clustered regularly interspaced short palindromic repeats (CRISPR). Analysis of single nucleotide polymorphisms (SNPs) among the 39 isolates yielded an average of 1,232 SNPs per isolate, ranging from 128 SNPs to 11,339 SNPs relative to the reference strain LT2. Phylogenetic analysis of the 39 isolates together with 66 publicly available genomes divided the 105 isolates into five clades and 19 lineages, with the majority of the isolates belonging to clades I and II. The composition of CRISPR profiles correlated well with the lineages, showing progressive deletion and occasional duplication of spacers. Prophage genes contributed nearly a quarter of the S. Typhimurium accessory genome. Prophage profiles were found to be correlated with lineages and CRISPR profiles. Three new variants of HP2-like P2 prophage, several new variants of P22 prophage and a plasmid-like genomic island StmGI_0323 were found. This study presents evidence of horizontal transfer from other serovars or species and provides a broader understanding of the global genomic diversity of S. Typhimurium.
Introduction
Salmonella enterica serovar Typhimurium is the most common Salmonella serovar causing foodborne infections in Australia and many other countries1. The phenotypic diversity of S. Typhimurium has been traditionally illustrated by the Anderson phage typing scheme with more than 200 phage types defined2. Since then, different molecular markers were used to assess its genetic diversity3, 4. S. Typhimurium is divided by multilocus sequence typing into more than 30 sequence types (STs). ST19 is the most prevalent ST internationally and the majority of the STs belong to the ST19 clonal complex5. By application of whole-genome-sequencing (WGS) and genomic analysis, our earlier study of six Australian S. Typhimurium strains from different phage types and 7 published genomes revealed three genomic clusters6. Hayden et al.7 analysed the genomic diversity of 35 US S. Typhimurium isolates together with 21 public genomes7 and found that the 56 S. Typhimurium strains could be divided into 3 clades and at least 10 lineages.
Genome sequencing showed that variation among S. Typhimurium strains was mainly due to the accumulation of single nucleotide polymorphisms (SNPs). The S. Typhimurium genome consists of a core genome of around 3,800 genes present in all strains and nearly 1,000 accessory genes which are variably present in one or more strains4, 7. The accessory genome contains mostly genes of prophages and genes of unknown function which have contributed to the genetic diversity of S. Typhimurium3, 4. However, the full depth of genomic diversity of S. Typhimurium remains to be explored.
Clustered regularly interspaced short palindromic repeats (CRISPR) belong to a family of unique repeat sequences and are possibly associated with adaptive resistance against invasive genetic elements such as phages. Salmonella generally possesses two CRISPR loci, which comprise conserved direct repeats separated by unique short sequences typically 32 bp long, called spacers8, 9. Variation in the spacer profiles of CRISPRs has been useful for subtyping Salmonella isolates10. Shariat et al.11 investigated CRISPR array composition in four major serovars, Enteritidis, Typhimurium, Newport and Heidelberg, and demonstrated serovar specificity of CRISPR array composition11. Other studies have also found that CRISPR variation is associated with serotype and sequence types, providing good phylogenetic signals for inferring strain relationships8, 12, 13. Hiley et al.14 performed CRISPR and variable number tandem repeat (VNTR) analyses on a diverse collection of 200 Australian S. Typhimurium isolates and 14 reference strains which separated them into 15 Repeats Groups (RGs)14. This presented an opportunity to apply genomic analysis to selected isolates from these RGs to capture a broad range of genetic diversity of S. Typhimurium.
In this study, we sequenced 39 strains, 37 of which were of Australian origin, representing 14 different RGs and we included in the analysis 66 publicly available S. Typhimurium genomes including 54 of international origins to provide a global overview of S. Typhimurium diversity. We constructed a core genome phylogeny for all 105 strains and examined the correlation of core genome relationships with CRIPSR array composition and prophage profiles.
Results and Discussion
Selection of S. Typhimurium isolates for genome sequencing
Strains were selected to represent 14 of the 15 RGs previously defined by Hiley et al.14 (Supplementary Figures S1 and Table S1). No RG15 strains were chosen for this study as only 2 RG15 strains were found at the time of study14. RG4, RG5, RG6, RG9, RG10 and RG12 had been further subtyped into 2 to 4 sub-categories based on one or more spacer differences in CRISPR1 and/or CRISPR2 so isolates from each sub-category were chosen. In all, 37 Australian and two UK S. Typhimurium isolates were selected for genome sequencing to represent the diversity of these 14 RGs (Table 1).
Table 1.
List of S. Typhimurium isolates sequenced in this study.
| Strain Name | Year of Collection | MLVA Euro | MLVA Aust Code | Phage Type |
|---|---|---|---|---|
| L1848 | 2001 | 02-11-12-09-212 | 03-13-13-10-523 | 29 |
| L1849 | 2004 | 04-12-20-07-211 | 05-14-21-08-490 | 12a |
| L1850 | 2005 | 03-09-NA-NA-111 | 04-11-00-00-463 | 44 |
| L1851 | 2006 | 04-14-11-NA-211 | 05-16-12-00-490 | 141 |
| L1852 | 2006 | 02-10-10-09-212 | 03-12-11-10-523 | 135a |
| L1853 | 2006 | 04-22-12-08-211 | 05-24-13-09-490 | 104 L |
| L1854 | 2007 | 02-11-10-09-212 | 03-13-11-10-523 | 135 |
| L1855 | 2007 | 03-13-08-08-211 | 04-15-09-09-490 | 197 |
| L1856 | 2007 | 02-10-10-09-212 | 03-12-11-10-523 | 135a |
| L1857 | 2007 | 02-23-12-10-212 | 03-25-13-11-523 | 9 |
| L1858 | 2007 | 04-14-14-11-211 | 05-16-15-12-490 | 193 |
| L1859 | 2007 | 02-26-15-11-212 | 03-28-16-12-523 | 8 |
| L1860 | 2007 | 03-12-14-14-311 | 04-14-15-15-517 | 12 |
| L1861 | 2007 | 02-09-09-07-212 | 03-11-10-08-523 | U302 |
| L1862 | 2007 | 04-13-08-08-211 | 05-15-09-09-490 | 6 |
| L1863 | 2007 | 03-17-10-15-210 | 04-19-11-16-457 | 179 |
| L1864 | 2008 | 02-11-11-08-212 | 03-13-12-09-523 | 102 |
| L1865 | 2008 | 02-10-11-09-212 | 03-12-12-10-523 | 135a |
| L1866 | 2008 | 03-14-12-NA-311 | 04-16-13-00-517 | 170 |
| L1867 | 2008 | 02-09-09-05-212 | 03-11-10-06-523 | 29 |
| L1868 | 2008 | 02-08-07-08-212 | 03-10-08-09-523 | 186 |
| L1869 | 2008 | 02-08-07-08-212 | 03-10-08-09-523 | 44 |
| L1870 | 2008 | 02-11-08-09-212 | 03-13-09-10-523 | 135 |
| L1871 | 2008 | 02-15-15-12-212 | 03-17-16-13-523 | 126 |
| L1872 | 2008 | 02-13-15-08-212 | 03-15-16-09-523 | 135 |
| L1873 | 2008 | 02-20-09-07-212 | 03-22-10-08-523 | 135 |
| L1874 | 2008 | 03-11-09-NA-211 | 04-13-10-00-490 | 193 |
| L1875 | 2008 | 03-13-14-NA-211 | 04-15-15-00-490 | 170 |
| L1876 | 2008 | 02-13-NA-NA-311 | 03-15-00-00-517 | 120 |
| L1877 | 2009 | 02-11-10-09-212 | 03-13-11-10-523 | 3 |
| L1878 | 2009 | 03-18-10-NA-111 | 04-20-11-00-463 | 41 |
| L1879 | 2011 | 07-13-NA-NA-211 | 08-15-00-00-490 | 197 |
| L1880 | 2011 | 02-12-10-10-212 | 03-14-11-11-523 | 135a |
| L1881 | 2011 | 02-07-06-11-212 | 03-09-07-12-523 | 170 |
| L1882 | 2011 | 02-10-11-10-212 | 03-12-12-09-523 | 135a |
| L1883 | 2011 | 02-07-07-11-212 | 03-09-08-12-523 | 170 |
| L818 | 2001 | 02-14-18-09-212 | 03-16-09-11-523 | 41 |
| L825 | 2002 | 05-10-NA-09-211 | 06-12-00-10-490 | 193 |
| L930 | 1997 | 02-08-07-08-212 | 03-10-08-09-523 | 44 |
An additional 66 strains, including 21 Salmonella reference collection A (SARA) strains4 and 45 other publicly available genomes (including 11 from Australia), were also included in the analysis (Supplementary Table S2). The CRISPR compositions of these strains were determined using genome data or by PCR sequencing. We found some strains had CRISPR1 or CRISPR2 composition similar to one RG but the other CRISPR composition was similar to another RG. We called them hybrid RGs and designated the genotype as RG CRISPR1/CRISPR2 to indicate the genotype difference in the two CRISPRs. For instance, strain SARA4 was a RG12D/10A hybrid. Genome sequencing statistics are listed in Table 2. The reads were assembled de novo. The number of contigs produced ranged from 214 to 378, with an average of 294 per genome. SNPs were discovered by mapping to the reference S. Typhimurium strain LT2 (NCBI GenBank Accession No. NC_003197). The number of SNPs ranged from 128 to 11,319 relative to the strain LT2 (Table 2).
Table 2.
General features of strains sequenced in this study.
| Strain Name | Total No. of reads | N50 | Contig Number | Total Length (bp) | Coverage (%) | No. of non-synonymous SNPs | No. of synonymous SNPs | No. of Intergenic SNPs | No. of Indels | Total No. of SNPs |
|---|---|---|---|---|---|---|---|---|---|---|
| L1848 | 832,934 | 270,522 | 252 | 4,878,662 | 22 | 276 | 189 | 83 | 45 | 593 |
| L1849 | 1,928,202 | 191,995 | 278 | 4,878,775 | 50 | 265 | 254 | 88 | 50 | 657 |
| L1850 | 1,918,013 | 217,086 | 366 | 4,912,563 | 69 | 250 | 217 | 104 | 55 | 626 |
| L1851 | 1,407,422 | 187,086 | 246 | 4,839,606 | 27 | 68 | 27 | 12 | 38 | 145 |
| L1852 | 2,262,488 | 225,499 | 360 | 4,935,680 | 46 | 345 | 301 | 139 | 68 | 853 |
| L1853 | 1,381,622 | 93,669 | 279 | 4,848,641 | 25 | 492 | 277 | 133 | 60 | 962 |
| L1854 | 2,458,806 | 172,518 | 271 | 4,873,430 | 40 | 272 | 209 | 144 | 69 | 694 |
| L1855 | 1,939,904 | 243,033 | 284 | 4,870,570 | 42 | 301 | 250 | 107 | 56 | 714 |
| L1856 | 2,289,358 | 191,815 | 287 | 4,911,441 | 53 | 297 | 216 | 137 | 66 | 716 |
| L1857 | 2,510,832 | 225,471 | 276 | 4,893,660 | 47 | 373 | 309 | 149 | 73 | 904 |
| L1858 | 1,784,738 | 461,717 | 214 | 4,942,127 | 31 | 59 | 21 | 12 | 36 | 128 |
| L1859 | 2,589,462 | 227,006 | 436 | 4,950,039 | 70 | 324 | 297 | 196 | 50 | 867 |
| L1860 | 1,091,774 | 204,354 | 292 | 4,972,319 | 42 | 321 | 283 | 116 | 60 | 780 |
| L1861 | 2,452,986 | 211,251 | 261 | 4,960,484 | 53 | 360 | 340 | 148 | 69 | 917 |
| L1862 | 2,271,550 | 247,127 | 346 | 4,997,225 | 37 | 270 | 209 | 83 | 55 | 617 |
| L1863 | 1,961,248 | 484,774 | 213 | 4,834,244 | 37 | 289 | 270 | 99 | 46 | 704 |
| L1864 | 1,382,398 | 250,313 | 250 | 4,920,225 | 25 | 280 | 190 | 120 | 55 | 645 |
| L1865 | 1,911,482 | 243,303 | 278 | 4,908,702 | 28 | 287 | 210 | 110 | 48 | 655 |
| L1866 | 1,981,270 | 207,403 | 255 | 4,853,339 | 38 | 162 | 112 | 83 | 51 | 408 |
| L1866 | 1,195,260 | 270,542 | 279 | 4,924,157 | 31 | 358 | 312 | 142 | 68 | 880 |
| L1868 | 1,943,952 | 170,885 | 279 | 4,969,999 | 28 | 335 | 297 | 128 | 60 | 820 |
| L1869 | 2,349,774 | 165,711 | 326 | 4,911,892 | 32 | 347 | 372 | 147 | 62 | 928 |
| L1870 | 1,280,956 | 242,937 | 275 | 4,881,193 | 30 | 301 | 211 | 126 | 66 | 704 |
| L1871 | 2,703,908 | 178,436 | 291 | 4,860,646 | 62 | 436 | 289 | 173 | 95 | 993 |
| L1872 | 2,557,384 | 219,635 | 370 | 4,882,236 | 69 | 303 | 322 | 125 | 72 | 822 |
| L1873 | 1,775,906 | 208,524 | 331 | 4,890,388 | 34 | 228 | 141 | 86 | 56 | 511 |
| L1874 | 1,911,482 | 271,008 | 347 | 5,052,975 | 35 | 248 | 201 | 95 | 63 | 607 |
| L1875 | 2,119,420 | 247,035 | 243 | 4,773,579 | 40 | 272 | 258 | 101 | 61 | 692 |
| L1876 | 1,348,454 | 188,257 | 266 | 4,773,760 | 27 | 1,831 | 8,183 | 1,236 | 89 | 11,339 |
| L1877 | 1,454,232 | 149,277 | 309 | 4,875,957 | 29 | 292 | 201 | 134 | 58 | 685 |
| L1878 | 1,259,628 | 266,717 | 304 | 4,748,690 | 60 | 308 | 232 | 166 | 72 | 778 |
| L1879 | 1,684,386 | 232,300 | 316 | 4,963,003 | 41 | 1,166 | 7,465 | 1,588 | 72 | 10,291 |
| L1880 | 1,338,944 | 256,474 | 285 | 4,913,050 | 32 | 265 | 212 | 152 | 64 | 693 |
| L1881 | 1,590,652 | 200,287 | 299 | 4,915,853 | 51 | 309 | 242 | 170 | 68 | 789 |
| L1882 | 2,646,186 | 232,306 | 274 | 4,914,415 | 32 | 266 | 215 | 157 | 56 | 694 |
| L1883 | 2,351,622 | 221,927 | 313 | 4,915,122 | 48 | 320 | 235 | 165 | 66 | 786 |
| L818 | 1,327,194 | 257,038 | 280 | 4,892,270 | 24 | 431 | 275 | 157 | 92 | 955 |
| L825 | 1,426,998 | 209,878 | 255 | 4,894,398 | 26 | 283 | 200 | 97 | 61 | 641 |
| L930 | 1,038,048 | 179,836 | 378 | 4,924,303 | 22 | 350 | 297 | 142 | 70 | 859 |
Core and accessory genomes of S. Typhimurium
Pan genome analysis showed that there were 8,849 genes, including 3,836 core genes, and 5,013 dispensable genes in S. Typhimurium. The core genome was smaller than in our previous report (3,846 genes)4 and the reports of Hayden et al. (3,910 genes)7 and Mather et al. (3,890 genes)15. This finding indicated that the current dataset had a strain coverage broad enough to achieve a stable core genome and adding more strains to the analysis would not substantially reduce the core genome size. Interestingly, prophage genes contributed up to 13.3% (1,175/8,849) of the pan genome and 23.4% (1,175/5,013) of the accessory genome, while plasmid and other mobile elements took up 13.3% (668/5,013) of the accessory genome. For the rest of the genes in the accessory genome, the majority (2,043 genes) encoded hypothetical proteins with currently unknown function.
Genomic relationships and their correlation with Repeats Groups (RGs)
A phylogeny of the 105 strains was constructed based on SNPs derived from the S. Typhimurium core genome4. The strains were separated into five clades and 19 lineages (Figs 1 and S1). Clade I contained RGs 10, 11, 12, 13 and 14 while Clade II contained RGs 1, 2, 4, 7, 8 and 9. Clades III, IV and V corresponded to RG3, RG6 and RG5 respectively. Clade V consisting of four strains L1876, L1879, SARA7 and SARA8 was the most distant clade being separated by more than 4,000 SNPs from the other clades as found in a previous study4. Clades III and IV were much closer to Clade II. Of the 19 lineages defined, 16 lineages contained more than one isolate and 3 lineages contained only one isolate. The strains falling within each lineage nearly always belonged to a single RG or combination of one RG and a CRISPR hybrid related to that RG and for the most part all the strains belonging to an RG fell into a single lineage. Exceptions were RG12A strains which were split into two lineages (lineages 10 and 8), separated by the RG14 lineage, and lineage 13 which contained strains belonging to RG4 and RG7 even though they are distinctly different by both CRISPR and VNTR profiles14 (Fig. 1).
Figure 1.
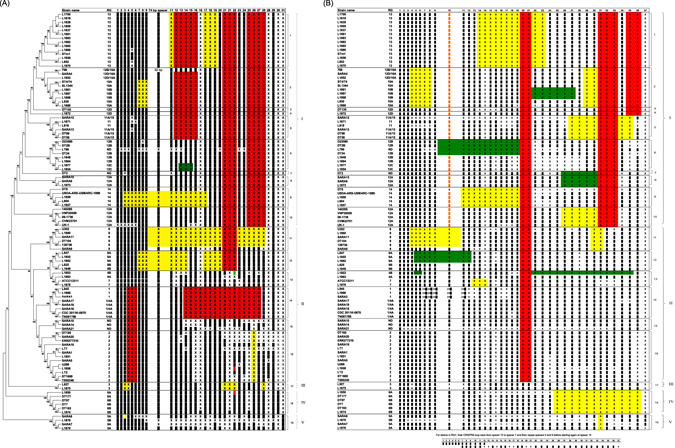
Phylogenetic relationship of 105 S. Typhimurium strains and its correlation with CRISPR1 (A) and CRISPR2 (B) patterns. The minimum evolution method was used to infer evolutionary relationships of the isolates based on their SNPs obtained from S. Typhimurium core genome. The Bootstrap was performed with 1,000 replicates. The bootstrap values (1,000 replicates, >50%) are shown next to the branches. The lineages (1 to 19) and clades (I to V) are indicated on the right side. CRISPR1 and CRISPR2 consist of 31 and 37 spacers respectively. The spacers are numbered in the order according to Table 6 from Fabre et al.8. The representation of CRISPR profiles is the presence (filled rectangular blocks) or absence (X) of the spacers in CRISPR1 and CRISPR2. In CRISPR1, the 74 bp spacer includes sp10. Sp22 contains a 6 bp tandem repeat; Green = 3 repeats, Black = 4 repeats, Red = 5 repeats, Blue = 6 repeats. In CRISPR2, sp10 variant that has one SNP is labelled in orange. The spacers lost in multiple RG, specific RG and sub-RG groups are highlighted in red, yellow and green, respectively.
Genomic typing resolved the phylogenetic relationships between RGs that were not clearly evident from the CRISPR profiles. Thus RG12D was the likely precursor for both RG10 and RG13 lineages. There was a loss of spacers from the RG12D CRISPR arrays specific to each of the RG10 and RG13 lineages. Three strains SARA10, SARA14 and SARA21 that could not be assigned to any recognised RG formed a separate lineage between RG1 and RG2.
Strains with hybrid RGs were genomically clustered with other strains which had the same or similar CRISPR1 RG or CRISPR2 RG. Five strains (i.e. TN061786, CDC 2011K-0870, SARA16, SARA17 and SARA18) shared similar CRISPR1 profiles with RG1 strains but their CRISPR2 profiles were like those in RG4A (RG1/4A in Fig. 1). They fell into the same lineage as RG1. Four strains, SARA12, SARA13, DT56 and DT99, were RG11A/15 hybrids and clustered with isolates belonging to RG11 genomically. SARA4 and L1852 were RG12D/10A hybrids which clustered with isolates belonging to RG10A. Likewise, strain 798 (NC _017046) which was a RG12B/10A hybrid was also genomically clustered with RG10A. These hybrids suggest that recombination has substituted CRISPR1 or CRISPR2 sequences of the respective strains in their common ancestors.
Our study revealed a higher genomic diversity of S. Typhimurium than that reported by Hayden et al.7, in which 10 lineages belonging to 3 clades were found among 35 sequenced US S. Typhimurium isolates and 21 published genomes. Examination of representative isolates in each lineage from Hayden et al.7 showed that only RG1, RG2, RG8, RG10, RG11A/15 and RG12 genotypes were included in their study, leaving an under-representation of the genomic diversity. Although genomes belonging to RG1, RG1/4A, RG3, RG5, RG6, RG9A, RG11A/15, RG12, RG13 and RG14, were also included in previous genome-sequencing studies4, 6, 16, 17, ours is the first such comprehensive study to also include RG4, RG7 and RG9B genomes. Further analysis of other published genomes did not uncover more RGs, suggesting that isolates included in this set represented the spectrum of RG diversity available to date (Table S3).
Comparison of Australian isolates and international isolates with published genomes showed that most RGs including RG1, RG2, RG5, RG6A and B, RG8, RG10A and B, RG11A and B, RG12A, B, C and D and RG14, contained isolates from Australia and other parts of the world, suggesting wide distribution of these RGs. Three RGs, RG3, RG9 and RG13, may be unique to Australia. However, more extensive sampling from other countries is required to ascertain whether these three RGs are restricted to Australia. RG8 and RG12A genotypes appear to be infrequently isolated in Australia14.
Progressive spacer deletion in the CRISPR evolution of S. Typhimurium
CRISPR arrays appear to evolve by the deletion or duplication of spacer-repeat units or by the rare acquisition of new spacers18. The divergence of the clades was associated with progressive losses of spacers (Fig. 1). For example, Clades I and II have lost sp20 and sp21 of CRISPR2 indicating an earlier divergence of Clade II from Clade III and Clade I has lost both sp25 to sp28 of CRISPR1 and sp31 to sp33 of CRISPR2 when Clades I and II diverged from their most recent common ancestor.
Generally, there were few single spacer deletions. Most deletions involved two or more spacers such as deletion of sp2 to sp32 of CRISPR2 in SARA21 that presumably occurred in one event. Many deletions were across lineages or RGs as single deletion events indicative of common ancestry rather than parallel loss (Fig. 1). There were many RG-specific spacer deletions. Sp31 of CRISPR1 and sp37 of CRISPR2 which were only present in Clade V, and sp30 in CRISPR1 which was only present in Clade IV may be more recent acquisitions.
Duplication of spacers can also now be better understood. Some duplications such as sp5, sp11 and sp21 in CRISPR1 arrays and sp14 in CRISPR2 arrays appeared to be rare events as they were only seen in single isolates14. Duplication of sp16 in CRISPR1 was only seen in RG9B and duplication of sp28 in CRISPR1 was only seen in some RG2 genotypes. However, duplication of sp15 in CRISPR2 arrays was much more widespread except for RG2, RG6, RG9A, RG12B and RG12C which have only one sp15. Therefore, this duplication may be an ancient event and has only been lost by relatively few genotypes as evolution has progressed.
Prophages found in the 105 S. Typhimurium genomes
Our previous studies have shown that much of the genomic diversity within S. Typhimurium was due to variation in prophage content4. This study extends that observation and presents a fuller characterisation of the S. Typhimurium prophages. Prophages in Salmonella are categorised into five groups, P27-like, P2-like, lambdoid, P22-like, and T7-like19. All except the last group were found in our S. Typhimurium strains. We also identified three new variants of HP2-like P2 prophages, several new variants of P22 prophages, including some in a novel insertion site STM0786 (LT2), a novel OLF-SE9-10012-like prophage and other outlier prophages. The prophage distribution is illustrated in Fig. 2.
Figure 2.
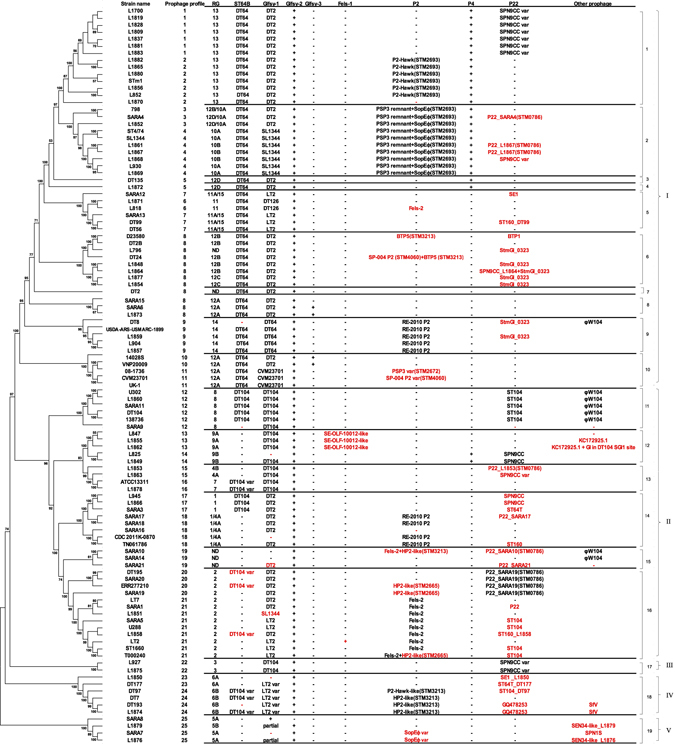
Phylogenetic tree of 105 S. Typhimurium strains and distribution of prophages and prophage profiles. “+” and “−” in the prophage profiles indicate the presence and absence of prophage. Additional insertion or deletion of prophages compared with universal patterns are labelled in red. Var is designated after the named phage indicating it is a variant of that phage. The insertion site that differs from the usual position is indicated in brackets. The lineage and clade number are indicated on the right-hand side of the figure.
Lambdoid prophages
The lambdoid prophages include Gifsy-1, Gifsy-2, Gifsy-3 and Fels-1. Gifsy-1 was previously subtyped into seven variants, Gifsy-1LT2, Gifsy-1DT104, Gifsy-1DT64, Gifsy-1SL1344, Gifsy-1DT2, Gifsy-1CVM23701 and Gifsy-1DT126 14. Gifsy-1LT2 was present in five of the 13 RG2 strains and in four RG11A/15 strains. A variant of Gifsy-1LT2 which has three segments different from LT2 occurred in five of the six RG6 strains. Gifsy-1DT104 was found in RG3, RG4, RG7 and RG8. Gifsy-1DT126 was exclusive to RG11. Gifsy-1DT64 was only found in RG14. Gifsy-1CVM23701 was exclusive to some RG12A strains. Gifsy-1SL1344 was found in RG10 and one strain from RG2. Gifsy-1DT2 was distributed widely in many RGs including RG13, RG12, RG1, RG2 and RG1/4A.
Gifsy-2 was found in all strains except SARA8 and is highly conserved with no variants as reported previously20. Gifsy-3 was found in SARA6, L1873, 14028 S and VNP20009 with >99% DNA sequence similarity to each other. Fels-1 was only found in LT2.
P27-like prophages
The P27-like prophage, ST64B has 2 variants, ST64BDT64 and ST64BDT104. The ST64BDT64 variant was found in all Clade I strains and not in any other clades (although a previous study by Hiley et al.14 showed that it was present in a minority of RG2A isolates in Clade II) while the ST64BDT104 variant was only found in some strains in Clades II and IV. Comparison of these ST64BDT104 prophage sequences in RG2, RG7 and RG6B to prototype ST64BDT104 in DT104 showed that some strains contained ST64BDT104 with varying degrees of coverage (range from 57.4% to 94.5%) (Supplementary Table S4).
P2 prophages, P2 variants and P4 prophage
Forty eight strains were found to contain one of the P2 prophages, Fels-2, RE-2010, PSP3, SopEφ, SP-004, 186-type, P2-Hawk and other HP2-like or variants of these. Three strains, DT24, SARA10 and T000240, had two P2 prophages in different locations. All RG10 strains had a remnant PSP3 P2 prophage.
Fels-2 was found in nine of the 13 RG2 strains as well as SARA10 (RG unassigned) and L818 (RG11). RE-201021, a variant of Fels-2, was found in two different lineages RG1/4A and RG14. SopEφ, another Fels-2 variant, always in tandem with a P4 prophage, was found in all RG10 strains as well as the three RG10A hybrids. P4 is a satellite phage using P2 as a helper phage20. A SopEφ variant was found in RG5A strains L1876 and SARA7 and was also inserted in the Fels-2 site. The SopEφ variant showed lower similarity to S. Typhimurium SopEφ, (Accession No. AY319521, 85%/97% coverage/identity for L1876) than to the SopEφ in S. Javiana 10721 (AOZA01000057) with 93%/98% (coverage/identity), indicating that it may have come from another serovar. A variant of SP-004 was found only in CVM23701 and DT24 while a variant of 186-type P2 was found in D23580 (BTP5) and DT24.
HP2 phage (NC_003315) was first found in Haemophilus influenzae 22, and a distantly related variant, P2-Hawk, was reported in S. Typhimurium strains in tandem with a P4 prophage17. In our study, we found 14 HP2-like prophages in various insertion sites. Six of these were identified as P2-Hawk and were all in RG13 strains.They were almost identical, differing by no more than five SNPs from each other and always in tandem with the P4 phage. A comparative analysis showed that a core genome of 10 kb was shared by HP2-like P2 prophages, but only 2.6 kb was shared by all the genomes of HP2-like, P2, Fels-2, RE-2010 and SopEΦ (Table S5). Phylogenetic analysis of the core genome of HP2-like P2 prophages showed that these prophages were divided into three variants all with considerable divergence from HP2 phage (NC_003315) (Figure S2). One variant was found in RG6B strains L1874, DT7 and DT193, inserted at the LT2 gene STM3213 which is the same insertion site as for BTP5, a coliphage 186-type P2 phage in strain D23580. This phage has also been found in strains of serovar Enteritidis and Newport with 99%/99% coverage/identity. The SARA10 HP2-like phage (RG unassigned) was also related to this variant but was closer to that found in serovar Heidelberg SL476. Strain DT97 (RG6B) had a variant which clustered with P2-Hawk but inserts at STM3213 instead of at STM2693. Another variant, which inserted at the LT2 gene STM2665 site, was found in ERR277210, T000240 and SARA19 all in RG2. P4 was present independently of P2 in RG9B, RG12D and more than half of the RG13 isolates.
P22 prophages and P22 variants
Forty six strains were found to contain a P22-like prophage or one of its variants. The P22-like prophages that are located in the STM0323 site have seven publicly available variants including P22 (NC_002371), SE-1 (DQ003260), SPN9CC (JF900176), ST160 (GU573886), ST104 (AAF75053), ST64T (AY052766) and BTP1 (D23580). We first used PHAST to assign the 46 P22-like prophages into the above categories. P22 (NC_002371) or its variant was found in SARA1, SARA17, and SARA21. There were other variants of P22 (NC_002371) in SARA4, L1861, L1867 (RG10), L1853 (RG4A), SARA10 (RG unassigned) and five strains from RG2 (SARA1, DT195, ERR277210, SARA19 and SARA20) but these were located in a novel insertion site, STM0786. SE-1-like prophages were found in SARA12 and L1850 with 100%/99% and 82%/99% coverage/identity, respectively. SPN9CC or one of its variants was found in 16 strains spread across six RGs. ST104 and one of its variants in DT97 were found in another 11 strains. ST160 was found in three strains, of which L1858 and DT99 each had a variant. Additionally, two strains had ST64T and strain D23580 had the BTP1 prophage.
We analysed the genetic diversity within P22 prophages. A phylogenetic tree of core genome sequences showed that the P22-related SPN9CC-like prophages could be divided into three variants (Figure S3). The first variant had only a few SNPs different from SPN9CC and was in RG1 strains L945 and L1866. The second variant contained a 1,213 bp deletion of the nin gene region in the position of 15268–16480 in phage SPN9CC (JF900176) and was in 13 strains from RG12, RG13 and RG10. The third variant had six unique sequences and was present only in L1864 from RG12B.
There was considerable diversity of gene content within the P22-like prophages. To better delineate the gene content variation of various P22-like prophages, we analysed the pan-genome of the 46 P22-like prophages found in this study as well as the genomes of seven publicly available P22 and its variants and obtained a 100 kb pan-genome. The pan-genome consisted of 193 DNA fragments, most of which were only shared by some prophage genomes (Supplementary Table S6). Only 11 fragments of 6.5 kb were shared by all, of which 3 kb belonged to capsid assembly genes and scaffold genes, indicating that the capsid assembly and scaffold genes can be conserved among the different P22-like branches. Based on the presence/absence of genes of the pan-genome (Supplementary Figure S4), SE-1, ST64T, ST160 and their variants were grouped into the same cluster, as were the ST104-like prophages and the SPN9CC-like prophages. P22-like prophages showed high genomic diversity as many variants are not closely related to any other P22 variants. Our pan-genome analysis also showed that some P22-like prophages were incorrectly categorised by PHAST. For example, a P22-like prophage in DT177 was initially identified as ST64T by PHAST but the pan-genome analysis showed that it was closer to SE1 with which 19 genes were shared.
We further explored the evolution of P22-like prophages by comparing their sequences with those from E. coli and other Salmonella serovars (Supplementary Table S6 and Supplementary Figure S4). The composition of these prophage genomes was mosaic with sequences from different sources, indicating frequent exchanges of DNA between subgroups of the P22-like phages as well as from E. coli and other Salmonella serovars. The majority of the fragments (137 out of 193) had high similarity to prophages in multiple serovars, while a few only had high similarity to one or two Salmonella serovars, including serovars closely related to S. Typhimurium such as S. Heidelberg, and more distantly related serovars like S. Newport and S. Paratyphi A. Twenty-four fragments had similarity to E. coli prophages, while 12 fragments were only found in S. Typhimurium prophages. Thus, there has been a considerable exchange of genetic information among diverging P22-like phage lineages, and the exchange appears to be randomly distributed throughout their genomes.
There was considerable sequence diversity for the P22-like prophages located at the STM0786 insertion site. The integrase for these prophages had 68% identity with that from Enterobacteria phage HK106 NC_019768. The P22 variant in SARA10 had 66% coverage/99% identity with P22 (NC_002371) but 99% coverage/99% identity with Paratyphi B ATCC 8759 (AOYE01000028.1). SARA4 which was a RG12D/10A hybrid had another variant with 50% coverage/99% identity with P22 (NC_002371). Another prophage, P22_SARA19, was found in SARA19, DT195, ERR277210 and SARA20 but also with considerable sequence divergence from other P22 variants (Table S7). Another variant, P22_L1867, was found in L1861 and L1867 belonging to RG10B. Strain L1853 had a P22 variant which had greater sequence coverage with BTP1 from D23580 (39% with 99% identity) than with P22 (NC_002371) (34% and 99% identity) and even less coverage with the other STM0786 prophages.
Other prophages
Five outlier prophages, SPN1S, OLF-SE9-10012-like prophage, a SfV-like prophage, φW10423, and SEN34-like which do not belong to any of the known S. Typhimurium prophage groups, were found. A SPN1S variant was found in SARA7 which was inserted at the same site (STM2510) as the SPN1S in S. Heidelberg strain 12-4374 (CP012924.1). A OLF-SE9-10012-like prophage of 36 kb was found to be inserted at the Fels-1 insertion site in RG9A strains L847, L1855 and L1862. A Shigella SfV-like prophage of 45 kb, which was inserted at STM4243, was found in RG6B strains DT193 and L1874. φW104 (belonging to the family Podoviridae) was detected in all but one RG8 strain and a variant of φW104 was found in SARA10 and SARA14 (unassigned RG) and DT8 (RG14). A 42 kb SEN34-like prophage (belonging to the family Podoviridae) in the STM2067 insertion site was found in strains L1876 and L1879.
Prophage profiles and correlation with core genome and CRISPR evolution
Based on the distribution of ST64B, Gifsy-1, Gifsy-3, Fels-1, P2, P4, P22 and φW104 prophages found in this study, the 105 S. Typhimurium strains were classified into 25 prophage profiles (Table 3). Each profile consisted of a set of shared prophages. Within these profiles other prophages may be variably present as shown in Fig. 2. There was a considerable level of correlation between prophage profile and lineages determined by genomic typing (Fig. 2). Prophage profiles, to a certain extent, also reflected the compositions of CRISPR arrays as the strains in many RGs or subsets within RGs have the same phage profile. Since the CRISPR-cas system is possibly involved in defence against phage invasion we investigated whether there is a link between the loss of spacers and gain of a phage. Loss of one or more spacers within an RG could provide a mechanism for phage invasion. A study in Cronobacter sakazakii showed that the CRISPR-cas system is active in that species where clinical strains carried few CRISPR spacers and had more phages, suggesting that gaining more prophage by clinical strains may offer an advantage to the host in survival and pathogenicity24. However, we did not find any loss of spacers that match the phage genome in corresponding RGs in this study. Shariat et al.11 have reported that the CRISPR-Cas systems in Salmonella are no longer active, arguing against a role in modulating phage invasion in S. Typhimurium in recent evolutionary history. Nevertheless, considering that some prophages carry virulence genes (see below), phages may have acted as a driving force in the evolution of S. Typhimurium, regardless of the mechanisms of phage acquisition25 , 26.
Table 3.
Prophage profiles for various strain clusters.
| RG | Prophage profile number | ST64B | Gifsy-1 | Gifsy-2 | Gifsy-3 | Fels-2 | P22 var | Other P2 | P4 | Other |
|---|---|---|---|---|---|---|---|---|---|---|
| 13 | 1 | DT64 | DT2 | + | − | − | SPN9CC var | − | + | − |
| 13 | 2 | DT64 | DT2 | + | − | − | − | P2-Hawk STM2693 | + | − |
| 12D(B)/10A | 3 | DT64 | DT2 | + | − | SopEφ | +/− | PSP3 remnant STM2693 | + | − |
| 10A, B | 4 | DT64 | SL1344 | + | − | SopEφ | +(RG10B)/− | PSP3 remnant STM2693 | + | − |
| 12D | 5 | DT64 | DT2 | + | − | − | − | − | + | − |
| 11 | 6 | DT64 | DT126 | + | − | − | − | − | − | − |
| 11A/15 | 7 | DT64 | LT2 | + | − | − | +/− | − | − | − |
| 12A, B, C | 8 | DT64 | DT2 | + | − | − | +/− | +/− | − | − |
| 14 | 9 | DT64/− | DT64 | + | − | RE-2010 P2 | +/− | − | − | − |
| 12A | 10 | DT64 | DT2 | + | + | − | − | − | − | − |
| 12A | 11 | DT64 | CVM23701 | + | − | − | − | +/− | − | − |
| 8 | 12 | DT104 | DT104 | + | − | − | ST104 | − | − | φW104/− |
| 9A | 13 | − | DT104 | + | − | − | − | − | − | SE-OLF-10012-like |
| 9B | 14 | − | DT104 | + | − | − | SPN9CC | − | + | − |
| 4 | 15 | − | DT104 | + | − | − | + | − | − | − |
| 7 | 16 | DT104 var | DT104 | + | − | − | − | − | − | − |
| 1 | 17 | DT104 | DT2 | + | − | − | + | − | − | − |
| 1/4A | 18 | − | DT2 | + | − | RE-2010 P2 | +/− | − | − | − |
| ND | 19 | − | − | + | − | − | +/− | +/− | − | φW104/− |
| 2 | 20 | DT104 var/− | DT2 | + | − | − | P22_SARA19 STM0786 | +/− | − | − |
| 2 | 21 | DT104 var/− | LT2/SL1344 | + | − | Fels-2 | +/− | − | − | − |
| 3 | 22 | − | DT104 | + | − | − | SPN9CC var | − | − | − |
| 6A | 23 | − | LT2 var | + | − | − | + | − | − | − |
| 6B | 24 | DT104 var/− | DT104 | + | − | − | + or GQ478263 | HP2-like STM3213 | − | − |
| 5 | 25 | − | − | − | − | − | − | − | − | − |
Prophages in S. Typhimurium genomes with similarity to prophages from other serovars and/or other species
The SPN1S variant had some additional genes which shared identity with phage SPC32N or the sequence from Klebsiella pneumoniae (Supplementary Table S8), indicating it was a hybrid prophage. The OLF-SE9-10012-like prophage in RG9A strains showed the highest similarity to a prophage in S. Enteritidis OLF-SE9-10012 (CP009091.1) with 70%/99% coverage/identity (Supplementary Table S9). This prophage was an unclassified member of the Myoviridae not related to P2 and with little similarity to other members of the Myoviridae family. Related prophages were also found in S. Muenchen BAA1674 (AOYT01000011.1), S. bongori N268-08 (CP006608.1), S. Hartford str 2012K-0272 (ARYS01000018) and S. Bovismorbificans 3114 (HF969015.2) as well as in multiple strains of S. Weltevreden and S. Bareilly. Close to the 3’ end of the prophage was the location of a variant form of the sopE gene (AF043239) which had been previously identified as a feature of all RG9A genotypes by PCR14.
The SfV-like prophage in two RG6B strains showed considerable divergence from SfV (AF339141) (Supplementary Table S10). It had a mosaic composition with some genes encoding hypothetical proteins coming from other species or other Salmonella serovars. These genes were inserted into different positions of the SfV-like prophage genome, indicating that multiple genetic exchanges have occurred (Figure S5). The SfV-like prophage lacked a 6 kb region present in SfV which carries the gtr genes for serotype conversion27. Additionally, the 5′ end region encoding phage packaging and structure, and right-hand side regions encoding replication and regulation, were partially replaced by sequences from other serovars with unknown functions. The SEN34-like prophage shared only 20% of its genome with phage SEN34 (KT630649.1). A SEN34-like prophage was also found in serovar Weltevreden 1655 (CP014996.1) and Paratyphi B SPB7 (NC_010102). L1879 had a 43.5 kb sequence in the same insertion site that had only 14% coverage with SEN34. The remaining sequence was found in a number of S. Typhimurium strains and in other Salmonella serovars.
Distribution of virulence genes carried on prophages
S. Typhimurium prophages may carry genes that enhance the virulence of the bacterial host28 thus the distribution of these genes deserves closer attention (Supplementary Table S11). The four virulence genes carried on Gifsy-1, gipA, gogB, gogD, and gogA, were present in nearly all except seven strains from four different RGs. Interestingly, these genes were mostly absent in the earliest diverged lineage (RG5). The artAB genes on Gifsy-1DT104 initially found in epidemic DT104 strains29 were found in other strains in six RGs. Similarly, the seven virulence genes carried by Gifsy-2 were present in most strains. However, sopE and sspH1 carried by a P2 and Gifsy-3 respectively were variably present in two different branches, while nanH carried by Fels-1 was only present in one strain (LT2). Some of these genes play a key role in virulence. Specifically, SopE activates RHO GTPases that lead to modification of cytoskeleton of the host cell for invasion and also induce caspase 1 to provoke inflammation30. SopE can also induce the production of nitrate by host cell so that Salmonella can use nitrate respiration in the gut31, which enhances the survival of S. Typhimurium inside the host and competition with gut microbiota. SseK, a T3SS effector, encoded on ST64B and shown to play a role in the inhibition of NF-κB activation32, 33, was found in five RGs. Some P22 prophages carry gtrABC genes which encode glucosyltransferases that glycosylate the galactose residues of the somatic O-antigen in S. Typhimurium34. Modification of the O-antigen may help evade host induced immunity. The distribution of the gtrABC carrying P22 prophages was random across the genome tree with only 3 RGs fully carrying these prophages. The variable carriage of these prophages and virulence genes suggest that S. Typhimurium strains can significantly differ in their pathogenic potential.
Genomic islands
A novel genomic island inserted in the STM0323 (thrW tRNA) site was found and named as StmGI_0323. It was present in some strains of RG12B and RG12C including L1848, L1854, L1864 and L1877 as well as L796. In L1864, StmGI_0323 occurred in tandem with a SPN9CC-related P22 prophage. The genomic island was also found in DT8 and L1859 in RG14. StmGI_0323 encodes 14 open reading frames (ORFs) (Table 4) and is clearly of plasmid origin as the majority of the ORFs, four of which encode conjugal transfer proteins, shared high similarity to E. coli plasmid sequences (>99% at protein level). It was noteworthy that another unrelated genomic island GQ478253 was inserted at this site in RG6B strains L1874 and DT193.
Table 4.
Detailed identity comparison of each gene in genomic island StmGI_0323.
| Open reading frame no. | Product/size (aa) | Best matches (BLASTp)* | Identity/coverage (%) |
|---|---|---|---|
| ORF 1 | Hypothetical protein (470) | E. coli UMEA 3318-1 | 100/100 |
| E.coli DORA_B_14 | 100/100 | ||
| ORF 2 | Hypothetical protein (149) | Cronobacter condimenti 1330 | 99/99 |
| E.coli DORA_B_14 | 98/99 | ||
| ORF 3 | Site-specific recombinase (202) | E. coli ( > 10) | 100/100 |
| E.coli DORA_B_14 | 99/99 | ||
| ORF 4 | Hypothetical protein (366) | E.coli DORA_B_14 | 99/99 |
| K. pneumoniae B1 | 99/98 | ||
| ORF 5 | HigA (antitoxin to HigB) | E.coli DORA_B_14 | 100/100 |
| E. coli (5), Enterobacter (2) | 98/98 | ||
| ORF 6 | Hypothetical protein (97) | E. coli D6-117.29 | 100/100 |
| ORF 7 | TraJ IncP-type conjugal transfer protein (121) | E. coli 907357 | 98/100 |
| E.coli DORA_B_14 | 98/100 | ||
| ORF 8 | TrbJ IncP-type conjugal transfer protein (264) | E. coli (3) | 99/99 |
| ORF 9 | Hypothetical protein (57) | E. coli (4) | 96/96 |
| Enterobacteriaceae bacterium FGI 57 | 93/94 | ||
| ORF 10 | TrbL IncP-type conjugal transfer protein (453) | E. aerogenes GN02286 | 98/99 |
| E. coli KTE171 | 98/98 | ||
| ORF 11 | Hypothetical protein (70) | E. coli (2), Enterobacter (2) | 100/100 |
| ORF 12 | Hypothetical protein | E. coli (5) | 100/100 |
| Enterobacteriaceae bacterium FGI 57 | 99/98 | ||
| ORF 13 | Putative membrane protein | E. aerogenes GN02286 | 100/100 |
| E. coli (4) | 99/99 | ||
| ORF 14 | Shufflon-specific DNA recombinase | E. aerogenes GN02286 | 99/99 |
*Numbers in brackets indicate the number of strains.
Conclusions
This study compared the genomic diversity of Australian and international strains of S. Typhimurium. The size of core genome in our set was slightly smaller than in previous reports, indicating that we have derived a stable core genome. The 105 strains could be divided into five clades and 19 lineages based on core genome variation. The strains represented 14 different RGs and the RGs primarily derived from CRISPR array composition correlated well with the lineages determined by core genomic typing. Previous studies also found CRISPR composition is correlated with genomic relationship in other Salmonella serovars35, suggesting this is a general phenomenon. The accessory genome of S. Typhimurium contained a fair proportion of prophage genes. Some prophages were widely present in S. Typhimurium while others were sporadic. There was a strong correlation of prophage profiles with lineages and CRISPR profiles. Acquisition of phages may have played an important role in the adaptation and virulence evolution of S. Typhimurium. There was high sequence diversity among related prophages with a considerable level of similarity with prophages from other serovars and/or other species, suggesting extensive horizontal gene transfer. Virulence genes such as sopE carried by prophages were variably present, indicating variation in pathogenicity among S. Typhimurium strains. These findings have extended our understanding of the genomic diversity and core genome evolution of S. Typhimurium, particularly its relationship with CRISPR evolution and prophage variation.
Materials and Methods
Bacterial strains and genomic DNA isolation
Thirty-nine human clinical isolates representing CRISPR diversity14 collected between 1997 and 2011 were selected for sequencing (Table 2). Thirty-seven isolates in this study had been referred by the laboratory of Queensland Department of Health, Forensic & Scientific Services in Brisbane, Australia. Two other isolates were obtained from the UK. The phenol/chloroform method was used to extract genomic DNA from each strain as described previously36. Sixty-six publicly available genomes were also used as shown in Supplementary Table S2. The plasmid and antibiotic resistance genes among the 105 strains were also analysed (Supplementary text).
CRISPR profiles
The CRISPR sequences of 36 strains (strain No. from L1849 to L1883) were determined in a previous study14. The CRISPR sequences from 35 S. Typhimurium strains sequenced in our previous studies4, 6, 16 were analysed in this study. The CRISPR1 and CRISPR2 sequences in each isolate were amplified using the primer pairs described by Hiley et al.14. PCR products were sequenced using the Applied Biosystems 3130 sequencer and BigDye Terminator v3.1 Cycle Sequencing Kit. ChromasPro was used to analyse the sequences. For 34 public genomes, the CRISPR finder program (http://crispr.u-psud.fr/) was used to locate the regular repeats and the intervening spacer sequences. Results were represented as filled rectangular blocks for ‘spacer present’ or an X for ‘spacer absent’ in the same order as for S. Typhimurium spacers in Table 6 in Fabre et al.8 (Supplementary Table S2).
Genome sequencing, de novo assembly and identification of Single nucleotide polymorphisms (SNPs)
Genomic DNA was sequenced using the Illumina Genome Analyzer (Illumina) with 250 bp paired end sequencing. Contigs were de novo assembled using the Velvet version 1.0.8 and VelvetOptimiser37. Large scaffolds and short contigs generated by Velvet were aligned to the S. Typhimurium LT2 genome (NC_003197) using progressiveMauve version 2.3.138. RAST was used to annotate the sequences from each NGS genome39. The number of coding sequences in the genomes was predicted based on RAST. For draft genomes, SNP calling was performed by Samtools (version 0.1.19) and followed the previously described criteria6. A custom script was used to determine whether a SNP in the genic region is synonymous SNP (sSNP) or non-synonymous SNP (nsSNP). For the complete genome, SNPs were determined by using the NUCmer program in the MUMmer package version 3.040.
Prophage analysis
The presence of prophages from the sequenced strains was screened using PHAST41. The prophages were confirmed by searching for integrase from annotated genomes. We subtyped the ST64B prophages and Gifsy-1 prophages into two and seven variants, respectively. ST64B prophage has two variants: ST64BDT104 and ST64BDT64. The sequence of ST64BDT64 (AY055382) and the sequence of ST64BDT104 obtained from DT104 (NC_022569) were used as reference sequences to confirm the variants in our studied strains. Gifsy-1 prophages were subtyped as either Gifsy-1LT2, Gifsy-1DT104, Gifsy-1DT126, Gifsy-1SL1344, Gifsy-1DT2, Gifsy-1CVM23701 or Gifsy-1DT64 based on the unique sequences among these variants as defined previously14. The core genome content of P2 prophage and HP2-like group of P2 prophage were obtained by analysing common shared regions of P2 prophages and HP2-like P2 prophages using progressiveMauve version 2.3.1, respectively38.
Phylogenetic analysis
Based on S. Typhimurium core genome SNPs we defined previously4 and core genome content of P2 prophage identified in this study, phylogenetic trees were constructed using the Minimum Evolution algorithms in MEGA 5.0 for 105 S. Typhimurium genomes and 15 P2 prophage genomes, respectively42. Bootstrap analysis was performed with 1,000 replicates. Based on the presence and absence of DNA segments in the P22 pan-genome, a UPGMA tree was constructed using the web-server DendroUPGMA (http://genomes.urv.cat/UPGMA).
Sequence data accession number
The raw sequencing data were submitted to GenBank (NCBI) under the BioProject No. PRJNA355598.
Electronic supplementary material
Acknowledgements
This work was supported by a grant from the National Health and Medical Research Council of Australia and a UNSW Early Career Research grant. SF was supported by a UNSW international postgraduate research award. MT was supported by an Australian Research Council Future Fellowship.
Author Contributions
Conceived and designed the experiments: S.F., L.H., S.O. and R.L. Performed the experiments: S.F., L.H. and S.O. Data analysis and draft of manuscript were performed by S.F., L.H., S.O., M.T., V.S. and R.L. All authors approved the final version of the manuscript for submission.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-06079-1
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Galanis E, et al. Web-based surveillance and global Salmonella distribution, 2000–2002. Emerg. Infect. Dis. 2006;12:381–388. doi: 10.3201/eid1205.050854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson ES, Ward LR, Saxe MJ, de Sa JD. Bacteriophage-typing designations of Salmonella typhimurium. J. Hyg. 1977;78:297–300. doi: 10.1017/S0022172400056187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pang S, et al. Genetic relationships of phage types and single nucleotide polymorphism typing of Salmonella enterica Serovar Typhimurium. J. Clin. Microbiol. 2012;50:727–734. doi: 10.1128/JCM.01284-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fu S, Octavia S, Tanaka MM, Sintchenko V, Lan R. Defining the core genome of Salmonella enterica serovar Typhimurium for genomic surveillance and epidemiological typing. J. Clin. Microbiol. 2015;53:2530–2538. doi: 10.1128/JCM.03407-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Achtman, M. et al. Multilocus sequence typing as a replacement forserotyping in Salmonella enterica. Plos Pathog. 8, doi:ARTN e1002776, doi:10.1371/journal.ppat.1002776 (2012). [DOI] [PMC free article] [PubMed]
- 6.Pang S, et al. Genomic diversity and adaptation of Salmonella enterica serovar Typhimurium from analysis of six genomes of different phage types. BMC Genomics. 2013;14:718. doi: 10.1186/1471-2164-14-718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayden HS, et al. Genomic analysis of Salmonella enterica serovar Typhimurium characterizes strain diversity for recent U.S. Salmonellosis cases and identifies mutations linked to loss of fitness under nitrosative and oxidative stress. MBio. 2016;7:e00154. doi: 10.1128/mBio.00154-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fabre L, et al. CRISPR typing and subtyping for improved laboratory surveillance of Salmonella infections. Plos One. 2012;7:e36995. doi: 10.1371/journal.pone.0036995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pettengill JB, et al. The evolutionary history and diagnostic utility of the CRISPR-Cas system within Salmonella enterica ssp. enterica. PeerJ. 2014;2:e340. doi: 10.7717/peerj.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhaya D, Davison M, Barrangou R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 2011;45:273–297. doi: 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- 11.Shariat N, Timme RE, Pettengill JB, Barrangou R, Dudley EG. Characterization and evolution of Salmonella CRISPR-Cas systems. Microbiology. 2015;161:374–386. doi: 10.1099/mic.0.000005. [DOI] [PubMed] [Google Scholar]
- 12.Liu F, et al. Novel virulence gene and clustered regularly interspaced short palindromic repeat (CRISPR) multilocus sequence typing scheme for subtyping of the major serovars of Salmonella enterica subsp. enterica. Appl, Environ, Microbiol. 2011;77:1946–1956. doi: 10.1128/AEM.02625-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu F, et al. Subtyping Salmonella enterica serovar enteritidis isolates from different sources by using sequence typing based on virulence genes and clustered regularly interspaced short palindromic repeats (CRISPRs) Appl, Environ, Microbiol. 2011;77:4520–4526. doi: 10.1128/AEM.00468-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hiley, L., Fang, N. X., Micalizzi, G. R. & Bates, J. Distribution of Gifsy-3 and of variants of ST64B and Gifsy-1 prophages amongst Salmonella enterica serovar Typhimurium Isolates: evidence that combinations of prophages promote clonality. Plos One.9, doi:ARTN e86203, doi:10.1371/journal.pone.0086203 (2014). [DOI] [PMC free article] [PubMed]
- 15.Mather AE, et al. Genomic Analysis of Salmonella enterica Serovar Typhimurium from wild passerines in England and Wales. Appl, Environ, Microbiol. 2016;82:6728–6735. doi: 10.1128/AEM.01660-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Octavia S, et al. Delineating community outbreaks of Salmonella enterica serovar Typhimurium by use of whole-genome sequencing: insights into genomic variability within an outbreak. J. Clin. Microbiol. 2015;53:1063–1071. doi: 10.1128/JCM.03235-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hawkey J, et al. Evidence of microevolution of Salmonella Typhimurium during a series of egg-associated outbreaks linked to a single chicken farm. BMC Genomics. 2013;14:800. doi: 10.1186/1471-2164-14-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barrangou R, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 19.Switt AIM, et al. Salmonella Phages and Prophages: Genomics, Taxonomy, and Applied Aspects. Methods, Mol, Biol. 2015;1225:237–287. doi: 10.1007/978-1-4939-1625-2_15. [DOI] [PubMed] [Google Scholar]
- 20.Canchaya C, Proux C, Fournous G, Bruttin A, Brussow H. Prophage genomics. Microbiol, Mol, Biol R. 2003;67:238–276. doi: 10.1128/MMBR.67.2.238-276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohammed M, Cormican M. Whole genome sequencing provides possible explanations for the difference in phage susceptibility among two Salmonella Typhimurium phage types (DT8 and DT30) associated with a single foodborne outbreak. BMC Res, Notes. 2015;8:728. doi: 10.1186/s13104-015-1687-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams BJ, et al. Bacteriophage HP2 of Haemophilus influenzae. J. Bacteriol. 2002;184:6893–6905. doi: 10.1128/JB.184.24.6893-6905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balbontin R, Figueroa-Bossi N, Casadesus J, Bossi L. Insertion hot spot for horizontally acquired DNA within a bidirectional small-RNA locus in Salmonella enterica. J. Bacteriol. 2008;190:4075–4078. doi: 10.1128/JB.00220-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeng H, et al. The driving force of prophages and CRISPR-Cas system in the evolution of Cronobacter sakazakii. Sci. Rep. 2017;7:40206. doi: 10.1038/srep40206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wagner, P. L. & Waldor, M. K. Bacteriophage control of bacterial virulence. Infect. Immun.70, 3985–3993 (2002). [DOI] [PMC free article] [PubMed]
- 26.Brussow H, Canchaya C, Hardt WD. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol. Mol.Biol. Rev. 2004;68:560–602. doi: 10.1128/MMBR.68.3.560-602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allison GE, Angeles DC, Huan P, Verma NK. Morphology of temperate bacteriophage SfV and characterisation of the DNA packaging and capsid genes: the structural genes evolved from two different phage families. Virology. 2003;308:114–127. doi: 10.1016/S0042-6822(03)00198-3. [DOI] [PubMed] [Google Scholar]
- 28.Figueroa-Bossi N, Bossi L. Inducible prophages contribute to Salmonella virulence in mice. Mol. Microbiol. 1999;33:167–176. doi: 10.1046/j.1365-2958.1999.01461.x. [DOI] [PubMed] [Google Scholar]
- 29.Saitoh M, et al. The artAB genes encode a putative ADP-ribosyltransferase toxin homologue associated with Salmonella enterica serovar Typhimurium DT104. Microbiology. 2005;151:3089–3096. doi: 10.1099/mic.0.27933-0. [DOI] [PubMed] [Google Scholar]
- 30.Hapfelmeier S, et al. Role of the Salmonella pathogenicity island 1 effector proteins SipA, SopB, SopE, and SopE2 in Salmonella enterica subspecies 1 serovar Typhimurium colitis in streptomycin-pretreated mice. Infect. Immun. 2004;72:795–809. doi: 10.1128/IAI.72.2.795-809.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lopez, C. A. et al. Phage-mediated acquisition of a type III secreted effector protein boosts growth of Salmonella by nitrate respiration. MBio. 3, doi:10.1128/mBio.00143-12 (2012). [DOI] [PMC free article] [PubMed]
- 32.Brown, N. F. et al. Salmonella phage ST64B encodes a member of the SseK/NleB effector family. Plos One. 6, doi:ARTN e17824, doi:10.1371/journal.pone.0017824 (2011). [DOI] [PMC free article] [PubMed]
- 33.Gao XF, et al. NleB, a bacterial effector with glycosyltransferase activity, targets GADPH function to inhibit NF-kappa B activation. Cell Host Microbe. 2013;13:87–99. doi: 10.1016/j.chom.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Villafane R, Zayas M, Gilcrease EB, Kropinski AM, Casjens SR. Genomic analysis of bacteriophage epsilon 34 of Salmonella enterica serovar Anatum (15+) BMC Microbiol. 2008;8:227. doi: 10.1186/1471-2180-8-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng X, et al. Comparative analysis of subtyping methods against a whole-genome-sequencing standard for Salmonella enterica serotype Enteritidis. J. Clin. Microbiol. 2015;53:212–218. doi: 10.1128/JCM.02332-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Octavia S, Lan R. Single nucleotide polymorphism typing of global Salmonella enterica serovar Typhi isolates by use of a hairpin primer real-time PCR assay. J. Clin. Microbiol. 2010;48:3504–3509. doi: 10.1128/JCM.00709-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Darling AE, Mau B, Perna N. T. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. Plos One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aziz RK, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delcher, A. L., Salzberg, S. L. & Phillippy, A. M. Using MUMmer to identify similar regions in large sequence sets. Curr. Protoc. Bioinformatics 10.13. 11-10.13. 18 (2003). [DOI] [PubMed]
- 41.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic. Acids. Res. 2011;39:W347–352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tamura K, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.