

Abstract
MicroRNAs (miRNAs) are small, non-coding RNAs which can serve as oncogenes or tumor suppressor genes in human cancers. Herein, the transcriptomic differences of miRNAs in ccRCC were globally assessed using publicly available microarray dataset (GSE71302) from Gene Expression Omnibus (GEO) and we identified miR-138 as a potential onco-suppressive miRNA. We further found that the expression of miR-138 was dramatically decreased in ccRCC cell lines and clinical ccRCC tissue samples, and the low miR-138 expression was closely correlated with tumor progression and prognosis in ccRCC patients. Overexpression of miR-138 inhibited, whereas downregulation of miR-138 promoted, the proliferation, migration and invasion of ccRCC cells in vitro, suggesting that miR-138 may function as a tumor suppressor in ccRCC. Furthermore, for the first time, we identified the EMT-related transcription factor SOX4 as a direct target gene of miR-138, evidenced by the direct binding of miR-138 with the 3’UTR of SOX4. Notably, the EMT marker E-cadherin or vimentin was also upregulated or downregulated upon miR-138 overexpression, and these effects were restored by SOX4 overexpression. We have also shown SOX4 overexpression reversed the attenuated migratory and invasive capacities mediated by miR-138. These results revealed that miR-138 functions as a tumor suppressor in ccRCC by targeting SOX4 and the EMT process and might represent a potential target in the treatment of human ccRCC.
Keywords: Clear cell renal cell carcinoma, miR-138, SOX4, epithelial-mesenchymal transition, tumorigenicity
Introduction
Kidney cancer, or renal cell carcinoma (RCC), which constitutes 2-3% of all adult malignancies, is the most prevailing urogenital tumor [1]. RCC morbidity and mortality rate has continuously elevated from 2003 to 2012 [2]. RCC is heterogeneous and comprises several histological subtypes. Clear cell renal cell carcinoma (ccRCC), also called conventional RCC, is the most common and lethal RCC histological subtype because of its high rates of local invasion, metastasis, and acquired chemoresistance [3]. Hence, a deep understanding of the molecular mechanisms involved in ccRCC development and progression is of great clinical significance for early diagnosis and treatment of this deadly disease.
MicroRNAs (miRNAs), which were discovered in recent years, are an abundant class of endogenous, short (21-24 nucleotides in length) non-coding RNAs that modulate gene expression in animals and plants by base pairing with target mRNAs in the 3’-untranslated region (3’-UTR), leading to translational repression or induction of mRNA degradation [4,5]. Dysregulation of miRNAs may contribute to alterations in several pivotal cellular activities of tumor cells, such as apoptosis, proliferation and migration [6]. An increasing number of studies have revealed a vital role of miRNAs in the pathogenesis and progression of ccRCC. For that matter, as a tumor suppressor in ccRCC, miR-206 overexpression inhibited ccRCC cell proliferation and invasion [7,8].
Previous studies have shown that miR-138 plays a crucial role in regulating cell functions, such as proliferation and migration, and is frequently downexpressed in a variety of malignant diseases, including hepatocellular carcinoma [9], gallbladder carcinoma [10], non-small cell lung cancer [11], osteosarcoma [12], and bladder cancer [13]. Despite several published reports that addressed the role of miR-138 in promoting cancer development, little is known about its involvement in ccRCC.
In human, SOX (sex-determining region Y (SRY)-related high-mobility-group (HMB) box transcription factor) gene family comprises at least 20 highly conserved members [14]. SOX4, a 47-kDa protein member of this family, serves a crucial role in the regulation of transcription during developmental processes including embryonic, cardiac, thymocytic, and nervous system development [15-17]. Recently, numerous studies reported altered expression of SOX4 in human malignancies.
In the current study, we examined the expression pattern of miR-138 in ccRCC tissues, as well as elucidated its role in regulating the phenotypes of ccRCC cells. Furthermore, miR-138 might function by directly binding to the 3’-UTR of SOX4. Our findings suggested that miR-138/SOX4 signaling might be a potential therapeutic target for patients with ccRCC.
Materials and methods
Data mining and analysis
The microarray data based on the Agilent-021827 Human miRNA Microarray (V3) (miRBase release 12.0 miRNA ID version), GSE71302 [18], was obtained from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). GSE71302 contain five pairs of ccRCC tissues and matched normal kidney tissues.
Tissue samples
67 pairs of ccRCC tissues and their adjacent non-cancerous tissues were acquired from patients who underwent partial or radical nephrectomy at Children’s Hospital (Chongqing, China). Diagnosis was based on pathological evidence, and collected tissue samples were immediately snap-frozen in liquid nitrogen until used. The study was approved by the Ethics Committee of Children’s Hospital, and all participants involved in this study signed informed consent forms. Clinicopathological characteristics of these patients were summarized in Table 1.
Table 1.
Correlation analysis between miR-138 expression and clinicopathological characteristics in 67 ccRCC patients
| Characteristics | Total number (n=67) | miR-138 expression | P value | |
|---|---|---|---|---|
|
| ||||
| High (n=37) | Low (n=30) | |||
| Age (years) | 0.102 | |||
| <55 | 32 | 21 | 11 | |
| ≥55 | 35 | 16 | 19 | |
| Gender | 0.457 | |||
| Male | 46 | 24 | 22 | |
| Female | 21 | 13 | 8 | |
| Tumor size (cm) | 0.046 | |||
| <4 | 38 | 25 | 13 | |
| ≥4 | 29 | 12 | 17 | |
| Distant metastasis | 0.027 | |||
| No | 51 | 32 | 19 | |
| Yes | 16 | 5 | 11 | |
| TNM stage | 0.003 | |||
| I+II | 46 | 31 | 15 | |
| III+IV | 21 | 6 | 15 | |
| Fuhrman grade | 0.103 | |||
| I+II | 49 | 30 | 19 | |
| III+IV | 18 | 7 | 11 | |
| Necrosis | 0.176 | |||
| Absent | 54 | 32 | 22 | |
| Present | 13 | 5 | 8 | |
Cell culture and transfection
The human kidney proximal tubular epithelial cell line HK-2, and the human ccRCC cell lines, including ACHN, 786-O, SN12-PM6, were obtained from the Cell Bank of the Chinese Academy of Science (Shanghai, China) and maintained in DMEM (Invitrogen, Carlsbad, CA, USA) with 10% FBS (Invitrogen) and 1% penicillin/streptomycin in a humidified atmosphere of 5% CO2 at 37°C.
The miR-138 mimics, antisense miR-138 inhibitor and corresponding negative control miR (miR-NC, anti-miR-NC) were purchased from RiboBio (Guangzhou, China). Transfection was performed by using Lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol.
Construction of plasmids
The 3’-untranslated region (3’-UTR) sequences of human SOX4, containing the putative miR-138 binding site, were amplified and inserted into the psiCHECK-2 vector (Promega, Madison, WI, USA), which was named psiCHECK-2-WT-SOX4. Point mutations in the putative miR-138 binding seed regions were created by Quick Change Site-Directed Mutagenesis Kit (Agilent, Roseville City, CA, USA). The resultant product was named psiCHECK-2-MUT-SOX4.
For SOX4 overexpression, the full-length cDNA of SOX4 was synthesized and inserted into the pcDNA3.1 (+) vector (Realgene, Shanghai, China), according to the manufacturer’s protocol. An empty pcDNA3.1 vector was used as a control.
Luciferase activity assay
Cells were seeded in 24-well plates 24 hours before transfection. The psiCHECK-2-WT-SOX4 or psiCHECK-2-MUT-SOX4 vectors were co-transfected with miR-138 mimics or miR-NC into ACHN cells by Lipofectamine 2000. After 48 hours, firefly and Renilla luciferase activities were assayed using the Dual Luciferase Reporter Assay Kit (Promega).
RNA extraction and quantitative RT-PCR assay
Total RNA was isolated with Trizol reagent (Invitrogen). Concentration and purification of RNA were assessed by NanoDrop 2000 Spectrophotometer (Thermo Scientific Wilmington, DE, USA). For miRNA quantification, cDNA was synthesized using One Step Prime script miRNA cDNA Synthesis Kit (Qiagen, Valencia, CA). Quantitative RT-PCR was performed using the miRNA-specific TaqMan® MiRNA Assay Kit (Applied Biosystems, Foster City, CA, USA) under ABI 7900 Fast system (Applied Biosystems). For mRNA detection, cDNA was synthesized using the PrimeScript RT reagent Kit (TaKaRa, Dalian, China). SYBR® Green (TaKaRa) was used to detect SOX4 expression levels, following the manufacturer’s instructions. Results were normalized to the expression of U6 snRNA or glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The specific primers used in reverse transcription and quantitative RT-PCR were listed in Table 2.
Table 2.
The sequences of primers
| Gene name | Primer sequences |
|---|---|
| miR-138-RT | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCGGCCT |
| U6-RT | AACGCTTCACGAATTTGCGT |
| miR-138 Forward primer | AGCTGGTGTTGTGAATC |
| miR-138 Reverse primer | GTGCAGGGTCCGAGGT |
| U6 Forward primer | CTCGCTTCGGCAGCACA |
| U6 Reverse primer | AACGCTTCACGAATTTGCGT |
| SOX4 Forward primer | GTGAGCGAGATGATCTCGGG |
| SOX4 Reverse primer | CAGGTTGGAGATGCTGGACTC |
| GAPDH Forward primer | ACAGTCAGCCGCATCTTCTT |
| GAPDH Reverse primer | GACAAGCTTCCCGTTCTCAG |
Western blot
Protein lysates were separated by SDS-PAGE and transferred to nitrocellulose membrane. After being blocked in PBS containing 5% non-fat milk, the membranes were plotted with primary antibodies against SOX4, E-cadherin, vimentin, and GAPDH, followed by incubation with appropriated horseradish peroxidase-conjugated secondary antibodies. After washing, the proteins of interest were visualised using ECL (Pierce, Rockford, IL, USA) and detected using BioImaging Systems (UVP Inc., Upland, CA, USA).
Colony formation assay
Approximately 200 viable cells were seeded in 6-well plates and incubated at 37°C. After two weeks, colonies were fixed with methanol, stained with crystal violet, and imaged.
MTT assay
Cells were seeded in 96-well plates at a density of 5×103 cells/well. 20 μL MTT (0.5 mg/mL; Sigma-Aldrich, St. Louis, Mo, USA) was added into each well at indicated time point (on day 1, 2, and 3) and incubated for another 4 h at 37°C. Then 150 μL DMSO was added to solubilize the crystals. The absorbance was detected using a multiwell spectrophotometer at a wavelength of 570 nm.
Transwell assay
The invasion assay was performed using Matrigel-coated transwell chambers (8 μm pore size; Corning, New York, NY, USA). The migration assay was performed using chambers without the Matrigel coating. 1×104 cells in serum-free medium were placed into the upper chamber, and the medium containing 10% FBS was placed to the lower chamber for use as a chemoattractant. After 48 hours of incubation, the cells remaining on the upper membrane were removed and then the cells on the bottom side of the membrane were fixed with methanol, stained with crystal violet, and photographed under a microscope.
Tumorigenicity assay in nude mice
Nude mice, 4-6 weeks old, were purchased from the Shanghai Lab Animal Research Center (Shanghai, China). All animal procedures were approved by the Animal Care and Use Committee of Children’s Hospital and performed strictly in accordance with institutional policies. 1×106 stably transfected cells in 0.2 ml PBS were injected subcutaneously into the right upper back of the mice. The tumor volume for each mouse was measured using an external caliper every 3 days. Five weeks after the injections, tumor-bearing mice were sacrificed by CO2 asphyxiation, and the tumors were dissected and weighted.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 6.0 software (GraphPad Software, San Diego, CA, USA) and SPSS 19 software package (IBM SPSS Inc; Chicago, IL, USA). Student’s t test was used to analyze the results expressed as mean ± SD. The χ2 test was used to analyze the correlation of miR-138 expression and clinicopathological characteristics of ccRCC patients. The survival curves were plotted by Kaplan-Meier analysis, and the survival differences were compared using the log-rank test. P<0.05 was regarded to be statistically significant.
Results
MiR-138 is down-regulated in ccRCC
To investigate the differential miRNA expression between ccRCC tissues and matched normal kidney tissues, we downloaded one group of data from GEO database (GSE71302) which included five pairs of ccRCC tissues and matched normal kidney tissues. We found 11 upregulated and 17 downregulated miRNAs (|fold change|≥2, P<0.05) (Figure 1A) in ccRCC tissues when compared with matched normal kidney tissues.
Figure 1.

MiR-138 is down-regulated in ccRCC tissues and cell lines. A. Heat Map representing color-coded expression levels of the 28 miRNAs differentially expressed between human ccRCC tissues and corresponding normal tissues. Red denotes high expression levels, whereas blue depicts low-expression levels. B. The expression of miR-138 was detected in 67 pairs of ccRCC tissues and their matched adjacent normal tissues by quantitative RT-PCR. C. Quantitative RT-PCR analysis of miR-138 expression levels in three ccRCC cell lines (ACHN, 786-O, SN12-PM6) compared with the human kidney proximal tubular epithelial cell line (HK-2). GAPDH was used as internal control. Data are presented as mean ± SD by at least three independent experiments. *P<0.05. D, E. Kaplan-Meier survival curve analysis showed that ccRCC patients with lower miR-138 expression had significantly shorter overall survival (OS) and disease-free survival (DFS). P value was calculated by log-rank test.
Subsequently, we validated the expression levels of miR-138 in both ccRCC tissues and corresponding adjacent normal tissues. We found that miR-138 expression was significantly decreased in ccRCC tissues compared with adjacent non-tumor tissue (P<0.05, Figure 1B), indicating that miR-138 might function as a tumor suppressor in ccRCC. According to the median value, we defined 30 patients with low miR-138 expression and 37 patients with high expression level. After analyzing the clinicopathological characteristics of patients, as recorded in Table 1, reduced expression level of miR-138 was associated with larger tumor size (P=0.046), distant metastasis (P=0.027) and advanced TNM stage (P=0.003) of ccRCC patients.
Next, the expression levels of miR-138 in human kidney proximal tubular epithelial cell line HK-2 and three human ccRCC cell lines including ACHN, 786-O, SN12-PM6 were determined by quantitative RT-PCR. As showed in Figure 1C, miR-138 expression was noticeably decreased in all the three ccRCC cell lines than in non-tumorigenic HK-2 cells (all P<0.05). Thus, these results revealed a possible link between decreased miR-138 expression and the progression of human ccRCC.
Kaplan-Meier survival analysis was performed to evaluate the clinical outcomes between two subgroups of patients divided by miR-138 expression. We found that ccRCC patients in low miR-138 group had worse OS (P=0.035, Figure 1D) and DFS (P=0.041, Figure 1E) than those in high miR-138 group.
MiR-138 impairs ccRCC cell proliferation, migration and invasion
To assess the biological functions of miR-138 in ccRCC, ACHN cells with relatively low levels of miR-138 were transfected with miR-138 mimics (miR-138) to achieve miR-138 overexpression, whereas 786-O cells with relatively high levels of miR-138 were transfected with miR-138 inhibitor (anti-miR-138) to achieve the depletion of miR-138 expression. Quantitative RT-PCR analysis showed that the transfection were successful (Data not shown). We observed that the ectopic overexpression of miR-138 in ACHN cells markedly impaired cell proliferation compared with the controls using colony formation assay (Figure 2A) and MTT assay (Figure 2B), and whereas miR-138 knockdown resulted in greater proliferation rate in 786-O cells.
Figure 2.
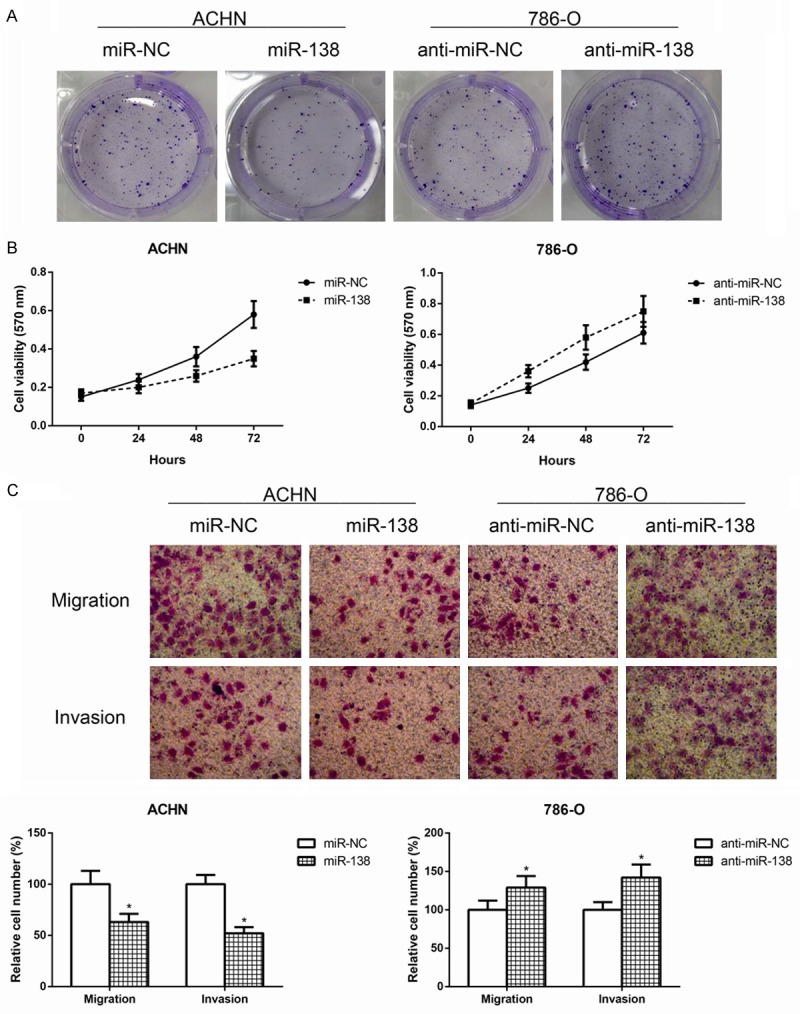
MiR-138 impairs ccRCC cell proliferation, migration and invasion. A. Colony formation assay was performed to detect the proliferation of ccRCC cells after transfection with miR-138 mimics or miR-138 inhibitor. B. MTT assay was performed to detect the proliferation of ccRCC cells after transfection with miR-138 mimics or miR-138 inhibitor. The difference of proliferation rate in 24, 48 and 72 hours between groups were compared. C. Transwell assay was performed to detect the migratory and invasive capacities of ccRCC cells after transfection with miR-138 mimics or miR-138 inhibitor. The cell numbers of migration and invasion were compared between groups. Data are representative images and presented as mean ± SD by at least three independent experiments. *P<0.05.
In addition to cell proliferation inhibition, the effect of miR-138 on cell migration and invasion was also determined using transwell assay in this study. As expected, forced miR-138 expression could markedly suppress the migratory and invasive capacities of ACHN cells, whereas miR-138 knockdown inhibited 786-O cells migration and invasion (Figure 2C).
MiR-138 inhibits tumor growth of ccRCC in vivo
We investigated whether ectopic expression of miR-138 repressed tumor growth in vivo. ACHN cells transfected with miR-138 or miR-NC were subcutaneously injected into nude mice. As demonstrated in Figure 3A, tumors overexpressing miR-138 grew more slowly and were smaller than control tumors. After five weeks, mice were killed, tumors were dissected and weighted. An approximately two-fold decrease in tumor weight was found in miR-138-overexpressing tumors compared to controls (P<0.05, Figure 3B). These results suggested that miR-138 inhibited ccRCC cell growth in vivo.
Figure 3.
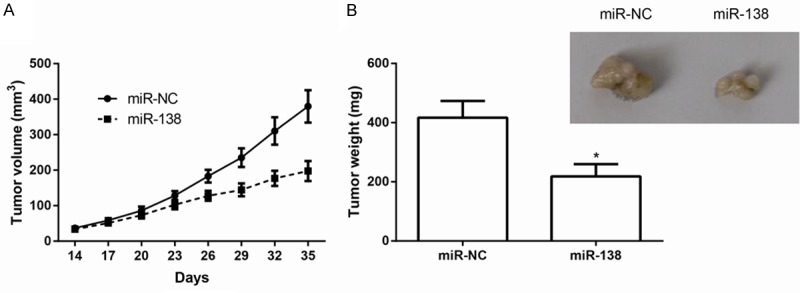
MiR-138 inhibits tumor growth of ccRCC in vivo. A. Growth curves for tumor volumes in xenografts of nude mice were plotted. B. Tumors were resected and weighed at five weeks after cell injection. Representative images of the xenograft tumors are shown. Data are representative images and presented as mean ± SD by at least three independent experiments. *P<0.05.
SOX4 is a target of miR-138 in ccRCC cells
To determine the molecular mechanism by which miR-138 regulates the aggressive phenotype of ccRCC cells, the TargetScan database (http://www.targetscan.org/) was used to predict the potential targets of miR-138. Of the target genes predicted by this database, the SOX4 gene was considered one of the most likely targets (Figure 4A) because it is a critical regulator of EMT in tumor cells.
Figure 4.
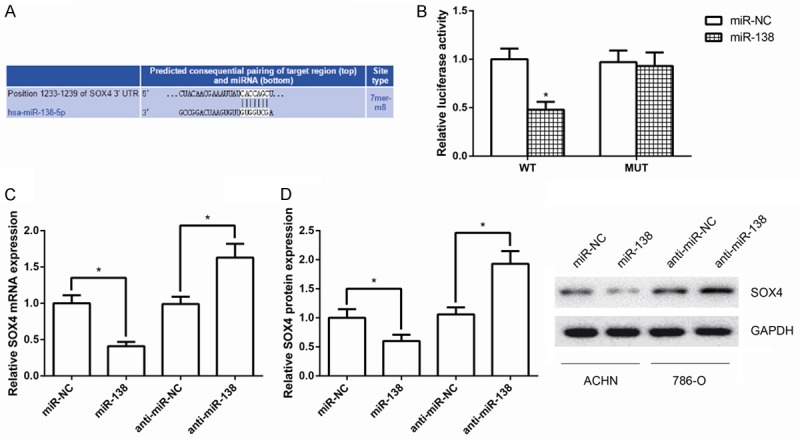
SOX4 is a target of miR-138 in ccRCC cells. A. Prediction of miR-138 binding sites on 3’-UTR of SOX4 mRNA. B. Luciferase activity in ACHN cells co-transfected with miR-138 mimics and luciferase reporters containing SOX4-WT or SOX4-MUT transcript. C. Quantitative RT-PCR analysis of SOX4 mRNA levels in ACHN cells after transfection with miR-138 mimics. D. Western blot analysis of SOX4 protein levels in ACHN cells after transfection with miR-138 mimics. GAPDH was used as internal control. Data are presented as mean ± SD by at least three independent experiments. *P<0.05.
To verify whether SOX4 is a bona fide target of miR-138, the 3’UTR of SOX4 cDNA was fused directly downstream of the luciferase reporter gene (psiCHECK-2-WT-SOX4-3’UTR). miR-138 mimics was then co-transfected with luciferase 3’UTR constructs into ACHN cells. As shown in Figure 4B, miR-138 markedly inhibited luciferase activity of the psiCHECK-2-WT-SOX4-3’UTR (P<0.05), but this inhibition was less changed for 3’UTR with mutated binding sites.
We next investigated whether miR-138 could regulate SOX4 at both mRNA and protein levels. miR-138 mimics and miR-NC were transfected into ACHN cells, and the levels of SOX4 mRNA and protein were monitored. Quantitative RT-PCR analysis and western blot analysis revealed that overexpression of miR-138 in ACHN cells led to reduced levels of endogenous SOX4 mRNA and protein compared with controls (Figure 4C, 4D). Besides, SOX4 mRNA and protein levels were remarkably up-regulated following transfection of 786-O cells with miR-138 inhibitor.
MiR-138 inhibits EMT in ccCRC by targeting SOX4
Rescue experiments were performed by overexpressing SOX4 in miR-138 mimics-transfected ACHN cells. The miR-138 induced downregulation of SOX4 was rescued upon the transfection of pcDNA-SOX4 (Data not shown). Since SOX4 is a critical regulator of EMT in tumor cells, as shown in Figure 5A, the expression of E-cadherin protein, an epithelial cell marker, was increased, whereas the mesenchymal marker vimentin was downexpressed in ACHN cells transfected with miR-138 mimics, and these effects were obviously restored by SOX4 overexpression, indicating that miR-138 inhibits EMT in ccCRC by targeting SOX4.
Figure 5.
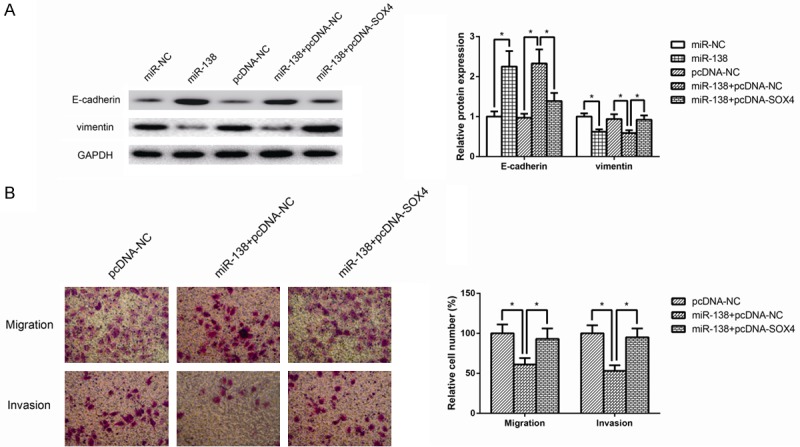
MiR-138 inhibits EMT in ccCRC by targeting SOX4. A. The protein expression levels of EMT-related markers, including E-cadherin and vimentin, were determined in ACHN cells by western bolt analysis. GAPDH was used as internal control. B. Transwell assay was performed to detect the migratory and invasive capacities of ACHN cells co-transfected with miR-138 mimics and pcDNA-SOX4. The cell numbers of migration and invasion were compared between groups. Data are representative images and presented as mean ± SD by at least three independent experiments. *P<0.05.
Moreover, as shown in Figure 5B, overexpression of SOX4 attenuated the inhibition of cell migration and invasion caused by miR-138. These observations suggested that SOX4 could partially recover the inhibitory effects of miR-138 on ccRCC cell migration and invasion.
Upregulation of SOX4 is inversely correlated with miR-138 expression in ccRCC
As miR-138 was down-expressed in ccRCC and targeted SOX4 by binding to its 3’UTR, we subsequently explored whether SOX4 expression was negatively associated with miR-138 levels in ccRCC tissue samples. Analysis of SOX4 expression level in 20 ccRCC tissues and corresponding normal tissues by quantitative RT-PCR revealed that SOX4 mRNA was evidently down-regulated in ccRCC (P<0.05, Figure 6A). Furthermore, Pearson’s correlation analysis revealed that low expression of SOX4 mRNA was more likely correlated with high levels of miR-138 (r=-0.467, P=0.038; Figure 6B), suggesting that the upregulation of SOX4 may be due to decreased miR-138 expression in ccRCC.
Figure 6.
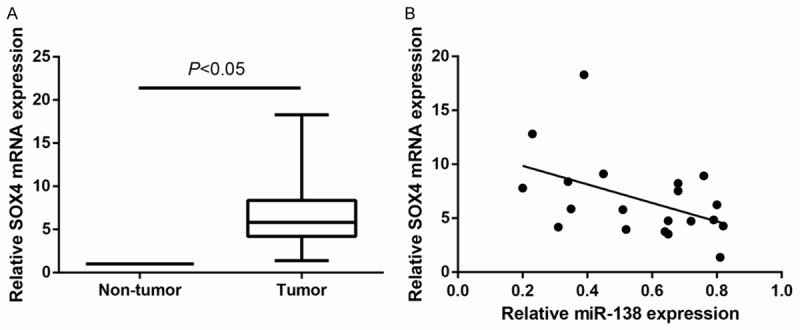
Upregulation of SOX4 is inversely correlated with miR-138 expression in ccRCC. A. SOX4 mRNA expression level was detected in 20 pairs of ccRCC tissues and adjacent noncancerous tissues by quantitative RT-PCR. GAPDH was used as internal control. Data are presented as mean ± SD by at least three independent experiments. *P<0.05. B. The correlation of the expression levels of SOX4 and miR-138 in 20 ccRCC tissue samples was detected by Pearson’s correlation analysis (r=-0.467, P=0.038).
Discussion
Until now, dysregulation (e.g., overexpression or loss of expression) of miRNAs has been linked to the initiation and progression of numerous human cancers, including ccRCC [19,20]. There are a few studies on miR-138 regulating proliferation and migration of ccRCC cells. A previous study indicated that miR-138 inhibits the expression of HIF-1α to regulate the apoptosis and migration of ccRCC cells [21]. Also, Liang et al. reported that miR-138 is a tumor-suppressor in ccRCC that induces ccRCC cell senescence by reducing EZH2 expression and increasing P16 expression [22].
In the present study, we found that miR-138 was downregulated in ccRCC cell lines and tissue specimens, and its expression was negatively associated with larger tumor size, distant metastasis, advanced TNM stage and unfavorable prognosis of ccRCC patients. More importantly, ectopic expression of miR-138 was found to cause a remarkable decline in the proliferative, migratory and invasive capacities of ccRCC cells. In vivo studies also showed that miR-138 overexpression evidently impaired xenograft tumor formation of ccRCC cells. These results promoted us to further explore the function of miR-138 in ccRCC.
Next, we explored the mechanisms by which miR-138 exerts its function in malignant ccRCC phenotypes. As miRNAs exerts functions in human cancer through mediating the expression of their target genes at a posttranscriptional level [23], we further focused on the target of miR-138 in ccRCC cells. Several miR-138 targets have been found in different cell context and organs, such as, HIF-1α was identified to be a target of miR-138, which mediated an effect on proliferation, invasion and glycolysis in malignant melanoma cells [24]. In the present study, SOX4 was predicted as a potential target of miR-138 by TargetScan, and its mRNA and protein levels were negatively mediated by miR-138 in ccRCC cells. Furthermore, a marked inverse correlation was found between the levels of miR-138 and SOX4 mRNA in the ccRCC tissues. This interaction between miR-138 and SOX4 mRNA has been previously reported in ovarian cancer [25]. As a facilitator of tumorigenesis and metastasis, SOX4 was also shown to be highly expressed in ccRCC [26]. Furthermore, our data demonstrated that miR-138 might act as a suppressor for EMT in ccRCC by targeting SOX4.
Metastasis is the primary cause of mortality in most cancer patients [27]. As a major hallmark of malignancy, metastasis is a complex process that includes decreased adhesion and increased motility, cell attachment, matrix dissolution and migration [28]. Epithelial-mesenchymal transition (EMT) is one of the most commonly accepted cellular transitioning events that drive tumor metastasis through which epithelial cells lose their characteristics, including cell-cell contacts and polarity, and adopt mesenchymal, fibroblast-like properties such as increased motility and decreased intercellular adhesion [29,30]. The expression of proteins that promote cell-to-cell contact, such as E-cadherin, may be lost, and the cells may acquire mesenchymal markers such as vimentin, fibronectin, and N-cadherin [31]. A number of recent studies have widely reported that SOX4 was involved in the promotion of EMT [32,33]. In the present study, increased SOX4 expression promoted migration and invasion, as well as EMT program, in ccRCC cells, yielding opposed function as that of miR-138. These results suggested that down-regulation of SOX4 may be a molecular mechanism by which miR-138 exerted its anti-tumor functions in ccRCC cells.
In summary, the findings from our study unveiled that miR-138 inhibits the migration and invasion of ccRCC cells as well as EMT program, at least in part, through its direct regulation of SOX4. Accordingly, it is plausible that our novel findings may improve our understanding of ccRCC tumorigenesis and aid in design of a potential therapeutic strategy for ccRCC patients in the clinic.
Acknowledgements
The work was supported by National Natural Science Foundation of China (Grant number: 81501220) and Chongqing Municipal Natural Science Foundation (Grant number: CSTC2015jcyjA10067).
Disclosure of conflict of interest
None.
References
- 1.Escudier B, Porta C, Schmidinger M, Algaba F, Patard JJ, Khoo V, Eisen T, Horwich A. Renal cell carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and followup. Ann Oncol. 2014;25(Suppl 3):iii49–56. doi: 10.1093/annonc/mdu259. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 3.Rini BI, Campbell SC, Escudier B. Renal cell carcinoma. Lancet. 2009;373:1119–1132. doi: 10.1016/S0140-6736(09)60229-4. [DOI] [PubMed] [Google Scholar]
- 4.Lui PY, Jin DY, Stevenson NJ. MicroRNA: master controllers of intracellular signaling pathways. Cell Mol Life Sci. 2015;72:3531–3542. doi: 10.1007/s00018-015-1940-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16:421–433. doi: 10.1038/nrg3965. [DOI] [PubMed] [Google Scholar]
- 6.Hwang HW, Mendell JT. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br J Cancer. 2006;94:776–780. doi: 10.1038/sj.bjc.6603023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai Y, Li H, Zhang Y. Downregulation of microRNA-206 suppresses clear cell renal carcinoma proliferation and invasion by targeting vascular endothelial growth factor A. Oncol Rep. 2016;35:1778–1786. doi: 10.3892/or.2015.4538. [DOI] [PubMed] [Google Scholar]
- 8.Xiao H, Xiao W, Cao J, Li H, Guan W, Guo X, Chen K, Zheng T, Ye Z, Wang J, Xu H. miR-206 functions as a novel cell cycle regulator and tumor suppressor in clear-cell renal cell carcinoma. Cancer Lett. 2016;374:107–116. doi: 10.1016/j.canlet.2016.01.032. [DOI] [PubMed] [Google Scholar]
- 9.Wang W, Zhao LJ, Tan YX, Ren H, Qi ZT. MiR-138 induces cell cycle arrest by targeting cyclin D3 in hepatocellular carcinoma. Carcinogenesis. 2012;33:1113–1120. doi: 10.1093/carcin/bgs113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma F, Zhang M, Gong W, Weng M, Quan Z. MiR-138 suppresses cell proliferation by targeting Bag-1 in gallbladder carcinoma. PLoS One. 2015;10:e0126499. doi: 10.1371/journal.pone.0126499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han LP, Fu T, Lin Y, Miao JL, Jiang QF. MicroRNA-138 negatively regulates non-small cell lung cancer cells through the interaction with cyclin D3. Tumour Biol. 2016;37:291–298. doi: 10.1007/s13277-015-3757-8. [DOI] [PubMed] [Google Scholar]
- 12.Jiang B, Mu W, Wang J, Lu J, Jiang S, Li L, Xu H, Tian H. MicroRNA-138 functions as a tumor suppressor in osteosarcoma by targeting differentiated embryonic chondrocyte gene 2. J Exp Clin Cancer Res. 2016;35:69. doi: 10.1186/s13046-016-0348-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang R, Liu M, Liang H, Guo S, Guo X, Yuan M, Lian H, Yan X, Zhang S, Chen X, Fang F, Guo H, Zhang C. miR-138-5p contributes to cell proliferation and invasion by targeting Survivin in bladder cancer cells. Mol Cancer. 2016;15:82. doi: 10.1186/s12943-016-0569-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowles J, Schepers G, Koopman P. Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev Biol. 2000;227:239–255. doi: 10.1006/dbio.2000.9883. [DOI] [PubMed] [Google Scholar]
- 15.Schilham MW, Oosterwegel MA, Moerer P, Ya J, de Boer PA, van de Wetering M, Verbeek S, Lamers WH, Kruisbeek AM, Cumano A, Clevers H. Defects in cardiac outflow tract formation and pro-B-lymphocyte expansion in mice lacking Sox-4. Nature. 1996;380:711–714. doi: 10.1038/380711a0. [DOI] [PubMed] [Google Scholar]
- 16.Schilham MW, Moerer P, Cumano A, Clevers HC. Sox-4 facilitates thymocyte differentiation. Eur J Immunol. 1997;27:1292–1295. doi: 10.1002/eji.1830270534. [DOI] [PubMed] [Google Scholar]
- 17.Cheung M, Abu-Elmagd M, Clevers H, Scotting PJ. Roles of Sox4 in central nervous system development. Brain Res Mol Brain Res. 2000;79:180–191. doi: 10.1016/s0169-328x(00)00109-1. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Chen X, Han W, Ruan A, Chen L, Wang R, Xu Z, Xiao P, Lu X, Zhao Y, Zhou J, Chen S, Du Q, Yang H, Zhang X. miR-200c targets CDK2 and suppresses tumorigenesis in renal cell carcinoma. Mol Cancer Res. 2015;13:1567–1577. doi: 10.1158/1541-7786.MCR-15-0128. [DOI] [PubMed] [Google Scholar]
- 19.Nikitina EG, Urazova LN, Stegny VN. MicroRNAs and human cancer. Exp Oncol. 2012;34:2–8. [PubMed] [Google Scholar]
- 20.Kuninty PR, Schnittert J, Storm G, Prakash J. MicroRNA targeting to modulate tumor microenvironment. Front Oncol. 2016;6:3. doi: 10.3389/fonc.2016.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song T, Zhang X, Wang C, Wu Y, Cai W, Gao J, Hong B. MiR-138 suppresses expression of hypoxia-inducible factor 1alpha (HIF-1alpha) in clear cell renal cell carcinoma 786-O cells. Asian Pac J Cancer Prev. 2011;12:1307–1311. [PubMed] [Google Scholar]
- 22.Liang J, Zhang Y, Jiang G, Liu Z, Xiang W, Chen X, Chen Z, Zhao J. MiR-138 induces renal carcinoma cell senescence by targeting EZH2 and is downregulated in human clear cell renal cell carcinoma. Oncol Res. 2013;21:83–91. doi: 10.3727/096504013X13775486749218. [DOI] [PubMed] [Google Scholar]
- 23.Breving K, Esquela-Kerscher A. The complexities of microRNA regulation: mirandering around the rules. Int J Biochem Cell Biol. 2010;42:1316–1329. doi: 10.1016/j.biocel.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 24.Chen Y, Cao KE, Wang S, Chen J, He B, He GU, Peng B, Zhou J. MicroRNA-138 suppresses proliferation, invasion and glycolysis in malignant melanoma cells by targeting HIF-1alpha. Exp Ther Med. 2016;11:2513–2518. doi: 10.3892/etm.2016.3220. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Yeh YM, Chuang CM, Chao KC, Wang LH. MicroRNA-138 suppresses ovarian cancer cell invasion and metastasis by targeting SOX4 and HIF-1alpha. Int J Cancer. 2013;133:867–878. doi: 10.1002/ijc.28086. [DOI] [PubMed] [Google Scholar]
- 26.Chen X, Ruan A, Wang X, Han W, Wang R, Lou N, Ruan H, Qiu B, Yang H, Zhang X. miR-129-3p, as a diagnostic and prognostic biomarker for renal cell carcinoma, attenuates cell migration and invasion via downregulating multiple metastasis-related genes. J Cancer Res Clin Oncol. 2014;140:1295–1304. doi: 10.1007/s00432-014-1690-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riihimaki M, Thomsen H, Hemminki A, Sundquist K, Hemminki K. Comparison of survival of patients with metastases from known versus unknown primaries: survival in metastatic cancer. BMC Cancer. 2013;13:36. doi: 10.1186/1471-2407-13-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yao D, Dai C, Peng S. Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumor formation. Mol Cancer Res. 2011;9:1608–1620. doi: 10.1158/1541-7786.MCR-10-0568. [DOI] [PubMed] [Google Scholar]
- 31.Moreno-Bueno G, Portillo F, Cano A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene. 2008;27:6958–6969. doi: 10.1038/onc.2008.346. [DOI] [PubMed] [Google Scholar]
- 32.Zhang J, Liang Q, Lei Y, Yao M, Li L, Gao X, Feng J, Zhang Y, Gao H, Liu DX, Lu J, Huang B. SOX4 induces epithelial-mesenchymal transition and contributes to breast cancer progression. Cancer Res. 2012;72:4597–4608. doi: 10.1158/0008-5472.CAN-12-1045. [DOI] [PubMed] [Google Scholar]
- 33.Wang L, Zhang J, Yang X, Chang YW, Qi M, Zhou Z, Han B. SOX4 is associated with poor prognosis in prostate cancer and promotes epithelial-mesenchymal transition in vitro. Prostate Cancer Prostatic Dis. 2013;16:301–307. doi: 10.1038/pcan.2013.25. [DOI] [PubMed] [Google Scholar]