

Abstract
Hepatocyte growth factor (HGF) is a multifunctional growth factor that plays important roles in promoting the invasion and metastasis of various tumor cells. However, there are few reports about the exact mechanisms of HGF involved in the regulation of cell invasion via the induction of COX2. In this study, we found that HGF could activate its receptor c-Met and up-regulate COX2 expression in a dose- and time-dependent manner, which resulted in an increase in MMP-9 expression and subsequent invasiveness of the breast cancer cell lines MDA-MB-231 and MCF-7. The HGF-induced expression of COX2 and MMP-9 and cell invasion were partially suppressed by COX2 gene silencing. The PI3K/Akt and p38 MAPK signaling pathways were activated by HGF in both cell lines. However, PI3K/Akt or p38 MAPK-specific inhibition alone partially attenuated HGF-induced COX2 and MMP-9 expression and the invasiveness of the two breast cancer cell lines, and these HGF-induced effects were almost completely abolished by simultaneous treatment with both inhibitors. Therefore, we concluded that HGF mediates the up-regulation of COX2 predominantly through the PI3K/Akt and p38 MAPK signaling pathways, leading to MMP-9 expression and the subsequent invasion of two breast cancer cell lines. This study improves our understanding of the signal transduction mechanisms in the HGF-induced invasion and progression of breast cancer.
Keywords: Hepatocyte growth factor (HGF), breast cancer, invasion, cyclooxygenase 2 (COX2), PI3K/Akt, p38 MAPK, metalloproteinase-9 (MMP9)
Introduction
Breast cancer is one of the most common malignancies in women worldwide. Because many breast cancer patients are diagnosed at an advanced stage of cancer in which invasion and metastasis have already occurred, the 5-year survival rate for patients with breast cancer has had limited improvement in the past twenty years. However, the invasion and metastasis mechanisms in breast cancer remain poorly understood, and it is necessary to gain a better understanding of their underlying causes to develop better targeted treatments.
Growth factors and their receptors are attractive targets for tumor therapy because their signaling pathways control tumor cell division and survival, both of which are imbalanced in tumorigenesis. It has been repeatedly demonstrated that numerous growth factors and tumor signaling pathways overlap and interact with each other. The pathogenesis of tumors is a multi-step and complicated process in which invasion is an essential step. There are many growth factors, and their receptors contribute to the stimulation of tumor cell growth, invasion, angiogenesis and metastasis [1,2]. Hepatocyte growth factor (HGF), which acts by binding to its tyrosine kinase receptor, c-Met, plays an important role in many solid cancers in humans including breast cancer [1]. The dysregulated expression of this ligand-receptor axis often leads to invasion, metastasis and poor prognosis [3]. HGF, a stromal-derived growth factor, is a multifunctional cytokine, and previous clinical studies have demonstrated that the high expression of HGF and/or c-Met correlates with an invasive phenotype for various tumors, including breast cancer [3-5]. HGF has been shown to be involved in the regulation of various cellular processes in vitro, including proliferation, apoptosis, angiogenesis and extracellular matrix invasion. However, the underlying mechanisms of HGF-induced cell invasion are not completely understood.
Cyclooxygenase 2 (COX2), an inducible form of cyclooxygenase, is the rate-limiting enzyme for the production of prostaglandins (PEGs), which are considered to be involved in inflammation and the progression of carcinoma [6]. The up-regulation of COX2 should lead to increased prostaglandin production, which in return activates the growth, invasion and suppression of apoptosis in cancer cells [6,7]. Nonsteroidal anti-inflammatory drugs (NSAIDs) are beneficial in breast cancer therapy. It has been reported that nimesulide and celecoxib, which are inhibitors of COX2, have been recently tested as therapeutic treatments for tumors [6-8]. Other research showed that nimesulide also inhibits the activity and expression of aromatase in some breast cancer cell lines [7]. It has been reported that HGF dramatically increased the expression of COX2 in colorectal carcinoma cells, which might be mediated by two major signaling pathways including the phosphatidylinositol 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) pathways, and it has been reported that COX2 is a vital downstream regulator of HGF [9].
In this study, we investigated whether HGF induces the up-regulation of COX2 via activation of the PI3K/Akt and p38 MAPK signaling pathways and facilitates the increased expression of MMP-9 and the subsequent invasion of MDA-MB-231 and MCF-7 cells. This study might provide valuable insight into the mechanism of the HGF signaling involved in the progression and development of breast cancer.
Material and methods
Cell lines and growth conditions
The human breast cancer cell lines, MDA-MB-231 and MCF-7, were obtained from the Department of Immunology, Zhongshan School of Medicine, Institute of Human Virology, Sun Yat-sen University, Guangzhou, PR China. The breast cancer cells were cultured in DMEM (Gibco, Grand Island, NY USA) supplemented with 10% FBS and a 1% penicillin/streptomycin solution (Invitrogen, Carlsbad, CA USA). Human recombinant HGF was obtained from PeproTech Inc. (Rocky Hill, NJ USA). The inhibitors of the p38 MAPK and Akt pathways were purchased from Cell Signaling Technology, Inc. (Beverly, MA USA).
RNA preparation and quantitative real-time PCR (qRT-PCR)
The breast cancer cells were deprived of serum for 24 h and then exposed to different concentrations of human HGF for the indicated times. Total RNA was isolated from the cells with the TRIzol reagent (Invitrogen, Carlsbad, CA USA) according to the manufacturer’s protocol. Reverse transcription polymerase chain reaction (RT/PCR) was performed. Then, quantitative real-time PCR (qRT-PCR) was performed to quantify the level of COX2 mRNA based on the general fluorescence detection of SYBR Green (TaKaRa, Osaka, Japan) using the Lightcycler thermocycler (BD, Bedford, MA USA). The procedure for quantitative real-time PCR were as follows: 30 s at 95°C for an initial denaturation followed by 40 cycles of 10 s at 95°C for denaturing, 22 s at 55°C for annealing, and 30 s at 72°C for extension. Measurements were performed in triplicate. The specific primers sets used for human COX2 and β-actin (control) were as follows: COX2: forward: 5’-TCA AAA CCG AGG TGT A-3’ and reverse: 5’-GTG GGT AAG TAT GTA GTG C-3’; β-actin: forward: 5’-AAG ATG ACC CAG ATC ATG TTT G-3’ and reverse: 5’-CTT TGC GGA TGT CCA CGT-3’.
Western blot analysis
Cells were treated with various concentrations of HGF or different inhibitors. All inhibitors were dissolved in DMSO. The working concentrations for various inhibitors were 40 μM LY294002 and 10 μM SB202190. The same volume of DMSO was used as a control. Different inhibitors and DMSO were added to cells 1 h before HGF stimulation. After treatment with a combination of HGF for the indicated times, cells were lysed with RIPA buffer consisting of 20 mM Tris/HCl (pH 7.5), 150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, 1 mM EDTA, 50 mM NaF, 1 mM sodium orthovanadate, and Complete Proteinase Inhibitor Mixture Tablets (Roche Applied Science, German). Protein concentrations were determined with the DC Protein Assay (Bio-Rad, Cambridge, MA USA). An equal amount of total protein was separated by 10% SDS polyacrylamide gel electrophoresis, and the proteins were transferred onto a polyvinylidene difluoride membrane. Membranes were incubated overnight in TBS containing 0.1% Tween-20 and 5% dried skim milk at 4°C to block nonspecific binding. The membranes were then incubated overnight with primary antibodies at 4°C and subsequently with the appropriate secondary antibodies for 2 hours at room temperature. Signals were detected using an enhance chemiluminescence (ECL) kit (Millipore, USA). Quantification of protein signals was performed with the Gel-Pro Analyzer software (Quantity One, USA). The primary antibodies used in the Western blot analysis included human COX2 (Santa Cruz, CA USA), c-Met, phosphor-c-Met (Santa Cruz, CA USA), β-actin (Santa Cruz, CA USA), Akt, phospho-Akt (Ser-473), p38 MAPK, phospho-p38 MAPK (Cell Signaling Technology, Danvers, MA USA), and MMP-9 (Bioworlde, USA).
Plasmids construction and transfection
The oligonucleotides (5’-GATCCCATGGAATTACCCAGTTTGTTCAAGAGACAAACTGGGTAATTCCATGTTTTTGTCGACA-3’ and 5’-AGCTTGTCGACAAAAACATGGAATTACCCAGTTTGTCTCTTGAACAAACTGGGTAATTCCATGG-3’), which encode the shRNA-specific targeting of human COX2, were cloned into the Hind III and Bam HI sites in the pGenesil-1 vector (Genesil, Wuhan, China), which carries a kanamycin-resistance gene. The recombinant interference and nonsense shRNA plasmids were named pshRNA-COX2 and pshRNA-HK, respectively. Breast cancer cells were transfected with pshRNA-COX2 or pshRNA-HK with LipofectamineTM 2000 (Invitrogen, Carlsbad, CA USA) following the manufacturer’s instructions.
Cell invasion assay
Invasion assays were performed with 8 μm-pore Transwell chambers (Corning, NY, USA) as described with modification [10,11]. The upper chamber was coated with 70 μl of Matrigel (BD Biosciences, Bedford, MA USA) diluted 1:3 in serum-deprived DMEM and incubated at 37°C for 2 h. The MDA-MB-231 and MCF-7 cells were suspended in serum-free DMEM and seeded in the upper wells of each transwell. The lower chambers were pro-coated with fibronectin filled with DMEM containing 10% FBS. HGF (60 ng/ml) plus various inhibitors or DMSO were added into the lower wells as indicated in the figures. After incubation at 37°C with 5% CO2 for 16 h, the invaded cells on the lower chamber membranes were fixed in methanol and stained with 0.2% crystal violet for 15 min. The number of invading cells was counted under a microscope in five random fields.
Statistical analysis
All values were expressed as the mean ± standard deviation (SD). The Student’s t test was used to compare data between two groups. Statistical analyses between three or more groups were performed using one-way ANOVA and Bonferroni correction. Values for P less than 0.05 were considered statistically significant.
Results
HGF facilitates the invasion of breast cancer cells
The positive expression of the HGF receptor c-Met and an HGF-induced increase in phosphorylated c-Met (p-c-Met) in the breast cancer cell lines MDA-MB-231 and MCF-7 as measured by We-stern blot suggested that both breast cancer cell lines could respond to HGF (Figure 1A). The effects of HGF on the invasion of breast cancer cells were determined by a transwell chamber assay. In the presence of various concentrations of HGF, the number of invasive cells for both breast cancer cell lines was significantly increased (Figure 1B), and HGF showed a concentration-dependent stimulation effect that became evident in the presence of 60 ng/ml HGF; thus, 60 ng/ml HGF was used in subsequent experiments.
Figure 1.
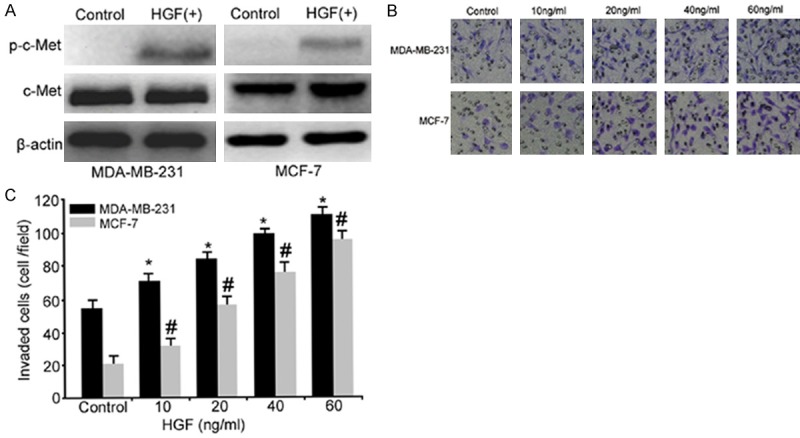
HGF induced the invasion of breast cancer cells through its receptor c-Met. A: Representative immunoblots of whole-cell lysates from the breast cancer cell lines MDA-MB-231 and MCF-7. Cell extracts were prepared, and equal amounts (30 μg) of each sample were analyzed by Western blotting as described in Materials and Methods. B: HGF induced breast cancer cell invasion. MDA-MB-231 and MCF-7 cells were serum-deprived overnight and then treated with various doses of HGF to detect their invasiveness. Cells were fixed in methanol and stained with crystal violet. The number of invasive cells was counted in five randomly selected fields. Micrographs demonstrate a random field in the lower chamber membrane under different concentrations of HGF (×100). C: Diagrams displaying the effect of different concentrations of HGF on invasion (*, P<0.05 vs. the MDA-MB-231 control group, #, P<0.05 vs. the MCF-7 control group).
HGF up-regulates COX2 expression
Given the important role of COX2 and HGF in cell invasion, proliferation, and survival during tumorigenesis, we hypothesized that HGF regulates COX2 in breast cancer cells. To examine this hypothesis, quantitative real-time PCR (qRT-PCR) was performed to determine whether HGF regulated COX2 mRNA expression in MDA-MB-231 and MCF-7 cells. As shown in Figure 2A, the mRNA level of COX2 was markedly increased by HGF in a dose-dependent manner (Figure 2A). Further analysis confirmed that COX2 mRNA was up-regulated by 60 ng/ml HGF in a time-dependent manner (Figure 2B). Consistent with the up-regulation of COX2 mRNA transcription, the protein expression level of COX2 as measured by Western blot showed similar dose- and time-dependent induction in HGF-stimulated cells (Figure 2C and 2D).
Figure 2.
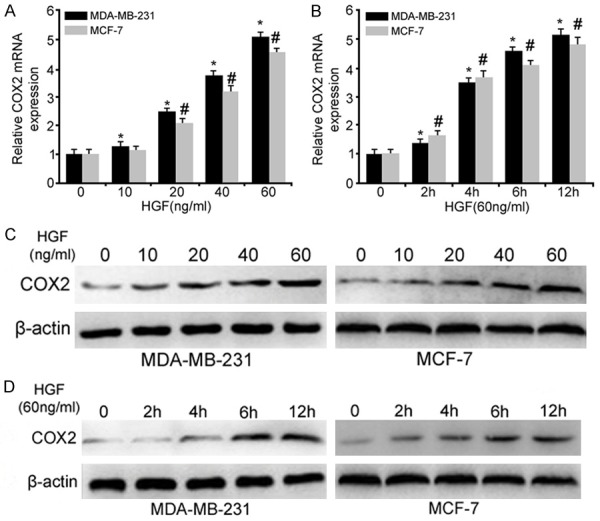
HGF-induced COX2 expression in breast cancer cells as analyzed by qRT-PCR and Western blot. A: HGF dose-response of COX2 mRNA expression in MDA-MB-231 and MCF-7 cells. Breast cancer cells were grown to 80% confluence and serum-deprived for 24 hours. Cells were then treated with the indicated concentrations, ranging from 10 to 60 ng/ml of HGF for 12 h. B: Time-dependent up-regulation of COX2 in breast cancer cells at the indicated time points. All values were normalized to the expression level of the control group, which was set to 1. C: Western blotting was performed for COX2, and β-actin confirmed equal protein loading. D: Western blot analysis for COX2 following 60 ng/ml HGF treatment for the indicated times in breast cancer cells (*, P<0.05 vs. the MDA-MB-231 control group and #, P<0.05 vs. the MCF-7 control group).
HGF-induced breast cancer cell invasion is partially abolished by COX2 gene silencing
We next investigated whether the breast cancer cell invasion induced by HGF was affected by COX2. MDA-MB-231 and MCF-7 cells were transfected with pshRNA-COX2, and the COX2 translation level in the HGF+pshRNA-COX2 group was lower than that in the HGF+pshRNA-HK and HGF groups (*P<0.05) but higher than control (*P<0.05). However, there was no significant difference in the COX2 expression between the HGF and HGF+pshRNA-HK groups (Figure 3A, 3B). These findings indicated that the pshRNA-COX2 plasmid could partially silence HGF-induced COX2 expression. The treatment of both breast cancer cell lines, which were transfected with the pshRNA-COX2 plasmid, with 60 ng/ml HGF showed less obvious HGF-induced invasiveness than those with HGF and HGF+pshRNA-HK treatment. However, there was no significant difference in the invasiveness of the HGF and HGF+pshRNA-HK transfected groups (Figure 3C). These results suggested that HGF-stimulated cell invasion was at least partially mediated by the up-regulation of COX2 expression in both breast cancer cell lines.
Figure 3.

The HGF-induced invasion of MDA-MB-231 and MCF-7 breast cancer cells is partially suppressed by COX2 gene silencing. A, B: MDA-MB-231 and MCF-7 cells were transfected with pshRNA-HK and pshRNA-COX2. Total cell lysates was used for Western blot analysis. A lower level of COX2 protein was detected in the HGF+pshRNA-COX2 group (*P<0.05), but it was higher than control (*P<0.05). C: Knockdown of the COX2 gene inhibited the HGF-induced cell invasion of MDA-MB-231 and MCF-7 cells (*P<0.05 vs. HGF and HGF+pshRNA-HK groups), and there was no statistically significant difference between the pshRNA-HK-transfected breast cancer cell lines and the HGF group (**P>0.05).
HGF-induced MMP-9 expression is partially restrained by COX2 gene knockdown
We further investigated the possible downstream effector mediating the invasive potential of breast cancer cell lines induced by HGF up-regulated COX2 expression. As shown in Figure 4, HGF could up-regulate the expression of the COX2 and MMP-9 protein levels in breast cancer cells. However, the HGF-induced MMP-9 up-regulation was reduced to 62% and 49% with COX2 gene target silencing in the MDA-MB-231/HGF+pshRNA-COX2 and MCF-7/HGF+pshRNA-COX2 groups compared with the MDA-MB-231/HGF and MCF-7/HGF groups, respectively (P<0.05). There was significantly decreased MMP9 expression in cells transfected with pshRNA-COX2 compared with control cells treated with or without HGF (P<0.05). These results were consistent with the invasion assay results (Figure 3).
Figure 4.
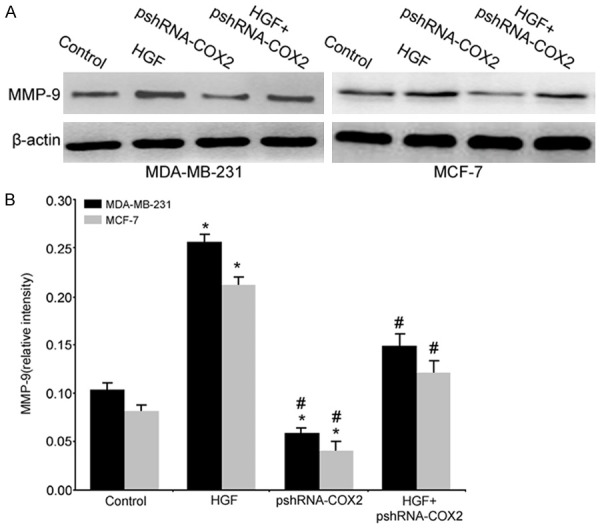
COX2 regulated expression of MMP-9 in HGF-stimulated MDA-MB-231 and MCF-7 cells. Total cell lysates were analyzed by Western blot analysis for MMP-9 (*, P<0.05 vs. the control group; #, P<0.05 vs. the HGF group).
HGF-induced COX2 up-regulation acts through the PI3K/Akt and p38 MAPK pathways
Having confirmed that COX2 expression is up-regulated by HGF, we further investigated the mechanism of COX2 up-regulation. Previous studies have demonstrated that HGF up-regulates COX2 via the MAPK signaling pathway in gastric and non-small cell lung cancer cells, and HGF activates COX2 expression in a PI3K-dependent fashion in endothelial cells; therefore, we determined whether HGF induced COX2 up-regulation via the PI3K/Akt and/or p38 MAPK pathways in breast cancer cells. The activation of c-Met by HGF was first confirmed by Western blot analysis for total and phosphorylated c-Met (p-c-Met) in MDA-MB-231 and MCF-7 cells. As shown in Figure 5A, phosphorylated Met was increased by HGF treatment in both breast cancer cell lines. Furthermore, Western blotting confirmed that the phosphorylation of Akt and p38 MAPK was increased by HGF (Figure 5A), and HGF led to the maximal induction of phosphorylated Akt (p-Akt) and phosphorylated p38 MAPK (p-p38 MAPK) within 15 min, and this stimulation persisted for 60 minutes (data not shown). In contrast, MDA-MB-231 and MCF-7 cells displayed little to no phosphorylated c-Met, Akt and p38 MAPK in the absence of HGF, and there were no obvious changes in the level of total c-Met, p38 MAPK and Akt with or without HGF treatment in both breast cancer cell lines (Figure 5A). These findings suggested that activation of the PI3K/Akt and p38 MAPK signaling pathways is involved in HGF-induced cell invasion.
Figure 5.
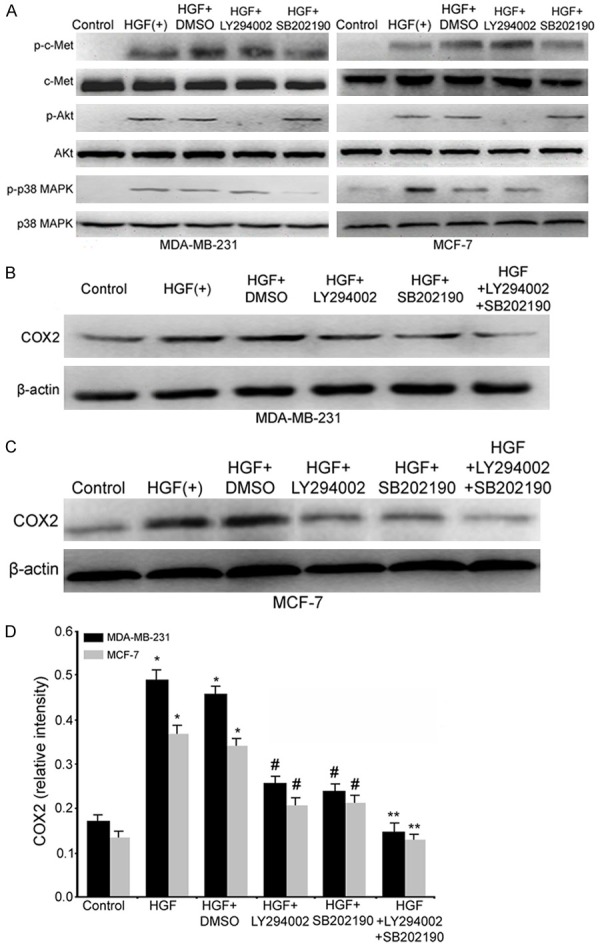
HGF up-regulates COX2 through the Akt/PI3K and p38 MAPK signaling pathways. A: HGF induces Akt and p38 MAPK phosphorylation in MDA-MB-231 and MCF-7 cells. Cells were serum-deprived for 24 h followed by treatment as indicated, and extracted proteins were analyzed by Western blot to assay the expression of Akt and p38 MAPK and phosphorylated Akt and p38 MAPK. B-D: The HGF-induced increase in the expression of the COX2 protein occurs via the Akt and p38 MAPK pathways. MDA-MB-231 and MCF-7 cells were serum-deprived for 24 h and then treated with signaling inhibitors for 1 h followed by 60 ng/ml HGF treatment for 15 min for p-Akt and p-p38 MAPK detection and 12 h for COX2 detection. (*, P<0.05 vs. the control group; #, P<0.05 vs. the HGF or HGF+DMSO groups; **, P<0.05 vs. the HGF+LY294002, HGF+SB202190, HGF or HGF+DMSO groups).
To examine whether the PI3K/Akt and/or p38 MAPK signaling pathways mediate HGF-induced COX2 up-regulation in breast cancer cells, MDA-MB-231 and MCF-7 cells were pretreated with the specific PI3-kinase inhibitor LY294002 and/or SB202190, a selective inhibitor of p38 MAPK phosphorylation, for 1 h followed by HGF treatment, and protein samples from the breast cancer cells were subjected to Western blot analysis. It was found that the p-c-Met levels were not obviously changed; however, the HGF-mediated phosphorylation of Akt and p38 MAPK was inhibited by LY294002 and SB202190, respectively (Figure 5A). The addition of either LY294002 or SB202190 partially suppressed HGF-induced COX2 up-regulation in both breast cancer cell lines (Figure 5A). Furthermore, in the presence of LY294002 and SB202190, HGF-mediated COX2 up-regulation was completed abolished (Figure 5B-D). These results indicate that HGF-driven COX2 up-regulation predominantly occurs through the PI3K/Akt and p38 MAPK signaling pathways.
HGF-driven COX2 up-regulation via the PI3K/Akt and p38 MAPK pathways results in increased MMP-9 expression and the subsequent invasion of MDA-MB-231 and MCF-7 cells
To further evaluate the potential role of the Akt/PI3-kinase and p38 MAPK pathways in HGF-induced invasion, the effects of LY294002 and SB202190 on the invasion of HGF-stimulated breast cancer cells were tested. Concentrations of 10 μmol/L for LY294002 and 10 μmol/L for SB202190 partially attenuated the HGF-induced increase in the invasion of both breast cancer cell lines. In the presence of simultaneous treatment with LY294002 and SB202190, the HGF-driven increase in invasion was completely prevented in MDA-MB-231 and MCF-7 cells (Figure 6A, 6B). Furthermore, the MMP-9 expression levels were significantly decreased in the LY294002/HGF or SB202190/HGF groups compared with the HGF group. Similarly, the HGF-induced expression of MMP-9 was completely inhibited in the presence of LY294002 and SB202190 as well (Figure 6C, 6D). Thus, we found that the Akt/PI3K and p38 MAPK pathways are involved in the HGF-mediated induction of COX2, leading to the up-regulation of MMP-9 and a subsequent increase in the invasion of MDA-MB-231 and MCF-7 cells.
Figure 6.
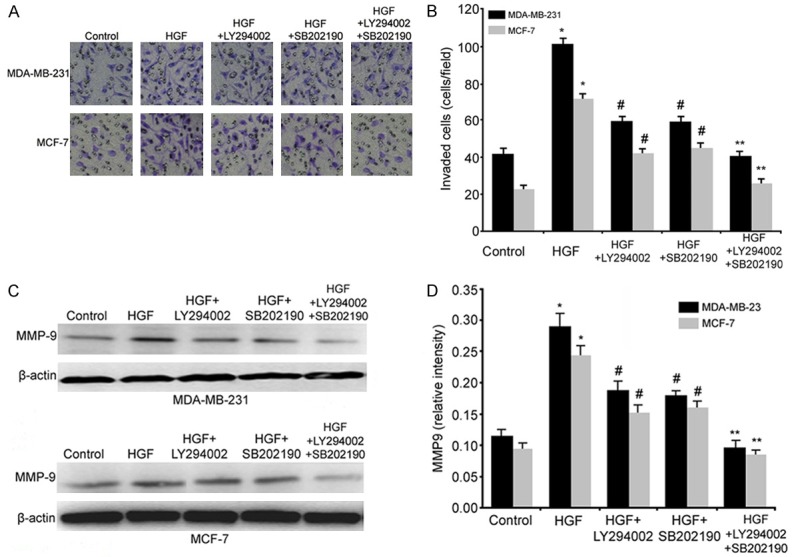
The Akt/PI3K and p38 MAPK pathways participate in COX2 regulation of HGF-induced invasion and MMP-9 production. A and B: MDA-MB-231 and MCF-7 cells were incubated in the presence or absence of 60 ng/ml HGF plus 10 μM LY294002 (Akt/PI3K inhibitor) or/and 10 me SB202190 (p38 inhibitor). Cells were used in invasion assays as described in Materials and Methods. Micrographs demonstrate a random field from the lower chamber membrane under different conditions (100×). C and D: LY294002 and SB202190 decreased the HGF-induced expression of the MMP-9 protein. (*, P<0.05 vs. the control group; #, P<0.05 vs. the HGF and control groups; **, P<0.05 vs. the HGF, HGF+LY294002 or HGF+SB202190 groups).
Discussion
It has been demonstrated that HGF could activate several biological functions in cancer cells including proliferation, migration, invasion, angiogenesis and morphogenesis through the tyrosine phosphorylation of its receptor c-Met [2,12]. HGF and/or its receptor c-Met are often dysregulated in breast cancer and other tumors, which is associated with poor prognosis [2,3,12]. Nevertheless, the exact mechanism of the HGF-induced increase in cell invasiveness is not fully understood. In previous studies, it has been reported that HGF increases the invasiveness of MBA-MB-231 cells in vitro [13-15]. In this report, we provide evidence that the HGF-stimulated increase in the invasiveness of breast cancer cells could be mediated via the up-regulation of COX2, and we identify the intracellular signal pathways that are potentially involved in this process.
We confirmed that the HGF receptor c-Met was positively expressed, and HGF could stimulate the production of phosphorylated c-Met (p-c-Met) and the invasion of both breast cancer cell lines in a dose-dependent manner (Figure 1). These findings have revealed that the HGF-induced invasion of breast cancer cells could be mediated through the HGF/c-Met pathway.
There are two classical isoenzymes of COX. COX1 is constitutively expressed in many cell types, whereas the inducible isoenzyme COX2 is expressed only in response to certain stimuli such as cytokines and various growth factors [16]. COX2 plays a role in the development of many cancers including breast cancer, which makes it a potential target for breast cancer prevention and therapy [17]. Singh et al. [18] demonstrated that COX2 expression in MCF-7 breast cancer cells stimulated genomic instability and doxorubicin resistance. Robertson et al. [19] found that the COX2 enzyme was abundant in invasive breast cancer. It was reported that IL-1β and TNF-α could up-regulate COX2 in a thyroid epithelial cell line and that COX2 was induced by pro-inflammatory cytokines via NF-κB transcription [20-22]. Evidence was provided demonstrating that COX2 expression could be induced by HGF in non-small cell lung cancer cells [6].
In this study, we investigated the effects of HGF on invasiveness and COX2 expression in the MDA-MB-231 and MCF-7 breast cancer cell lines. It was found that HGF increased the invasion of both breast cancer cell lines in a dose-dependent manner. It was also demonstrated that the levels of COX2 were up-regulated in MDA-MB-231 and MCF-7 cells after HGF treatment in a dose- and time-dependent manner. To further examine the role of COX2 in HGF-induced cell invasion, breast cancer cells were transfected a COX2-silencing vector and observed for their response to HGF. Our findings demonstrate that HGF-induced MDA-MB-231 and MCF-7 cell invasion was partially inhibited by COX2 gene silencing. This finding further supported the hypothesis that the HGF-induced up-regulation of COX2 expression contributes to the HGF-stimulated invasion of breast cancer cells.
It is known that the primary component of the extracellular matrix (ECM), a meshwork-like substance found within the extracellular space that is in association with the basement membranes of cell surfaces, includes collagen, glucosamine, glycoprotein, and proteoglycan [23]. Tumor cells can generate proteolytic enzymes that act through autocrine or paracrine mechanisms to degrade the ECM so that invasion occurs [24]. MMP-9, a member of matrix metalloproteinases (MMPs) family, is considered to play an important role in tumor cell invasion [25]. Our results revealed that HGF could up-regulate MMP-9 expression and induce cell invasion, and targeting the COX2 gene by silencing could down-regulate MMP-9 expression and reduce the invasiveness of breast cancer cell lines (Figures 3, 4). These findings indicate that MMP-9 is a downstream effector mediating the increased invasion of breast cancer cells induced by HGF-up-regulated COX2 expression. Taken together, these results have suggested that HGF might induce the invasion of breast cancer cells through a COX2-dependent pathway, and HGF mediated COX2 up-regulation could lead to increased MMP-9 expression and the subsequent invasion of MDA-MB-231 and MCF-7 cells.
It was reported that various signaling pathways are involved in the HGF-induced regulation of COX2 expression in different tumors. In colorectal cancer cells, the ERK and PI3K/Akt signaling pathways were required for HGF-induced COX2 protein up-regulation [26]. Siegfried et al. found that the MEK1/2 and p38 pathways and not the PI3K/Akt pathway participate in the HGF-mediated induction of COX2 in non-small cell lung cancer [6]. Furthermore, HGF could induce MMP-9 expression and the subsequent invasion of SKOV-3 ovarian cancer cells through the MAPK signaling pathway [10]. Evidence was provided that the HGF/c-Met/stat3 signaling pathway is involved in the invasion of skin tumor cells [27]. Moreover, it was reported that HGF promoted MDA-MB-231 cell invasion via a PI3K dependent mechanism [28].
We next investigated which signaling pathway is involved in the HGF-driven regulation of COX2 expression. Our results demonstrated that HGF induced the expression of COX2, whereas LY294002 (an inhibitor of PI3k that blocks Akt activation) or SB202190 (an inhibitor of p38 MAPK) could partially inhibit the HGF-induced up-regulation of COX2, subsequent MMP-9 expression and cell invasion. Furthermore, in the presence of LY294002 and SB202190, the HGF-mediated COX2 up-regulation, MMP-9 expression and cell invasion were almost completed abolished. These results have indicated that the PI3K/Akt and p38 MAPK pathways are the predominant signaling pathways that mediate HGF-induced COX2 up-regulation in breast cancer cells, which further result in an increase in MMP-9 expression and cell invasion.
Given that there is strong evidence for the involvement of HGF/c-Met-induced COX2 up-regulation in the oncogenesis of human cancers including breast cancer, inhibitors targeting this pathway have been in research and development [29-32]. For example, NK4, an HGF mutant that is a competitive antagonist of HGF, showed obvious anticancer effects in various carcinomas [1,33,34]. In addition, it has been reported that the selective COX2 inhibitors nimesulide and celecoxib were studied as therapeutic agents against tumors [6-8], and the tumor c-Met level and circulating HGF levels could be used as criteria for COX2 inhibitor use for gastric cancer treatment [35]. All of the above reports and our current research have indicated that HGF/Met-induced COX2 up-regulation is a valuable, potential target for the prevention and treatment of cancer invasion and metastasis.
In conclusion, our study has demonstrated that HGF could up-regulate COX2 expression predominantly through the PI3K/Akt and p38 MAPK signaling pathways and result in the increased production of MMP-9 and the subsequent invasiveness of breast cancer cells.
Acknowledgements
This work was supported by Shenzhen Science and Technology plan project (JCYJ20160428135756806); Guangdong Medical Science Foundation (A2016543); National Natural Science Foundation of China (81601382).
Disclosure of conflict of interest
None.
References
- 1.Nakamura T, Sakai K, Nakamura T, Matsumoto K. Anti-cancer approach with NK4: bivalent action and mechanisms. Anticancer Agents Med Chem. 2010;10:36–46. doi: 10.2174/1871520611009010036. [DOI] [PubMed] [Google Scholar]
- 2.Wang S, Liu Q, Zhang Y, Liu K, Yu P, Liu K, Luan J, Duan H, Lu Z, Wang F, Wu E, Yagasaki K, Zhang G. Suppression of growth, migration and invasion of highly-metastatic human breast cancer cells by berbamine and its molecular mechanisms of action. Mol Cancer. 2009;8:81. doi: 10.1186/1476-4598-8-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang ZX, Lu BB, Yang JS, Wang KM, De W. Adenovirus-mediated siRNA targeting c-Met inhibits proliferation and invasion of small-cell lung cancer (SCLC) cells. J Surg Res. 2011;171:127–135. doi: 10.1016/j.jss.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 4.Giubellino A, Linehan WM, Bottaro DP. Targeting the Met signaling pathway in renal cancer. Expert Rev Anticancer Ther. 2009;9:785–793. doi: 10.1586/era.09.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jankowski K, Kucia M, Wysoczynski M, Reca R, Zhao D, Trzyna E, Trent J, Peiper S, Zembala M, Ratajczak J, Houghton P, Janowska-Wieczorek A, Ratajczak MZ. Both hepatocyte growth factor (HGF) and stromal-derived factor-1 regulate the metastatic behavior of human rhabdomyosarcoma cells, but only HGF enhances their resistance to radiochemotherapy. Cancer Res. 2003;63:7926–7935. [PubMed] [Google Scholar]
- 6.Siegfried JM, Gubish CT, Rothstein ME, Queiroz de Oliveira PE, Stabile LP. Signaling pathways involved in cyclooxygenase-2 induction by hepatocyte growth factor in non small-cell lung cancer. Mol Pharmacol. 2007;72:769–779. doi: 10.1124/mol.107.034215. [DOI] [PubMed] [Google Scholar]
- 7.Su B, Chen S. Lead optimization of COX-2 inhibitor nimesulide analogs to overcome aromatase inhibitor resistance in breast cancer cells. Bioorg Med Chem Lett. 2009;19:6733–6735. doi: 10.1016/j.bmcl.2009.09.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris RE, Beebe-Donk J, Doss H, Burr Doss D. Aspirin, ibuprofen, and other non-steroidal anti-inflammatory drugs in cancer prevention: a critical review of non-selective COX-2 blockade (review) Oncol Rep. 2005;13:559–583. [PubMed] [Google Scholar]
- 9.Day RM, Cioce V, Breckenridge D, Castagnino P, Bottaro DP. Differential signaling by alternative HGF isoforms through c-Met: activation of both MAP kinase and PI 3-kinase pathways is insufficient for mitogenesis. Oncogene. 1999;18:3399–3406. doi: 10.1038/sj.onc.1202683. [DOI] [PubMed] [Google Scholar]
- 10.Wei W, Kong B, Yang Q, Qu X. Hepatocyte growth factor enhances ovarian cancer cell invasion through downregulation of thrombospondin-1. Cancer Biol Ther. 2010;9:79–87. doi: 10.4161/cbt.9.2.10280. [DOI] [PubMed] [Google Scholar]
- 11.Marra M, Santini D, Meo G, Vincenzi B, Zappavigna S, Baldi A, Rosolowski M, Tonini G, Loeffler M, Lupu R, Addeo SR, Abbruzzese A, Budillon A, Caraglia M. Cyr61 downmodulation potentiates the anticancer effects of zoledronic acid in androgen-independent prostate cancer cells. Int J Cancer. 2009;125:2004–2013. doi: 10.1002/ijc.24648. [DOI] [PubMed] [Google Scholar]
- 12.Nakamura T, Mizuno S. The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc Jpn Acad Ser B Phys Biol Sci. 2010;86:588–610. doi: 10.2183/pjab.86.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu J, Shao S, Song Y, Zhao J, Dong Y, Gong L, Yang P. Hepatocyte growth factor induces invasion and migration of ovarian cancer cells by decreasing the expression of E-cadherin, beta-catenin, and caveolin-1. Anat Rec (Hoboken) 2010;293:1134–1139. doi: 10.1002/ar.21147. [DOI] [PubMed] [Google Scholar]
- 14.Nakashiro K, Hayashi Y, Oyasu R. Immunohistochemical expression of hepatocyte growth factor and c-Met/HGF receptor in benign and malignant human prostate tissue. Oncol Rep. 2003;10:1149–1153. [PubMed] [Google Scholar]
- 15.Lengyel E, Prechtel D, Resau JH, Gauger K, Welk A, Lindemann K, Salanti G, Richter T, Knudsen B, Vande Woude GF, Harbeck N. C-Met overexpression in node-positive breast cancer identifies patients with poor clinical outcome independent of Her2/neu. Int J Cancer. 2005;113:678–682. doi: 10.1002/ijc.20598. [DOI] [PubMed] [Google Scholar]
- 16.Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56:387–437. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- 17.Cronin-Fenton DP, Pedersen L, Lash TL, Friis S, Baron JA, Sorensen HT. Prescriptions for selective cyclooxygenase-2 inhibitors, non-selective non-steroidal anti-inflammatory drugs, and risk of breast cancer in a populationbased case-control study. Breast Cancer Res. 2010;12:R15. doi: 10.1186/bcr2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh B, Cook KR, Vincent L, Hall CS, Martin C, Lucci A. Role of COX-2 in tumorospheres derived from a breast cancer cell line. J Surg Res. 2011;168:e39–49. doi: 10.1016/j.jss.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robertson FM, Mallery SR, Bergdall-Costell VK, Cheng M, Pei P, Prosperi JR, Ferrari M. Cyclooxygenase-2 directly induces MCF-7 breast tumor cells to develop into exponentially growing, highly angiogenic and regionally invasive human ductal carcinoma xenografts. Anticancer Res. 2007;27:719–727. [PubMed] [Google Scholar]
- 20.Smith TJ, Jennings TA, Sciaky D, Cao HJ. Prostaglandin-endoperoxide H synthase-2 expression in human thyroid epithelium. Evidence for constitutive expression in vivo and in cultured KAT-50 cells. J Biol Chem. 1999;274:15622–15632. doi: 10.1074/jbc.274.22.15622. [DOI] [PubMed] [Google Scholar]
- 21.Berg J, Stocher M, Bogner S, Wolfl S, Pichler R, Stekel H. Inducible cyclooxygenase-2 gene expression in the human thyroid epithelial cell line Nthy-ori3-1. Inflamm Res. 2000;49:139–143. doi: 10.1007/PL00000204. [DOI] [PubMed] [Google Scholar]
- 22.Zeng Q, McCauley LK, Wang CY. Hepatocyte growth factor inhibits anoikis by induction of activator protein 1-dependent cyclooxygenase-2. Implication in head and neck squamous cell carcinoma progression. J Biol Chem. 2002;277:50137–50142. doi: 10.1074/jbc.M208952200. [DOI] [PubMed] [Google Scholar]
- 23.Li HW, Shan JX. Effects of hepatocyte growth factor/scatter factor on the invasion of colorectal cancer cells in vitro. World J Gastroenterol. 2005;11:3877–3881. doi: 10.3748/wjg.v11.i25.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tandon A, Sinha S. Structural insights into the binding of MMP9 inhibitors. Bioinformation. 2011;5:310–314. doi: 10.6026/97320630005310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jovanovic M, Stefanoska I, Radojcic L, Vicovac L. Interleukin-8 (CXCL8) stimulates trophoblast cell migration and invasion by increasing levels of matrix metalloproteinase (MMP)2 and MMP9 and integrins alpha5 and beta1. Reproduction. 2010;139:789–798. doi: 10.1530/REP-09-0341. [DOI] [PubMed] [Google Scholar]
- 26.Moore AE, Greenhough A, Roberts HR, Hicks DJ, Patsos HA, Williams AC, Paraskeva C. HGF/Met signalling promotes PGE(2) biogenesis via regulation of COX-2 and 15-PGDH expression in colorectal cancer cells. Carcinogenesis. 2009;30:1796–1804. doi: 10.1093/carcin/bgp183. [DOI] [PubMed] [Google Scholar]
- 27.Syed ZA, Yin W, Hughes K, Gill JN, Shi R, Clifford JL. HGF/c-met/Stat3 signaling during skin tumor cell invasion: indications for a positive feedback loop. BMC Cancer. 2011;11:180. doi: 10.1186/1471-2407-11-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trusolino L, Cavassa S, Angelini P, Ando M, Bertotti A, Comoglio PM, Boccaccio C. HGF/scatter factor selectively promotes cell invasion by increasing integrin avidity. Faseb J. 2000;14:1629–1640. doi: 10.1096/fj.14.11.1629. [DOI] [PubMed] [Google Scholar]
- 29.Yue D, Wang Y, Ma P, Li YY, Chen H, Wang P, Ren CS. Effects of transferred NK4 gene on proliferation, migration, invasion and apoptosis of human prostate cancer DU145 cells. Asian J Androl. 2010;12:381–389. doi: 10.1038/aja.2010.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saimura M, Nagai E, Mizumoto K, Maehara N, Minamishima YA, Katano M, Matsumoto K, Nakamura T, Tanaka M. Tumor suppression through angiogenesis inhibition by SUIT-2 pancreatic cancer cells genetically engineered to secrete NK4. Clin Cancer Res. 2002;8:3243–3249. [PubMed] [Google Scholar]
- 31.Kuba K, Matsumoto K, Date K, Shimura H, Tanaka M, Nakamura T. HGF/NK4, a fourkringle antagonist of hepatocyte growth factor, is an angiogenesis inhibitor that suppresses tumor growth and metastasis in mice. Cancer Res. 2000;60:6737–6743. [PubMed] [Google Scholar]
- 32.Matsumoto K, Nakamura T. NK4 gene therapy targeting HGF-Met and angiogenesis. Front Biosci. 2008;13:1943–1951. doi: 10.2741/2813. [DOI] [PubMed] [Google Scholar]
- 33.Ueda K, Iwahashi M, Matsuura I, Nakamori M, Nakamura M, Ojima T, Naka T, Ishida K, Matsumoto K, Nakamura T, Yamaue H. Adenoviral-mediated gene transduction of the hepatocyte growth factor (HGF) antagonist, NK4, suppresses peritoneal metastases of gastric cancer in nude mice. Eur J Cancer. 2004;40:2135–2142. doi: 10.1016/j.ejca.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki Y, Sakai K, Ueki J, Xu Q, Nakamura T, Shimada H, Nakamura T, Matsumoto K. Inhibition of Met/HGF receptor and angiogenesis by NK4 leads to suppression of tumor growth and migration in malignant pleural mesothelioma. Int J Cancer. 2010;127:1948–1957. doi: 10.1002/ijc.25197. [DOI] [PubMed] [Google Scholar]
- 35.Chen JH, Wu CW, Kao HL, Chang HM, Li AF, Liu TY, Chi CW. Effects of COX-2 inhibitor on growth of human gastric cancer cells and its relation to hepatocyte growth factor. Cancer Lett. 2006;239:263–270. doi: 10.1016/j.canlet.2005.08.026. [DOI] [PubMed] [Google Scholar]