

Abstract
T7 RNA polymerase presents a very simple model system for the study of fundamental aspects of transcription. Some time ago it was observed that in the presence of only GTP as a substrate, on a template encoding the initial sequence GGGA…, T7 RNA polymerase will synthesize a ‘ladder’ of poly-G RNA products. At each step, the ratio of elongation to product release is consistently ∼0.75 until the RNA reaches a length of ∼13–14 nt, at which point this ratio drops precipitously. One model to explain this drop in complex stability suggests that the nascent RNA may be structurally hindered by the protein; the RNA may be exiting via a pathway not taken by normally synthesized RNA and therefore becomes sterically destabilized. The fact that the length of RNA at which this occurs is close to the length at which the transition to a stably elongating complex occurs might have led to other mechanistic proposals. Here we show instead that elongation falls off due to the cooperative formation of structure in the nascent RNA, most likely an intramolecular G-quartet structure. Replacement of GTP by 7-deaza-GTP completely abolishes this transition and G-ladder synthesis continues with a constant efficiency of elongation beyond the limit of detection. The polymerase–DNA complex creates no barrier to the growth of the nascent (slippage) RNA, rather termination is similar to that which occurs in rho-independent termination.
INTRODUCTION
T7 RNA polymerase possesses all of the fundamental features of an RNA polymerase. It initiates transcription at a unique position in its promoter, it is characterized by an early less-processive abortive cycling phase, it then proceeds to a stably elongating complex and it terminates at specific sequences. As such, it serves as an ideal model system in which to study functional mechanisms of transcription.
Some time ago it was noted that in the presence of GTP as the sole substrate, on a template encoding GGGA…, T7 RNA polymerase synthesizes a ‘ladder’ of transcription products ranging from 2 to ∼14 nt in length, at a rate comparable to normal abortive synthesis (1). Addition of ATP eliminates the ladder and restores normal abortive synthesis, indicating that this effect arises from the (imposed) inability of the system to proceed normally to incorporation at position +4. The ladder is substantially reduced if the template encodes a run of only two Gs and is eliminated if only one G is encoded at the start site. The latter result led to the conclusion that the synthesis occurs via a slippage mechanism, as illustrated below, and that a minimum of 2 bp is necessary in the slipped product in order to achieve efficient ladder synthesis (1). Slippage synthesis has also been shown to occur in Escherichia coli RNA polymerase, synthesizing long runs of U or A (dependent on template) (2–4). In those cases, an abrupt decrease in products of a certain length was not observed.
As shown in Figure 1, at each round in the cycle, either the RNA can slip back, re-exposing the templating C at position +3 and allowing another round of elongation, or the complex can release the product RNA. The complex is stalled at position +3 of the template throughout the entire cycle, and so presumably never loses contact with the upstream promoter elements (5). At each round in the cycle from a slippage product length of 4 to ∼13 , the ratio of elongation to product dissociation was determined to be ∼0.75, consistent with the otherwise uniform nature of the complex. However, at an RNA length of ∼14 nt, elongation efficiency drops off dramatically and most RNAs dissociate within a few bases.
Figure 1.

Minimal mechanism for slippage transcription. The efficiency of elongation at each step is the ratio of the elongation velocity to the sum of the velocities of the dissociation steps.
The observation of a sharp fall-off in elongation (and/or increase in dissociation) associated with an RNA length near 14 bases is surprising. The length is comparable to the lengths at which RNA polymerase converts from a less processive abortive cycling phase to the more processive phase characteristic of elongation, typically 8–10 bases (1,6). This latter transition has been attributed to a variety of effects. One model suggests that once the product RNA has reached a minimum length, it can interact with an RNA binding site on the enzyme (possibly amino acids 172–180), providing extra stability to the complex (6). More recent studies have reported that a polymerase mutated in a different region (amino acid 148), but which has also lost the RNA-binding ability, nevertheless produces the G-ladder exactly as does the wild-type enzyme (7). In any case, the behavior of the G-ladder slippage products is opposite to the behavior predicted by a model in which the G-ladder interacts favorably with an RNA binding site, the complex apparently becomes less stable. This might suggest that slippage RNA follows a different path out of the enzyme.
Another model for abortive cycling associates abortive release with a build-up of stress in the system as the active site translocates along the DNA, while the enzyme retains promoter contact (8–11). This model has received support from the recent crystal structure of a paused ternary complex in the T7 RNA polymerase system (5). This structure shows that a complex containing a GGG trinucleotide, with a fourth non-hydrolyzable NTP, retains promoter contacts almost indistinguishable from the pre-initiation complex (12). The structure shows that the DNA may accumulate, or ‘scrunch’, to allow movement of the DNA relative to the active site. In this refined model, the accumulation of more and more DNA within a pocket in the enzyme ultimately leads to release of the upstream promoter contacts, and to a transition to the non-promoter bound elongation complex (5). This model would also not predict the behavior seen with the slippage products; throughout the ladder synthesis, the DNA only moved three bases relative to the active site, equivalent to the crystallized ternary complex.
Since the exiting of RNA from the active site without translocation along the DNA is an unnatural process, perhaps the growing ladder RNA exits such that it accumulates as does the proposed scrunched DNA in normal RNA synthesis, rather than following the normal path of nascent RNA (see above). This would predict that a maximal length of RNA would be tolerated before the complex becomes unstable and dissociates, exactly the behavior observed.
Finally, a very different mechanism to explain the abrupt termination of slippage synthesis at position +14 is that the RNA cooperatively adopts a structure that interferes with the stable interaction of the RNA near the active site (RNA–protein and/or RNA–DNA contacts), much as formation of a hairpin in the RNA is thought to facilitate rho-independent termination (13–15). Indeed, runs of G in both RNA and DNA are known to form G-quartet structures in solution (16,17). The requirement for the incorporation of 13 or 14 guanosines into the RNA before the onset of termination suggests the cooperative formation of structure, possibly analogous to the structures of the thrombin-binding DNA aptamer (18,19) or of the flavin-binding RNA aptamer (20). Formation of this structure would disrupt the ternary complex, removing the RNA from the active site.
MATERIALS AND METHODS
RNA polymerase
T7 RNA polymerase was prepared from E.coli strain BL21 carrying the overproducing plasmid pAR1219 (kindly supplied by F. W. Studier, Brookhaven National Laboratory, NY), which contains the T7 RNA polymerase gene under the inducible control of the lacUV5 promoter. The enzyme was purified and concentration determined (ɛ280 = 1.4 × 105 M–1 cm–1) as described previously (21). Purity of the enzyme was verified by SDS–PAGE.
Oligonucleotides
Oligonucleotides were synthesized by the phosphoramidite method on an Applied Biosystems Expedite 8909 DNA synthesizer. Single strands from a 1 µmol scale synthesis were purified trityl-on using an Amberchrom CG-161cd reverse phase resin (TosoHaas Inc.) as described (22). Purity of the oligonucleotides was confirmed by denaturing (urea) gel electrophoresis of 5′-end-labeled single strands.
Double-stranded DNA was made by heating complementary single strands to 90°C and allowing the resulting mixture to cool to room temperature over 2 h.
Kinetic assays
Steady-state assays of slippage transcription were carried out in a total volume of 20 µl at 37°C. The resulting mixture contained 30 mM HEPES pH 7.8, 15 mM magnesium acetate, 25 mM potassium glutamate, 0.25 mM EDTA, 0.05% (v/v) Tween-20 (Calbiochem, protein grade), 0.4 mM GTP or 7-deaza-GTP, and <0.06 µM [α-32P]GTP (NEN Life Sciences) as a label, 0.2 µM DNA promoter and 0.2 µM T7 RNA polymerase. Reactions were incubated at 37°C for 10 min and stopped by addition of a 95% formamide, 20 mM EDTA pH 7.8 gel-loading buffer. The 3.0 µl aliquots were loaded onto a 7 M urea/18% polyacrylamide sequencing gel. After 2.5 h electrophoresis at 2000 V/50 W, gels were dried and quantified using a Molecular Dynamics Storm 840 PhosphorImager. The percent fall-off was calculated for each band by taking the ratio of intensity of the band In corrected for the number of radioactive labels incorporated (In/n), dividing by the sum of corrected intensities of all the bands length n and longer, and multiplying by 100%.

Quench-flow experiments were performed on a KinTek RQF-3 quench-flow apparatus under the same reaction conditions as in steady-state assays, except that the concentrations of DNA promoter and T7 RNA polymerase were 3.0 and 5.0 µM, respectively.
We note that the enzyme might show a small preference for the incorporation of G versus 7-deaza-G, so that quantitative comparisons between lanes in Figure 3 should be done with caution. Comparisons of band intensities within a lane should not be influenced by this (but see below regarding intensities of very short RNA products).
Figure 3.
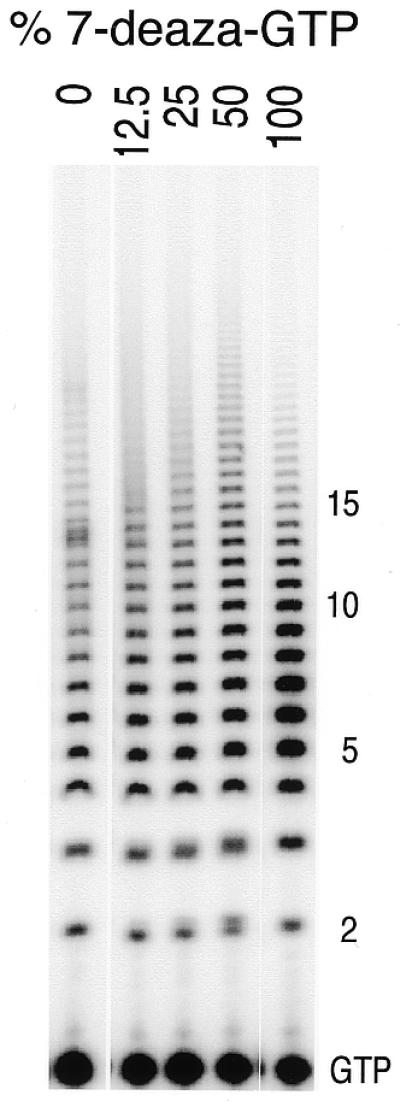
Effect of different fractional concentrations of 7-deaza-GTP on the termination of G-ladder synthesis. Conditions were as in Figure 2, except that varying amounts of 7-deaza-GTP replace GTP. In each case, the total concentration of nucleoside triphosphate (GTP plus 7-deaza-GTP) was 400 µM. Trace amounts (<0.06 µM) of [α-32P]GTP were present in all lanes for detection.
RESULTS AND DISCUSSION
The observation that under conditions which limit the next nucleotide on a template encoding an initial sequence of GGGX, T7 RNA polymerase synthesizes an array of products longer than 3 bases is understandable in terms the mechanistic model in Figure 1 (1). The uniform ratio of slippage incorporation to product release is similarly expected, given a cycling 2–3 base heteroduplex. The observation that this ratio decreases abruptly and dramatically at an RNA length of ∼14 nt is not at all expected in terms of that simple model. The fact that the transition from an abortive cycling to a stably elongating complex occurs near this length suggested that the transitions might reflect a common mechanism. In particular, since in slippage synthesis the enzyme has not translocated more than 3 bases from the promoter, this might suggest that the transition to a stably elongation complex is related not to distance from the promoter (along the DNA), but to the length of the nascent RNA.
An alternative explanation is that the abrupt increase in termination of slippage synthesis arises from formation of structure in the nascent RNA. Although poly-G RNA cannot form a hairpin, as occurs in rho-independent termination, it can form structures based on the G-quartet. Such structures could arise either unimolecularly (as for hairpin formation), or might arise by intermolecular interactions with free poly-G RNAs.
Incorporation of 7-deaza-G abolishes abrupt termination near +14
The formation of G-quartet structures involves hydrogen bonding to the N7 position of each guanine residue, as shown in Figure 2B. In order to test the proposal that the abrupt termination of G-ladder slippage near position +14 occurs as a result of the formation of G-quartet-like structures, we have carried out transcription reactions replacing GTP by 7-deaza-GTP, which replaces the nitrogen and its lone pair at position 7 by a CH group. The 7-position is thus completely incapable of serving as a hydrogen bond acceptor, such that quartet structures should lose four of the eight stabilizing hydrogen bonds per quartet and become unstable. It has been shown previously that poly(7-deazaguanylic acid) does not form non-canonical multi-stranded structures in solution (23).
Figure 2.

Comparison of the effects of 7-deaza-GTP on the synthesis of ‘G–-adder’ slippage products. (A) RNA products in a 10 min reaction at 37°C, containing 0.2 µM promoter DNA, 0.2 µM T7 RNA polymerase and 400 µM GTP (left) or 7-deaza-GTP (right), in a buffer of 20 mM HEPES pH 7.8, 25 mM potassium glutamate, 0.025% Tween-20, 2.5 mM Tris, 15 mM Mg(OAc)2, 0.25 mM EDTA. Both reactions contained trace amounts (<0.06 µM) of [α-32P]GTP for detection. The part of the gel showing the ‘regular’ G-ladder has been overexposed to make the difference between the two more apparent. (B) Structures of a G-quartet and of guanine and its 7-deaza analog. Note that replacement of guanine in the quartet structure by 7-deaza-guanine destroys the stability of the quartet. (C) Plots of percent fall-off as a function of the transcript length.
The results presented in Figure 2 show that incorporation of the 7-deaza analog completely destroys the transition near position +14. Moreover, the RNA ladder continues to follow a pattern of 70% elongation/30% fall-off for as far as can be reliably detected (to an RNA length of more than 25 bases). This result demonstrates that structural interactions involving the 7-nitrogen lone pair on guanine are key to the increased termination at an RNA length of ∼14 bp, fully consistent with the G-quartet model for termination. Other models involving transitions triggered by a certain length of RNA should be abandoned.
The formation of the G-quartet based structures in both RNA and DNA is favored by the presence of monovalent ions of appropriate size, such as K+ and Na+ (the optimal transcription buffer contains 25 mM K+), but not by divalent metal ions and monovalent ions of inappropriate size, such as Li+ (24,25). With this in mind, we attempted to completely replace Na+ and K+ ions by Li+ in our reaction buffers. Unfortunately, the buffer exchange led to almost complete elimination of initiation in the system, an interesting result in its own right.
Structures based on G-quartets are expected to form with high cooperativity, and the incorporation of only a few 7-deaza-guanine nucleotides per RNA should be sufficient to weaken the RNA structure substantially, effectively abolishing the transition. The results presented in Figure 3 show that almost complete abolition of the transition near position +14 occurs for a reaction mixture with ratios of 7-deaza-GTP to GTP as low as 1:3. Doping of the RNA with 12.5% 7-deaza-G does not destroy the transition, consistent with the retention of a stable quartet structure at this low level of doping (about 1 out of 8). The energetics/kinetics of formation of the structured RNA must balance against that of processive elongation.
Evidence that the RNA does not interact stably with the protein
Previous studies using a self-cleaving RNA transcript showed that during normal elongation the nascent RNA must reach a length of at least 13 nt before the RNA can be self-cleaved, and concluded that the RNA is sequestered (or in a heteroduplex) at least 10 bases distant from its 5′-end (26). Since recent evidence points to an ∼8 bp heteroduplex, the RNA ought to be protected for an additional 2 bases (27–29). Does the RNA coming from a slippage complex interact stably with the protein, or does it clear the protein immediately past the cycling 2–3 base heteroduplex?
To date, the minimum length of polynucleotide that has been shown to unimolecularly form a stable quartet structure appears to be ∼14–15 nt, two stacked tetramers with three connections of two bases each (18,19). Multimeric structures, in contrast, could form with much less than 14 nt exposed to solution. In the steady state experiments above, we cannot distinguish between the formation of monomeric or multimeric RNA structures as the cause of transcription termination.
If the structured RNA forms via a cooperative intermolecular mechanism, then at very short times, when relatively low concentrations of free oligomeric RNAs are present, the transcribing complex should be better able to proceed beyond RNA lengths of 14–15 nt. However, measurements of the early time course of slippage synthesis, shown in Figure 4A, demonstrate that the abrupt termination of slippage transcription occurs as soon as products of that length can be detected (5–10 s). The quantification of individual bands in Figure 4A, shown in Figure 4B, reveals quantitatively that by 10 s, the concentration of 14mer (free plus bound) is only 0.06 µM, while the concentration of the most abundant product larger than dinucleotide (4mer) is ∼4.2 µM. At this point, the total molar concentration of oligomers 3–14 nt in length is only 14 µM, and a fraction of these (up to 3 µM) is expected to be bound in enzyme–DNA complexes. Thus, it seems unlikely that the disruptive structure forms intermolecularly with short RNA products. It seems most likely that the disruptive structure forms intramolecularly. Indeed, recent studies have shown that G-rich oligonucleotides longer than 15 nt in length, and which have two or more clusters of three or more contiguous Gs, readily associate intramolecularly but not intermolecularly (30).
Figure 4.

(A) Time course of slippage synthesis with GTP as substrate. Conditions were as in Figure 2, except that in order to detect RNA at low turnover, concentrations of polymerase and promoter DNA were 5.0 and 3.0 µM, respectively. (B) Product concentrations at limited turnover. Data correspond to the reaction quenched at 10 s.
These results suggest that the abrupt termination beyond position +14 at least partially mimics normal (rho-independent) termination of transcription (13–15,31,32). In this case, formation of a G-quartet structure might serve the same function as the formation of an RNA hairpin in the simplest models of termination (but in the case of slippage, the heteroduplex anchoring the RNA to the complex can be no longer than 3 bp). The fact that cooperative quartet-like structures form readily suggests that the enzyme does not sterically hinder the RNA beyond this short heteroduplex; the RNA is not exiting via a well-protected channel.
Product recycling
Interestingly, Figure 2 shows that the apparent percent fall-off for 2mer and 3mer drops dramatically with 7-deaza-GTP as substrate. This is, in fact, misleading. This effect almost certainly arises from the fact that although 7-deaza-GTP is a good substrate for elongation, it is a poor substrate for initiation (incorporation at position +1) (I.Kuzmine, P.A.Gottlieb and C.T.Martin, unpublished results). In contrast, T7 RNA polymerase initiates very well with the dinucleotide pppGpG (and with the trimer as well), the Km for this substrate being near 5 µM (33). Consequently, in the presence of 7-deaza-GTP, the enzyme will initially use the analog as an initiating substrate, but as the concentrations of dimer and trimer build in the reaction and as the GTP pool is depleted, these products will be consumed in producing longer polymers. The result is a reduction in the amounts of the shorter products, leading to an apparent reduction in the percent fall-off of short products. This effect is more dramatic with 7-deaza-GTP as substrate because slippage transcription appears to be more rapid (and the products longer) in the presence of 7-deaza-GTP.
CONCLUSIONS
These results show clearly that the abrupt decrease in slippage synthesis, which occurs near 14 nt, arises from the formation of G-quartet-like structure in the nascent RNA. The results indicate that this structure is unimolecular, so that termination occurs in a manner analogous to rho-independent termination of transcription. It would seem that the close correspondence between this length and the length of RNA at which the complex, during normal transcription, transitions to a stably elongating complex is fortuitous. Finally, the results suggest that the nascent RNA does not interact strongly with the protein, as is expected for RNA exiting a (longer) heteroduplex in stably elongating complexes.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by grants 1R01GM55002 from the National Institutes of Health and MCB-9630447 from the National Science Foundation
References
- 1.Martin C.T., Muller,D.K. and Coleman,J.E. (1988) Processivity in early stages of transcription by T7 RNA polymerase. Biochemistry, 27, 3966–3974. [DOI] [PubMed] [Google Scholar]
- 2.Harley C.B., Lawrie,J., Boyer,H.W. and Hedgpeth,J. (1990) Reiterative copying by E.coli RNA polymerase during transcription initiation of mutant pBR322 tet promoters. Nucleic Acids Res., 18, 547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jacques J.P. and Susskind,M.M. (1990) Pseudo-templated transcription by Escherichia coli RNA polymerase at a mutant promoter. Genes Dev., 4, 1801–1810. [DOI] [PubMed] [Google Scholar]
- 4.Jin D.J. and Turnbough,C.L.,Jr (1994) An Escherichia coli RNA polymerase defective in transcription due to its overproduction of abortive initiation products. J. Mol. Biol., 236, 72–80. [DOI] [PubMed] [Google Scholar]
- 5.Cheetham G.M. and Steitz,T.A. (1999) Structure of a transcribing T7 RNA polymerase initiation complex. Science, 286, 2305–2309. [DOI] [PubMed] [Google Scholar]
- 6.Muller D.K., Martin,C.T. and Coleman,J.E. (1988) Processivity of proteolytically modified forms of T7 RNA polymerase. Biochemistry, 27, 5763–5771. [DOI] [PubMed] [Google Scholar]
- 7.He B., Rong,M., Durbin,R.K. and McAllister,W.T. (1997) A mutant T7 RNA polymerase that is defective in RNA binding and blocked in the early stages of transcription. J. Mol. Biol., 265, 275–288. [DOI] [PubMed] [Google Scholar]
- 8.Carpousis A.J. and Gralla,J.D. (1980) Cycling of ribonucleic acid polymerase to produce oligonucleotides during initiation in vitro at the lac UV5 promoter. Biochemistry, 19, 3245–3253. [DOI] [PubMed] [Google Scholar]
- 9.Carpousis A.J. and Gralla,J.D. (1985) Interaction of RNA polymerase with lacUV5 promoter DNA during mRNA initiation and elongation. Footprinting, methylation and rifampicin-sensitivity changes accompanying transcription initiation. J. Mol. Biol., 183, 165–177. [DOI] [PubMed] [Google Scholar]
- 10.Straney D.C. and Crothers,D.M. (1987) A stressed intermediate in the formation of stably initiated RNA chains at the Escherichia coli lac UV5 promoter. J. Mol. Biol., 193, 267–278. [DOI] [PubMed] [Google Scholar]
- 11.Krummel B. and Chamberlin,M.J. (1989) RNA chain initiation by Escherichia coli RNA polymerase. Structural transitions of the enzyme in early ternary complexes. Biochemistry, 28, 7829–7842. [DOI] [PubMed] [Google Scholar]
- 12.Cheetham G.M., Jeruzalmi,D. and Steitz,T.A. (1998) Transcription regulation, initiation and ‘DNA scrunching’ by T7 RNA polymerase. Cold Spring Harb. Symp. Quant. Biol., 63, 263–267. [DOI] [PubMed] [Google Scholar]
- 13.Adhya S., Sarkar,P., Valenzuela,D. and Maitra,U. (1979) Termination of transcription by Escherichia coli RNA polymerase: influence of secondary structure of RNA transcripts on rho-independent and rho-dependent termination. Proc. Natl Acad. Sci. USA, 76, 1613–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reynolds R., Bermudez-Cruz,R.M. and Chamberlin,M.J. (1992). Parameters affecting transcription termination by Escherichia coli RNA polymerase. I. Analysis of 13 rho-independent terminators. J. Mol. Biol., 224, 31–51. [DOI] [PubMed] [Google Scholar]
- 15.Lyakhov D.L., He,B., Zhang,X., Studier,F.W., Dunn,J.J. and McAllister,W.T. (1998) Pausing and termination by bacteriophage T7 RNA polymerase. J. Mol. Biol., 280, 201–213. [DOI] [PubMed] [Google Scholar]
- 16.Sen D. and Gilbert,W. (1990) A sodium–potassium switch in the formation of four-stranded G4-DNA. Nature, 344, 410–414. [DOI] [PubMed] [Google Scholar]
- 17.Sen D. and Gilbert,W. (1992) Guanine quartet structures. Methods Enzymol., 211, 191–199. [DOI] [PubMed] [Google Scholar]
- 18.Macaya R.F., Schultze,P., Smith,F.W., Roe,J.A. and Feigon,J. (1993) Thrombin-binding DNA aptamer forms a unimolecular quadruplex structure in solution. Proc. Natl Acad. Sci. USA, 90, 3745–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schultze P., Macaya,R.F. and Feigon,J. (1994) Three-dimensional solution structure of the thrombin-binding DNA aptamer d(GGTTGGTGTGGTTGG). J. Mol. Biol., 235, 1532–1547. [DOI] [PubMed] [Google Scholar]
- 20.Lauhon C.T. and Szostak,J.W. (1995) RNA aptamers that bind flavin and nicotinamide redox cofactors. J. Am. Chem. Soc., 117, 1246–1257. [DOI] [PubMed] [Google Scholar]
- 21.King G.C., Martin,C.T., Pham,T.T. and Coleman,J.E. (1986) Transcription by T7 RNA polymerase is not zinc-dependent and is abolished on amidomethylation of cysteine-347. Biochemistry, 25, 36–40. [DOI] [PubMed] [Google Scholar]
- 22.Schick C. and Martin,C.T. (1993) Identification of specific contacts in T3 RNA polymerase-promoter interactions: kinetic analysis using small synthetic promoters. Biochemistry, 32, 4275–4280. [DOI] [PubMed] [Google Scholar]
- 23.Seela F., Tran-Thi,Q.H. and Franzen,D. (1982) Poly(7-deazaguanylic acid), the homopolynucleotide of the parent nucleoside of queuosine. Biochemistry, 21, 4338–4343. [DOI] [PubMed] [Google Scholar]
- 24.Kim J., Cheong,C. and Moore,P.B. (1991) Tetramerization of an RNA oligonucleotide containing a GGGG sequence. Nature, 351, 331–332. [DOI] [PubMed] [Google Scholar]
- 25.Lu M., Guo,Q. and Kallenbach,N.R. (1993) Thermodynamics of G-tetraplex formation by telomeric DNAs. Biochemistry, 32, 598–601. [DOI] [PubMed] [Google Scholar]
- 26.Tyagarajan K., Monforte,J.A. and Hearst,J.E. (1991) RNA folding during transcription by T7 RNA polymerase analyzed using the self-cleaving transcript assay. Biochemistry, 30, 10920–10924. [DOI] [PubMed] [Google Scholar]
- 27.Huang J. and Sousa,R. (2000) T7 RNA polymerase elongation complex structure and movement. J. Mol. Biol., 303, 347–358. [DOI] [PubMed] [Google Scholar]
- 28.Temiakov D., Mentesana,P.E., Ma,K., Mustaev,A., Borukhov,S. and McAllister,W.T. (2000) The specificity loop of T7 RNA polymerase interacts first with the promoter and then with the elongating transcript, suggesting a mechanism for promoter clearance. Proc. Natl Acad. Sci. USA, 97, 14109–14114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen H. and Kang,C. (2001) Two-site contact of elongating transcripts to phage T7 RNA polymerase at C-terminal regions. J. Biol. Chem., 276, 4080–4084. [DOI] [PubMed] [Google Scholar]
- 30.Cheng A.J. and Van Dyke,M.W. (1997) Oligodeoxyribonucleotide length and sequence effects on intramolecular and intermolecular G-quartet formation. Gene, 197, 253–260. [DOI] [PubMed] [Google Scholar]
- 31.Macdonald L.E., Zhou,Y. and McAllister,W.T. (1993) Termination and slippage by bacteriophage T7 RNA polymerase. J. Mol. Biol., 232, 1030–1047. [DOI] [PubMed] [Google Scholar]
- 32.Macdonald L., Durbin,R., Dunn,J. and McAllister,W. (1994) Characterization of two types of termination signal for bacteriophage T7 RNA polymerase. J. Mol. Biol., 238, 145–158. [DOI] [PubMed] [Google Scholar]
- 33.Kuzmine I. and Martin,C.T. (2001) Pre-steady-state kinetics of initiation of transcription by T7 RNA polymerase: a new kinetic model. J. Mol. Biol., 305, 559–566. [DOI] [PubMed] [Google Scholar]