


Abstract
N.BstNBI is a nicking endonuclease that recognizes the sequence GAGTC and nicks the top strand preferentially. The Type IIs restriction endonucleases PleI and MlyI also recognize GAGTC, but cleave both DNA strands. Cloning and sequencing the genes encoding each of these three endonucleases discloses significant sequence similarities. Mutagenesis studies reveal a conserved set of catalytic residues among the three endonucleases, suggesting that they are closely related to each other. Furthermore, PleI and MlyI contain a single active site for DNA cleavage. The results from cleavage assays show that the reactions catalyzed by PleI and MlyI are sequential two step processes. The double-stranded DNA is first nicked on one DNA strand and then further cleaved on the second strand to form linear DNA. Gel filtration analysis shows that MlyI dimerizes in the presence of a cognate DNA and Ca2+ whereas N.BstNBI remains a monomer, implicating dimerization as a requisite for the second strand cleavage. We suggest that N.BstNBI, MlyI and PleI diverged from a common ancestor and propose that N.BstNBI differs from MlyI and PleI in having an extremely limited second strand cleavage activity, resulting in a site-specific nicking endonuclease.
INTRODUCTION
Restriction endonucleases are enzymes that recognize and cleave specific DNA sequences. Usually there is a corresponding DNA methyltransferase that methylates and thereby protects the endogenous host DNA from digestion by the accompanying restriction endonuclease. More than 3000 restriction endonucleases with over 200 different specificities have been isolated (1), and are classified into three groups: Types I, II and III. Type II and Type IIs restriction enzymes cleave DNA at a specific position, and therefore are useful in genetic engineering and molecular cloning (2). Type II enzymes, such as EcoRI and EcoRV, recognize palindromic sequences and cleave both strands symmetrically within their recognition sequences. Type IIs R–M systems differ from the Type II systems in that they recognize an asymmetric sequence, and the Type IIs endonucleases cleave DNA on one side of the recognition sequence at a defined distance usually >20 bp (3).
FokI is the only well-characterized Type IIs endonuclease. Proteolytic studies showed that FokI contains two separate domains, one for DNA recognition and one for cleavage (4). Based on the two-domain theory, several FokI-based chimeric endonucleases have been constructed by linking the DNA binding domains of other proteins to the FokI cleavage domain, and these chimeric endonucleases can introduce double strand breaks into DNA near the recognition sequence specified by the DNA binding protein (5). FokI has been shown to exist as a monomer in solution (6) and to bind DNA as a monomer (7). Researchers speculated that FokI has two catalytic centers and that each cleaves one DNA strand. However, mutational analysis (4) and the three-dimensional structure of FokI with bound DNA shows that there is only one catalytic center (8). How does monomeric FokI cleave both DNA strands using a single catalytic center? A more recent crystal structure of FokI reveals a dimerization interface between the catalytic domains (9), and biochemical data strongly suggest that dimerization of FokI endonuclease is required for DNA cleavage (10). It is proposed that FokI binds to DNA as a monomer and dimerizes with a second FokI molecule when it reaches its recognition sequence (10). Double-stranded cleavage occurs following dimerization.
There are some proteins that cleave only one DNA strand and therefore introduce a nick into DNA. Most of those proteins are involved in DNA replication, DNA repair and other aspects of DNA metabolism (11). For example, the gpII protein of bacteriophage fI recognizes and binds to the origin of replication. It introduces a nick in the plus strand, which initiates rolling circle replication, and is also involved in circularizing the plus strand to generate single-stranded circular phage DNA (12,13). A second example is the MutH protein, involved in DNA mismatch repair in Escherichia coli. MutH binds the sites of dam methylation (GATC), where it forms a protein complex with a nearby MutS, which has bound to a mismatch. The MutL protein facilitates this interaction and triggers single-stranded cleavage by MutH at the 5′ end of the unmethylated GATC site. The nick is then translated by an exonuclease to remove the mismatched nucleotide (14). Another example is E.coli endonuclease V, which recognizes and introduces nicks into DNA containing deoxyinosine, deoxyuridine and base mismatches (15).
Recently, we have purified a nicking endonuclease, N.BstNBI, from the thermophilic bacterium Bacillus stearothermophilus (16). N.BstNBI is an isoschizomer of N.BstSEI, which has not been cloned (17). Unlike gpII, MutH and endonuclease V, N.BstNBI behaves like a restriction endonuclease. It recognizes a simple asymmetric sequence, 5′-GAGTC-3′, and cleaves only one DNA strand, 4 bases away from the 3′ end of its recognition site (Fig. 1A). Two Type IIs restriction endonucleases, PleI (Fig. 1B) and MlyI (Fig. 1C), recognize the same GAGTC sequence. These three endonucleases seem to be related to each other. They all act as a single polypeptide (unpublished observations). They all recognize the same DNA sequence. Their cleavage patterns are also similar: N.BstNBI and PleI cleave at the same position on the top strand and PleI and MlyI cleave at the same site on the bottom (Fig. 1). To further investigate the unique properties of this nicking endonuclease, we have cloned and sequenced the genes encoding N.BstNBI as well as the two related Type IIs endonucleases, PleI and MlyI. We have also identified amino acid residues involved in DNA cleavage in each endonuclease and explored the DNA cleavage mechanism of these enzymes.
Figure 1.

Recognition sequences and gene organizations of N.BstNBI, PleI and MlyI R–M systems. The recognition sequence, GAGTC, is highlighted in bold, and cleavage sites are indicated by small arrows. Open boxes represent the ORFs and their directions are indicated by arrows. Endonuclease genes are shown in black and methyltransferase genes are shown in white. The size of each gene is indicated under the gene.
MATERIALS AND METHODS
Bacterial strains
The nicking endonuclease N.BstNBI was originally isolated from the thermophilic bacterium B.stearothermophilus (NEB 928). Type IIs restriction endonucleases PleI and MlyI were isolated from the mesophilic bacteria Pseudomonas lemoignei and Micrococcus lylae, respectively. The E.coli strains ER2566 and ER2502 were obtained from E.Raleigh and M.Sibley (New England Biolabs, Beverly, MA) and used as host strains for cloning and expressing endonuclease genes.
Purification of nicking endonuclease N.BstNBI
The N.BstNBI endonuclease protein was purified to near homogeneity by seven chromatography steps. (i) Bacillus stearothermophilus cells (177 g) were resuspended in 530 ml of buffer A (20 mM KPO4, 7 mM 2-mercaptoethanol, 0.1 mM EDTA, 5% glycerol, pH 6.9) supplemented with 100 mM NaCl. The cells were broken with a Manton–Gaulin homogenizer. The extract was centrifuged at 22 000 g for 10 min at 4°C. All of the following procedures were performed on ice or at 4°C. The supernatant was loaded onto a 275 ml XK 50/14 fast flow Phosphocellulose column (Whatman International Ltd, Kent, UK) equilibrated with buffer A1 (buffer A plus 100 mM NaCl). The column was washed with 550 ml of Buffer A1, and then eluted with a 2750 ml linear gradient of 0.1–1 M NaCl in buffer A. N.BstNBI restriction activity was assayed by digestion of T7 phage DNA at 55°C in 1× N.BstNBI buffer [150 mM KCl, 10 mM Tris–HCl, 10 mM MgCl2, 1 mM dithiothreitol (DTT), 100 µg/ml bovine serum albumin (BSA), pH 8.0]. N.BstNBI activity eluted around 0.2 M NaCl. (ii) Active fractions were pooled and dialyzed against 100 mM NaCl-supplemented buffer B (20 mM Tris–HCl, 0.1 mM EDTA, 7 mM 2-mercaptoethanol and 5% glycerol, pH 8.0). The dialyzed pool was then diluted with buffer B to a final concentration of 50 mM NaCl and applied to a 22 ml HR 16/10 Source™ 15Q column (Pharmacia Biotech, Piscataway, NJ) equilibrated in buffer B1 (buffer B plus 50 mM NaCl). The column was washed with 44 ml of buffer B1 followed by a 220 ml linear gradient of 50–800 mM NaCl in buffer B. The majority of the restriction enzyme activity flowed through the column. (iii) The active fractions in the flowthrough and wash fractions were combined and loaded onto a 23 ml HR 16/10 Heparin TSK-guard gel 5PW (20 µm) column (TosoHaas, Montgomeryville, PA), which had been equilibrated with buffer B2 (buffer B plus 100 mM NaCl). The column was washed with 46 ml of buffer B2 and then eluted with a 230 ml linear gradient of 0.1–1 M NaCl in buffer B. Activity was found in the fractions containing ∼550 mM NaCl. (iv) Active fractions were loaded onto an 8 ml HR 10/10 Source™ 15Q column equilibrated with buffer B1. The column was washed with 16 ml buffer B1 and then eluted with a 120 ml linear gradient of 50–800 mM NaCl in buffer B. The majority of the activity was detected in the flowthrough, wash and the beginning of the gradient. (v) The flowthrough and wash fractions were combined and applied to a 23 ml HR 16/10 Heparin TSK column in buffer B2 and eluted as discussed in step iii. (vi) Active fractions were applied to a third 23 ml HR 16/10 Heparin TSK column equilibrated with buffer A2 (buffer A plus 50 mM NaCl). The column was washed with 46 ml of buffer A2 followed by a 460 ml linear gradient of 0.05–1 M NaCl in buffer A. The peak of the enzyme activity eluted at ∼630 mM NaCl. (vii) Active fractions were diluted to 50 mM NaCl in buffer A and then loaded onto a 1 ml Resource™ 15S (Pharmacia Biotech, Piscataway, NJ), which had been previously equilibrated with buffer A2. The column was washed with 2 ml buffer A2 and eluted with a 20 ml linear gradient of 0.05–1 M NaCl in buffer A. Twenty fractions were collected. The majority of the activity was found in fractions 13–19 with the most activity being in fraction 15. N.BstNBI was purified to ∼80% homogeneity.
Determination of protein sequence of N.BstNBI
Purified N.BstNBI was subjected to electrophoresis and electroblotted onto a membrane (18). The membrane was stained with Coomassie blue R-250 and a protein band of ∼70 kDa was excised and subjected to sequential Edman degradation (18). The first 31 residues of the 72 kDa protein band corresponded to M-A-K-K-V-N-W-Y-V-S-C-S-P-W-S-P-E-K-I-Q-P-E-L-K-V-L-A-N-F-E-G. A 6 kDa polypeptide fragment was purified after digesting the 72 kDa N.BstNBI endonuclease with cyanogen bromide. The amino acid sequence from the N-terminus of the 6 kDa polypeptide fragment was M-X-I-P-Y-E-D-F-A-D-L-G.
Cloning the N.BstNBI R–M system genes
Degenerate primers (5′-TGGCNAARAARGTNAAYTGGTA-3′ and 5′-TCNGCRAARTCYTCRTA-3′) were designed based on both the N-terminal and internal amino acid sequences. These primers were used to amplify the N.BstNBI endonuclease gene (n.bstNBIR) using PCR. The resulting 1.4 kb PCR fragment was cloned into plasmid pCAB16 (L.Greenough and W.Jack, New England Biolabs) and sequenced. To clone the entire n.bstNBIR gene, a chromosome walking technique via inverse PCR was adopted to amplify DNA adjacent to the original 1.4 kb endonuclease gene fragment (19,20). After two rounds of inverse PCR, the entire n.bstNBIR gene and its corresponding methyltransferase gene, n.bstNBIM, were obtained.
Cloning the PleI R–M system genes
The M gene of PleI R–M system was cloned on a 1 kb Sau3AI partially digested fragment of P.lemoignei DNA by selecting for protectively modified, PleI-resistant plasmid recombinants using the methylase selection method (21). The adjacent R gene, pleIR, was cloned by chromosome walking as described above.
Cloning the MlyI R–M system genes
The M gene of the MlyI R–M system was cloned in a 1.4 kb ApoI partially digested fragment using the methylase selection method similar to the PleI. The adjacent R gene, mlyIR, was also cloned by chromosome walking.
Sequence comparison
The deduced amino acid sequences of PleI, MlyI and N.BstNBI endonucleases were compared using GAP, PILEUP and PRETTYBOX programs (Genetic Computer Group, Madison, WI). Gap creation penalty was 8 and gap extension penalty was 2.
Construction of cleavage-deficient endonucleases
Based on sequence comparisons of PleI, MlyI and N.BstNBI endonucleases, seven conserved negatively charged residues (Fig. 2) were identified as potential catalytic residues and were changed into alanine by PCR site-directed mutagenesis (22). The primers are listed in Table 1.
Figure 2.

Sequence comparison between the three endonucleases in this study. Identical residues are shown as white on black, and conservative replacements are black on gray. 1, PleI (GenBank accession no. AF355461); 2, MlyI (GenBank accession no. AF355462); 3, N.BstNBI (GenBank accession no. AF329098). Conserved, negatively charged residues are indicated by circles.
Table 1. Oligodeoxyribonucleotides used for PCR-mediated mutagenesis.
| Enzyme |
Residue |
Forward oligonucleotides |
| N.BstNBI | D389A | AACTTGAAGAAGTCATTGCTCTTCTTGAGGTATATCATGA |
| E392A | AGTCATTGACCTTCTTGCAGTATATCATGAGAAAAAGAAT | |
| E418A | AAATACTGTATTTGCCTGGCTTACGTGGAATGGCTTC | |
| D441A | AAACAACTTCGTTATTGCAGAAGAGTTACAACCAGTTA | |
| D456 | GCCGCAGGTAACCAGCCTGCCATGGAAATTATATATGAAG | |
| E469A | ACTTTATTGTTCTTGGTGCCGTAACAACTTCTAAGGGAGC | |
| E482A | AACCCAGTTTAAGATGGCGTCAGAACCAGTAACAAGGCAT | |
| PleI | D355A | GTTATACGAAGCCATTCAAGAGGTTTTTG |
| E358A | GAAGACATTCAAGCGGTTTTTGAGAAG | |
| E377A | ATTGATGCTTGCCTGGAATACATGG | |
| D399A | AAATTTGAAATTTGCGGATTTTGGAAGT | |
| D414A | GGAAATATGCCAGCTATAGTGTGTG | |
| E422A | AGTTGTCTGTAGCTGTCACAATGGC | |
| E440A | AATATGAAATGGCAGGTGAGCCAGTC | |
| MlyI | D359A | CTTAGATCTGATATTGAAGCTATTTTAGACGTCTTTGC |
| D363A | GATATTTTAGCCGTCTTTGCAAAAA | |
| E380A | GTTCCTTTATTCCTTGCATGGAATATATGG | |
| D403A | GGAACTTCATTGTAGCTTTAGATGGAAT | |
| D418A | CAGGTAAGAAGCCTGCTATAGAAATTGGT | |
| E430A | ATCTTTTTCATGCATTGTTGCAGTAA CTATGTCATCAGGG | |
| E444A | CTCAATTTAATATGGCGGGGTCTTCTGTT′ |
Both strands were synthesized and the forward sequences are shown in the 5′→3′ orientation. The targeted codons are underlined.
DNA binding assay
This experiment was carried out by incubating DNA substrates with the catalytic mutants of MlyI and PleI as well as the wild-type MlyI and PleI as controls. The DNA substrate was prepared by digestion of plasmid pNB1 with restriction endonucleases BssS1 (two sites) and BsrFI (one site). Three DNA fragments, 584, 794 and 1067 bp, were produced from the digestion of pNB1. Only the middle 794 bp fragment contains the recognition sequence for MlyI and PleI. Crude cell extracts (1 µl) which contain various amounts of N.BstNBI, PleI, MlyI and their mutant variants (Figs 3 and 4) were incubated with ∼0.25 pmol of the BssSI–BsrFI digested pNB1 at 37°C for 5 min in 50 mM potassium acetate, 20 mM Tris–acetate pH 7.9, 10 mM CaCl2, 1 mM DTT. The samples were analyzed on an agarose gel. The same experiment was carried out with N.BstNBI at 55°C in 10 mM Tris–HCl, 150 mM KCl, 10 mM CaCl2, 1 mM DTT.
Figure 3.
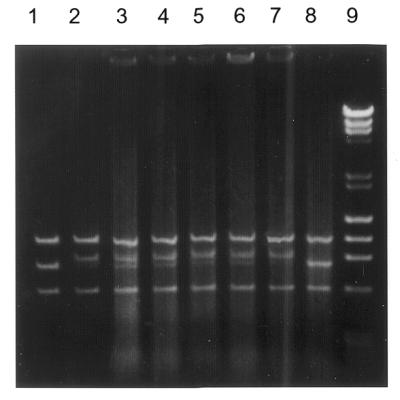
Gel retardation assay with the nicking enzyme N.BstNBI and its variants. Lane 1, three DNA fragments, 1067, 794 and 584 bp, generated by digestion of plasmid pNB1, a plasmid derived from pUC19 containing a single GAGTC site (16), with BssSI and BsrFI. Only the middle 794 bp fragment contains the GAGTC recognition sequence. The digested pNB1 substrate (0.25 pmol) was incubated with 0.75 pmol of purified N.BstNBI (lane 2), and 1 µl of the crude cell extract containing ∼9.5 pmol of N.BstNBI (lane 3), N.BstNBI-D456A (lane 4), N.BstNBI-E418A (lane 5), N.BstNBI-E469A (lane 6), N.BstNBI-E482A (lane 7). Lane 8 is the control plasmid only. Lane 9 is the size standard of λ DNA digested by HindIII and φ174 DNA digested by HaeIII (New England Biolabs).
Figure 4.
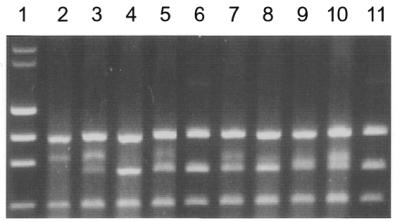
Gel retardation assay with MlyI, PleI and their variants. Conditions are described in the Figure 3 legend. Lane 1 is the size standard of λ DNA digested by HindIII, φ174 DNA digested by HaeIII. 0.25 pmol of the BssSI–BsrFI digested pNB1 was incubated with 1 µl of crude cell extracts containing ∼0.3 pmol of wild-type MlyI (lane 2) and its variants E380A (lane 3), D403A (lane 4), D418A (lane 5). Same DNA substrate (0.25 pmol) was also incubated with 1 µl of crude cell extracts containing ∼0.45 pmol of wild-type PleI and its variants D399A, D414A, D427A, shown in lanes 7, 8, 9 and 10, respectively. Lanes 6 and 11 are the controls of plasmid DNA only.
DNA cleavage assay
The DNA cleavage activities of nicking endonuclease N.BstNBI and its mutant variants were assayed by incubating various amounts of protein at 55°C in 50 µl of N.BstNBI reaction buffer (150 mM KCl, 10 mM Tris–HCl pH 7.5, 10 mM MgCl2, 1 mM DTT) containing 1 µg of bacteriophage T7 DNA or plasmid pNB1 (16). The DNA cleavage activities of the PleI or MlyI endonucleases and of their mutant variants were assayed by incubating various amounts of protein in the standard MlyI–PleI reaction buffer (50 mM potassium acetate, 20 mM Tris–acetate, 10 mM magnesium acetate, 1mM DTT, 100 µg/ml BSA, pH 7.9) containing 1 µg of (N6-methyladenine free) bacteriophage λ DNA (λ DNA) or plasmid pNB1, for 1 h at 37°C. The reactions were then terminated by addition of 12.5 µl of stop solution [1% sodium dodecyl sulfate (SDS), 0.05 mM EDTA, 30% glycerol, 0.025% bromophenol blue], and reaction products were analyzed by electrophoresis on a 1% agarose gel.
Gel filtration
The gel filtration experiment was performed on a Amersham Pharmacia Akta FPLC system using a Pharmacia Superdex 200 HR 10/30 column. The column was equilibrated with a buffer containing 20 mM Tris–HCl pH 7.5, 150 mM NaCl, 10 mM CaCl2, 0.1 mM EDTA, 7 mM 2-mercaptoethanol, 5% glycerol. Protein elution was detected by recording the absorption at λ = 280 nm. The calibration curve was generated by using the calibration kits (low and high molecular weight) from Pharmacia. Purified MlyI (4 nmol) was mixed with 40 nmol of either non-specific or specific DNA duplexes, which were generated by annealing two complementary oligodeoxynucleotides (27mer) in a buffer containing 50 mM potassium acetate, 20 mM Tris–acetate pH 7.9, 10 mM calcium chloride, 1 mM DTT. The samples were incubated for 5 min at 37°C before being loaded onto the column. As a control the protein was loaded alone as well as the oligodeoxynucleotides. A similar experiment was carried out with N.BstNBI with a few changes: a buffer containing 10 mM Tris–HCl, 150 mM KCl, 10 mM CaCl2, 1 mM DTT was used and the samples were incubated for 5 min at 55°C.
RESULTS
Cloning of the N.BstNBI, PleI and MlyI genes
To clone the N.BstNBI nicking endonuclease gene, the methylase selection strategy was tried and one clone, containing plasmid DNA resistant to N.BstNBI endonuclease digestion, was isolated. The 4.3 kb insert in the plasmid was sequenced and a small m6-β adenine methyltransferase (M) gene was identified based on sequence similarity to other methyltransferases (23). This methyltransferase was able to methylate DNA at GASTC (S = G or C) sites and therefore was able to protect DNA from endonucleolytic digestion by N.BstNBI, PleI or MlyI. However, the flanking ORFs did not encode the N.BstNBI nicking enzyme, suggesting that this m6-β adenine methyltransferase was not associated with that R–M system. A protein-based cloning strategy was adopted. The N.BstNBI nicking endonuclease protein was purified to near homogeneity by column chromatography as detailed in the Materials and Methods. The purified protein was used to determine its N-terminal sequence as well as a short segment of its internal amino acid sequence. The nicking endonuclease gene, n.bstNBIR, was cloned based on reverse translation of these amino acid sequences using degenerated-primer PCR, and the entire gene was cloned by several rounds of inverse PCR (19,20). The n.bstNBIR gene encodes a 604 amino acid protein with a deduced molecular weight of 70 769, comparable to the Mr value of 70 000 kDa estimated by SDS–PAGE. Downstream from the n.bstNBIR ORF, two ORFs are identified (Fig. 1). One is a putative gene, ORF1, encoding a 189 amino acid hypothetical protein that shares 38% sequence similarity with the carboxyl half of the nicking endonuclease. The function of ORF1 is still unknown, but it is not required for the nicking activity. The other is the n.bstnbIM gene, which encodes a 301 amino acid adenine methyltransferase (Fig. 1). The specificity of this methyltransferase is the same as that of the orphan methyltransferase but they belong to different sub-families (23): m6A-α for the N.BstNBIM and m6A-β for the orphan methyltransferase.
Both the PleI and the MlyI R–M system genes were cloned using the methylase selection method (21). The nucleotide sequences of the R and M genes were determined. The gene organization of the PleI and MlyI R–M systems is shown in Figure 1. They both consist of a large endonuclease gene and a small adenine methyltransferase gene. The methyltransferase encoded by the pleIM gene belongs to the m6-α methyltransferase family like N.BstNBI methyltransferase and the methyltransferase encoded by mlyIM gene belongs to the m6-β methyltransferase family like the orphan methyltransferase. Type IIs restriction endonucleases recognize asymmetric sequences, therefore a typical Type IIs R–M system usually contains two methyltransferases, one to methylate each strand. However, all of the methyltransferases in our study recognize the degenerate, symmetric sequence GASTC (S = G or C). Thus a single methyltransferase is capable of methylating both DNA strands.
Sequence comparison of restriction endonuclease genes
The deduced protein sequence of N.BstNBI was compared to the deduced protein sequences of PleI and MlyI using a multiple-sequence alignment program (Fig. 2). The nicking enzyme shares significant sequence similarities with both PleI and MlyI, suggesting that it is related to these Type IIs restriction endonucleases. These large endonucleases could be divided into two domains based on sequence alignment: a more conserved N-terminal domain and a less conserved C-terminal domain (Fig. 2). Since all three endonucleases recognize the same DNA sequence but cleave DNA differently, the conservation in the N-terminal domains may reflect their common function of recognizing the same GAGTC sequence. The well-characterized Type IIs enzyme FokI also has two separate structural domains: N-terminal for DNA binding and C-terminal for DNA cleavage (8). Only a marginal sequence identity (26.1%) was observed when N.BstNBI was compared with FokI using the GAP program (gap weight = 5; length weight = 2). We speculate that N.BstNBI, PleI and MlyI may also have two domain structures like FokI: an N-terminal half for DNA binding and a C-terminal half for DNA cleavage.
Mapping the DNA cleavage domain via mutagenesis
Based on earlier crystal structures, all Type II and Type IIs restriction endonucleases have a structurally similar catalytic core, which spatially brings together three essential charged residues, typically two acidic residues (D or E) and one lysine residue (K), forming a P-D/E-XN-D/E-X-K motif (24). One exception is BamHI, which has three acidic residues (D94, E111, E113) in its active site (25). The other exception is BglII, which has an asparagine rather than lysine in the D/E-X-K motif (26). Potential catalytic residues were chosen for each endonuclease in this study based on the following three criteria: (i) they are located in the C-terminal half (because these endonucleases share marginal sequence similarity with FokI and the C-terminal domain of FokI is the DNA cleavage domain); (ii) they are acidic, D and E, residues, or basic K residues; (iii) they are conserved among all three endonucleases. Although sequence alignment did not identify the conserved lysine residue, it did reveal seven acidic residues indicated by circles in Figure 2.
The potential catalytic residues were changed to alanine residues (Ala, A) using a PCR-based mutagenesis method (22). The mutant variants were sequenced to verify that no additional change had been introduced. The DNA cleavage activities of the mutated endonucleases in crude cell extract were tested and the results are summarized in Table 3. No DNA cleavage activity was detected when residues E418 and D456 of N.BstNBI were changed to Ala, and <1% cleavage activity remained when E469 and E482 were changed to Ala (Table 3). Substitutions of D399A, D414A, E427A in PleI and E380, D403A, D418A in MlyI completely abolished cleavage activity (Table 3). A decrease of activity was observed for E377A in PleI and for E430A and E444A in MlyI. The parallel effects of analogous mutants support the view that these residues perform similar functions in the respective endonucleases, and may reside in structurally similar regions.
Table 3. The effect of amino acid substitutions on the DNA cleavage activity of N.BstNBI, PleI and MlyI.
| N.BstNBI | D389A | E392A | E418A | D441A | D456A | E469A | E482A |
| N, 100% | N, 100% | C, 0% | N, 100% | C, 0% | C, 1% | C, 1% | |
| Ple1 | D355A | E358A | E377A | D399A | D414A | E427A | E440A |
| N, 100% | N, 100% | C, 10% | S, 0% | C, 0% | C, 0% | N, 100% | |
| Mly1 | D359A | D363A | E380A | D403A | D418A | E430A | E444A |
| N, 100% | N, 100% | C, 0% | S, 0% | C, 0% | C, 10% | C, 1% |
N, non-essential residues and substitution of these residues to alanine did not affect DNA cleavage activity. C, catalytic residues and changing these residues to alanine abolished or significantly reduced the DNA cleavage activity, but not the DNA binding activity, of the endonuclease. S, structurally important residues, which when changed to alanine affected both DNA cleavage and binding activities. The percentage represents the remaining activity and is shown under each residue. 0%, no cleavage activity can be detected in the crude cell extract. Conserved catalytic residues among the three endonucleases are shown in bold.
DNA binding assays
To eliminate residues such as those involved in maintaining overall fold of the protein, DNA retardation experiments were carried out to test whether the created mutants could still bind DNA. DNA substrates were prepared by digesting plasmid pNB1 with BssSI and BsrFI, which resulted in three fragments: 584, 794 and 1067 bp (Fig. 3, lane 1). Only the 794 bp fragment in the middle contained the sequence GAGTC. When purified N.BstNBI nicking enzyme was incubated with the three DNA fragments, the mobility of the 794 bp fragment was retarded (Fig. 3, lane 2), suggesting that only the DNA containing the recognition site for the enzyme was bound specifically by N.BstNBI. The same mobility shift of the middle band was also observed when crude N.BstNBI endonuclease from a cell extract was used in the assay (Fig. 3, lane 3). Crude cell extracts of cleavage-deficient N.BstNBI endonucleases were also used in DNA binding assays. All four mutated endonucleases were able to shift the middle DNA fragment containing GAGTC (Fig. 3, lanes 4–7), which suggests that they can still bind to DNA specifically, like the wild-type enzyme. No DNA retardation was observed when an E.coli crude cell extract prepared from cells containing the cloning vector was used as a control in the DNA binding assay (Fig. 3, lane 8).
Similar binding assays were performed to test the specific DNA binding activity of cleavage-deficient MlyI and PleI variants. Two mutated MlyI enzymes, MlyI-E380A and MlyI-D418A could still bind the 794 bp middle fragment just like wild-type MlyI (Fig. 4). The other two DNA fragments were not retarded. Again this suggested that those two MlyI mutants were still able to bind DNA specifically. None of the DNA fragments were retarded in the presence of the MlyI-D403A mutant (Fig. 4, lane 4). The mutation at this position abolished both DNA cleavage and binding activities, suggesting that either the D403 residue was involved in both cleavage and binding activities or it was important in supporting protein structure. We think that D403 in MlyI is probably required for structural integrity for two reasons. The first is that Type IIs endonucleases usually consist of two structural domains, one for cleavage and the other for binding, so it is unlikely that a residue would be involved in both functions. Secondly, the corresponding D403 residue in the N.BstNBI endonuclease is D441 (Table 3), and this residue is not a catalytic residue for DNA cleavage. The cleavage-deficient PleI variants behaved similarly to the MlyI variants. The middle-size band was retarded by PleI-D414A and PleI-E427A but not by PleI-D399A (Fig. 4, lanes 9, 10 and 8, respectively).
DNA cleavage assays
The results from sequence alignments, mutagenesis experiments and DNA binding assays showed that N.BstNBI is very similar to its Type IIs counterparts MlyI and PleI. To further investigate the differences between these endonucleases, the time course of cleavage reactions with each enzyme were studied. Plasmid pNB1, which contains a single GAGTC recognition sequence, was used in the cleavage assay. The undigested supercoiled form of pNB1 is converted into the nicked closed-circular form when one strand is nicked by an endonuclease or into linear form when both strands are broken within a close distance. When plasmid pNB1 was incubated with MlyI, the supercoiled form was first converted into the nicked form (Fig. 5A). The nicked form peaked at 5 min and disappeared after 45 min. The amount of linear DNA increased during the time course and the digestion was complete at 60 min. The existence of an intermediate nicked form suggests that double strand cleavage by MlyI endonuclease is probably a two step process: the supercoiled substrate was nicked on one strand first, and then the other strand was cleaved in a subsequent step. The same sequential progression was observed with PleI (Fig. 5B). However in the case of PleI, some nicked intermediate remained even after 120 min incubation, suggesting that PleI has a lower efficiency than MlyI in cleaving the two strands of pNB1 DNA. When a similar digestion time course was performed using the nicking endonuclease N.BstNBI, all of the supercoiled pNB1 was converted into the nicked form in <12 min (Fig. 5C). However, to our surprise, a small amount of linear DNA (∼5–10%) was also observed after 105 min of digestion, suggesting that N.BstNBI was also capable of cleaving both DNA strands. We have tested the double strand cleavage activity of N.BstNBI in four different restriction enzyme digestion buffers (buffers 1, 2, 3 and 4; New England Biolabs), and the double strand cleavage activity was not enhanced in any of these buffers (data not shown). The linearized DNA was purified from an agarose gel after electrophoresis and the break points on that DNA fragment were determined by dideoxynucleotide sequencing reactions. The cleavage sites were mapped on both strands at ∼5–6 nt on the 3′ side of the GAGTC recognition sequence (data not shown). This suggested that the double-stranded cleavage sites of N.BstNBI were located at approximately the same locations as its Type IIs counterparts.
Figure 5.

Digestion reactions of plasmid pNB1 with MlyI, PleI and N.BstNBI. Plasmid pNB1 (7.8 nM) was incubated with (A) MlyI (2 nM), (B) PleI (14.2 nM) and (C) N.BstNBI (0.8 nM). Aliquots were removed at different time intervals. M, DNA size marker.
Gel filtration assays
The structure of the Type IIs endonuclease FokI reveals a dimer, in which the dimerization interface is mediated by the cleavage domain (9), and biochemical studies on FokI also provide evidence that FokI catalytic domain must dimerize for DNA cleavage to occur (10). To obtain evidence of dimerization, a gel filtration was carried out to measure the molecular mass of MlyI and N.BstNBI protein complexes with or without DNA. The buffer used contained Ca2+ instead of Mg2+. DNA binding experiments demonstrated that under these conditions both endonucleases retained their binding specificity but were unable to cleave the DNA (Fig. 4). Purified MlyI protein was loaded onto the Superdex-200 column without DNA, and its molecular mass was calculated to be ∼65 kDa (Fig. 6), suggesting that MlyI (Mr = 63 865) is a monomer in solution in the absence of DNA. When a mixture of MlyI protein and excess specific DNA duplex was loaded to the column, two peaks of absorption were observed. The low molecular mass peak had a molecular mass of 17.8 kDa, which corresponded to the DNA duplex alone (17 476). The high molecular mass peak had a molecular mass of 159 kDa based on the standard linear curve generated from molecular weight standards (Fig. 6). The molecular masses of the possible MlyI complexes are as follows: 1 MlyI + 1 DNA = 81.3 kDa; 2 MlyI = 127.7 kDa; 2 MlyI + 1 DNA = 145.2 kDa; 2 MlyI + 2 DNA =162.7 kDa. The observed molecular mass of MlyI complex has the best match to a complex containing two molecules of MlyI plus two DNA duplexes. No high molecular mass complex was formed when MlyI was mixed with non-specific DNA lacking the MlyI recognition sequence, suggesting that the MlyI–DNA complex was sequence dependent.
Figure 6.

Determination of molecular mass of enzyme–DNA complex by gel filtration results. The Kav value for each protein standard (on the linear scale) was plotted against the corresponding molecular weight (on the logarithmic scale). Standard proteins (ovalbumine, 43 kDa; albumine, 67 kDa; aldolase, 158 kDa; catalase, 232 kDa) are represented by squares. The Kav values of the proteins of interest (diamonds) were located on this calibration curve and the corresponding molecular weights were calculated using calibration curve equation: Kav = –0.266 log(molecular weight) + 1.658.
The same gel filtration experiment was performed with N.BstNBI. The molecular mass of N.BstNBI alone was determined as 60 kDa based on molecular weight standards (Fig. 6). This suggests that, like MlyI, N.BstNBI (deduced Mr = 70 769) exists as a monomer in solution in the absence of DNA. When mixed with specific DNA duplex, a slightly larger complex was observed with a molecular mass of 99 kDa, which might correspond to a complex composed of one N.BstNBI molecule bound to one DNA (70 769 + 17 476 = 88 245). These results suggest that MlyI and N.BstNBI are both monomers in solution and that MlyI apparently dimerizes in presence of specific DNA, whereas N.BstNBI remains as a monomer.
DISCUSSION
We have cloned and sequenced the genes encoding the N.BstNBI nicking endonuclease and the Type IIs restriction endonucleases PleI and MlyI. The nicking enzyme shares significant sequence similarities with both PleI and MlyI, suggesting that it is related to these Type IIs restriction endonucleases. These large endonucleases could be divided into two domains based on sequence alignments: a more conserved N-terminal domain and a less conserved C-terminal domain (Table 2). Since all three endonucleases recognize the same DNA sequence but cleave DNA differently, the conservation noted in the N-terminal domains may reflect their common function of recognizing the GAGTC sequence. The well-characterized Type IIs enzyme FokI has two separate structural domains: an N-terminal domain for DNA binding and the C-terminal for DNA cleavage (8). We speculate that N.BstNBI, PleI and MlyI may also have two domains like FokI: the N-terminal half for DNA binding and the C-terminal half for DNA cleavage.
Table 2. Percent sequence identity for the whole enzymes against each other, as well as for their two putative domains, N- and C-terminal.
| Entire enzyme | N-terminal half | C-terminal half | |||||||
| |
PleI |
MlyI |
N.Bst |
PleI |
MlyI |
N.Bst |
PleI |
MlyI |
N.Bst |
| PleI | – | – | – | ||||||
| MlyI | 37.5 | – | 43.5 | – | 32.2 | – | |||
| N.Bst | 33.1 | 32.1 | – | 34.3 | 34.5 | – | 30.3 | 30.1 | – |
N.Bst, N.BstNBI.
Based on sequence alignment, seven conserved acidic residues were chosen as potential catalytic residues (Fig. 2 and Table 3). We changed all those candidate residues to alanine and then assayed the DNA cleavage and binding activities of these mutated endonucleases. Several likely catalytic residues have been assigned because changing them to alanine abolished DNA cleavage activity but not DNA binding activity (Table 3). Two residues, D399 in PleI and D403 in MlyI, are excluded from being catalytic residues because changing them to alanine abolished both DNA cleavage and DNA binding activities, suggesting that these residues are probably important for the overall structural integrity of the PleI and MlyI endonucleases. It is possible that catalytic residues can be important to both catalysis and DNA binding and the binding results can be influenced by buffer conditions (27,28). This is particularly true for Type II restriction endonucleases because they cleave DNA within their DNA recognition sequences and have integrated DNA recognition and DNA cleavage domains (2,24). In the case of Type IIs restriction endonucleases, their catalytic residues are less likely to be involved in DNA binding due to the fact that Type IIs endonucleases contain separate DNA binding and DNA cleavage domains (4,9). The corresponding residue D441 in N.BstNBI seems to be less important for protein structure because changing it to alanine had no effect on DNA binding or DNA cleavage activity. Most of these catalytically important residues are conserved among N.BstNBI, PleI and MlyI (Fig. 2 and Table 3), which further supports the idea that these endonucleases are related to each other. Identification of catalytic residues in the C-terminal domains, such as D456 (N.BstNBI), D414 (PleI) and D418 (MlyI), supports the notion that Type IIs endonucleases in this study, like FokI, have two structural domains: N-terminal for DNA binding and C-terminal for DNA cleavage.
In addition, our mutagenesis results suggest that PleI and MlyI contain a single active site for DNA cleavage. This is because if two active sites were required for cleavage, one for each strand, then disabling one catalytic center by mutation should generate an enzyme with strand-specific ‘nicking’ activity like N.BstNBI. However, catalytic mutants showing a complete loss of double-stranded cleavage activity displayed no nicking activity on T7 DNA or pNB1 DNA. We thus believe a single active site is used to cleave both DNA strands. To cleave two DNA strands using one catalytic center, the endonuclease could use the following strategies. One possibility is that the endonucleases act as monomers and cleavage on both strands of DNA is accomplished by cleavage of one strand first, translocation of its active site to the scissile phosphate on the second DNA strand, and then cleavage of the second strand. The movement might involve a conformational change in the protein to rearrange its single catalytic center or the protein might dissociate and re-associate with the substrate DNA. A second possibility is that the endonucleases form dimers to achieve double-stranded DNA cleavage like FokI (9,10). In this model, the first strand is cleaved by one enzyme molecule and the second strand is cleaved by the active site of another enzyme. Results from this study favor the second model. We found that additional sites on the DNA substrates stimulated the cleavage rates of PleI and MlyI (unpublished observations). This is consistent with two recognition sites interacting with each other. When the two sites are located in the same DNA molecule, the interaction is a trimolecular reaction, which is more efficient than the tetramolecular reaction which occurs on DNA with a single site. Stronger evidence of dimerization came from gel filtration experiments. Although MlyI and N.BstNBI endonucleases behave like monomers in solution, MlyI dimerizes in presence of cognate DNA and divalent metal ion, whereas N.BstNBI remains a monomer under similar conditions (Fig. 6).
Figure 7 is a schematic diagram showing the current model for DNA cleavage by the model Type IIs endonuclease, FokI, and the MlyI, PleI and N.BstNBI endonucleases. FokI binds to DNA at its recognition site, and the sequence-specific binding event exposes its dimerization interface (Fig. 7A). Two DNA-bound FokI molecules then dimerize through their cleavage domains, forming the catalytically active dimer, which then cleaves the two DNA strands in a concerted manner (Fig. 7A). When low FokI concentrations were used in the reaction, a slight accumulation of nicked DNA was observed (10). This does not seem to be the case for PleI and MlyI. A significant excess of the nicked intermediate over the linear DNA was observed in the early stage of the time course (Fig. 5). It seems that MlyI and PleI cleave two strands of DNA in a sequential fashion with the DNA first being nicked and then further digested to the final linear form. We propose that monomers of PleI and MlyI can bind to DNA substrates and nick one strand but not the other because the catalytic residues probably cannot reach the scissile phosphodiester bond on the second strand (Fig. 7B). At this point two possible pathways might occur. The monomeric enzyme might dissociate from the nicked DNA, forming the nicked intermediate species observed in Figure 5A and B, or the monomeric enzyme might stay attached to its nicked substrate and dimerize with another monomer which then cleaves the second strand (Fig. 7B). In the case of the nicking endonuclease N.BstNBI, the dimerization may be diminished by mutations and thus nicked DNA could not be efficiently converted into the linear DNA by N.BstNBI resulting in the accumulation of nicked DNA under normal digestion conditions (Fig. 7C). Support for this model came from gel filtration experiments which showed that MlyI formed dimer in the presence of DNA and divalent metal ion, but no dimer of N.BstNBI was observed under similar conditions (Fig. 6).
Figure 7.

Schematic diagram showing the hypothesized DNA cleavage models of Type IIs restriction endonucleases FokI (A), MlyI and PleI (B) and the N.BstNBI nicking enzyme (C). Ellipses indicate the DNA binding domain and the gray circles represent DNA cleavage domains of the Type IIs restriction endonucleases. Double strand DNA is shown as two solid lines and the GAGTC recognition sequence is indicated by the black-filled box between the two lines.
Figure 7 also illustrates a fundamental difference in activation between FokI and our enzymes (MlyI and PleI). FokI appears to be catalytically inactive prior to dimerization and does not introduce nicks into DNA substrates at a significant rate (Fig. 7A) (10). In contrast, MlyI and PleI do introduce nicks into DNA substrates at a much high rate prior to double-stranded cleavages (Fig. 5A and B). They appear to be catalytically active before dimerization and able to cleave one DNA strand first (Fig. 7B).
We suggest that the nicking endonuclease N.BstNBI has probably evolved from a Type IIs endonuclease based on the results from this study and that the biological function of this nicking endonuclease is probably still the restriction of foreign DNA. Although N.BstNBI only nicks DNA under normal digestion conditions, double-strand breaks still take place when two nicks, located on two different strands of the same DNA molecule, are close enough together (<12–15 bp apart) to permit the termini to melt and give fragments. There are six such sites in bacteriophage T7 DNA, and the T7 DNA was cleaved into six fragments by N.BstNBI in vitro (16). Thus, like other restriction endonucleases, N.BstNBI can introduce double-stranded breaks into unmodified foreign DNA. In addition, there is a corresponding DNA methylase gene, n.bstNBIM, located close to the N.BstNBI endonuclease gene, just like other R–M systems (Fig. 1). This corresponding methylase methylates GAGTC and GACTC sequences and thus protects both DNA strands from the endonucleolytic digestion of N.BstNBI (H.Kong, unpublished results).
Acknowledgments
ACKNOWLEDGEMENTS
We thank Michael Dalton for assistance in protein purification, and Rafael Agra, Megan Holden and Nicole Porter for helping with some of the experiments. We thank Dr Aneel Aggarwal for sharing unpublished FokI results. We thank Dr Jack Benner for protein sequencing. We are very grateful to Drs Richard Roberts, Ira Schildkraut, William Jack and Elisabeth Raleigh for fruitful discussions and critical reading of this manuscript. We thank Dr Donald Comb for his support. Part of this work was supported by National Institutes of Health Grant GM60057 (to H.K.).
DDBJ/EMBL/GenBank accession nos+ To whom correspondence should be addressed. Tel: +1 978 927 5054; Fax: +1 978 921 1350; Email: AF355461, AF355462, AF329098
References
- 1.Roberts R.J. and Macelis,D. (2000) REBASE-restriction enzymes and methylases. Nucleic Acids Res., 28, 306–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilson G.G. and Murray,N.E. (1991) Restriction and modification systems. Annu. Rev. Genet., 25, 585–627. [DOI] [PubMed] [Google Scholar]
- 3.Szybalski W., Kim,S.C., Hasan,N. and Podhajska,A.J. (1991) Class-IIS restriction enzymes—a review. Gene, 100, 13–26. [DOI] [PubMed] [Google Scholar]
- 4.Li L., Wu,L.P. and Chandrasegaran,S. (1992) Functional domains in FokI restriction endonuclease. Proc. Natl Acad. Sci. USA, 89, 4275–4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chandrasegaran S. and Smith,J. (1999) Chimeric restriction enzymes: what is next? Biol. Chem., 380, 841–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaczorowski T., Skowron,P. and Podhajska,A.J. (1989) Purification and characterization of the FokI restriction endonuclease. Gene, 80, 209–216. [DOI] [PubMed] [Google Scholar]
- 7.Skowron P., Kaczorowski,T., Tucholski,J. and Podhajska,A.J. (1993) Atypical DNA-binding properties of class-IIS restriction endonucleases: evidence for recognition of the cognate sequence by a FokI monomer. Gene, 125, 1–10. [DOI] [PubMed] [Google Scholar]
- 8.Wah D.A., Hirsch,J.A., Dorner,L.F., Schildkraut,I. and Aggarwal,A.K. (1997) Structure of the multimodular endonuclease FokI bound to DNA. Nature, 388, 97–100. [DOI] [PubMed] [Google Scholar]
- 9.Wah D.A., Bitinaite,J., Schildkraut,I. and Aggarwal,A. (1998) Structure of FokI has implications for DNA cleavage. Proc. Natl Acad. Sci. USA, 95, 10564–10569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bitinaite J., Wah,D., Aggarwal,A. and Schildkraut,I. (1998) FokI dimerization is required for DNA cleavage. Proc. Natl Acad. Sci. USA, 95, 10570–10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kornberg A. and Baker,T.A. (1992) DNA Replication, 2nd Edn. W.H. Freeman and Company, New York, NY.
- 12.Geider K., Baumel,I. and Meyer,T. (1982) Intermediate stages in enzymatic replication of bacteriophage fd duplex DNA. J. Biol. Chem., 257, 6488–6493. [PubMed] [Google Scholar]
- 13.Higashitani A., Greenstein,D., Hirokawa,H., Asano,S. and Horiuchi,K. (1994) Multiple DNA conformational changes induced by an initiator protein precede the nicking reaction in a rolling circle replication origin. J. Mol. Biol., 237, 388–400. [DOI] [PubMed] [Google Scholar]
- 14.Modrich P. (1989) Methyl-directed DNA mismatch correction. J. Biol. Chem., 264, 6597–6600. [PubMed] [Google Scholar]
- 15.Yao M. and Kow,Y.W. (1997) Further characterization of Escherichia coli endonuclease V. Mechanism of recognition for deoxyinosine, deoxyuridine and base mismatches in DNA. J. Biol. Chem., 272, 30774–30779. [DOI] [PubMed] [Google Scholar]
- 16.Morgan R.D., Calvet,C., Demeter,M., Agra,R. and Kong,H. (2000) Characterization of the specific DNA nicking activity of restriction endonuclease N.BstNBI. Biol. Chem., 381, 1123–1125. [DOI] [PubMed] [Google Scholar]
- 17.Abdurashitov M.A., Belichenko,O.A., Shevchenko,A.V. and Degtyarev,S.K. (1996) N.BstSE—site-specific nuclease from Bacillus stearothermophilus SE-589—restriction endonuclease production. Mol. Biol. (Mosk.), 30, 1261–1267. [PubMed] [Google Scholar]
- 18.Waite-Rees P.A., Keating,C.J., Moran,L.S., Slatko,B.E., Hornstra,L.J. and Benner,J.S. (1991) Characterization and expression of the Escherichia coli Mrr restriction system. J. Bacteriol., 173, 5207–5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ochman H., Gerber,A.S. and Hartl,D.L. (1988) Genetics, 120, 621–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Triglia T., Peterson,M.G. and Kemp,D.J. (1988) A procedure for in vitro amplification of DNA segments that lie outside the boundaries of known sequences. Nucleic Acids Res., 16, 8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lunnen K.D., Barsomian,J.M., Camp,R.R., Card,C.O., Chen,S.-Z., Croft,R., Looney,M.C., Meda,M.M., Moran,L.S., Nwankwo,D.O., Slatko,B.E., Van Cott,E.M. and Wilson,G.G. (1988) Cloning Type II restriction and modification genes. Gene, 74, 25–32. [DOI] [PubMed] [Google Scholar]
- 22.Morrison H. and Desrosiers,R. (1993) A PCR-based strategy for extensive mutagenesis of a target DNA sequence. Biotechniques, 14, 454–457. [PubMed] [Google Scholar]
- 23.Wilson G.G. (1992) Amino acid sequence arrangements of DNA-methyltransferases. Methods Enzymol., 216, 259–279. [DOI] [PubMed] [Google Scholar]
- 24.Kovall R.A. and Matthews,B.W. (1999) Type II restriction endonucleases: structural, functional and evolutionary relationships. Curr. Opin. Chem. Biol., 3, 578–583. [DOI] [PubMed] [Google Scholar]
- 25.Newman M., Strzelecka,T., Dorner,L.F., Schildkraut,I. and Aggarwal,A.K. (1995) Structure of Bam HI endonuclease bound to DNA: partial folding and unfolding on DNA binding. Science, 269, 656–663. [DOI] [PubMed] [Google Scholar]
- 26.Lukacs C.M., Kucera,R., Schildkraut,I. and Aggarwal,A.K. (2000) Understanding the immutability of restriction enzymes: crystal structure of BglII and its DNA substrate at 1.5Å resolution. Nat. Struct. Biol., 7, 134–140. [DOI] [PubMed] [Google Scholar]
- 27.Martin A.M., Horton,N.C., Luseti,S., Reich,N.O. and Perona,J.J. (1999) Divalent metal dependence of site-specific DNA binding by EcoRV endonuclease. Biochemistry, 38, 8430–8439. [DOI] [PubMed] [Google Scholar]
- 28.Engler L.E., Welch,K.K. and Jen-Jacobson,L. (1997) Specific binding by EcoRV endonuclease to its DNA recognition site GATATC. J. Mol. Biol., 269, 82–101. [DOI] [PubMed] [Google Scholar]