


Abstract
The selective alteration of the genome using Cre recombinase to target the rearrangement of genes flanked by LOX recognition sequences has required the use of two separate genetic constructs in trans, one containing cre and the other containing the gene of interest flanked by LOX sites. We have developed a strategy in which both the cre recombinase gene and LOX recombination sites may be cloned within a single vector in cis. This method uses a modified form of Cre (CREM) that contains alterations to the 5′ region including the introduction of a Kozak consensus sequence and insertion of a functional intron. This system allows for the inducible, tissue-specific activation or inactivation of gene expression in a single vector and can be utilized for the 300-fold amplification of gene expression from a weak promoter. This approach can be applied to targeting strategies for generating genetically altered mice and gene therapy.
INTRODUCTION
Recent advances in transgenic technology provide a means to fundamentally alter the mouse genome by either over-expressing a gene of interest (1–4), or deleting or modifying the endogenous allele of a gene in a tissue-specific and temporal manner. (5–7) Several of these approaches utilize technology based upon the Cre/LOX system where the P1 bacteriophage recombinase is used to act on target LOX sites (8) in order to modify a genetic locus or activate transgene expression in a tissue-specific manner. In this system, two distant LOX sites recombine resulting in the excision and loss of intervening nucleotide sequences. This technology has been particularly useful for conditionally altering genetic loci in mice where the expression of the Cre recombinase is under the transcriptional control of a tissue-specific and temporally regulated promoter (9–26). A major limitation of this approach, however, is that the genetically engineered constructs used to achieve these manipulations cannot contain both the sequences for cre recombinase and the LOX sites. Cre will be expressed when present in a plasmid transformed into bacteria which will lead to the recombination of LOX sites contained within the same plasmid and excision of genetic material contained between them. For this reason, two separate lines of transgenic animals must be generated and bred together in order to effect Cre recombination of genes flanked by LOX sites (fLOXed genes). Similarly, the use of Cre-LOX technology for gene therapy applications requires the use of two separate constructs, which makes its use inefficient for such purposes.
Another major limitation of transgenic technology and gene therapy approaches has been that many tissue-specific promoters are transcriptionally weak and lead to relatively low levels of heterologous gene expression. A method to maintain tissue-specificity but amplify levels of transcription of a gene of interest would be highly desirable.
In order to address these limitations, we have developed a novel system in which cre recombinase has been modified (CREM) such that it is not expressed in prokaryotic cells during the cloning process, but is actively expressed in eukaryotic cells. Based upon this strategy, we have generated single vectors containing both the modified cre recombinase and fLOXed gene of interest. In addition, we have developed novel reporter systems to assess Cre activity. By using Cre/LOX strategies in combination with very strong transcriptional promoters, we can achieve high levels of inducible, tissue-specific gene expression using a single vector. These strategies can be applied to manipulating the mouse genome and gene therapy approaches to greatly improve the targeted expression of genes of interest.
MATERIALS AND METHODS
Construction of plasmids
Plasmid constructs were generated using standard cloning techniques as described below. For all PCR reactions, the following conditions were used: 10–100 ng of DNA template, 1 mM MgSO4, 0.3 mM dNTPs, 0.3 µM primers, and 1.25 U of PFX-polymerase (Life Technologies, Rockville, MD) in a total reaction volume of 50 µl. Thermocycle parameters were: 94°C for 2 min, 50°C for 30 s, 68°C for 1 min for 1 cycle, followed by 94°C for 15 s, 55°C for 30 s and 68°C for 1 min for a total of 34 cycles.
Generation of gene-switch reporter constructs
Generation of an SV40 promoter-driven gene switch plasmid containing floxed enhanced green fluorescent protein (EGFP) inserted into the β-gal reading frame. Plasmid pEGFP-CI (Clontech, Palo Alto, CA) was digested with BglII/BamHI and religated. Synthetic oligonucleotides OLN 111 (5′-CTA-GCG-GTA-CCG-ATA-ACT-TCG-TAT-AGC-ATA-CAT-TAT-ACG-AAG-TTA-TCA-3′) and OLN 112 (5′-CCG-GTG-ATA-ACT-TCG-TAT-AAT-GTA-TGC-TAT-ACG-AAG-TTA-TCG-GTA-CCG-3′), designed to contain a LOX site flanked by 5′ NheI and KpnI sites and a 3′ AgeI site, were cloned into the NheI and AgeI sites of the modified pEGFP-CI vector resulting in plasmid p138. A second set of synthetic oligonucleotides OLN 113 (5′-CGC-GTA-TAA-CTT-CGT-ATA-GCA-TAC-ATT-ATA-CGA-AGT-TAT-CGG-TAC-CAC-TAC-3′) and OLN 114 (5′-GTG-GTA-CCG-ATA-ACT-TCG-TAT-AAT-GTA-TGC-TAT-ACG-AAG-TTA-TA-3′) containing the recognition sequence for LOX flanked by MluI at the 5′ portion and KpnI and DraIII at the 3′ portion were inserted into the MluI and DraIII sites of p138 resulting in the generation of p142 in which EGFP is flanked by LOX sites. The floxed EGFP cassette was removed from p142 by digestion with KpnI and cloned into the KpnI site of pSV-β-Galactosidase (Promega, Madison, WI) resulting in pSV-EGFP/β-gal (p144) (Fig. 1B).
Figure 1.
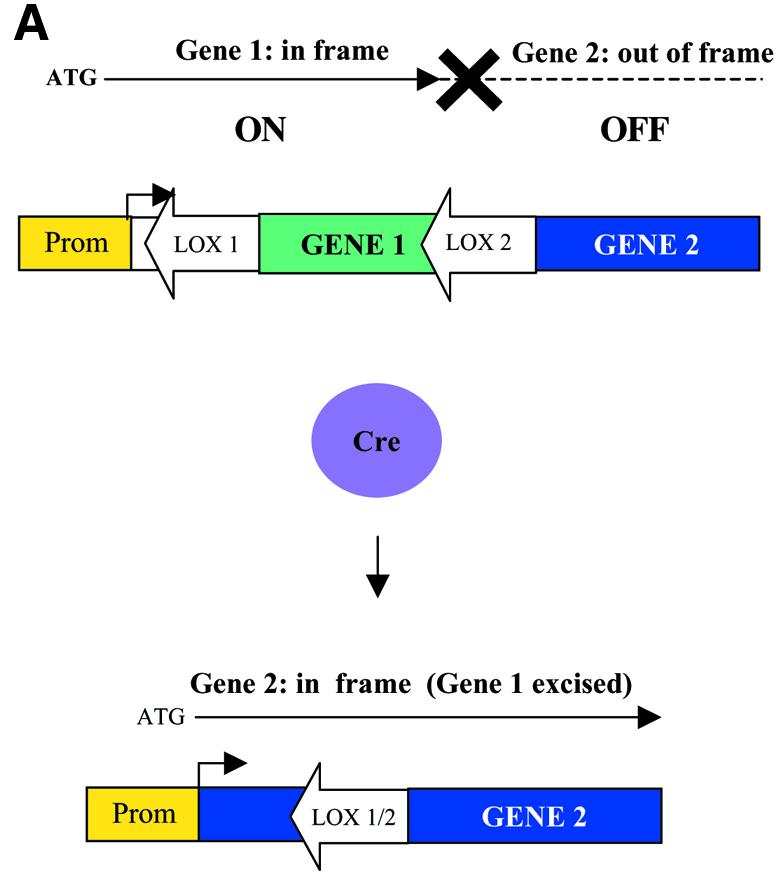


Gene-switch strategy using cre/LOX induced translational frameshift. (A) Gene 1 flanked by LOX sites is inserted in frame within the open reading frame of gene 2 resulting in appropriate translation of Gene 1, but not Gene 2. In the presence of Cre recombinase Gene 1 is excised and the correct reading frame of Gene 2 is restored which is now expressed. (B) EGFP/β-gal gene-switch under the transcriptional control of the SV40 early promoter. Translational start site and first 18 amino acids of E.coli gtp are upstream of the floxed EGFP followed by E.coli gtp amino acids 19–47 fused in-frame to β-gal. The insertion of a cytosine at the 3′ end of LOX I results in a disruption of the reading frame of the β-gal gene. The Cre-mediated excision of the floxed EGFP restores the correct reading frame for β-gal. (C–F) Transfection of CHO-K1 cells with pSV-EGFP/β-gal results in expression of only EGFP without cre whereas co-transfection with pCMV-cre results in loss of EGFP expression but gain of β-gal expression. (C and D) Fluorescent microscopy, GFP filter; (E and F) bright field (X 320). Assays performed 48 h post-transfection.
Generation of plasmids containing a gene switch cassette under the transcriptional regulation of CMV. The StuI/SalI fragment from pSV-EGFP/β-gal containing the floxed EGFP was removed and ligated into the SmaI/SalI sites of of pCMV-β-gal (Clontech) resulting in the generation of pCMV-EGFP/β-gal (p155) containing the floxed EGFP gene switch cassette driven by the CMV. A second construct utilizing the CMV enhancer/chicken β-actin promoter was also generated. The StuI/SalI fragment containing the floxed EGFP/β-gal from pSV-EGFP/β-gal (p144) was inserted into the XhoI site of pCAGGS (27) resulting in pCMV/βAc-EGFP/β-gal (p169).
Generation of an EGFP-RFP gene switch plasmid. The KpnI fragment from pSV-EGFP/β-gal (p144) containing the floxed EGFP cassette was cloned into the KpnI site of pDSRed1-N1 (Clontech) upstream of the coding sequence for red fluorescent protein resulting in the generation of pCMV-EGFP/RFP (p212) (Fig. 2A).
Figure 2.

Gene-switch using pCMV-EGFP/RFP vector. (A) Generation of gene-switch reporter vector pCMV-EGFP/RFP containing the floxed EGFP gene upstream of RFP. (B–G) CHO-K1 cells were transfected with pCMV-EGFP/RFP either in the absence (B and E) or presence of pCMV-cre (C, D, F and G). (B and C) Fluorescent microscopy using GFP filter; (E and F) using RFP filter; (D) bright field; (G) using dual GFP/RFP filter (X 400). Assays performed 48 h post-transfection.
Generation of a floxed-stop cassette-luciferase reporter construct
Generation of SV40 early promoter-STOP-luc. The oligonucleotides OLN 01 (5′-CTA-GGA-TAA-CTT-CGT-ATA-GCA-TAC-ATT-ATA-CGA-AGT-TAT-A-3′) and OLN 02 (5′-AGC-TTA-TAA-CTT-CGT-ATA-ATG-TAT-GCT-ATA-CGA-AGT-TAT-C-3′) containing a LOX site flanked by AvrII and HindIII sites were ligated to the HindIII–AvrII fragment of pGL-3 Control (Promega). A 2.2 kb SalI–StuI fragment and 3 kb SalI–AvrII fragment were separately isolated from the resulting plasmid. The HindIII (blunted)–SpeI fragment of plasmid p302 containing the STOP cassette (Life Technologies) was isolated. The three individual fragments as described above were ligated to generate pSV-STOP-luc (p133) (Fig. 3A).
Figure 3.
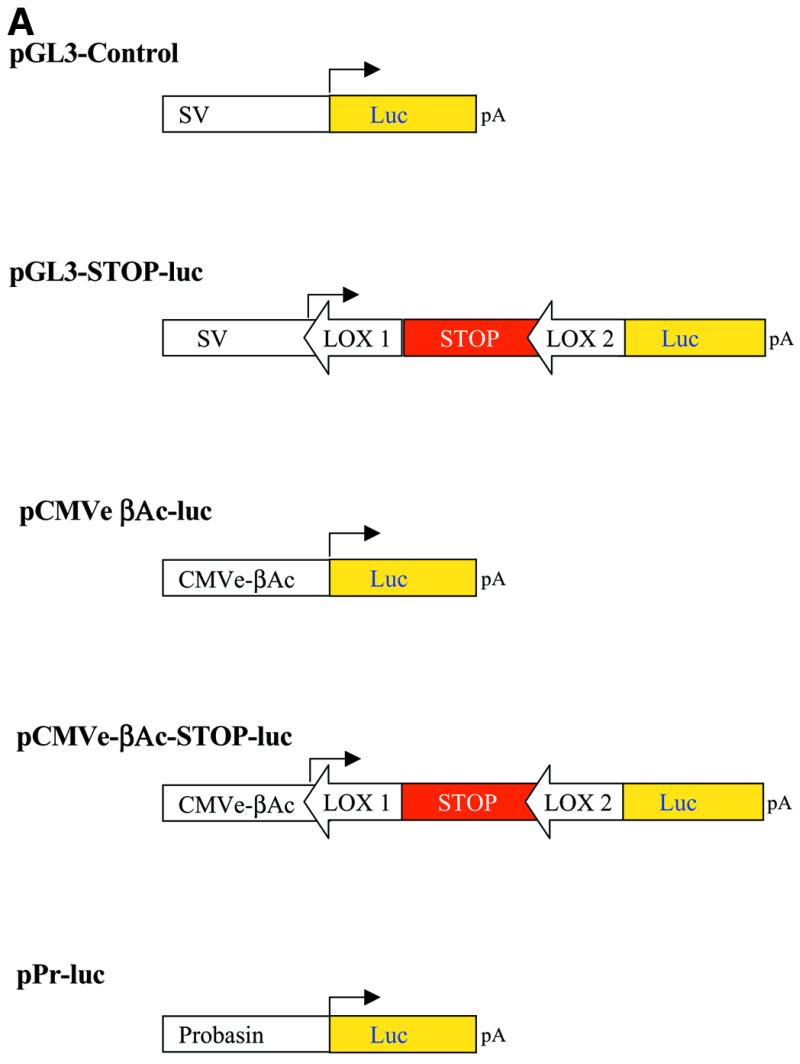
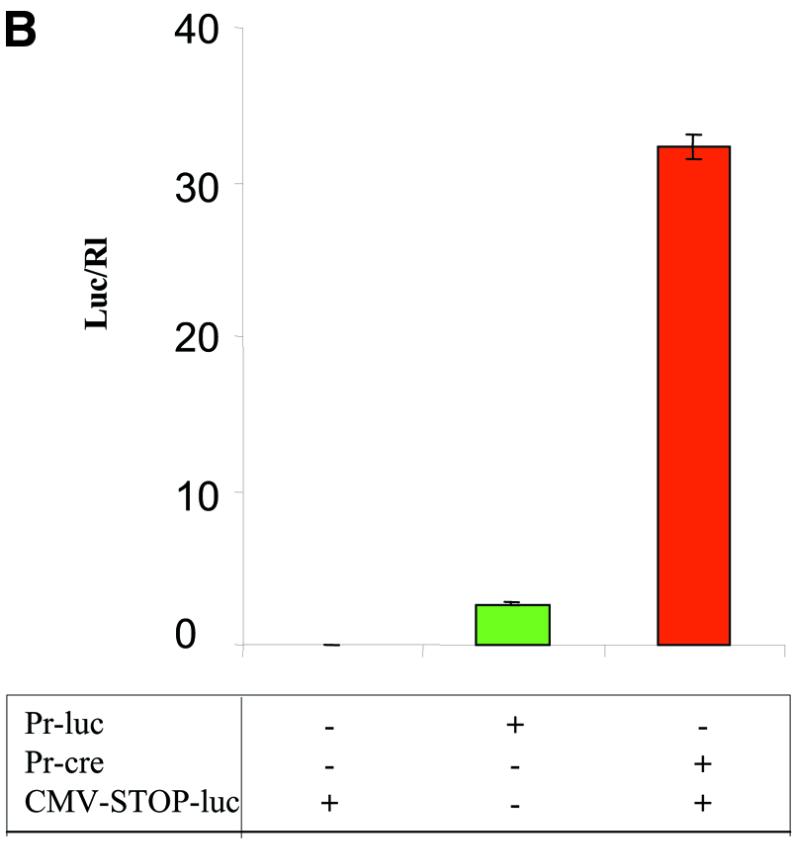
Amplification of gene expression using a weak, tissue-specific promoter to activate transcription from a strong promoter. (A) Luciferase reporter gene vectors. SV, SV40 early promoter; CMVe-βAc, CMV enhancer, β-actin promoter. (B) PC-3 cells were transfected with the reporter constructs pPr-luc, pCMV-STOP-luc or pCMV-STOP-luc with pPr-cre. Luciferase expression increased 15-fold using this amplification strategy. Assays performed 48 h post-transfection.
Generation of a CMV-enhancer-βAc driven luciferase reporter gene. The SalI and XhoI fragment of pCAGGS containing the CMVe-βAc transcriptional control unit was inserted into the XhoI site of pGL3-Basic (Promega) to generate pCMVe-βAc-luc (p230) (Fig. 3A).
Generation of a CMV-enhancer-βAc driven luciferase reporter gene containing a floxed stop-cassette. pCMVe-βAc-luc was digested with BglII, blunt-ended, and digested with HindIII. pSV-STOP-luc was digested with StuI/HindIII and the fragment containing the floxed STOP cassette was isolated and cloned into the HindIII site of pCMVe-βAc-luc in which the BglII site had been previously removed by blunt-ending. This resulted in generation of pCMVe-βAc-STOP-luc (p 231) (Fig. 3A).
Generation of a probasin–luciferase reporter construct. Plasmid prPb-tva950 (kindly provided by Pam Schwartzenberg, NCI) was digested with NdeI, blunt-ended, and digested with KpnI. The fragment containing the probasin promoter was isolated and cloned into the SmaI/BglII sites of pGL-3 Basic, resulting in pPr-luc (p159) (Fig. 3A).
Generation of modified cre-recombinase
Alteration of the 5′ region and introduction of a Kozak consensus translational start site, LOX sites and intron. Plasmid pBS185 (Life Technologies) containing the CMV enhancer/promoter driving expression of wild-type cre was digested with EcoRI, partially digested with HindIII, blunt-ended and religated to remove a 1.0 kb fragment containing a KpnI site generating pCMV-cre-del (p205) (Fig. 4A). Plasmid p205 was digested with XhoI and BssHII to remove a 5′ region of cre. A 5′ portion of cre was PCR amplified using OLN C (5′-CGC-CAG-AAT-TCC-AAA-ATT-TGC-CTG-CAT-TAC-CGG-TCG-ATG-CAA-CG-3′) and OLN D (5′-GAC-CGC-GCG-CCT-GCA-GAT-ATA-GAA-GAT-AAT-CGC-GAA-CAT-CTT-3′), which contain an internal PstI restriction site generated by a silent mutation and digested with EcoRI and BssHII. The p205 XhoI/BssHII fragment, cre amplification product and the synthetic double-stranded oligonucleotides OLN A (5′-CTG-AGC-GGC-CGC-CTA-GGC-CCA-TGG-CGA-ATT-TAC-TGA-CGG-TAC-CAG-3′) and OLN B (5′-AAT-TCT-GGT-ACC-GTC-AGT-AAA-TTC-GCC-ATG-GCC-TAG-GCGGCC-GC-3′) were ligated together to form pCMV-cre-K (p209) (Fig. 4A). These modifications resulted in the significant shortening of the 5′ untranslated region from 484 to 19 bp, an optimized Kozak translational start site (28) and the introduction of an EcoRI site at nucleotide 509 and a PstI restriction site through a silent T→G mutation at nucleotide 718 within the coding region of cre to allow for additional alterations of cre. These alterations resulted in the changes in the amino acid sequence of Cre as depicted in Figure 4A. The second amino acid was changed from serine to alanine, the eighth amino acid was changed from histidine to proline and codons for glutamic acid and phenylalanine were inserted at amino acid positions 9 and 10. The human β-globin intron from plasmid pCI (Promega) was altered by PCR mutagenesis to place a PstI site at the 5′ end and a BssHII site at the 3′ end using PCR primers OLN E (5′-GCG-ATC-TGC-AGG-TAA-GTA-TCA-AGG-TTA-CAA-GAC-AGG-3′) and OLN F (5′-ATA-TGC-GCG-CCT-GTG-GAG-AGA-AAG-GCA-AAG-TGG-AT-3′). The 5′ primer E contains a PstI restriction site and the first 35 nt of the β-globin intron (position 953 to 881 in pCI) and the 3′ primer F contains a BssHII restriction site and 26 nt of the 3′ portion of the β-globin intron (position 890 to 965 in pCI). The modified human β-globin intron was cloned into the PstI/BssH II sites of pCMV-cre-K to generate pCMV-CREM (p210) (Fig. 4A).
Figure 4.
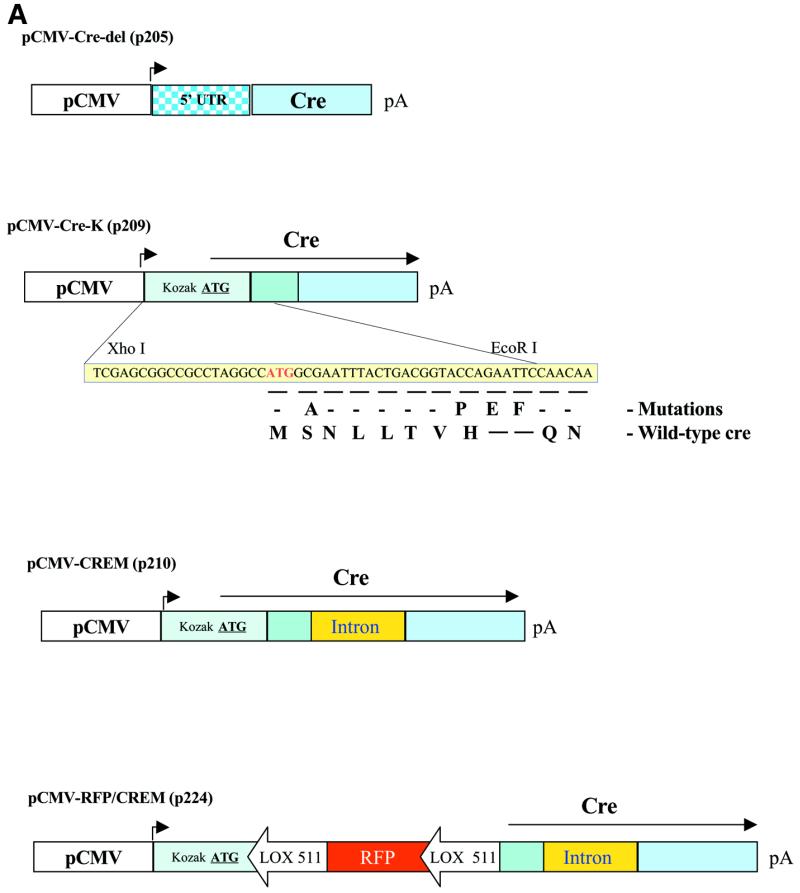

Modification of Cre recombinase. (A) pCMV-Cre-del is the backbone of subsequent constructs. pCMV-Cre-K contains the modified 5′ untranslated region and amino acid changes as indicated. pCMV-CREM contains the chimeric/β-globin intron within the cre coding sequence. pCMV-RFP/CREM contains a floxed RFP gene within the Cre coding sequence. (B–G) Demonstration of functional activity of modified Cre. Co-transfection of pCMV-CREM with reporter plasmid pCMV-EGFP/RFP in CHO-KI cells results in the switch from EGFP to RFP expression. (B–D) Reporter pCMV-EGFP/RFP alone; (E–G) reporter with pCMV-CREM (p210). (B and E) Fluorescence with GFP filter; (C and F) fluorescence with RFP filter; (D and G) fluorescence using dual GFP/RFP filters. Assays performed 48 h post-transfection.
Generation of a plasmid containing modified cre recombinase and a single LOX 511 site inserted into the cre coding region. An aliquot of plasmid pCMV-CREM (p210) was digested with KpnI and another aliquot was digested with EcoRI. Both aliquots were treated with calf intestinal alkaline phosphatase and subsequently digested with ScaI. The ScaI/KpnI fragment containing the CMV promoter and 5′ region of Cre and the ScaI/EcoRI fragment containing the remaining portion of cre were isolated and ligated with a phosphorylated oligonucleotides OLN 135 (5′-CAA-TAA-CTT-CGT-ATA-ATG-TAT-ACT-ATA-CGA-AGT-TAT-TCG-3′) and OLN 136 (5′-AAT-TCG-AAT-AAC-TTC-GTA-TAG-TAT-ACA-TTA-TAC-GAA-GTT-ATT-GGT-AC-3′) containing KpnI and EcoRI sites at the 5′ and 3′ ends of the LOX 511 sequence to generate pCMV-CREM-L (p218) (Fig. 4A).
Generation of a plasmid containing floxed RFP inserted into the coding sequence of cre recombinase. Plasmid pDsRed1-N1 (Clontech) was digested with DraIII. An aliquot of this digest was cut with KpnI and the 3.5 kb DraIII/KpnI fragment was isolated. Another aliquot was digested with AgeI and the 1.1 kb DraIII/AgeI fragment was isolated. The two isolated fragments were ligated with oligonucleotides OLN 03 (5′-CAA-TAA-CTT-CGT-ATA-ATG-TAT-ACT-ATA-CGA-AGT-TAT-CTA-GA-3′) and OLN 04 (5′-CCG-GTC-TAG-ATA-ACT-TCG-TAT-AGT-ATA-CAT-TAT-ACG-AAG-TTA-TTG-GTA-C-3′) containing the LOX 511 sequence (29) flanked by KpnI and AflII restriction sites resulting in the generation of p221. p221 was digested with KpnI and AflII to isolate the fragment containing a single LOX 511 recognition sequence upstream of the RFP gene. pCMV-CREM-L (p218) was digested with KpnI and EcoRI to remove the LOX 511 site. These two fragments were ligated together with the synthetic oligonucleotides OLN 05 (5′-TTA-AGA-ATA-ACT-TCG-TAT-AAT-GTA-TAC-TAT-ACG-AAG-TTA-TTC-G-3′) and OLN 06 (5′-AAT-TCG-AAT-AAC-TTC-GTA-TAG-TAT-ACA-TTA-TAC-GAA-GTT-ATT-C-3′) containing a LOX 511 site flanked by AflII and EcoRI. The final plasmid product containing the floxed RFP inserted within the coding sequence of cre recombinase is designated pCMV-RFP/CREM (p224) (Fig. 4A).
Generation of cre expression vectors using the androgen-responsive, prostate-specific probasin promoter
Generation of probasin driven Cre recombinase. The 428 bp probasin promoter fragment was isolated from prPb-tva950 (kindly provided by Pam Schwartzenberg, NCI) following digestion and blunt-ending of the NdeI site, and digestion with KpnI. This fragment was cloned into the SmaI/KpnI site of pβ-gal-Basic (Clontech) resulting in pPr-β-gal (p150). pPr-β-gal was cut with ClaI, filled-in, and cut with XhoI in order to remove the coding sequence for β-gal. The MluI (blunt-ended)-XhoI fragment from pBS185 (Life Technologies) containing the coding sequence for cre recombinase was isolated and inserted into the XhoI/ClaI (blunt-ended) fragment of p150 resulting in the generation of pPr-cre (p153) (Fig. 5A).
Figure 5.
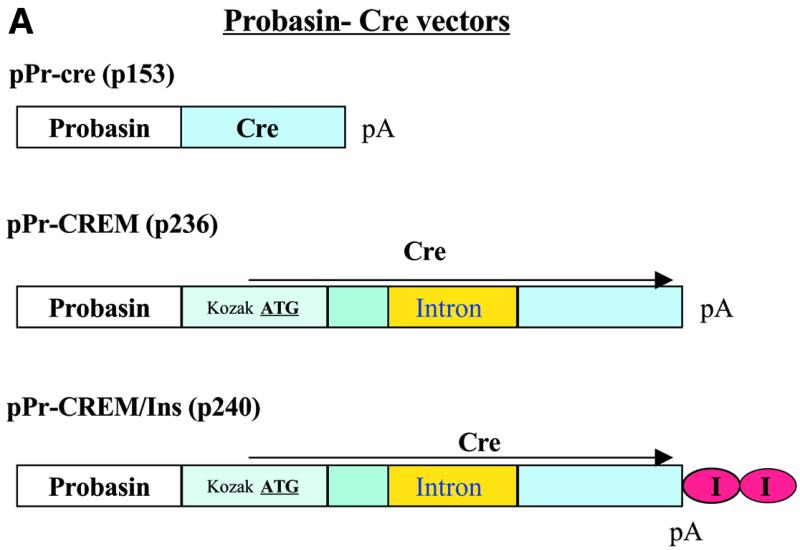
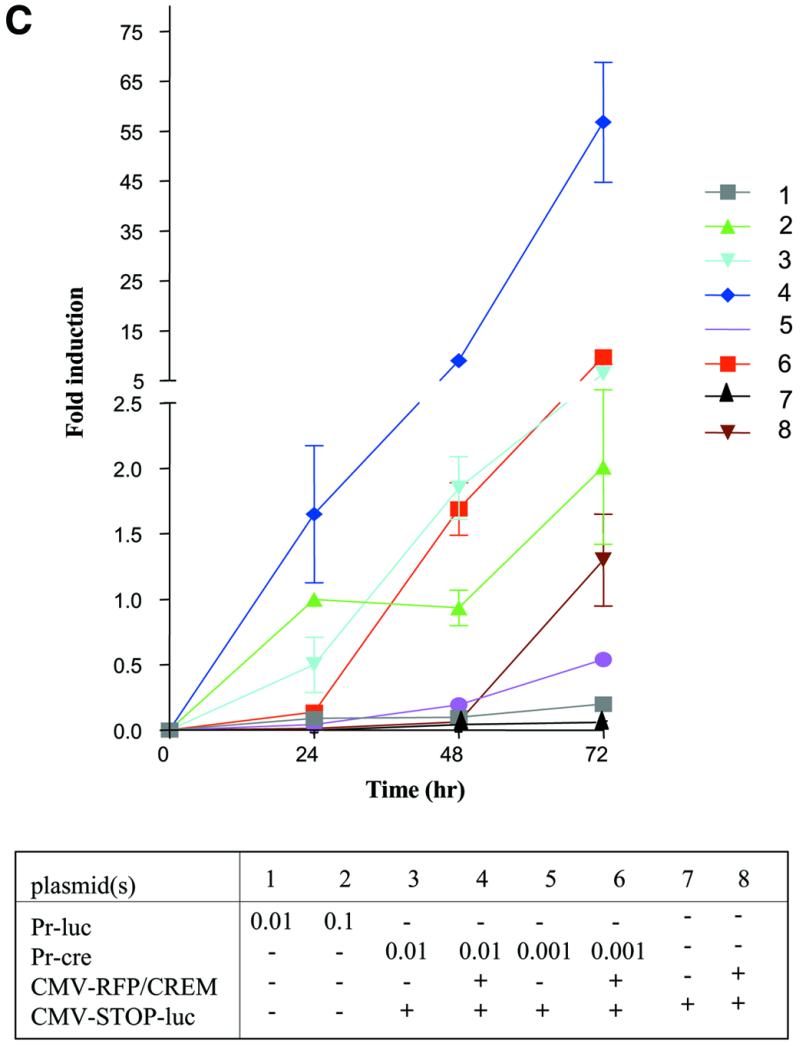
Generation of probasin-cre vectors and enhanced amplification of gene expression using a second Cre. (A) Probasin driven Cre recombinase expression vectors. (B) PC-3 cells were transfected with the murine androgen receptor producing-plasmid pCMV-mAR (0.06 pmol) in combination with the indicated plasmids (0.1 pmol) in the presence or absence of 4 nM mibolerone. The presence of the second conditional cre vector led to the additional 8-fold amplification of luciferase activity. Assays performed 48 h post-transfection. (C) Time course of gene amplification using a second conditionally expressed Cre recombinase. PC-3 cell were transfected with pCMV-mAR (0.06 pmol) and the indicated plasmids. Except as indicated, 0.1 pmol of each plasmid was used in transfections. pTK-RL (0.1 pmol) (Promega) was used to normalize for transfection efficiency.
Generation of probasin-modified cre recombinase plasmid. The XhoI/EcoRV fragment from plasmid pCMV-CREM containing the modified cre recombinase was substituted for the XhoI and EcoRV fragment of plasmid Pr-cre (p153) generating the plasmid pPr-CREM (p236) (Fig. 5A).
Generation of a plasmid containing probasin-modified Cre flanked by insulators. The vector backbone of plasmid pPr-CREM (p236) was isolated following digestion with DraIII and partially digested with SalI. The DraIII and BamHI fragment of pPr-CREM (p210) containing the probasin promoter and modified cre recombinase was isolated separately. Plasmid pJC13.1 (kindly provided by G.Felsenfeld, NCI) was digested with SalI and BamHI and the fragment containing the pair of chicken globin insulators (29) was isolated. All three isolated fragments were ligated together to produce pPr-CREM/Ins (p240) (Fig. 5A).
Generation of single vector plasmids containing modified cre and a reporter gene conditionally activated using LOX recombination sites. The DraIII and EcoICRI fragment containing the CMV-STOP-luc cassette was isolated from pCMVe-βAc-STOP-luc (p231) and ligated to the DraIII and Eco47III fragment of plasmid pPr-CREM (p236) containing probasin-modified cre to create plasmid pPr-CREM/CMV-STOP-luc (p252) (Fig. 6A). A second construct was made in which chicken globin insulators (29) were placed between the Pr-CREM and CMV-STOP-luc cassettes. The region containing the probasin-modified cre with insulators was isolated from pPr-CREM/Ins following digestion with DraIII and Eco47III and ligated into the EcoRICI/DraIII fragment of pCMVe-βAc-STOP-luc (p231). This resulted in the generation of a single vector pPr-CREM/Ins/CMV-STOP-luc (p255) containing the modified cre under the transcriptional regulation of probasin separated from the Cre-dependent CMV-STOP-luc cassette by insulators (Fig. 6A).
Figure 6.

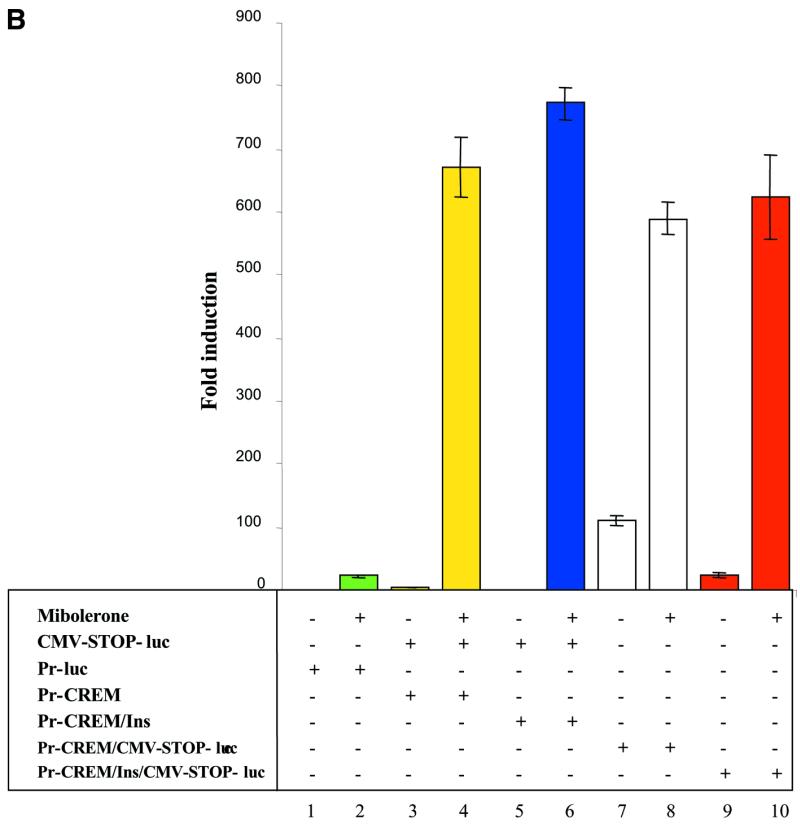
Construction and functional testing of single vectors containing both modified Cre recombinase and LOX-dependent conditional reporter cassettes. (A) pPr-CREM/CMV-STOP-luc vector is a fusion of pPr-CREM and CMV-STOP-luc cassettes. pPr-CREM/Ins/CMV-STOP-luc contains pPr-CREM/Ins separated from CMV-STOP-luc by two chicken globin insulators. (B) Evaluation of inducible and amplified gene expression using single vector constructs. PC-3 cells were transfected with pCMV-mAR in the presence or absence of 4 nM mibolerone and additional plasmids (0.1 pmol) as indicated. Assays were performed 48 h post-transfection.
pCMV-mAR. The cDNA for mouse androgen receptor was modified by PCR mutagenesis to place a BglII site 9 bp upstream of the ATG and the 2.8 kb BglII–PstI fragment was inserted into the BglII–PstI sites of pEGFP-C1 vector (Clontech) after which the EGFP coding sequences were removed by digestion with AgeI and BglII, which were blunt-ended and religated.
pTK-RL. PTK-RL (Promega) uses the TK promoter to direct expression of Renilla reniformis luciferase and was used to normalize for transfection efficiency.
Tissue culture
CHO-KI and PC-3 cells (ATCC, Rockville, MD) were propagated in RPMI-1640 medium supplemented with 10% fetal calf serum (FCS) (Life Technologies), 1 mM sodium pyruvate, 10 mM HEPES buffer, 2 mM glutamine, 50 U/ml of penicillin G sodium and 50 µg/ml of streptomycin sulfate. Charcoal stripped FCS (HyCLONE, Logan, UT) was used for all studies that required hormone induction.
Transfection of cells with DNA
Cells were plated at a density of 1.5 × 105 cells/well in 3 ml of culture media 24 h prior to transfection using 6-well plates (Costar, Corning, NY). For hormonal induction studies, PC-3 cells were trypsinized (Life Technologies), washed in culture media and centrifuged at 190 g for 5 min at room temperature. Cells pellets were resuspended in 1× PBS, centrifuged and resuspended in RPMI 1640 without phenol red and supplemented with charcoal/dextran-treated FCS (HyCLONE). On the morning of transfection, 2 ml of fresh media supplemented with 4 nM of mibolerone (NEN, Boston, MS) was added to plates. Transfections were performed 1 h later with a total of 2 µg of plasmid DNA per 6 µl of FuGENETM 6 (Roche, Indianapolis, IN) in a total of 100 µl of transfection solution containing serum-free culture medium per well. Fresh medium with mibolerone was added daily. Transfections were performed in triplicate and experiments were repeated at least three times. Cells were visualized using a Zeiss-G436 fluorescent microscope. GFP was visualized using a 436 nm excitation, 510 nm emission filter. RFP was visualized using a 546 nm excitation, 580 nm emission filter. Dual fluorescence was observed using a 460–490 m excitation, 510 emission filter.
Luciferase assay
The Dual-LuciferaseTM Reporter assay system (Promega) was used to determine activity of firefly luciferase (Photinus pyralisus) and sea pansy Renilla luciferase (R.reniformis) according to the manufacturer’s instructions. Cells were washed in 1× PBS and 500 µl passive lysis solution was added per well. Lystate was collected after 15 min and centrifuged in an Eppendorf 5415C centrifuge at 13 000 r.p.m. for 30 s. An aliqout of 20 µl of supernatant was used for luciferase assays.
Statistical analyses
InStatR (GraphPad Software Inc, San Diego, CA) statistical package program was used for statistical analysis.
RESULTS
Development of LOX/frameshift vectors for selective gene expression
The regulation of gene expression using Cre/LOX technology has relied on the use of either a floxed ‘STOP cassette’ inserted into the 5′ untranslated region of the gene of interest (14,24) or a floxed gene cassette upstream of a second gene to be expressed (12,15,18). In the latter case, the second gene of interest is out of frame for proper translation and therefore not expressed. In the presence of Cre recombinase, the floxed gene or STOP cassette will be removed allowing for expression of the downstream gene of interest. However, due to occasional read-through of transcription and protein translation despite the STOP cassette, there may be detectable background levels of expression of the downstream gene in the absence of Cre (C.Couldrey, unpublished observations).
To address this issue, we designed a novel expression cassette where expression of gene A could be selectively turned off with the simultaneous activation of gene B by placing the floxed gene A within the coding sequence of gene B, whose nucleotide sequence is out of frame to gene A (Fig. 1A and B). This strategy results in an undetectable background level of expression of gene B in the absence of Cre (Fig. 1C–F). Our expression vector pSV-EGFP/βgal uses the SV40 early region promoter to drive the expression of one of two genes depending upon whether cre is expressed or not. To demonstrate the feasibility of this approach, the coding sequence for EGFP flanked by LOX sites was inserted in frame into the 5′ region of the Escherichia coli gpt gene downstream of the translational start site but out of frame with the coding sequence for the β-gal gene placed downstream (Fig. 1A). Similar constructs were made using the CMV enhancer promoter (pCMV-EGFP/βgal) or the CMV enhance/β-actin promoter (pCMV/βAc-EGFP/βgal). In order to maintain the open reading frame between gtp and EGFP, but place the β-gal gene out of frame, an additional cytosine was added after the LOX I recognition sequence (Fig. 1B). The insertion of this additional cytosine also resulted in the generation of a translational stop codon upstream of the lacZ reading frame within the LOX II recognition sequence (Fig. 1B).
Results of transfection experiments demonstrated that this strategy of gene switching worked as predicted (Fig. 1C–F). Transfection of pSV-EGFP/β-gal alone resulted only in the expression of EGFP, whereas co-transfection with pCMV-cre led to the excision of EGFP and the functional expression of the LacZ gene (Fig. 1C–F). Similar constructs utilizing the CMV enhancer-promoter derived from pCMV β-gal (Clontech) or the CMV enhancer β-Actin promoter from pCAGGS (27) were also generated and gave similar results (data not shown). However, because of the high level of GFP expression under the transcriptional regulation of the CMV promoter/enhancer, it took longer for all GFP protein to be eliminated compared to that using the weaker SV40 promoter (data not shown). Nonetheless, the gene-switch occurred using the CMV promoter/enhancer. These results demonstrate that the LOX/frame-shift strategy is effective for the tight regulation of switching gene expression.
Since the assay for β-gal activity cannot easily be performed in vivo, we also developed the reporter construct pCMV-EGFP/RFP for Cre recombinase activity in which the EGFP gene is expressed without Cre, but the RFP gene is expressed following excision of the EGFP gene by Cre (Fig. 2). Both the EGFP and RFP reporter genes can be assayed in vivo.
Amplification of gene expression using alternate promoters in combination with Cre/LOX
Although tissue-specific promoters are expressed in a limited range of cell types, their level of transcription may often be relatively low and limiting (30). In order to increase the levels of expression but maintain tissue-specificity, we developed a strategy which utilized the tissue-specific expression of Cre in combination with the gene-switch approach as well as a gene activation approach under the transcriptional control of a strong, ubiquitously expressed promoter. The 428 bp tissue-specific, hormone-responsive probasin promoter was used to drive the expression of Cre (pPr-cre). The gene-switch expression vectors (pSV-EGFP/β-gal (Fig. 1B), pCMV-EGFP/β-gal and pCMVe/βAc- EGFP/β-gal direct the expression of a specific reporter gene only in the presence of Cre. This strategy maintains the tissue-specific expression of the desired reporter gene since Cre is produced using the tissue-specific probasin promoter, but greatly amplifies the expression of the desired reporter gene, which is under the transcriptional control of the CMV promoter.
In order to quantitatively assess the level of amplification by switching transcriptional regulation from the probasin promoter to the CMV enhancer/β-actin promoter, a series of luciferase reporter constructs were generated (Fig. 3A). PC-3 cells were co-transfected with pCMV-mAR to express the murine androgen receptor and the reporter construct probasin-luciferase (Pr-luc) (Fig. 3A) in the presence of the androgen analog mibolerone (Fig. 3B). This was compared to the levels of luciferase activity generated by the second reporter construct pCMV-STOP-luc in the presence or absence of the Cre producing plasmid probasin-cre. The results of these experiments demonstrated that luciferase activity was amplified at least 15-fold when the CMV-STOP-luc was activated in the presence of Pr-cre compared to the level of luciferase activity generated directly under the transcriptional regulation of the probasin promoter by pPr-luc (Fig. 3B).
Modification of cre recombinase for eukaryotic but not prokaryotic expression
In order to clone in bacteria a plasmid construct containing both the coding sequence for cre recombinase and the LOX target sequences, we designed modifications to the cre recombinase that prevented its translation in bacteria, but allowed for its translation in eukaryotic cells. This required the modification of the 5′ region of cre and insertion of an artificial intron within the cre coding sequence (Fig. 4A). Using mutagenesis strategies, the 5′ untranslated region of cre was removed, the translational start site was optimized for eukaryotic translation by using a Kozak consensus sequence (28) and unique KpnI and EcoRI restriction sites were introduced downstream of the ATG. As further described in the Materials and Methods, an artificial intron based upon a β-actin intron was inserted into a newly generated restriction site within the cre gene to create the plasmid pCMV-CREM (Fig. 4A).
To determine whether recombinase activity was maintained following these extensive modifications to cre, pCMV-CREM was co-transfected with the reporter plasmid pCMV-EGFP/RFP. As demonstrated in Figure 4B–G, the functional Cre recombinase activity was maintained with these alterations of cre.
Addition of a second cre amplification step further enhances reporter gene expression
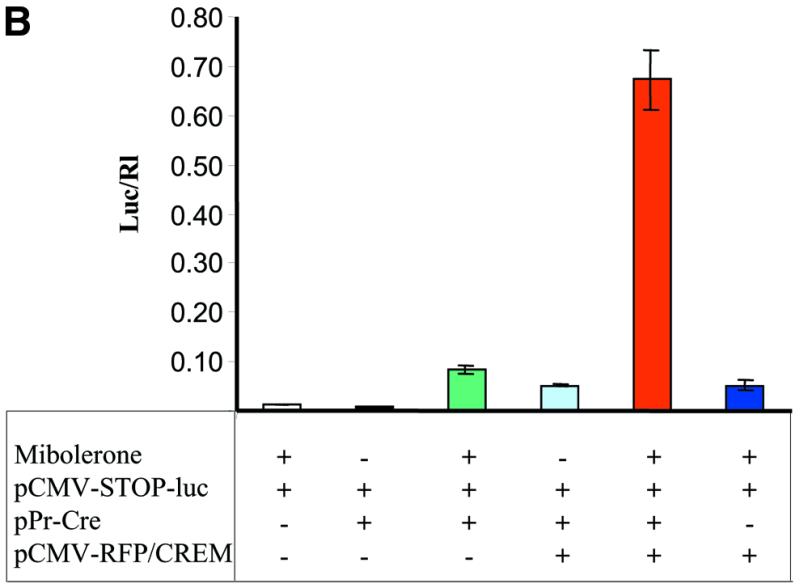
In order to further amplify the expression of the reporter gene, we developed an additional construct in which cre expression would also be conditionally amplified using a strong promoter. This would occur following the initial expression of Cre by the relatively weak, tissue-specific and hormone-inducible probasin promoter using the plasmid Pr-cre (Fig. 5A). An intermediate construct pCMV-CREM-L was first generated by placing a LOX 511 sequence (31) into the 5′ region of cre in pCMV-CREM. Cre activity was maintained despite the presence of the LOX 511 sequence within the cre coding region as demonstrated in co-transfection experiments with pCMV-EGFP/RFP (data not shown). The generation of the construct pCMV-RFP-CREM (p224) is depicted in Figure 4A. This construct uses the same frame-shift principles as those described previously for the EGFP/β-gal reporter constructs except that the LOX 511 target sequences flank the RFP gene. LOX 511 sequences were chosen to avoid potential recombination with LOX P sequences used in the other constructs. Without the expression of Cre, RFP is expressed and the modified cre is out of frame and not expressed. In the presence of Cre, RFP is excised and the modified cre is correctly translated.
Transfection experiments using PC-3 cells demonstrated that the presence of the second conditionally expressed modified cre-containing plasmid pCMV-RFP/CREM resulted in a 7-fold increase in luciferase activity compared to transfections in the absence of the second conditionally expressed Cre (Fig. 5B). In order to more clearly demonstrate the effect of Cre amplification by pCMV-RFP-CREM on luciferase expression using pCMV-STOP-luc, transfections were performed over several time points using varying amounts of Pr-cre (from 0.01 to 0.001 pmol) alone or with the additional inducible Cre recombinase using pCMV-RFP-CREM (Fig. 5C). A 32-fold increase in luciferase activity was observed using the gene expression amplification approach (Pr-cre and CMV-STOP-luc) compared to an equimolar amount of Pr-luc alone (Fig. 5C, 3 versus 1). The addition of pCMV-RFP-CREM further increased the level of luciferase activity by 9-fold (Fig. 5C, 4 versus 3). Thus, there was an ∼300-fold amplification of luciferase activity using the gene amplification approach with the second conditional cre construct compared to luciferase activity from Pr-cre alone. These results strongly suggest that the addition of a second conditionally active Cre could significantly increase the effective expression from other weak promoters.
Generation of a single vector containing a modified cre regulated by an inducible, tissue-specific promoter and conditionally expressed reporter gene
Having established that the modified cre maintained full activity for recombination of LOX target sequences, the modified cre, placed under the transcriptional control of the tissue-specific, androgen-inducible probasin promoter, was placed in cis with the expression reporter construct to generate pPr-CREM/CMV-STOP-luc (Fig. 6A). In order to prevent the strong enhancer activity of the CMV regulatory region from activating the upstream probasin promoter, an additional construct was made by placing a set of chicken globin insulators (pPr-CREM/Ins/CMV-STOP-luc). Both vectors were assayed in transient transfection experiments to determine whether the expression of the reporter genes was inducible by mibolerone and whether the insulators reduced potential background activity that might otherwise occur through the action of the CMV enhancer on the probasin promoter (Fig. 6B).
In order to determine whether chicken globin insulators might alter the level of expression from Pr-cre, insulators were added to flank Pr-cre to generate pPr-CREM/Ins (Fig. 5A) and co-transfected with pCMV-STOP-luc. No difference in luciferase activity was observed whether insulators did or did not flank Pr-cre (Fig. 6B, columns 4 and 6; P = 0.161, Mann–Whitney test). When the single construct containing pPr-CREM/CMV-STOP-luc was transfected in the absence of mibolerone, a significant basal level of luciferase activity was observed compared to co-transfection of the separate constructs Pr-CREM and CMV-STOP-luc (Fig. 6B, columns 3 and 7). This was presumably due to the strong transcriptional activating effect of the CMV enhancer on the probasin promoter. However, when the set of insulators was placed between the Pr-CREM and CMV-STOP-luc cassettes (pPr-CREM/Ins/CMV-STOP-luc), the uninduced level of luciferase activity was reduced ∼5-fold compared to the single construct without insulators (Fig. 6B, columns 7 and 9; P = 0.0001, Mann–Whitney test). The levels of mibolerone-induced luciferase activities were similar whether or not the insulators were present (Fig. 6B, columns 8 and 10; P = 0.297, Mann–Whitney test). No significant difference in inducible luciferase activities were observed using the single Pr-CREM/CMV-STOP-luc or Pr-CREM/Ins/CMV-STOP-luc vectors comparing to the co-transfection of the individual constructs (Fig. 6B, columns 4, 8 and 10; P = 0.681, Kruskal–Wallis test). These results demonstrate that the single vector containing the modified cre under an inducible promoter and the reporter cassette in cis worked equally as well as separate constructs in trans.
DISCUSSION
The results of this work demonstrate several novel modifications of Cre-LOX technology that can be applied to both human gene therapy and transgenic animal manipulations including the Cre-inducible activation or switching of gene expression, tissue-specific gene expression amplification and the development of single vectors containing both a modified cre recombinase and LOX recognition sequences.
Although the tissue-specific expression of Cre activation of gene transcription has been previously reported (11,20,24–26) these approaches have generally relied upon the use of a STOP cassette to prevent transcriptional read-through flanked by LOX recombination sites (14,24). This method is adequate for certain experimental approaches such as β-gal expression, but is greatly limited by the fact that the STOP cassette does not absolutely prevent transcriptional read-through (C.Couldrey, unpublished data). Thus, highly transforming genes or genes whose biological activity only require very low levels of expression may still be transcribed despite the presence of the STOP cassette leading to unwanted biological activities. In order to overcome this technical limitation, we developed a strategy in which the switch in expression from one gene to another relied upon the design of altering the translational reading frame upon recombination of LOX sites in the presence of Cre (Fig. 1A). We have demonstrated the utility of this approach using several reporter genes including lacZ, GFP, RFP and luciferase.
Although numerous tissue-specific expression vectors exist, levels of transcriptional activity are often much lower than is desired to effect a biological response when applied to transgenic or gene therapy approaches. In order to overcome this technical limitation, we have demonstrated a method in which the level of gene expression from a relatively weak but tissue-specific promoter can be amplified through the Cre-induced transcriptional activation using a very strong promoter. Using this technical approach, we have shown that the level of transcription using the prostate-specific probasin promoter can be amplified 30-fold. When the use of a second, conditionally expressed Cre was employed, the amplification of the reporter increased an additional 10-fold resulting in a total amplification of reporter gene expression of 300-fold. This method can readily be applied to other tissue-specific but relatively weak promoters in transgenic applications and gene therapy approaches when high levels of expression are warranted. However, depending upon the experimental questions, low levels of tissue-specific expression of a particular gene may be more appropriate using existing approaches.
A limiting feature of Cre-LOX technology has been the fact that it has not been possible to develop single constructs in which an active Cre recombinase could be contained in cis with a pair of LOX recombination sites since the LOX sites would be combined when grown in bacteria. This has necessitated the use of two separate genetic constructs in which cre and LOX sites exist in trans. This requires the generation of two separate lines of transgenic animals or the less efficient introduction of two separate constructs in in vitro or gene therapy strategies. We have demonstrated that our modifications to cre recombinase allow for the placement of a modified cre in the same construct containing LOX recombination sites. The addition of chicken globin insulators reduces potential enhancer effects from a strong transcriptional regulatory unit on the relatively weak but tissue-specific promoter driving the expression of Cre. Although background levels of expression remain somewhat elevated, the addition of flanking insulators may overcome this problem.
The strategies and modifications to cre recombinase presented in this work will have broad applications in developing tissue-specific inducible gene expression systems.
References
- 1.Le Menuet D., Zennaro,M.C., Viengcharen,S. and Lombes,M. (2000) Transgenic mouse models to study human mineralocorticoid receptor function in vivo. Kidney Int., 57, 1299–1306. [DOI] [PubMed] [Google Scholar]
- 2.Heintz N. (2000) Analysis of mammalian central nervous system gene expression and function using bacterial artificial chromosome-mediated transgenesis. Hum. Mol. Genet., 9, 937–943. [DOI] [PubMed] [Google Scholar]
- 3.Green J.E., Shibata,M.A., Yoshidome,K., Liu,M.L., Jorcyk,C., Anver,M.R., Wigginton,J., Wiltrout,R., Shibata,E., Kaczmarczyk,S., Wang,W., Liu,Z.Y., Calvo,A. and Couldrey,C. (2000) The C3(1)/SV40 T-antigen transgenic mouse model of mammary cancer: ductal epithelial cell targeting with multistage progression to carcinoma. Oncogene, 19, 1020–1027. [DOI] [PubMed] [Google Scholar]
- 4.Stern M.H. (1999) Transgenic models of T-cell prolymphocytic leukaemia. Haematologica, 84, 64–66. [PubMed] [Google Scholar]
- 5.Aguzzi A., Brandner,S., Sure,U., Ruedi,D. and Isenmann,S. (1994) Transgenic and knock-out mice: models of neurological disease. Brain Pathol., 4, 3–20. [DOI] [PubMed] [Google Scholar]
- 6.Hocker M. and Wiedenmann,B. (1998) Molecular mechanisms of enteroendocrine differentiation. Ann. N. Y. Acad. Sci., 859, 160–174. [DOI] [PubMed] [Google Scholar]
- 7.Pich E.M. and Epping-Jordan,M.P. (1998) Transgenic mice in drug dependence research. Ann. Med., 30, 390–396. [DOI] [PubMed] [Google Scholar]
- 8.Sternberg N. and Hamilton,D. (1981) Bacteriophage P1 site-specific recombination. I. Recombination between loxp sites. J. Mol. Biol., 150, 467–486. [DOI] [PubMed] [Google Scholar]
- 9.Akagi K., Sandig,V., Vooijs,M., Vander,V., Giovannini,M., Strauss,M. and Berns,A. (1997) Cre-mediated somatic site-specific recombination in mice. Nucleic Acids Res., 25, 1766–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gu H., Marth,J.D., Orban,P.C., Mossmann,H. and Rajewsky,K. (1994) Deletion of a DNA polymerase β gene segment in T cells using cell type-specific gene targeting. Science, 265, 103–106. [DOI] [PubMed] [Google Scholar]
- 11.Kaczmarczyk S.J. (1997) The role of peripheral insulin resistance in the aetiology of non-insulin dependent diabetes (type 2 diabetes). PhD Thesis, University of Melbourne, Melbourne, Australia.
- 12.Kawamoto S., Niwa,H., Tashiro,F., Sano,S., Kondoh,G., Takeda,J., Tabayashi,K. and Miyazaki,J. (2000) A novel reporter mouse strain that expresses enhanced green fluorescent protein upon Cre-mediated recombination. FEBS Lett., 470, 263–268. [DOI] [PubMed] [Google Scholar]
- 13.Kuhn R., Schwenk,F., Aguet,M. and Rajewsky,K. (1995) Inducible gene targeting in mice. Science, 269, 1427–1429. [DOI] [PubMed] [Google Scholar]
- 14.Lakso M., Sauer,B., Mosinger,B.,Jr, Lee,E.J., Manning,R.W., Yu,S.H., Mulder,K.L. and Westphal,H. (1992) Targeted oncogene activation by site-specific recombination in transgenic mice. Proc. Natl Acad. Sci. USA, 89, 6232–6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lobe C.G., Koop,K.E., Kreppner,W., Lomeli,H., Gertsenstein,M. and Nagy,A. (1999) Z/AP, a double reporter for cre-mediated recombination. Dev. Biol., 208, 281–292. [DOI] [PubMed] [Google Scholar]
- 16.Mao X., Fujiwara,Y. and Orkin,S.H. (1999) Improved reporter strain for monitoring Cre recombinase-mediated DNA excisions in mice. Proc. Natl Acad. Sci. USA, 96, 5037–5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagy A., Moens,C., Ivanyi,E., Pawling,J., Gertsenstein,M., Hadjantonakis,A.K., Pirity,M. and Rossant,J. (1998) Dissecting the role of N-myc in development using a single targeting vector to generate a series of alleles. Curr. Biol., 8, 661–664. [DOI] [PubMed] [Google Scholar]
- 18.Orban P.C., Chui,D. and Marth,J.D. (1992) Tissue- and site-specific DNA recombination in transgenic mice. Proc. Natl Acad. Sci. USA, 89, 6861–6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rajewsky K., Gu,H., Kuhn,R., Betz,U.A., Muller,W., Roes,J. and Schwenk,F. (1996) Conditional gene targeting. J. Clin. Invest., 98, 600–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rucker E.B. and Piedrahita,J.A. (1997) Cre-mediated recombination at the murine whey acidic protein (mWAP) locus. Mol. Reprod. Dev., 48, 324–331. [DOI] [PubMed] [Google Scholar]
- 21.Sauer B. (1993) Manipulation of transgenes by site-specific recombination: use of Cre recombinase. Methods Enzymol., 225, 890–900. [DOI] [PubMed] [Google Scholar]
- 22.Sauer B. (1998) Inducible gene targeting in mice using the Cre/lox system. Methods, 14, 381–392. [DOI] [PubMed] [Google Scholar]
- 23.St Onge L., Furth,P.A. and Gruss,P. (1996) Temporal control of the Cre recombinase in transgenic mice by a tetracycline responsive promoter. Nucleic Acids Res., 24, 3875–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsien J.Z., Chen,D.F., Gerber,D., Tom,C., Mercer,E.H., Anderson,D.J., Mayford,M., Kandel,E.R. and Tonegawa,S. (1996) Subregion- and cell type-restricted gene knockout in mouse brain. Cell, 87, 1317–1326. [DOI] [PubMed] [Google Scholar]
- 25.Utomo A.R., Nikitin,A.Y. and Lee,W.H. (1999) Temporal, spatial, and cell type-specific control of Cre-mediated DNA recombination in transgenic mice. Nat. Biotechnol., 17, 1091–1096. [DOI] [PubMed] [Google Scholar]
- 26.Wagner K.U., Wall,R.J., St Onge,L., Gruss,P., Wynshaw-Boris,A., Garrett,L., Li,M., Furth,P.A. and Hennighausen,L. (1997) Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res., 25, 4323–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niwa H., Yamamura,K. and Miyazaki,J. (1991) Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene, 108, 193–199. [DOI] [PubMed] [Google Scholar]
- 28.Kozak M. (1987) An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res., 15, 8125–8148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chung J.H., Whiteley,M. and Felsenfeld,G. (1993) A 5′ element of the chicken β-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell, 74, 505–514. [DOI] [PubMed] [Google Scholar]
- 30.Yan Y., Sheppard,P.C., Kasper,S., Lin,L., Hoare,S., Kapoor,A., Dodd,J.G., Duckworth,M.L. and Matusik,R.J. (1997) Large fragment of the probasin promoter targets high levels of transgene expression to the prostate of transgenic mice. Prostate, 32, 129–139. [DOI] [PubMed] [Google Scholar]
- 31.Hoess R.H., Wierzbicki,A. and Abramski,A. (1986) The role of the loxP spacer region in P1 site-specific recombination. Nucleic Acids Res., 14, 2287–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]