

Abstract
Parkinson's disease (PD) is a progressive neurodegenerative movement disorder primarily affecting the nigrostriatal dopaminergic system. The link between heightened activity of glycogen synthase kinase 3β (GSK3β) and neurodegene‐rative processes has encouraged investigation into the potential disease‐modifying effects of novel GSK3β inhibitors in experimental models of PD. Therefore, the intriguing ability of several anesthetics to readily inhibit GSK3β within the cortex and hippocampus led us to investigate the effects of brief isoflurane anesthesia on striatal GSK3β signaling in naïve rats and in a rat model of early‐stage PD. Deep but brief (20‐min) isoflurane anesthesia exposure increased the phosphorylation of GSK3β at the inhibitory Ser9 residue, and induced phosphorylation of AKTThr308 (protein kinase B; negative regulator of GSK3β) in the striatum of naïve rats and rats with unilateral striatal 6‐hydroxydopamine (6‐OHDA) lesion. The 6‐OHDA protocol produced gradual functional deficiency within the nigrostriatal pathway, reflected as a preference for using the limb ipsilateral to the lesioned striatum at 2 weeks post 6‐OHDA. Interestingly, such motor impairment was not observed in animals exposed to four consecutive isoflurane treatments (20‐min anesthesia every 48 h; treatments started 7 days after 6‐OHDA delivery). However, isoflurane had no effect on striatal or nigral tyrosine hydroxylase (a marker of dopaminergic neurons) protein levels. This brief report provides promising results regarding the therapeutic potential and neurobiological mechanisms of anesthetics in experimental models of PD and guides development of novel disease‐modifying therapies.
Keywords: anesthesia, dopamine, neurodegeneration, phosphorylation, sensorimotor test
Abbreviations used
- 6‐OHDA
6‐hydroxydopamine
- AKT
protein kinase B
- anova
analysis of variance
- AP
anterior‐posterior
- DA
dopamine
- DV
dorsal‐ventral
- ECT
electroconvulsive therapy
- EEG
electroencephalogram
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- GSK3β
glycogen synthase kinase 3β
- L
lateral
- NF‐kB
nuclear factor kappa‐light‐chain‐enhancer of activated B cells
- O2
oxygen
- PD
Parkinson's disease
- PI3K
phosphoinositide 3‐kinase
- SEM
standard error of mean
- Ser
serine
- SN
substantia nigra
- Thr
threonine
- TH
tyrosine hydroxylase
Parkinson's disease (PD) is a multifactorial neurodegenerative disease with no curative treatment. Gradual degeneration of dopaminergic neurons in the midbrain substantia nigra and resulting dopamine (DA) deficiency in the striatum are pathological hallmarks and causes of the progressive motor symptoms of PD (Schapira 2009; Meissner et al. 2011). Drugs compensating striatal DA deficiency initially ameliorate motor symptoms associated with PD; however, their efficacy eventually subsides because of progressive neurodegeneration (Schapira 2009; Meissner et al. 2011). Disease‐modifying therapies that prevent further neurodegeneration and/or bring adaptive neurorestorative changes are thus desperately needed for the management of PD symptomologies (Meissner et al. 2011).
The established link between dysregulation of glycogen synthase kinase 3β (GSK3β), a promiscuous and multifunctional serine‐threonine kinase (Jope and Johnson 2004), and neurodegenerative processes has recently encouraged studies on the potential disease‐modifying effects of novel GSK3β inhibitors in experimental models of PD with promising results (Credle et al. 2015; Golpich et al. 2015). In addition to pharmacological inhibition, the activity and efficacy of GSK3β can be dynamically regulated through specific post‐translational phosphorylation modifications (Jope and Johnson 2004). Phosphorylation of the inhibitory serine 9 residue within the N‐terminus is considered among the most important mechanisms reducing GSK3β activity (Ilouz et al. 2008). Intriguingly, diverse anesthetics, including the commonly used volatile anesthetic isoflurane, strongly and rapidly increases GSK3βSer9 phosphorylation in the adult rodent cortex and hippocampus (Li et al. 2005; Kohtala et al. 2016). This prompted us to hypothesize that brief exposure(s) to isoflurane induces similar GSK3βSer9 phosphorylation in the striatum, and results in improved motor performance in a rat model of early‐stage PD.
Materials and methods
Animals
Adult male Wistar rats (Laboratory Animal Center of the University of Eastern Finland, Kuopio, Finland; Charles River, Sulzfeld, Germany) were used. The rats weighed 200–300 g at the beginning of the experiments. Unless otherwise stated, the rats were group‐housed (3–4 rats/cage) in stainless steel cages and kept on a 12/12 h light/dark cycle (lights on at 07:00 AM) at an ambient temperature of 22 ± 1°C. Pelleted food (Teklad 2016S; Harlan Inc., Indianapolis, IN, USA) and tap water were freely available. The experiments were carried out during the light phase. The procedures were performed in compliance with the European Communities Council Directive of 24 November 1986 (86/609/EEC) and were approved by the County Administrative Board of Southern Finland (ESAVI/1350/04.10.03/2011, ESAVI‐2014‐701; ESAVI/10527/04.10.07/2014). All efforts were made to minimize animal suffering.
Experimental design
The experiments were performed in three cohorts. In the first cohort, the acute effect of a single isoflurane anesthesia exposure (20‐min) on GSK3βSer9 and AKTThr308 phosphorylation in the cortex and striatum was investigated (N = 4/group). The second cohort addressed whether GSK3βSer9 and AKTThr308 phosphorylation is regulated within the striatum after repeated isoflurane administrations (20‐min exposure delivered every 48 h for five times) in naïve rats (N = 6/group). In the third cohort, the effects of repeated isoflurane exposures on GSK3βSer9 and AKTThr308 signaling, tyrosine hydroxylase (TH) protein expression, and motor performance in a rat model of early‐stage PD (N = 9/group) were evaluated as follows: (i) unilateral intrastriatal injection of dopaminergic neurotoxin 6‐OHDA (6‐hydroxydopamine) was performed on day 0; (ii) isoflurane anesthesia (20‐min) was first administered on days 7, 9, 11, and 13 after 6‐OHDA lesion; (iii) motor performance was tested on day 14 (24 h after the previous anesthesia exposure); (iv) animals received a final 20‐min isoflurane anesthesia exposure on day 15, after which striatal and nigral samples were collected for western blot analyses. This experimental setup allowed us to analyze both motor performance and molecular changes within the same animals. In each experiment, rats were assigned arbitrarily to treatment groups. Group sizes were determined on the basis of expected effect size and variation observed in our previous experiments (Kohtala et al. 2016; Leikas et al. 2017). Power analyses were conducted using online tools (www.powerandsamplesize.com) to determine a sufficient sample size using an alpha of 0.05, a power of 0.80, and estimated effect size of two in biochemical assays and 1.5 in behavioral tests as calculated by Cohen's d value (estimated mean difference between groups/pooled standard deviation). Based on the aforementioned assumptions, the desired minimum sample size for biochemical assays was four and for behavioral tests, seven. In the behavioral studies, the observer was blinded to the treatment and lesioning protocols.
Drugs
6‐OHDA·HCl (Sigma‐Aldrich, St Louis, MO, USA) was dissolved in 0.9% NaCl containing 0.2 mg/mL ascorbic acid. Pentobarbital (Mebunat vet®, Orion Pharma, Espoo, Finland) and buprenorphine (Temgesic®, Reckitt & Colman Products Ltd, Hull, UK) were dissolved in saline. Lidocaine (Xylocain®, Astra Zeneca Oy, Espoo, Finland) was obtained from AstraZeneca and isoflurane from Virbac (Vetflurane®, Virbac Animal Health Ltd. Bury St Edmunds, Suffolk, UK). The doses of drugs refer to the free bases.
Deep isoflurane anesthesia
After anesthesia induction (3–4% ~ 2 min), the anesthetic concentration was gradually reduced to ~ 2% for 20 min (O2 flow: 0.3−0.5 L/min). Control animals (sham anesthesia) were kept in the induction chamber for 2 min with flowing O2 and no anesthetic. Animals were randomly assigned for the treatments. The depth of anesthesia set by the employed isoflurane administration protocol in rats was pre‐determined using electroencephalogram (EEG) (Kohtala et al. 2016) (Fig. 1a). Briefly, EEG electrodes were implanted under isoflurane anesthesia. Tetracaine and bupivacaine were used for local analgesia and buprenorphine (0.045 mg/kg, i.m., Temgesic®, Reckitt & Colman Products Ltd, Hull, UK) for post‐operative care. Electrodes were placed bilaterally above the frontal cortex (Take out AP +2.7 mm; L ± 4.0 mm) (Paxinos and Watson 2007). A reference electrode was placed above the parietal cortex (AP −2.0 mm; L −2.0 mm). Screw electrodes were fixed at one and a half turn into the skull, so that the tip of the screw sat just above the cortex. Recovery time after surgery was 2 weeks. The EEG signals were amplified and filtered (high/low pass: 1/100 Hz) with one‐channel AC amplifier (ADInstruments Ltd., Bella Vista, New South Wales, Australia) and PowerLab/800s analog‐digital converter (ADInstruments), and recorded using PowerLab (ADInstruments). The EEG data was processed using Spike2 (version 6; Cambridge Electronic Design Ltd., Milton, UK).
Figure 1.
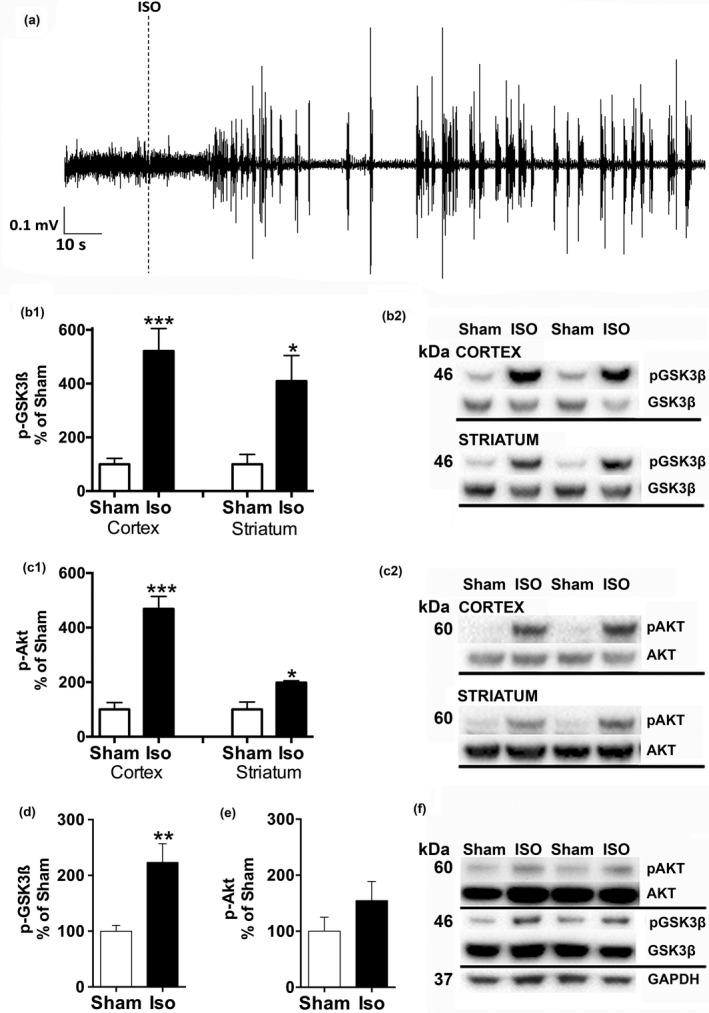
Isoflurane anesthesia exposure increases GSK3βSer9 and AKTThr308 phosphorylation in the adult rat cortex and striatum. Representative cortical EEG spectrogram demonstrating the burst suppression pattern during isoflurane administration (4% induction for 2 min, 2% maintenance) (a). Effect of a single brief isoflurane anesthesia exposure (4% induction for 2 min, 2% maintenance for 20 min; N = 4/group) on GSK3βSer9 phosphorylation (b) and AKTThr308 phosphorylation (c) in the adult rat cortex and striatum. Striatal GSK3βSer9 and AKTThr308 phosphorylation after repeated (20‐min anesthesia exposure every 48 h for total of five consecutive times; N = 6/group) isoflurane administration (d–f). *p < 0.05; **p < 0.01; ***p < 0.005, Student t‐test. EEG, electroencephalogram; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; GSK3β, glycogen synthase 3β; AKT, protein kinase B; ISO, isoflurane; Sham, rats in the induction chamber for 2 min with continuous O2 flow (with no anesthetics).
Dissection of brain samples and immunoblotting
Rats were killed by decapitation immediately after a single isoflurane/sham anesthesia, or after the last of a train of consecutive anesthesia treatments. The brains were removed and cortex, both striata, and bilateral substantia nigra areas were dissected on ice, frozen and stored at −80°C until analysis. The samples were homogenized in NP buffer [137 mM NaCl, 20 mM Tris, 1% NP‐40, 10% glycerol, 48 mM NaF, H2O, Complete inhibitor mix (Roche Molecular Biochemicals, Indianapolis, IN, USA), PhosphoStop (Roche)], incubated on ice, and centrifuged (16 000 g, 15 min, +4°C), with the resulting supernatant collected for analysis. Sample protein concentrations were measured using Bio‐Rad DC assay (Bio‐Rad Laboratories, Hercules, CA, USA). Western blotting was performed as previously described (Kohtala et al. 2016). The following primary antibodies were used: anti‐pAKTThr308 (1 : 500; Cell Signaling Technology, Beverly, MA, USA Cat# 9275, RRID:AB_329828), anti‐AKT (1 : 1000; Cell Signaling Technology Cat# 9272, RRID:AB_329827), anti‐p‐GSK3βSer9 (1 : 1000; Cell Signaling Technology Cat# 9336, RRID:AB_331405), anti‐GSK3β (1 : 1250; Cell Signaling Technology Cat# 9315, RRID:AB_490890), anti‐TH (1 : 1000; Millipore Corporation, Bedford, MA, USA Cat# MAB318, RRID:AB_2201528; kindly provided by Dr. Timo Myöhänen), anti‐glyceraldehyde 3‐phosphate dehydrogenase (1 : 5000; Santa Cruz Biotechnology, Santa Cruz, CA, USA Cat# sc‐25778, RRID:AB_10167668). Horseradish peroxidase‐conjugated secondary antibodies (1 : 10 000 in non‐fat dry milk, 1 h at 21°C temperature; Bio‐Rad) and enhanced chemiluminescence (Clarity™ Western ECL Substrate; Bio‐Rad Laboratories) were used for detection by ChemiDoc MP camera (Bio‐Rad Laboratories, Vantaa, Finland). Optical densities were calculated with ImageJ 1.5 (National institutes of Health, Washington DC, USA). The expression levels of AKT, GSK3β, and TH were normalized to glyceraldehyde 3‐phosphate dehydrogenase.
Unilateral 6‐OHDA lesion model of early‐stage Parkinson′s disease
Lesions were performed as described (Leikas et al. 2017). Briefly, the rats were anesthetized with pentobarbital (50 mg/kg, i.p.). Lidocaine was applied to the scalp and to the surface of the skull and a single dose of buprenorphine (0.02 mg/kg, s.c.) was given to relieve post‐operative pain. The 6‐OHDA (10 μg/4 μL) was infused into the right striatum (AP +1.0; L −2.7; DV −5.0) (Paxinos and Watson 2007) at a flow rate 0.5 μL/min. The needle was retained in position for 4 min to prevent backflow. The animals were housed individually for 3 days after the surgery.
Sensorimotor tests
6‐OHDA‐lesioned animals were administered isoflurane (or sham) anesthesia on days 7, 9, 11, and 13. On day 14 (24 h after a preceding isoflurane exposure) the nigrostriatal motor functions of the animals were evaluated using sensorimotor behavioral tests with a combined ipsilateral limb use score for each animal. The overall limb use asymmetry score was previously utilized by O'Dell et al. (2007), and this approach has been shown to be more sensitive in detecting motor deficits in partially 6‐OHDA‐lesioned rats compared to individual tests alone (Leikas et al. 2017). The battery consisted of sensorimotor tests with proven ability to detect diverse and varying levels of motor deficits caused by 6‐OHDA lesion. In each test, the ipsilateral limb use was assessed by calculating successful limb placings with the following equation: (ipsilateral/ipsilateral + contralateral) − (contralateral/ipsilateral + contralateral). Thus, the calculated combined ipsilateral score values ranged from +1.0 (exclusive use of ipsilateral forelimb to the lesion), through 0 (no asymmetric limb use) to −1.0 (exclusive use of contralateral forelimb). The tests were performed by an experimenter, blind to treatments and lesioning protocols, consecutively in the following order (i) the vibrissae test, where the rat was held by its torso allowing the forelimbs to move freely. The vibrissae were brushed against the surface of a table, and the ability of the rat to place its forelimbs on the surface was monitored in 10 consecutive trials. (ii) The cylinder test, where the rat was placed into a clear cylinder (20 cm diameter, 30 cm height) for 3 min. Animal′s exploratory behavior was video‐recorded, and the use of ipsilateral and contralateral forelimbs were scored during vertical exploration along the cylinder walls. (iii) The movement initiation test, where the rat was held by its torso above a table surface so that one forelimb bore the weight of the animal. The average number of self‐initiated steps in eight consecutive 10‐second trials for each forelimb was monitored. (iv) The adjusting steps test, where the rat was held by its torso above a table surface so that one forelimb bore the weight of the animal. The rat was moved laterally (~ 1 m) on the table top, first in the forehand and then in the backhand direction, and the number of stepping adjustments were calculated. The average number of adjusting steps taken in eight consecutive trials was monitored for each forelimb.
Statistical analysis
Results are expressed as a percentage of the Sham (intact side) anesthesia administration or intact side as mean ± SEM (standard error of mean). Statistical analyses were done in GraphPad Prism 5.03 software (GraphPad Software; GraphPad Software Inc., San Diego, CA, USA) using two‐sample two‐tailed Student t‐test or, when applicable (two categorical independent variables), two‐way anova (analysis of variance) followed by Newmann–Keuls post hoc test. No data were excluded from the analyses. The criterion for statistical significance was set at p < 0.05.
Results
Isoflurane anesthesia increases GSK3βSer9 and AKTThr308 phosphorylation in the cortex and striatum
Isoflurane administration protocol consisting of ~ 4% induction phase and ~ 2% maintenance readily and rapidly induces the burst suppression pattern in the EEG (a representative EEG graph shown in Fig. 1a). Such deep isoflurane anesthesia has been shown to increase GSK3βSer9 phosphorylation in the adult rodent hippocampus (Kohtala et al. 2016). To investigate whether isoflurane anesthesia produces similar phosphorylation of GSK3β in the striatum and cortex, we exposed naïve rats to a 20‐min isoflurane anesthesia exposure, and obtained cortical and striatal tissues for western blot analyses. Indeed, the phospho‐GSK3βSer9 levels were significantly up‐regulated in both cortex and striatum (p < 0.005 and p < 0.05, respectively; Student t‐test) of anesthetized animals (Fig. 1b). The phosphorylation state of AKTThr308, one of the upstream regulators of GSK3β, was also significantly increased in both brain regions (p < 0.005 and p < 0.05, respectively; Student t‐test) (Fig. 1c). The ability of isoflurane to regulate striatal GSK3βSer9 phosphorylation was also observed following repeated administration (20‐min anesthesia exposure every 48 h for a total of five times) (p < 0.001; Student t‐test) although phosphorylation of AKTThr308 was not significantly increased (p = 0.22, Student t‐test) (Fig. 1d).
Isoflurane regulates the striatal AKT‐GSK3β pathway and improves motor deficits in an animal model resembling early‐stage Parkinson′s disease
Slowly developing degeneration of DA neurons within the nigrostriatal pathway is the pathological hallmark of PD. Although this pathology cannot be fully recapitulated in animals, intrastriatal injection of neurotoxin 6‐OHDA produces gradual degeneration of nigrostriatal DA neurons and motor impairments resembling PD phenotypes in rats. We utilized our recently validated unilateral 6‐OHDA rat model of early‐stage PD (Leikas et al. 2017) to determine whether repeated brief isoflurane anesthesia exposures (20‐min anesthesia exposure every 48 h for a total of four times; starting 7 days after 6‐OHDA infusion) influences the emergence of sensorimotor impairments induced by intrastriatal 6‐OHDA (Fig. 2a). To reduce the potential acute influence of anesthesia motor performance, the sensorimotor test battery was performed 24 h after the preceding isoflurane administration. As expected, rats subjected to repeated sham anesthesia (O2) preferred the use of the ipsilateral limb corresponding to the lesion site, as revealed by the combined ipsilateral limb use score from the behavioral tests (Fig. 2b). Importantly, these sensorimotor deficits were completely absent in animals exposed to brief isoflurane anesthesia delivered every second day for a total of four consecutive times (Fig. 2b; O2 vs. isoflurane: p < 0.05; Student t‐test). To assess locomotor activity of the animals, the amount of spontaneous exploratory rearings in the cylinder test were monitored. We found no statistical difference between treatment groups (sham 17.7 ± 1.6; Isoflurane 15.8 ± 2.2; p = 0.500; Student t‐test), indicating normal locomotor activity in isoflurane‐treated animals.
Figure 2.
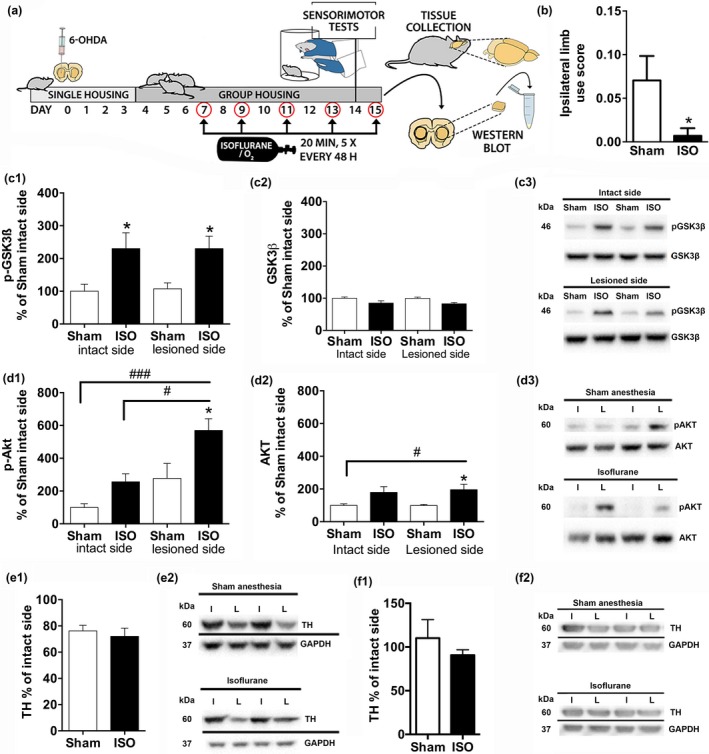
Isoflurane anesthesia exposure regulates striatal AKT‐GSK3β signaling and ameliorates motor deficits in an animal model of early‐stage Parkinson′s disease. (a) Experimental workflow. Unilateral intrastriatal 6‐hydroxydopamine (10 μg) administration was performed on day 0. Animals were exposed to isoflurane (4% induction for 2 min, 2% maintenance for 20 min; N = 9) or sham conditions (rats in the induction chamber for 2 min with O2 flow on; N = 9) on days 7, 9, 11, and 13. On day 14 (24 h after previous anesthesia exposure), animals were subjected to behavioral tests, assessing sensorimotor functions (cylinder, vibrissae, movement initiation, and adjusting step tests). The animals were exposed to an additional isoflurane/sham anesthesia treatment on day 15, after which the striata were collected for analyses. (b) Effect of repeated isoflurane anesthesia exposure on motor functions (combined ipsilateral limb use score of sensorimotor performance); (c) Phospho‐GSK3βSer9/total‐GSK3β ratio and total GSK3β normalized to GAPDH; (d) Phospho‐AKTThr308/total‐AKT ratio and total AKT normalized to GAPDH; (e) The ratio of TH protein in lesioned versus intact striatum. (f) The ratio of TH protein in lesioned versus intact substantia nigra area. #/*p < 0.05; ###/***p < 0.005, Student t‐test (b, e–f) or two‐way anova followed by Newmann–Keuls post hoc test (c–d). GAPDH, Glyceraldehyde 3‐phosphate dehydrogenase; GSK3β, glycogen synthase 3β; AKT, protein kinase B; ISO, isoflurane; I, intact hemisphere; L, lesioned hemisphere; TH, tyrosine hydroxylase.
To confirm that the ability of isoflurane to regulate GSK3β phosphorylation is not altered by progression of the 6‐OHDA‐induced lesion, we exposed the animals to one more anesthesia treatment and collected striata for analysis. The ratio of phospho‐GSK3βSer9/total‐GSK3β was similarly up‐regulated in the striatum of both lesioned and non‐lesioned animals following anesthesia exposure (Fig. 2c; p < 0.05 on intact side, p < 0.01 on lesioned side; Newmann–Keuls post hoc test). Isoflurane also increased phospho‐AKTThr308 levels to the same degree in both striata (Fig. 2d; p < 0.01 on intact side, p < 0.05 on lesioned side, Newmann–Keuls post hoc test). However, since basal phospho‐AKTThr308 levels tend to be elevated in the lesioned side, the effect of isoflurane appears more prominent in this side. Isoflurane also increased AKT protein levels in both intact and lesioned striatum although this effect was statistically significant only in the latter (p < 0.05, sham vs. isoflurane on lesioned side; Newmann–Keuls post hoc test) (Fig. 2d). The total protein levels of GSK3β remained unaltered by the lesion or anesthesias (Fig. 2c).
To investigate whether isoflurane directly influences the progression of 6‐OHDA‐induced lesion, we measured the total levels of TH, a marker of dopaminergic neurons, in the striatum. TH levels were similarly down‐regulated in the lesioned striatum compared to intact side of both sham and isoflurane‐treated rats (p < 0.05 and p < 0.01, respectively; Student t‐test) (Fig. 2e). This result indicates that the apparent beneficial effects of isoflurane anesthesia exposure against sensorimotor impairment in 6‐OHDA‐lesioned rats is not directly attributed to neuroprotection. Determination of striatal TH immunoreactivity in brain sections is, however, required to confirm this hypothesis.
Intrastriatal low‐dose 6‐OHDA infusion initially affects dopaminergic projections in the striatum and gradual loss of TH (and neuroapoptosis) within the substantia nigra is observed several weeks thereafter (Penttinen et al. 2016). Indeed, TH levels in substantia nigra were not significantly different between the non‐lesioned and lesioned samples at 2‐ weeks after toxin delivery in both the sham and isoflurane‐treated rats (Fig. 2f).
Discussion
GSK3β gene polymorphism, as well as GSK3 hyperactivity are associated with PD, and GSK3β inhibitors are expected to provide therapeutic benefits against PD pathologies (Kalinderi et al. 2011; Credle et al. 2015; Golpich et al. 2015). This study was set forth to investigate whether the ability of isoflurane, a commonly used volatile anesthetic, to regulate GSK3β signaling within the hippocampus (Kohtala et al. 2016) can be recapitulated in the rat striatum, where DA depletion is the underlying cause of motor deficits in PD. Indeed, phosphorylation of the inhibitory Ser9 residue of GSK3β was significantly increased in striatum after a single and repeated exposure to a brief isoflurane anesthesia in adult naïve rats. Concomitant phosphorylation of AKT, a negative regulator of GSK3β activity (Cross et al. 1995) was also observed indicating that isoflurane renders GSK3β inactive via AKT, although other kinases may also be involved (Pap and Cooper 1998; Jope and Johnson 2004). Guided by these results, we studied the effects of isoflurane on striatal AKT‐GSK3β signaling and motor deficits in a rat model of early‐stage PD. Our recently described intrastriatal unilateral 6‐OHDA infusion protocol reproducibly produces 40–60% depletion of striatal DA in the lesioned hemisphere, and causes motor deficits that can be quantified by a sensitive sensorimotor test battery (Leikas et al. 2017). Importantly, isoflurane anesthesia readily regulated AKT‐GSK3β signaling in the 6‐OHDA‐lesioned striatum and ameliorated motor deficits observed in this model.
Increased oxidative stress, mitochondrial dysfunction, and/or neuroinflammation may contribute to dopaminergic neurodegeneration associated with PD (Schapira 2009). Inactivation of GSK3β by the phosphoinositide 3‐kinase/AKT signaling pathway regulates inflammatory pathways; GSK‐3β inhibition reduces the production of pro‐inflammatory cytokines (like IL‐β, IL‐6, and IFN‐γ) and increases the release of anti‐inflammatory cytokines (like IL‐10) by modulating a crucial pro‐inflammatory pathway through NF‐κB (Martin et al. 2005; Zhang et al. 2014). Moreover, activation of phosphoinositide 3‐kinase/AKT signaling and GSK3β inhibition bring anti‐apoptotic effects (Pap and Cooper 1998) and exert neuroprotection in both in vitro and in vivo models of PD (Kozikowski et al. 2006; Wang et al. 2007; Gong et al. 2012). Yet, we did not find any effect of isoflurane on TH levels in striatum or substantia nigra after consecutive treatments. As isoflurane has been shown to modulate extracellular levels of DA (Irifune et al. 1997; Votaw et al. 2003; Adachi et al. 2005) – a functional and clinical biomarker of PD (Haas et al. 2012) – the observed improvement in motor performance after consecutive exposures to isoflurane anesthesia could be related to adaptive mechanisms leading into increased DA signaling in striatum rather than direct neuroprotection in the nigrostriatal dopaminergic pathway. Further studies with different isoflurane administration protocols and assessment of both cellular (DAT, TH+ cells) and functional (TH activity, DA levels) markers of the nigrostriatal dopaminergic pathway at different stages of 6‐OHDA‐induced lesions are needed to test this hypothesis. Notably, and as observed in some previous studies (Bychkov et al. 2007), phospho‐AKTThr308 tends to be elevated in the striatum in response to 6‐OHDA. Interestingly, however, this was not reflected as an increased phosphorylation of GSK3βSer9. Isoflurane also significantly increased the total levels of AKT.
With dozens of putative substrates, GSK3β orchestrates a plethora of cellular functions (Beurel et al. 2015). Indeed, dysregulation of GSK3β has been associated with numerous neurological and psychiatric conditions such as Alzheimer's disease, mood disorders, multiple sclerosis, and PD. Likewise, the beneficial effects of many of the currently available treatment interventions, including the mood‐stabilizer lithium and the rapid‐acting antidepressant ketamine, are related, in part, to their ability to inhibit GSK3β (O'Brien and Klein 2009; Zarate and Machado‐Vieira 2016). In addition, lithium has been shown to produce neuroprotective effects in some animal models of PD (Lieu et al. 2014; however, see Yong et al. 2011). Interestingly, the electroconvulsive therapy (ECT), an intervention used to treat drug‐resistant depression and schizophrenia, readily inhibits GSK3β (Roh et al. 2003; Kang et al. 2004), and provides anti‐parkinsonian effects in animal models (Nomikos et al. 1991; Strome et al. 2007; Tsen et al. 2013). Intriguingly, ECT has been shown to ameliorate severe motor deficits in non‐depressed patients with advanced PD (Andersen et al. 1987; Fall et al. 1995). Our observations support the hypothesis that GSK3β inhibition may provide beneficial effects in animal models of PD and provide evidence suggesting that clinically used anesthetics may function through this pathway. While general anesthesia (like ECT) cannot be considered as convenient therapeutic intervention for the management of PD, this brief report suggests that the therapeutic potential and molecular mechanisms of diverse anesthetics, in addition to administration protocols, should be evaluated in experimental models of PD to find potential new leads for the development of novel disease‐modifying therapies.
Author contributions
T.R. developed the original hypothesis for the study; A.J., M.F., T.R. designed the experiments; J.L., S.K., W.T., A.J. performed the experiments; J.L., S.K., A.J. performed statistical analyses; J.L., S.K., T.R. prepared the figures; J.L., A.J., M.F., T.R. wrote the paper.
Acknowledgments and conflict of interest disclosure
The authors thank Dr. Claudia Brandt and Virpi Perko for technical assistance. Dr. Giuseppe P. Cortese is thanked for language editing and his constructive comments on the manuscript. This study has been supported by grants from the Academy of Finland (T.R.; grants: 276333, 284569). Authors declare no conflict of interest related to this manuscript.
All experiments were conducted in compliance with the ARRIVE guidelines.
References
- Adachi Y. U., Yamada S., Satomoto M., Higuchi H., Watanabe K. and Kazama T. (2005) Isoflurane anesthesia induces biphasic effect on dopamine release in the rat striatum. Brain Res. Bull. 67, 176–181. [DOI] [PubMed] [Google Scholar]
- Andersen K., Balldin J., Gottfries C. G., Granérus A. K., Modigh K., Svennerholm L. and Wallin A. (1987) A double‐blind evaluation of electroconvulsive therapy in Parkinson's disease with “on‐off” phenomena. Acta Neurol. Scand. 76, 191–199. [DOI] [PubMed] [Google Scholar]
- Beurel E., Grieco S. F. and Jope R. S. (2015) Glycogen synthase kinase‐3 (GSK3): regulation, actions, and diseases. Pharmacol. Ther. 148, 114–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bychkov E., Ahmed M. R., Dalby K. N. and Gurevich E. V. (2007) Dopamine depletion and subsequent treatment with l‐DOPA, but not the long‐lived dopamine agonist pergolide, enhances activity of the Akt pathway in the rat striatum. J. Neurochem. 102, 699–711. [DOI] [PubMed] [Google Scholar]
- Credle J. J., George J. L., Wills J. et al (2015) GSK‐3β dysregulation contributes to parkinson's‐like pathophysiology with associated region‐specific phosphorylation and accumulation of tau and α‐synuclein. Cell Death Differ. 22, 838–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross D. A. E., Alessi D. R., Cohen P., Andjelkovich M. and Hemmings B. A. (1995) Inhibition of glycogen synthase kinase‐3 by insulin mediated by protein kinase B. Nature 378, 785–789. [DOI] [PubMed] [Google Scholar]
- Fall P. A., Ekman R., Granérus A. K., Thorell L. H. and Wålinder J. (1995) ECT in Parkinson's disease. Changes in motor symptoms, monoamine metabolites and neuropeptides. J. Neural. Transm. Park. Dis. Dement. Sect. 10, 129–140. [DOI] [PubMed] [Google Scholar]
- Golpich M., Amini E., Hemmati F., Ibrahim N. M., Rahmani B., Mohamed Z., Raymond A. A., Dargahi L., Ghasemi R. and Ahmadiani A. (2015) Glycogen synthase kinase‐3 beta (GSK‐3β) signaling: implications for Parkinson's disease. Pharmacol. Res. 97, 16–26. [DOI] [PubMed] [Google Scholar]
- Gong L., Zhang Q.‐L., Zhang N. et al (2012) Neuroprotection by urate on 6‐OHDA‐lesioned rat model of Parkinson's disease: linking to Akt/GSK3β signaling pathway. J. Neurochem. 123, 876–885. [DOI] [PubMed] [Google Scholar]
- Haas B. R., Stewart T. H. and Zhang J. (2012) Premotor biomarkers for Parkinson's disease – a promising direction of research. Transl. Neurodegener. 1, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilouz R., Pietrokovski S., Eisenstein M. and Eldar‐Finkelman H. (2008) New insights into the autoinhibition mechanism of glycogen synthase kinase‐3beta. J. Mol. Biol. 383, 999–1007. [DOI] [PubMed] [Google Scholar]
- Irifune M., Sato T., Nishikawa T., Masuyama T., Nomoto M., Fukuda T. and Kawahara M. (1997) Hyperlocomotion during recovery from isoflurane anesthesia is associated with increased dopamine turnover in the nucleus accumbens and striatum in mice. Anesthesiology 86, 464–475. [DOI] [PubMed] [Google Scholar]
- Jope R. S. and Johnson G. V. W. (2004) The glamour and gloom of glycogen synthase kinase‐3. Trends Biochem. Sci. 29, 95–102. [DOI] [PubMed] [Google Scholar]
- Kalinderi K., Fidani L., Katsarou Z., Clarimón J., Bostantjopoulou S. and Kotsis A. (2011) GSK3β polymorphisms, MAPT H1 haplotype and Parkinson's disease in a Greek cohort. Neurobiol. Aging 32, 546.e1–546.e5. [DOI] [PubMed] [Google Scholar]
- Kang U. G., Roh M.‐S., Jung J.‐R., Shin S. Y., Lee Y. H., Park J.‐B. and Kim Y. S. (2004) Activation of protein kinase B (Akt) signaling after electroconvulsive shock in the rat hippocampus. Prog. Neuropsychopharmacol. Biol. Psychiatry 28, 41–44. [DOI] [PubMed] [Google Scholar]
- Kohtala S., Theilmann W., Suomi T., Wigren H.‐K., Porkka‐Heiskanen T., Elo L. L., Rokka A. and Rantamäki T. (2016) Brief isoflurane anesthesia produces prominent phosphoproteomic changes in the adult mouse hippocampus. ACS Chem. Neurosci. 7, 749–756. [DOI] [PubMed] [Google Scholar]
- Kozikowski A. P., Gaisina I. N., Petukhov P. A., Sridhar J., King L. T., Blond S. Y., Duka T., Rusnak M. and Sidhu A. (2006) Highly potent and specific GSK‐3beta inhibitors that block tau phosphorylation and decrease alpha‐synuclein protein expression in a cellular model of Parkinson's disease. ChemMedChem 1, 256–266. [DOI] [PubMed] [Google Scholar]
- Leikas J. V., Kääriäinen T. M., Jalkanen A. J., Lehtonen M., Rantamäki T. and Forsberg M. M. (2017) Combined ipsilateral limb use score as an index of motor deficits and neurorestoration in parkinsonian rats. J. Neurosci. Res. https://doi.org/10.1002/jnr.24022. [DOI] [PubMed] [Google Scholar]
- Li X., Friedman A. B., Roh M.‐S. and Jope R. S. (2005) Anesthesia and post‐mortem interval profoundly influence the regulatory serine phosphorylation of glycogen synthase kinase‐3 in mouse brain. J. Neurochem. 92, 701–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieu C. A., Dewey C. M., Chinta S. J., Rane A., Rajagopalan S., Batir S., Kim Y.‐H. and Andersen J. K. (2014) Lithium prevents parkinsonian behavioral and striatal phenotypes in an aged parkin mutant transgenic mouse model. Brain Res. 1591, 111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M., Rehani K., Jope R. S. and Michalek S. M. (2005) Toll‐like receptor‐mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 6, 777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner W. G., Frasier M., Gasser T. et al (2011) Priorities in Parkinson's disease research. Nat. Rev. Drug Discov. 10, 377–393. [DOI] [PubMed] [Google Scholar]
- Nomikos G. G., Zis A. P., Damsma G. and Fibiger H. C. (1991) Electroconvulsive shock produces large increases in interstitial concentrations of dopamine in the rat striatum: an in vivo microdialysis study. Neuropsychopharmacology 4, 65–69. [PubMed] [Google Scholar]
- O'Brien W. T. and Klein P. S. (2009) Validating GSK3 as an in vivo target of lithium action. Biochem. Soc. Trans. 37, 1133–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dell S. J., Gross N. B., Fricks A. N., Casiano B. D., Nguyen T. B. and Marshall J. F. (2007) Running wheel exercise enhances recovery from nigrostriatal dopamine injury without inducing neuroprotection. Neuroscience 144, 1141–1151. [DOI] [PubMed] [Google Scholar]
- Pap M. and Cooper G. M. (1998) Role of glycogen synthase kinase‐3 in the phosphatidylinositol 3‐Kinase/Akt cell survival pathway. J. Biol. Chem. 273, 19929–19932. [DOI] [PubMed] [Google Scholar]
- Paxinos G. and Watson C. D. (2007) The Rat Brain in Stereotaxic Coordinates, 6th edn Elsevier, London. [DOI] [PubMed] [Google Scholar]
- Penttinen A.‐M., Suleymanova I., Albert K., Anttila J., Voutilainen M. H. and Airavaara M. (2016) Characterization of a new low‐dose 6‐hydroxydopamine model of Parkinson's disease in rat. J. Neurosci. Res. 94, 318–328. [DOI] [PubMed] [Google Scholar]
- Roh M.‐S., Kang U. G., Shin S. Y., Lee Y. H., Jung H. Y., Juhnn Y.‐S. and Kim Y. S. (2003) Biphasic changes in the Ser‐9 phosphorylation of glycogen synthase kinase‐3beta after electroconvulsive shock in the rat brain. Prog. Neuropsychopharmacol. Biol. Psychiatry 27, 1–5. [DOI] [PubMed] [Google Scholar]
- Schapira A. H. V. (2009) Neurobiology and treatment of Parkinson's disease. Trends Pharmacol. Sci. 30, 41–47. [DOI] [PubMed] [Google Scholar]
- Strome E. M., Zis A. P. and Doudet D. J. (2007) Electroconvulsive shock enhances striatal dopamine D1 and D3 receptor binding and improves motor performance in 6‐OHDA‐lesioned rats. J. Psychiatry Neurosci. 32, 193–202. [PMC free article] [PubMed] [Google Scholar]
- Tsen P., El Mansari M. and Blier P. (2013) Effects of repeated electroconvulsive shocks on catecholamine systems: electrophysiological studies in the rat brain. Synapse 67, 716–727. [DOI] [PubMed] [Google Scholar]
- Votaw J., Byas‐Smith M., Hua J., Voll R., Martarello L., Levey A. I., Bowman F. D. and Goodman M. (2003) Interaction of isoflurane with the dopamine transporter. Anesthesiology 98, 404–411. [DOI] [PubMed] [Google Scholar]
- Wang W., Yang Y., Ying C. et al (2007) Inhibition of glycogen synthase kinase‐3beta protects dopaminergic neurons from MPTP toxicity. Neuropharmacology 52, 1678–1684. [DOI] [PubMed] [Google Scholar]
- Yong Y., Ding H., Fan Z., Luo J. and Ke Z.‐J. (2011) Lithium fails to protect dopaminergic neurons in the 6‐OHDA model of Parkinson's disease. Neurochem. Res. 36, 367–374. [DOI] [PubMed] [Google Scholar]
- Zarate C. A. and Machado‐Vieira R. (2016) GSK‐3: a key regulatory target for ketamine's rapid antidepressant effects mediated by enhanced AMPA to NMDA throughput. Bipolar Disord. 18, 702–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Wang W., Fang H. et al (2014) GSK‐3β inhibition attenuates CLP‐induced liver injury by reducing inflammation and hepatic cell apoptosis. Mediators Inflamm. 2014, 629507. [DOI] [PMC free article] [PubMed] [Google Scholar]