

Abstract
Type 2 diabetes (T2D) and adiposity associate with increased risk of several cancers, but the impact of competing risk of noncancer deaths on these associations is not known. We prospectively examined participants in the Malmö Diet and Cancer Study aged 44–73 years with no history of cancer at baseline (n = 26,953, 43% men). T2D was ascertained at baseline and during follow‐up, and body mass index (BMI) and waist circumference (WC) at baseline. Multivariable cause‐specific hazard ratios (HR) and subdistribution hazard ratios (sHR), taking into account noncancer deaths, were estimated using Cox‐ and competing risk regression. During follow‐up (mean 17 years), 7,061 incident cancers (3,220 obesity‐related cancer types) and 2,848 cancer deaths occurred. BMI and WC were associated with increased risk of obesity‐related cancer incidence and cancer mortality. In T2D subjects, risk of obesity‐related cancer was elevated among men (HR = 1.31, 95% CI: 1.12–1.54; sHR = 1.29, 95% CI: 1.10–1.52), and cancer mortality among both men and women (HR = 1.34, 95% CI: 1.20–1.49; sHR = 1.30, 95% CI: 1.16–1.45). There was no elevated actual risk of cancer death in T2D patients with long disease duration (sHR = 1.00, 95% CI: 0.83–1.20). There was a significant additive effect of T2D and adiposity on risk of obesity‐related cancer and cancer mortality. In conclusion, detection bias may partially explain the increased risk of cancer morbidity among T2D patients. Both excess risk of competing events among patients with T2D and depletion of susceptibles due to earlier cancer detection will lower the actual risk of cancer, particularly with longer diabetes duration and at older ages.
Short abstract
What's new?
Type 2 diabetes (T2D) and obesity are associated with increased risk of cancer. However, the impacts on mortality and causal pathways for cancer remain unclear, owing to the multitude of genetic, environmental and behavioral factors that influence T2D and obesity. Here, in an investigation of mortality dynamics, T2D patients were found to suffer significantly higher risk of cancer death than individuals without the condition. Obesity‐related cancer incidence was higher in men with T2D. The findings also suggest, however, that competing noncancer events, such as increased cardiovascular mortality, can lower actual cancer risk among T2D patients.
Prevalence of type 2 diabetes (T2D) is increasing worldwide due to aging populations and societal changes, leading to obesogenic environments and lifestyles.1, 2 Many studies suggest that T2D patients and obese subjects are at higher risk of several cancers.3, 4 Mendelian randomization studies point to a causal positive association between body mass index (BMI) and some cancers that have shown consistent associations with adiposity in observational studies.5, 6, 7, 8, 9 The causal pathways for the T2D‐cancer association are, however, far from established. It is not clear whether it is the metabolic changes (e.g., hyperglycemia or hyperinsulinemia) associated with T2D that affect cancer risk or whether the association is due to nonbiological factors or potentially shared risk factors for T2D and cancer. The within‐disease heterogeneity and multifactorial nature of these diseases make the disentanglement of individual aspects difficult. Proposed nonbiological factors include primarily different health‐seeking behavior and medical surveillance among T2D patients that may result in detection bias.10 Shared risk factors or potential mediators of the association include antidiabetic drug treatments and genetic and/or environmental factors, particularly adiposity.3 As most common cancers are age‐dependent and diabetes and adiposity are associated with other disease outcomes and early mortality, dependent censoring of competing events may also result in a selection bias.11 While there is no way to test the independent censoring assumption (i.e., that censoring occurs at random within subgroups of interest) in conventional survival analysis, which estimates the cause‐specific hazard, competing risk analysis does not assume independent censoring. Competing risk methods are useful for prediction and risk communication since they provide an estimate of the actual (or absolute) risk in scenarios where competing events occur.14, 15 A competing event can be defined as any event that prevents (e.g., death) or alters the probability of the main event (e.g., cancer) to occur.12 To correct for this bias, observational studies need to account not only for confounders associated with the exposure and the main outcome, but also factors that associate with the exposure and competing events.13 To determine the relative influence of T2D and adiposity on the risk of obesity‐related cancers and cancer death, we explored the mortality dynamics in T2D patients compared to individuals without diabetes using a competing risk framework with noncancer deaths as competing events. The aim of this study was to assess both the etiological risk (i.e., cause‐specific hazard ratio) and actual risk (i.e., hazard ratio taking into account competing risk of noncancer death) of cancer morbidity (particularly obesity‐related cancers) and mortality among subjects with T2D, and for general and central adiposity. As it is still unclear to what extent T2D and adiposity are independent risk factors for cancer, we further aimed to assess the interaction between T2D and adiposity on cancer risk to examine the level of independence between these factors.
Subjects and Methods
Study population
The Malmö Diet and Cancer Study (MDCS) is a prospective cohort study established between 1991 and 1996.16 The MDCS was approved by the Ethics Committee at Lund University (LU 51–90), and informed written consent was obtained from all participants. Men and women living in Malmö who were born from 1923 to 1950 constituted the source population and were invited to join. With a participation rate of approximately 40%, the cohort consists of 30,446 participants aged 44–73 years at baseline. Follow‐up time was accrued from entry into the study until the date of death from any cause, emigration, or until 31 December 2014. For this study, we excluded all subjects with a missing start date, a history of cancer at baseline, or with other types of diabetes than T2D; after excluding subjects, the study population was 26,953 (11,449 men and 15,504 women). A description of the study population with exclusions is shown in Supporting Information, Figure S1.
Outcome ascertainment
Causes of death and the vital status of participants were identified by linkage to the Swedish Cause of Death registry and subjects lost to follow‐up due to emigration identified by linkage to the Swedish Tax Agency (<0.5%). Cancer diagnoses were ascertained by linkage to the Swedish Cancer Registry. Subjects with no history of cancer were followed from baseline examinations until their first cancer diagnosis, death, emigration or until 31st December 2014, whichever occurred first. The main outcome was a composite of cancer types created based on the World Cancer Research Fund's updated meta‐analyses of cancers showing consistent positive associations with adiposity. These included stomach (ICD7 = 151), colorectal (ICD7 = 153–154), pancreas (ICD7 = 157), postmenopausal breast (invasive only) (ICD7 = 170), endometrial (ICD7 = 172), ovary (ICD7 = 175), advanced prostate (ICD7 = 177), kidney (ICD7 = 180) and hepatocellular (including gallbladder) (ICD7 = 155–156). Data on esophageal adenocarcinoma were not available for the current study. Postmenopausal breast cancer included women with invasive breast cancer diagnosed at age 50 or older. Advanced prostate cancer included localized but high‐risk tumors and was identified by linkage to the Swedish National Prostate Cancer Registry and defined as tumor stage T3 or T4, presence of metastasis (N1 or M1), Gleason score ≥8, or prostate‐specific antigen (PSA) >50 ng/L. Risk of total cancer incidence (excluding cervix in situ) and cancer mortality (based on ICD‐9 codes 140–239 or ICD‐10 codes D00–D48) was also examined.
Diabetes mellitus ascertainment
Diabetes status at baseline and during follow‐up and information on date of diabetes diagnosis was identified from 7 registers as well as baseline and re‐examination screenings of the MDCS and the Malmö Preventive Project, a population‐based intervention study including some participants from the MDCS.17 The National Diabetes Register18 and the regional Diabetes 2000 register19 required a proven diagnosis by a physician at the hospital based on international standards for diagnosis (i.e., fasting plasma glucose concentration ≥7.0 mmol/l measured twice). For cases not diagnosed at a hospital, the primary source was the local HbA1c register from the Department of Clinical Chemistry, Skåne University Hospital, Malmö, was used,20, 21 where individuals with two HbA1c values ≥6% (not on the same day) were classified as having diabetes. Other registries used to identify diabetes cases included the National Patient Register, the Swedish Cause of Death Register (ICD10 codes E10–E14 and O244–O249) and the Prescribed Drug Register (ATC code A10). The different sources of case ascertainment were overlapping. Specified information on diabetes type was available for 37% of diabetes cases. To include only T2D, all cases that were specified as type 1, LADA, secondary diabetes or other were excluded from the analysis (n = 162). Furthermore, we excluded 81 subjects diagnosed with diabetes before the age of 40 years (Supporting Information, Fig. S1). In total, there were 1,161 prevalent T2D diagnoses and 3,852 incident T2D diagnoses.
Other variables
Information on age and sex was obtained through the participants' unique personal identification number. Calendar year of study entry was included as a categorical variable to account for the potential bias caused by the recruitment of slightly older individuals during the last two years of study recruitment. Weight (kg) and height (m) were measured by trained nurses with subjects wearing light indoor clothing and no shoes. Body mass index (BMI) was defined as kg/m2. BMI was examined as a continuous variable in the categories normal‐weight (<25 kg/m2), overweight (25‐<30 kg/m2) and obese (≥30 kg/m2). Waist circumference was measured between the lowest rib and the iliac crest and was used to categorize subjects by central obesity according to the World Health Organization cut‐points for increased risk of metabolic complications (<94, 94–102 and >102 cm for men and <80, 80–88 and >88 for women). The subjects were considered hypertensive if their blood pressure was ≥140/90 mmHg or if they reported taking antihypertensive drugs at baseline. Current use of lipid‐lowering drugs was dichotomized as yes/no. Educational level was categorized as elementary, primary and secondary, upper secondary, further education without degree or university/college degree. Smoking status was defined as never, former or current (including irregular). Leisure‐time physical activity was assessed by questionnaire adapted from the Minnesota Leisure Time Physical Activity Questionnaire22, 23 and the physical activity score was categorized into sex‐specific quartiles. Alcohol consumption was categorized as sex‐specific quartiles of alcohol intake as a percentage of total energy intake based on reported intake in the MDCS diet history assessment.24 Past food habit change (yes/no) was defined using the baseline questionnaire item “Have you substantially changed your food habits in the past due to illness or other reason?” Current use of menopausal hormone replacement therapy (HRT) was defined as yes/no.
Statistical analysis
We examined the baseline characteristics of the study population by T2D status at baseline and during follow‐up, and we formally tested for differences (χ 2 test and t‐test) between never/ever T2D status and prevalent/incident T2D status. We used sex‐stratified Cox proportional hazards regression with attained age as the underlying time‐metric to estimate HRs and 95% confidence intervals (CI) for total and obesity‐related cancer incidence and cancer mortality. HRs were estimated for prevalent, incident and total T2D compared to subjects without T2D. Furthermore, we estimated the HRs for general adiposity (as HR per standard deviation (SD)) and central adiposity (as HR per SD). Subjects with prevalent diabetes were excluded from analyses of incident diabetes in relation to cancer risk, and incident T2D was modeled as a time‐varying covariate. When combining prevalent and incident T2D patients, those with prevalent T2D were considered as exposed at the age of their baseline enrollment. We estimated the actual risk of total and obesity‐related cancer incidence and cancer mortality by estimation of the subdistribution HR (sHR), which is based on the cumulative incidence function in the presence of competing risks.25 All noncancer deaths were considered as competing events for cancer incidence and mortality. A directed acyclic graph depicting competing risk bias in the current setting is shown in Supporting Information, Figure S2. Potential confounders were identified based on literature review of known and putative risk factors and were evaluated using directed acyclic graphs. The main multivariable model was adjusted for the calendar year of study entry, height, educational level, physical activity, alcohol consumption, past food habit change, smoking status, hypertension, use of lipid‐lowering drugs, family history of cancer, BMI and current HRT use (women only). Subjects with missing values for any of the included covariates were excluded from the multivariable analysis (n = 2,455). The proportional hazards assumption was evaluated graphically and was tested based on the scaled Schoenfeld residuals. Cox–Snell's residuals were examined to assess model fit. No deviations from the proportional hazards assumption were observed in the presented models. To assess the joint impact of T2D and obesity on the risk of cancer incidence and mortality, we cross‐classified subjects by obesity and diabetes status at baseline. Multiplicative interaction was tested by including the cross‐product between BMI or WC and T2D status in the models. The relative excess risk of interaction was calculated as HR11‐HR10‐HR01 + 1,26 and 95% CIs were obtained using the bootstrap percentile method with 1,000 bootstrap samples. Additive interaction was calculated on the basis of adjusted HRs for the following groups: prevalent T2D, BMI < 30 (HR10); no diabetes, BMI ≥ 30 (HR01); and prevalent T2D, BMI ≥ 30 (HR11), where no diabetes and BMI < 30 were used as the reference group (HR00). Similar analysis was carried out to assess the relative excess risk of interaction for prevalent T2D and high waist circumference. To further understand the potential interdependence of T2D and adiposity on cancer risk, we performed two sets of mediation analysis (Supporting Information, Fig. S3). We calculated the percent excess risk according to the following: (HRunadj − HRadj)/(HRunadj − 1) × 100, where HRadj refers to the multivariable HR adjusted for the potential mediator and HRunadj refers to the multivariable HR with no adjustment for the potential mediator. For prevalent T2D, we considered BMI and waist circumference as potential mediators of the association, and for adiposity and cancer risk, we considered incident T2D as a potential mediator (after excluding prevalent T2D from analyses).
In sensitivity analyses, to investigate the effects of preclinical cancer or detection bias on our associations, we (1) excluded cancers diagnosed within three months from diabetes diagnosis or baseline examinations and (2) excluded cases diagnosed within two years of diabetes diagnosis or baseline examinations. The effect of competing risks is likely to increase over time due to increased mortality rates and depletion of susceptibles. We therefore carried out a sensitivity analysis, which limited follow‐up until the age of 80 (or age at death/censoring if before this age). Furthermore, for prevalent T2D at study start, we examined the potential effect of diabetes severity by stratifying by current diabetes treatment as a proxy for diabetes severity, that is, diet or metformin only versus other oral antihyperglycemic drugs (alone or in combination with metformin) and/or insulin. Stata/SE 14.2 (StataCorp LP, College Station, TX) was used for all statistical analyses. All tests were two‐sided, and P < 0.05 was considered statistically significant.
Results
Description of study population
During a mean follow‐up of 16.9 years, there were in total 7,061 incident cancer cases, of which 3,220 were obesity‐related cancers. The incidence rate for total cancer per 1000 person‐years was 15.5 (95% CI: 15.2–15.9), and for obesity‐related cancer, it was 7.1 (95% CI: 6.8–7.3). At the end of follow‐up, 28.8% of the study participants had died (n = 7,761), of which 37% were from cancer‐related causes. Baseline characteristics of the study population by T2D status at baseline and during follow‐up are shown in Table 1. The mean duration of diabetes at the end of the follow‐up was 20.7 years (mean age at onset 56.7 years) for those with prevalent T2D, and it was 8.1 years (mean age at onset 69.2 years) for those who were diagnosed with T2D during follow‐up. At baseline, patients with prevalent T2D were more likely to have hypertension, use lipid‐lowering drugs, and report past food habit changes, while they were less likely to use menopausal HRT (women only), be current smokers, and have high alcohol consumption, compared to those who were diagnosed with T2D during follow‐up.
Table 1.
Baseline characteristicsa of MDCS by type 2 diabetes (T2D) status at baseline (prevalent) and during follow‐up until 31 December 2014 (incident)
| Characteristic | All | No diabetes | Prevalent T2D | Incident T2D | p valued | p valuee |
|---|---|---|---|---|---|---|
| Number of subjects | 26,953 | 21,940 | 1,161 | 3,852 | ||
| Demographics and disease history | ||||||
| Male sex (n, %) | 11,449 (42.5) | 8,788 (40.1) | 650 (56.0) | 2,011 (52.2) | <0.0001 | 0.02 |
| Age at baseline screening (years) | 57.9 (7.5) | 57.8 (7.6) | 61.4 (6.5) | 57.9 (7.0) | <0.0001 | <0.0001 |
| Age at diabetes onset (years) | – | – | 56.7 (8.1) | 69.2 (7.9) | – | <0.0001 |
| Diabetes duration until end of follow‐up (years) | – | – | 20.7 (7.6) | 8.1 (5.2) | – | <0.0001 |
| Family history of cancer (n, %) | 11,488 (45.6) | 9,375 (45.5) | 481 (45.6) | 1,632 (45.8) | 0.78 | 0.88 |
| Hypertension (n, %) | 15,482 (61.5) | 11,940 (58.1) | 856 (81.6) | 2,686 (75.5) | <0.0001 | <0.0001 |
| Use of lipid‐lowering drugs (n, %) | 778 (3.1) | 498 (2.4) | 106 (10.1) | 174 (4.9) | <0.0001 | <0.0001 |
| Current HRT use (n, %) women only | 2,836 (20.9) | 2,493 (21.6) | 50 (11.3) | 293 (18.4) | <0.0001 | <0.0001 |
| Anthropometric factors b | ||||||
| Height (cm) | 168.9 (9.0) | 169.0 (0.04) | 168.4 (0.18) | 168.3 (0.10) | <0.0001 | 0.63 |
| Weight (kg) | 74.0 (13.8) | 72.6 (0.08) | 80.2 (0.35) | 80.0 (0.19) | <0.0001 | 0.20 |
| BMI (kg/mb) | 25.9 (4.0) | 25.3 (0.03) | 28.2 (0.11) | 28.2 (0.06) | <0.0001 | 0.08 |
| Waist circumference | 84.8 (13.1) | 83.3 (0.07) | 92.3 (0.29) | 91.1 (0.16) | <0.0001 | <0.0001 |
| Lifestyle factors | ||||||
| Current smokers (n, %) | 6,859 (27.6) | 5,600 (27.5) | 246 (24.0) | 1,013 (29.0) | <0.0001 | 0.001 |
| High alcohol consumption (n, %)c | 6,221 (25.0) | 5,231 (25.7) | 186 (18.1) | 804 (23.0) | <0.0001 | <0.0001 |
| Low physical activity (n, %)c | 6,205 (25.1) | 4,852 (24.0) | 295 (29.1) | 1,058 (30.6) | <0.0001 | 0.30 |
| University/college degree (n, %) | 3,562 (14.4) | 3,093 (15.2) | 101 (9.9) | 368 (10.6) | <0.0001 | 0.76 |
| Past food habit change (n, %) | 5,956 (24.0) | 4,308 (21.2) | 722 (70.3) | 926 (26.5) | <0.0001 | <0.0001 |
Data are presented as mean (standard deviation, SD) for continuous variables and frequency (%) for categorical variables, unless otherwise noted. Variables with missing data included family history of cancer (n = 1,744), hypertension (n = 1,779), use of lipid‐lowering drugs (n = 1,755), current HRT use (n = 1,948; women only), height (n = 46), weight (n = 47), BMI (n = 47), smoking status (n = 2,075), alcohol consumption (n = 2,065), physical activity (n = 2,227), educational level (n = 2,127) and past food habit change (n = 2,099).
Age‐ and sex‐adjusted means (standard error, SE) in subgroups by diabetes status.
High alcohol consumption defined has the highest sex‐specific quartile of alcohol consumption (energy percentage) and low physical activity defined as the lowest sex‐specific quartile of leisure‐time physical activity score.
p values for difference between subjects with diabetes (prevalent and incident) and those with no diabetes. Differences in anthropometrics tested with adjustment for age and sex.
p values for difference between subjects with prevalent and incident T2D. Differences in anthropometrics tested with adjustment for age and sex.
Type 2 diabetes and risk of cancer morbidity and mortality
The incidence rate of all‐cause mortality was 15.6 deaths per 1000 person‐years, for which the contribution of noncancer deaths was stronger (incidence rate 9.9 per 1000 person years) than cancer deaths (Supporting Information, Table S1). After adjustment for potential confounders, prevalent T2D was associated with an increased risk of noncancer mortality (HR = 2.01, 95% CI: 1.80–2.24) but was not associated with cancer mortality (HR = 1.17, 95% CI: 0.97–1.39) (Supporting Information, Table S1).
Taking into account the competing risk of noncancer mortality, there was no indication of an increase in the actual risk of cancer mortality among subjects with prevalent T2D (sHR = 1.00, 95% CI: 0.83–1.20) (Supporting Information, Table S1 and Fig. 1). The HRs and sHRs for incidence of total cancer and obesity‐related cancer incidence and cancer mortality by T2D status at baseline and after follow‐up are shown in Figure 1 and Supporting Information, Table S2. Prevalent T2D was associated with higher risk of cancer mortality among men (HR = 1.35, 95% CI: 1.08–1.70; sHR = 1.16, 95% CI: 0.92–1.46) but not among women (p heterogeneity = 1.3 × 10−3). Incident T2D was associated with higher risk of total (HR = 1.10, 95% CI: 1.00–1.20; sHR = 1.13, 95% CI: 1.03–1.24) and obesity‐related cancer incidence (HR = 1.15, 95% CI: 1.00–1.32; sHR = 1.19, 95% CI: 1.04–1.37) (Fig. 1). There was, however, a significant difference between men and women for risk of obesity‐related cancer incidence with incident T2D (p heterogeneity = 2.1 × 10−10). Incident T2D was associated with significantly higher risk of cancer mortality among both men (HR = 1.31, 95% CI: 1.12–1.54; sHR = 1.35, 95% CI: 1.15–1.60) and women (HR = 1.50, 95% CI: 1.24–1.82; sHR = 1.51, 95% CI: 1.24–1.85). Combining prevalent and incident T2D cases, there was an elevated risk of cancer mortality among both men and women, but an increased risk of obesity‐related cancer incidence was only observed among men (Fig. 1 and Supporting Information, Table S2).
Figure 1.
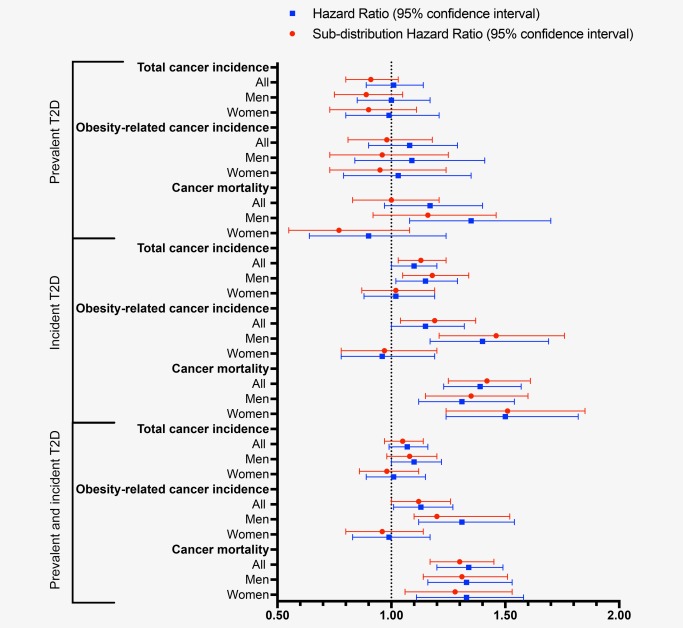
Cause‐specific and subdistribution hazard ratios for total and obesity‐related cancer incidence and cancer mortality among participants in the Malmö Diet and Cancer Study (MDCS) with type 2 diabetes (T2D) at baseline and follow‐up compared to those without diabetes mellitus at baseline. Models were adjusted for age as the time‐scale, sex, calendar year of study entry, height, smoking status, physical activity level, alcohol consumption, educational level, past food habit change, hypertension, use of lipid‐lowering drugs, family history of cancer and body mass index. Analyses performed for women were further adjusted for the current use of HRT. [Color figure can be viewed at wileyonlinelibrary.com]
Adiposity and risk of cancer morbidity and mortality
Both general and central adiposity were associated with higher risk of total and obesity‐related cancer incidence and cancer mortality (Fig. 2 and Supporting Information, Table S3). No significant gender‐differences were observed. BMI was associated with a 3%, 7% and 5% risk increase per SD increase for total cancer incidence, obesity‐related cancer incidence and cancer mortality, respectively. Waist circumference was associated with a 5%, 9% and 12% risk increase per SD increase for total cancer incidence, obesity‐related cancer incidence and cancer mortality, respectively. In general, estimates from the competing risk regression model did not substantially differ from the HRs. The most notable difference between the two models was observed for men in relation to waist circumference and cancer mortality (HR = 1.12, 95% CI: 1.03–1.20); sHR = 1.06, 95% CI: 0.99–1.14).
Figure 2.
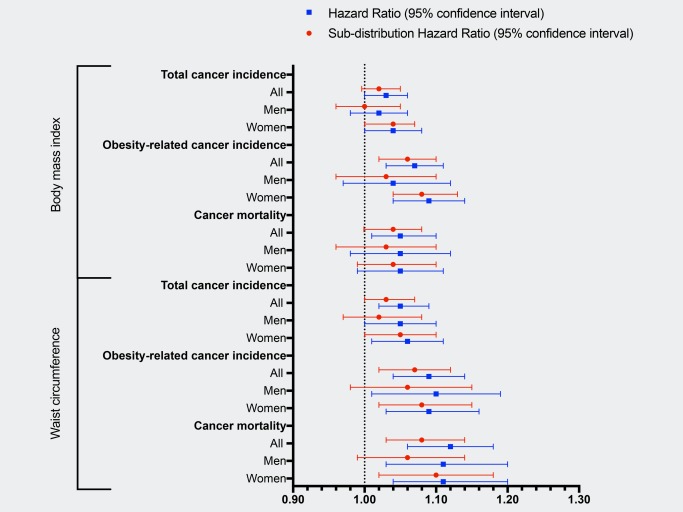
Cause‐specific and subdistribution hazard ratios for total and obesity‐related cancer incidence and cancer mortality per standard deviation (SD) increment in body mass index (1 SD = 4.0 kg/m2) and waist circumference (1 SD = 13.1 cm) among participants of the Malmö Diet and Cancer Study (MDCS). Models were adjusted for attained age as the time‐scale, sex, calendar year of study entry, height, smoking status, physical activity level, alcohol consumption, educational level, past food habit change, hypertension, use of lipid‐lowering drugs and family history of cancer. Analyses performed for women were further adjusted for the current use of HRT. [Color figure can be viewed at wileyonlinelibrary.com]
The combined and independent effects of general or central adiposity and T2D status at baseline on cancer risk
We observed no significant multiplicative interaction between measures of adiposity and T2D status at baseline on the risk of cancer incidence or mortality (Table 2). However, we observed a borderline significant interaction (p = 0.05) between BMI and T2D on cancer mortality. The relative excess risk of interaction estimates for obesity‐related cancer incidence and cancer mortality indicated moderate additive interactions (Table 2). For cancer mortality, the relative excess risk of interaction estimate for BMI and prevalent T2D was 0.71 (95% CI: 0.15, 1.26; p = 0.013). Compared to their normal‐weight, nondiabetic counterparts, subjects with BMI ≥ 30 kg/m2 and prevalent T2D had an HR of 1.81 (95% CI: 1.37–2.40) for cancer mortality. Similar excess risks were seen for obesity‐related cancer incidence and for combined abdominal obesity and prevalent T2D in relation to both obesity‐related cancer incidence and cancer mortality (Table 2). In a competing risk model taking into account noncancer deaths (data not tabulated), the sHR for cancer mortality among subjects with prevalent T2D and BMI ≥ 30 kg/m2 was 1.50 (95% CI: 1.13–2.00) compared to subjects without diabetes and BMI < 25.
Table 2.
Combined effecta of type 2 diabetes (T2D) and adiposity on cause‐specific risk of cancer incidence and mortality among participants (n = 24,498) of the MDCS (1991–2014)
| Total cancer incidence | Body mass index | Relative excess risk of interactionb | HR per SD increment | |||
|---|---|---|---|---|---|---|
| Diabetes status | <25 | 25–29.9 | ≥30 | p trend | ||
| No diabetes | 1.00 (ref) | 1.03 (0.98–1.09) | 1.09 (1.00–1.18) | 0.039 | 1.03 (1.00–1.06) | |
| Prevalent T2D | 1.11 (0.87–1.42) | 0.93 (0.77–1.12) | 1.21 (0.98–1.50) | 0.54 | 0.15 (–0.14, 0.44) | 1.07 (0.95–1.20) |
| p interaction = 0.89 | p = 0.31 | |||||
| Obesity‐related cancer incidence | Body mass index | HR per SD increment | ||||
| Diabetes status | <25 | 25–29.9 | ≥ 30 | p trend | ||
| No diabetes | 1.00 (ref) | 1.10 (1.02–1.20) | 1.17 (1.04–1.31) | 3.2 × 10−3 | 1.06 (1.02–1.10) | |
| Prevalent T2D | 1.21 (0.84–1.73) | 0.87 (0.64–1.19) | 1.65 (1.25–2.18) | 0.096 | 0.52 (0.05, 1.00) | 1.21 (1.04–1.41) |
| p interaction = 0.29 | p = 0.032 | |||||
| Cancer mortality | Body mass index | HR per SD increment | ||||
| Diabetes status | <25 | 25–29.9 | ≥ 30 | p trend | ||
| No diabetes | 1.00 (ref) | 1.06 (0.97–1.15) | 1.10 (0.97–1.25) | 0.091 | 1.04 (1.00–1.09) | |
| Prevalent T2D | 1.09 (0.75–1.59) | 0.97 (0.73–1.30) | 1.81 (1.37–2.40) | 4.8 × 10−3 | 0.71 (0.15, 1.26) | 1.22 (1.04–1.45) |
| p interaction = 0.05 | p = 0.013 | |||||
| Total cancer incidence | Waist circumference | HR per SD increment | ||||
| Diabetes status | <80/<94 | 80–88/94–104 | >88/>104 | p trend | ||
| No diabetes | 1.00 (ref) | 1.03 (0.97–1.10) | 1.09 (1.01–1.17) | 0.030 | 1.05 (1.02–1.09) | |
| Prevalent T2D | 1.10 (0.90–1.35) | 0.80 (0.61–1.04) | 1.19 (0.99–1.43) | 0.32 | 0.15 (−0.12, 0.41) | 1.09 (0.94–1.25) |
| p interaction = 0.96 | p = 0.28 | |||||
| Obesity‐related cancer incidence | Waist circumference | HR per SD increment | ||||
| Diabetes status | <80/<94 | 80–88/94–104 | >88/>104 | p trend | ||
| No diabetes | 1.00 (ref) | 1.08 (0.98–1.18) | 1.12 (1.00–1.24) | 0.028 | 1.07 (1.02–1.13) | |
| Prevalent T2D | 0.97 (0.70–1.36) | 0.86 (0.58–1.28) | 1.50 (1.17–1.92) | 8.6 × 10−3 | 0.47 (0.03, 0.90) | 1.30 (1.08–1.57) |
| p interaction = 0.09 | p = 0.035 | |||||
| Cancer mortality | Waist circumference | HR per SD increment | ||||
| Diabetes status | <80/<94 | 80–88/94–104 | >88/>104 | p trend | ||
| No diabetes | 1.00 (ref) | 1.11 (1.01–1.22) | 1.20 (1.07–1.34) | 7.5 × 10−4 | 1.10 (1.04–1.16) | |
| Prevalent T2D | 1.08 (0.77–1.50) | 0.93 (0.63–1.38) | 1.66 (1.29–2.14) | 4.4 × 10−3 | 0.48 (−0.01, 0.94) | 1.34 (1.10–1.63) |
| p interaction = 0.17 | p = 0.054 | |||||
Hazard ratios (HRs) and 95% confidence intervals (CIs) from a sex‐stratified cox regression model with attained age as the time‐metric adjusting for calendar year of study entry, height, smoking status, physical activity level, alcohol consumption, educational level, past food habit change, hypertension, lipid‐lowering drugs and family history of cancer. p interaction estimated by inclusion of the cross‐product for T2D and obesity status (body mass index or waist circumference categories) in the model. Standard deviation (SD) for body mass index was 4.0 kg/m2 and for waist circumference 13.1 cm.
Relative excess risk of interaction for obesity (body mass index ≥ 30 or waist circumference >88/>104 cm) and prevalent T2D on cancer risk. Confidence intervals obtained by the bootstrap percentile method with 1,000 bootstrap samples.
We explored two sets of mediation analyses (Supporting Information, Fig. S3). Adjustment for BMI had only small effects on the associations between prevalent T2D and cancer outcomes, suggesting weak mediation of BMI on these associations after accounting for potential confounders (Supporting Information, Table S8). The percent excess risk of prevalent T2D on cancer mortality explained by BMI was 5%. For the association between adiposity and cancer risk, there appeared to be indirect effects mediated by incident T2D, particularly for cancer mortality (Supporting Information, Tables S9 and S10). The percent excess risk of BMI ≥ 30 kg/m2 on cancer mortality risk explained by incident T2D was 70% and the corresponding percent excess risk of high waist circumference on cancer mortality explained by incident T2D was 35%.
Sensitivity analyses for the association between T2D and cancer risk
We carried out two main sensitivity analyses to assess the impact of detection bias on the association between T2D and cancer incidence. After exclusion of subjects diagnosed with cancer within three months of baseline examinations or after their incident T2D diagnosis, the findings were unchanged (Supporting Information, Table S4). When we excluded all subjects diagnosed with cancer within 2 years of their T2D diagnosis or baseline examinations (Supporting Information, Table S5), only prevalent T2D was associated with increased risk of obesity‐related cancer (HR = 1.21, 95% CI: 1.00–1.47). For analyses including incident cases of T2D, the increased risk of obesity‐related cancers was greatly attenuated. To examine the plausibility of an attenuating effect on risk estimates at older ages, we limited the analysis time to 80 years of age. Overall, the results indicated stronger associations when restricting the follow‐up until 80 years of age (Supporting Information, Table S6). Among subjects with prevalent T2D at baseline we used current antidiabetes medications as a proxy for diabetes severity by classifying patients according to treatment at baseline examinations (Supporting Information, Table S7). Results suggested a potential association between prevalent T2D and obesity‐related cancer incidence among subjects with severe T2D at baseline. The association between prevalent T2D and cancer mortality was similarly only observed among subjects with severe T2D at baseline (multivariable HR = 1.58, 95% CI: 1.22–2.05), with no excess risk among patients treated with lifestyle or metformin only. The excess risk of cancer mortality was stronger among men with severe T2D (multivariable HR = 1.88, 95% CI: 1.39–2.55) (Supporting Information, Table S7). In a competing risk model accounting for the excess risk of noncancer death, the multivariable sHR for cancer mortality among subjects with severe T2D was 1.26 (95% CI: 0.97–1.64) (data not tabulated).
Discussion
Principal findings
Individuals with T2D had a significantly higher risk of cancer death compared to the population without T2D, and men with T2D had a higher incidence of obesity‐related cancers. The increased risk for incident cancer diagnoses seemed to be particularly elevated during the first years following T2D diagnosis, which could indicate detection bias. Both detection bias and competing risk of noncancer deaths would lead to a depletion of individuals susceptible to cancer, which could attenuate the association between T2D and cancer risk over time. Our competing risk analyses showed that, particularly among subjects with long‐term/severe T2D, the actual risk of cancer may be considerably overestimated if failing to account for competing events. However, the risk estimates from the two models did not differ significantly. Finally, we found that individuals with comorbid T2D and adiposity at baseline had a more pronounced increased risk of obesity‐related cancer incidence and cancer mortality, compared to the population without T2D and with general or central adiposity within the recommended ranges, with significant interactions on an additive scale. Mediation analysis further suggested that the association between prevalent T2D and cancer risk was largely independent of BMI after accounting for other potential confounders, but that the association between adiposity and cancer risk may in part be mediated by an increased risk of incident T2D.
Potential mechanisms and comparisons with other studies
It is plausible that the metabolic and hormonal changes associated with T2D and adiposity could explain the associations with obesity‐related cancers and cancer mortality observed in our study and previous studies.3, 27 While adjustment for BMI slightly attenuated the observed associations, obesity did not explain the observed associations between T2D and cancer incidence and mortality. Our findings further suggest that subjects with pre‐existing T2D and obesity may have a substantially higher risk of obesity‐related cancer incidence and total cancer mortality compared to their normal‐weight counterparts without T2D. When accounting for competing events, the actual risk increase was however only moderate. Numerous studies have examined the role of T2D and obesity in cancer risk, while the combined impact of both has remained less studied. If T2D and obesity would indeed contribute to the risk of cancer by primarily different mechanisms, obese T2D patients may, in particular, be at higher risk for cancer morbidity and mortality. Further studies are needed to assess whether potential synergistic effects of T2D and adiposity exist across specific cancer sites. We observed gender differences regarding cancer mortality and risk of obesity‐related cancer incidence among subjects with prevalent and incident T2D, respectively. Results suggested positive associations among men only. These differences could plausibly be reflective of differences in cancer detection among men and women or reflect the heterogeneity in included cancer sites for men and women, respectively. The lack of association between incident T2D and obesity‐related cancer incidence among women may in part be reflected by a lack of association between incident T2D and risk of postmenopausal breast cancer in this population (which accounted for the largest part of these cancers in women; n cases = 1,064). The HR for postmenopausal breast cancer was 0.89 (95% CI: 0.65–1.22) in a fully adjusted model (data not tabulated). Screening practices among men and women in this cohort may thus differ, particularly related to the time interval close to T2D diagnosis.
Prospective cohort studies have mostly studied prevalent T2D status (ever/never) at study start in relation to cancer risk. Subjects with prevalent T2D are likely to be represented by those with earlier disease onset, and inclusion in the study is conditional on them being alive and event‐free at the start of the study. This could lead to a selection bias in which T2D patients with certain characteristics and/or with potential survival benefits are included in cohort studies with a prevalent exposure design.28 Most notably, in this study, subjects with prevalent T2D appear to have made several lifestyle changes, including reducing their alcohol consumption, changing their food habits and quitting smoking. They were also significantly more likely to have hypertension and be users of lipid‐lowering drugs at baseline. Such changes may either affect future risk of cancer, or result in inadequate adjustment for potentially shared risk factors for both T2D and cancer and for competing events (Supporting Information, Fig. S2). An incident T2D design has the advantage of covariate assessment prior to diabetes diagnosis. The main disadvantage is that incident T2D will more heavily reflect short‐term diabetes and may therefore be more prone to detection bias.
A few studies have examined temporal association between T2D and cancer risk. Three studies found substantially elevated risk of most cancer types during the first 3 months after diabetes diagnosis 29, 30, 31. We did not observe any major differences in cause‐specific HRs after exclusion of those diagnosed within three months. While previous studies suggest that the associations between T2D and site‐specific cancers appear to increase sharply in the period following diabetes diagnosis, it appears to level off around 2 years after diagnosis of diabetes 10, 31, a finding that is in line with our results. With the exclusion of subjects diagnosed with cancer within two years from baseline examinations or incident T2D diagnosis, the observed associations with obesity‐related cancer incidence were greatly attenuated. Only prevalent T2D was associated with an increased risk of obesity‐related cancer incidence in this analysis. Prevalent T2D (ever/never) at baseline reflects long‐term exposure to T2D (mean duration 21 years until the end of follow‐up) and thus generally excludes the time window for detection bias close to the diagnosis of T2D. A detailed analysis of the temporal association between T2D and obesity‐related cancer incidence was not possible with the current study design. It is, however, unclear and virtually impossible to address whether cases diagnosed within the first 2 years after T2D diagnosis or study enrollment are solely due to increased screening or health‐care seeking behavior in this subpopulation, or if they also have some increased underlying susceptibility. The time‐dependent attenuation of risk observed in studies examining the temporal association between T2D and cancer may also be due to the intensified early screening and detection, which may result in a depletion of susceptibilities in the first years after diabetes diagnosis.
While long‐term diabetes (i.e., prevalent) may to a lesser extent be affected by detection bias, both a depletion of susceptibles and the increasing impact of competing events on the likelihood of cancer diagnosis and mortality may be more of a concern. We noted that associations were strengthened when limiting the follow‐up up to 80 years of age. Indeed, the increased risk of overall mortality at older ages may deflate the observed risk of cancer incidence. With both T2D and cancer typically being age‐dependent diseases, the competing risk of death needs to be taken into account when assessing the actual cancer risk.32 As patients with T2D are at increased risk of cardiovascular morbidity and mortality, they may be at lower actual risk of cancer by simply being less likely to stay alive until an age when cancer diagnoses are more common. Our competing risk analysis suggests that among subjects with long‐term T2D (i.e., prevalent T2D), particularly severe T2D, the actual risk of cancer morbidity and mortality may be only very modestly elevated compared to subjects without T2D. However, competing events appeared to have only marginal effects on the risk estimates associated with adiposity. One notable exception was for waist circumference among men, where the sHRs were closer to null compared to the cause‐specific HRs. This may be reflective of waist circumference being a stronger predictor for noncancer death compared to general adiposity in particular among men.
Strengths and weaknesses
The main strengths of this study include the large population‐based cohort design with complete prospective registration of exposures and outcomes, the access to information on important covariates, and virtually no loss to follow‐up (<0.5%). Weaknesses include the clinical nature of both T2D and cancer, both being diseases that rarely demonstrate acute clinical onset. Both T2D and cancer may therefore be underdiagnosed, and the date of diagnosis may not reflect the true onset of these diseases. An important strength of this study is, however, the use of nation‐wide disease registries with virtually complete coverage. Any residual misclassification of exposure or outcome would likely result in attenuation of the observed associations. We used broad cancer endpoints (total or obesity‐related) in this study to assure sufficient statistical power for subgroup analysis. It is however well known that the associations between T2D and adiposity in relation to cancer risk differ by cancer site and, for some cancers, by subtypes within sites. Therefore, additional studies on the potential synergistic or the independent effects of T2D and adiposity for site‐specific cancers are warranted. In addition, the results from the competing risk analysis may also differ depending on cancer site, as the influence of competing risks is likely to be higher for strongly age‐dependent cancers. In this study, we lacked data on esophageal adenocarcinoma to include in the composite end‐point of obesity‐related cancers; however, this cancer is rare and the number of cases is very low in this cohort. In addition, the International Agency for Research on Cancer (IARC) Working Group recently published a compilation of adiposity‐associated cancer sites, which differ slightly from the WCRF's conclusions.33 There is thus not complete consensus on which cancers are conclusively associated with adiposity. Confounder assessment was carried out at baseline, which for most prevalent cases did not coincide with T2D diagnosis. As such, there are limitations in addressing the confounding of the observed associations. The possibility of residual confounding is potentially greater when using a prevalent exposure design as the assessment of confounders is performed after the onset of T2D. We had incomplete information on the type and duration of T2D treatment and cannot rule out the possibility that the observed associations could be related to use of antidiabetic drugs. For prevalent T2D cases information on current use (at baseline examinations) of antidiabetic medications was available and used as a proxy for T2D severity. The representability of the MDCS has previously been reported.34 In general, the cohort was found to be representative of the source population. For a better understanding of the potential role of T2D in cancer, it would be ideal to investigate T2D cases and controls matched on age and date of T2D diagnosis. Studies also require substantial follow‐up time to fully account for duration effects and detailed data with repeated measurements of T2D‐related phenotypes, potential confounders and drug treatments over time.
Conclusions
In this study, we found that T2D and obesity are associated with a higher risk of cancer mortality and an increased risk of obesity‐related cancer incidence. T2D and obesity may increase the risk in an additive fashion, as suggested by the finding that comorbid T2D and obesity were associated with a more markedly increased risk of obesity‐related cancers and cancer mortality. Initially, increased risk of cancer incidence among T2D patients is likely to be inflated by detection bias. It is uncertain and difficult to ascertain whether the increased detection rates are fully independent of an underlying increased susceptibility. With a longer T2D duration and at older ages, both depletion of susceptibles due to increased detection in the years following T2D diagnosis and competing risk of noncancer deaths are likely to lower the probability of cancer diagnosis. There remain questions related to the least‐biased time interval to assess cancer risk among T2D patients.
Supporting information
Supporting Information
References
- 1. Danaei G, Finucane MM, Lu Y, et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country‐years and 2.7 million participants. Lancet 2011; 378:31–40. [DOI] [PubMed] [Google Scholar]
- 2. Hu FB. Globalization of diabetes: the role of diet, lifestyle, and genes. Diabetes Care 2011; 34:1249–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Giovannucci E, Harlan DM, Archer MC, et al. Diabetes and cancer: a consensus report. Diabetes Care 2010; 33:1674–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Renehan AG, Tyson M, Egger M, et al. Body‐mass index and incidence of cancer: a systematic review and meta‐analysis of prospective observational studies. Lancet 2008; 371:569–78. [DOI] [PubMed] [Google Scholar]
- 5. Painter JN, O'Mara TA, Marquart L, et al. Genetic risk score Mendelian randomization shows that obesity measured as body mass index, but not waist:hip ratio, is causal for endometrial cancer. Cancer Epidemiol Biomarker Prevent 2016; 25:1503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gao C, Patel CJ, Michailidou K, et al. Mendelian randomization study of adiposity‐related traits and risk of breast, ovarian, prostate, lung and colorectal cancer. Int J Epidemiol 2016; 45:896–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dixon SC, Nagle CM, Thrift AP, et al. Adult body mass index and risk of ovarian cancer by subtype: a Mendelian randomization study. Int J Epidemiol 2016; 45:884–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thrift AP, Shaheen NJ, Gammon MD, et al. Obesity and risk of esophageal adenocarcinoma and Barrett's esophagus: a Mendelian randomization study. J Natl Cancer Inst 2014; 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guo Y, Warren Andersen S, Shu XO, et al. Genetically predicted body mass index and breast cancer risk: Mendelian randomization analyses of data from 145,000 women of European descent. PLoS Med 2016; 13:e1002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Harding JL, Shaw JE, Peeters A, et al. Cancer risk among people with type 1 and type 2 diabetes: disentangling true associations, detection bias, and reverse causation. Diabetes Care 2015; 38:264–70. [DOI] [PubMed] [Google Scholar]
- 11. Thompson CA, Zhang ZF, Arah OA. Competing risk bias to explain the inverse relationship between smoking and malignant melanoma. Eur J Epidemiol 2013; 28:557–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lau B, Cole SR, Gange SJ. Competing risk regression models for epidemiologic data. Am J Epidemiol 2009; 170:244–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lesko CR, Lau B. Bias due to confounders for the exposure‐competing risk relationship. Epidemiology 2017; 28:20–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Andersen PK, Geskus RB, de Witte T, et al. Competing risks in epidemiology: possibilities and pitfalls. Int J Epidemiol 2012; 41:861–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haggstrom C, Stattin P, Stocks T, et al. Interpretation of conventional survival analysis and competing‐risk analysis: an example of hypertension and prostate cancer. BJU Int 2016; 118:850–2. [DOI] [PubMed] [Google Scholar]
- 16. Berglund G, Elmstahl S, Janzon L, et al. The Malmo Diet and Cancer Study . Design and feasibility. J Intern Med 1993; 233:45–51. [DOI] [PubMed] [Google Scholar]
- 17. Berglund G, Nilsson P, Eriksson KF, et al. Long‐term outcome of the Malmo preventive project: mortality and cardiovascular morbidity. J Intern Med 2000; 247:19–29. [DOI] [PubMed] [Google Scholar]
- 18. Cederholm J, Eeg‐Olofsson K, Eliasson B, et al. Risk prediction of cardiovascular disease in type 2 diabetes: a risk equation from the Swedish National Diabetes Register. Diabetes Care 2008; 31:2038–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lindholm E, Agardh E, Tuomi T, et al. Classifying diabetes according to the new WHO clinical stages. Eur J Epidemiol 2001; 17:983–9. [DOI] [PubMed] [Google Scholar]
- 20. Hanas R, John G, International HBAcCC . 2010 consensus statement on the worldwide standardization of the hemoglobin A1C measurement. Diabetes Care 2010; 33:1903–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hoelzel W, Weykamp C, Jeppsson JO, et al. IFCC reference system for measurement of hemoglobin A1c in human blood and the national standardization schemes in the United States, Japan, and Sweden: a method‐comparison study. Clin Chem 2004; 50:166–74. [DOI] [PubMed] [Google Scholar]
- 22. Richardson MT, Leon AS, Jacobs DR, Jr. , et al. Comprehensive evaluation of the Minnesota Leisure Time Physical Activity Questionnaire. J Clin Epidemiol 1994; 47:271–81. [DOI] [PubMed] [Google Scholar]
- 23. Li C, Aronsson CA, Hedblad B, et al. Ability of physical activity measurements to assess health‐related risks. Eur J Clin Nutr 2009; 63:1448–51. [DOI] [PubMed] [Google Scholar]
- 24. Wirfalt E, Mattisson I, Johansson U, et al. A methodological report from the Malmo Diet and Cancer study: development and evaluation of altered routines in dietary data processing. Nutr J 2002; 19:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fine JP, Gray R. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc 1999; 94:496–509. [Google Scholar]
- 26. Knol MJ, & VanderWeele TJ. Recommendations for presenting analyses of effect modification and interaction. Int J Epidemiol 2012; 41:514–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gallagher EJ, LeRoith D. Epidemiology and molecular mechanisms tying obesity, diabetes, and the metabolic syndrome with cancer. Diabetes Care 2013; 36:S233–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Danaei G, Tavakkoli M, Hernan MA. Bias in observational studies of prevalent users: lessons for comparative effectiveness research from a meta‐analysis of statins. Am J Epidemiol 2012; 175:250–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lega IC, Wilton AS, Austin PC, et al. The temporal relationship between diabetes and cancer: a population‐based study. Cancer 2016; 122:2731–8. [DOI] [PubMed] [Google Scholar]
- 30. De Bruijn KM, Ruiter R, de Keyser CE, et al. Detection bias may be the main cause of increased cancer incidence among diabetics: results from the Rotterdam Study. Eur J Cancer 2014; 50:2449–55. [DOI] [PubMed] [Google Scholar]
- 31. Johnson JA, Bowker SL, Richardson K, et al. Time‐varying incidence of cancer after the onset of type 2 diabetes: evidence of potential detection bias. Diabetologia 2011; 54:2263–71. [DOI] [PubMed] [Google Scholar]
- 32. Berry SD, Ngo L, Samelson EJ, et al. Competing risk of death: an important consideration in studies of older adults. J Am Geriatrics Soc 2010; 58:783–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lauby‐Secretan B, Scoccianti C, Loomis D, et al. Body fatness and cancer – viewpoint of the IARC Working Group. N Engl J Med 2016; 375:794–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Manjer J, Carlsson S, Elmstahl S, et al. The Malmo Diet and Cancer Study: representativity, cancer incidence and mortality in participants and non‐participants. Eur J Cancer Prevent 2001; 10:489–99. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information