

Abstract
A 40‐year‐old Japanese woman presented with slowly progressing parkinsonism in adulthood. She had a history of epilepsy with intellectual disability in childhood. In a head magnetic resonance scan, T2‐weighted imaging showed low signal intensity areas in the globus pallidus and the substantia nigra; T1‐weighted imaging showed a halo in the nigra. Because the patient's symptoms and history were similar to those of patients with neurodegeneration with brain iron accumulation, we ran an exome analysis to investigate neurodegeneration with brain iron accumulation‐associated genes. We identified a c.700 C>T (p.Arg 234*) mutation in exon 9 of the WDR45 gene, which had not been reported in Japanese patients with beta‐propeller protein‐associated neurodegeneration (a neurodegeneration with brain iron accumulation subtype). Sanger sequencing confirmed a heterozygous mutation in this patient that was absent in both her parents, so it was judged to be a de novo nonsense mutation.
Keywords: autophagy, basal ganglia, beta‐propeller protein‐associated neurodegeneration, iron accumulation, parkinsonism
Introduction
Neurodegeneration with brain iron accumulation (NBIA) is a rare disorder characterized by iron deposition in areas such as the globus pallidus, resulting in progressive extrapyramidal symptoms and intellectual impairment.1 Advances in genomics have allowed various NBIA subtypes to be classified. Recently, mutations in the autophagy gene WDR45 (WD repeat domain 45, Xp 11.23) were found to cause beta‐propeller protein‐associated neurodegeneration (BPAN).2, 3 BPAN, a subtype of NBIA‐5, is characterized by a biphasic regression. Here, we describe a patient with a de novo WDR45 gene mutation whose diagnosis was confirmed by exome analysis.
Case report
The patient was a 40‐year‐old woman without a notable family history, except for her maternal grandparents being second cousins. She had no perinatal abnormalities and was delivered normally. At the age of 1 year and 7 months, she was shouting repeatedly with a squawking noise and hugging her mother. A local pediatrician identified a possible seizure‐like electroencephalographic abnormality. She was referred to Kobe University Hospital, Kobe, Hyogo, Japan, at age 1 year and 11 months, and started an oral anticonvulsant. At age 3 years, she still spoke two words at most. The static seizure was initially refractory, but gradually became well‐controlled with valproic acid and phenobarbital after the age of 11 years. She graduated from a school for special needs children, and began working at a vocational facility.
Around the age of 30 years, she presented with neurological symptoms including memory loss, aggression, irritability and anhedonia. At the age of 33 years, another hospital carried out a genetic examination, which was negative for dentatorubral‐pallidoluysian atrophy, spinocerebellar ataxia type 3 and spinocerebellar ataxia type 6. Endocrine and amino acid analyses were also negative. At the age of 35 years, she developed progressive bradykinesia, and by the age of 36 years she was unable to speak or to walk unaided. T2‐weighted magnetic resonance imaging (MRI) of the head showed low signal intensity in the globus pallidus and substantia nigra; notably, serum ceruloplasmin was normal. She was tentatively diagnosed with NBIA, although genetic tests of the NBIA‐associated genes pantothenate kinase (PANK2), ferritin light chain, ATP13A2, and PLA2G6 carried out elsewhere yielded negative results.
At the age of 38 years, she presented with parkinsonism, including left dominant limb rigidity, bradykinesia, frozen gait, forward‐bent posture and pulsion; she also showed parasomnia. Her posture improved and her gait became smooth with L‐DOPA (400 mg/day) therapy; her parasomnia was controlled with flunitrazepam.
At the age of 40 years, she was referred to our hospital, presenting with cognitive impairment, frontal sign, left dominant parkinsonism, left dominant hyperreflexia and right arm myoclonus. Laboratory data showed mild iron‐deficiency anemia. Electroencephalography identified diffuse 5–6 Hz theta waves and small, sharp fronto‐parietal spikes. Head MRI are shown in Figure 1a. We diagnosed her with BPAN based on: (i) progressive parkinsonism in adulthood; (ii) childhood seizures and psychogenic developmental delay; and (iii) our head MRI findings. Given her previous genetic findings, we investigated the WDR45 gene through exome analysis (Fig. 1b), which identified a c.700 C>T (p.Arg 234*) mutation in exon 9. This confirmed the BPAN diagnosis. Sanger sequencing (Fig. 1c) confirmed a heterozygous mutation of the same site in the patient not present in the biological parents (parentage confirmed by microsatellite analysis: data not shown), and thus it was classified as a de novo mutation.
Figure 1.
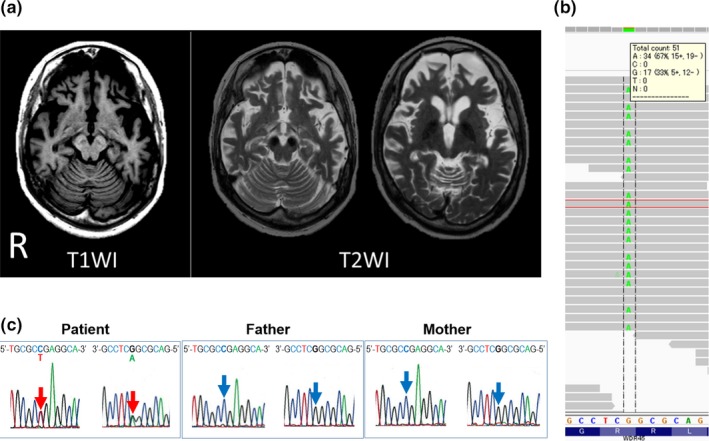
Clinical and genetic data in a Japanese case of WDR45 gene mutation. (a) Magnetic resonance imaging identified a halo in the substantia nigra in T1‐weighted imaging (T1WI); and low signal intensity in the globus pallidus and substantia nigra in T2‐weighed images (T2WI). Note also the diffuse cerebral and cerebellar atrophy. (b) Exome analysis identified a c.700 C>T (p.Arg 234*) nonsense mutation in WDR45. In this region, heterogeneity of adenine (A) and guanine (G) was shown with sufficient accuracy. (c) Sanger sequencing electropherograms in exon 9 of the WDR45 gene. The c. 700 position was identified by red (patient) or blue arrows (parents). DNA sequence traces showed the presence of the de novo heterozygous nonsense mutation, c.700C>T (p.Arg 234*), in the patient.
Discussion
When referred to our department, the present patient was suspected to be BPAN with typical disease progression and MRI findings. Our patient showed the symptom of sleep problems, which was not reported in a patient with the same mutation c.700C>T.4 The frequency of sleep problems was reported to be relatively low in BPAN (26.1–28.6%).4, 5
In the early phase, it is still difficult to confirm such a diagnosis. As more than 40 mutation sites in the WDR45 gene have been reported in BPAN, exome analysis was the most effective means of identifying the mutation in the present case.6 According to the study by Fieremans et al., the nonsense mutation c.700C>T was seen in only one out of 51 patients (cumulative total number of people). Thus, this position was not considered to be a mutation hot spot. However, in five patients (9.8%), a codon CGA for arginine was replaced by a stop codon TGA, meanwhile the other mutations were not fixed.6 Although exon 9 of the WDR45 gene, including this mutation c.700 C>T, is not embedded in CpG islands according to the UCSC genome browser database, methylated CpG dinucleotides was known to have hypermutability of C to T.7 Therefore, this nonsense mutation that changed CGA for arginine to TGA for the stop codon, could be relatively frequent in BPAN.2, 3, 4, 5, 6
In diagnosing NBIA, it is helpful to recognize the progressive extrapyramidal symptoms and low signal intensity of the globus pallidus in T2‐weighted MRI. These are quite rare diseases, though, and at present, it is difficult to confirm such a diagnosis using only clinical information. However, exome analysis is expected to propel the practical application of genomic medicine, so active genetic testing to diagnose and differentiate NBIA and its subtypes has quickly become an important procedure.
Acknowledgments
This work was supported by Grants‐in‐Aid from the Research Committee of CNS Degenerative Diseases, the Ministry of Health, Labor and Welfare to TT, and a Grant‐in‐Aid for Young Scientists (25713015) to WS. We thank Akiko Hosoya for providing the data of microsatellite analysis. The authors disclose no conflict of interest.
References
- 1. Schneider SA. Neurodegeneration with brain iron accumulation. Curr. Neurol. Neurosci. Rep. 2016; 16: 9. [DOI] [PubMed] [Google Scholar]
- 2. Haack TB, Hogarth P, Kruer MC et al Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X‐linked dominant form of NBIA. Am. J. Hum. Genet. 2012; 91: 1144–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Saitsu T, Nishimura K, Muramatsu H et al De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 2013; 45: 445–9. [DOI] [PubMed] [Google Scholar]
- 4. Hayflick SJ, Kruer MC, Gregory A et al β‐Propeller protein‐associated neurodegeneration: a new X‐linked dominant disorder with brain iron accumulation. Brain 2013; 136: 1708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nishioka K, Oyama G, Yoshino H et al High frequency of beta‐propeller protein‐associated neurodegeneration (BPAN) among patients with intellectual disability and young‐onset parkinsonism. Neurobiol. Aging 2015; 36: 2004.e9‐15. [DOI] [PubMed] [Google Scholar]
- 6. Fieremans N, Van Esch H, Holvoet M et al Identification of intellectual disability genes in female patients with a skewed X‐inactivation pattern. Hum. Mutat. 2016; 37: 804–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ehrlich M, Norris KF, Wang RY, Kuo KC, Gehrke CW. DNA cytosine methylation and heat‐induced deamination. Biosc. Rep. 1986; 6: 387–93. [DOI] [PubMed] [Google Scholar]