


Abstract
To study the role of the poly(A) tail length during the replication of poly(A)-containing plus-strand RNA virus, we have developed a simple reverse transcription polymerase chain reaction (RT–PCR)-based method that substantially improves the previously reported PAT [poly(A) test] assay. In contrast to the PAT assay, the new method is based on the enzymatic 3′ elongation of mRNA with guanosine residues, thus immediately preserving the 3′ end of the RNA and creating a unique poly(A)–oligo(G) junction. The oligo(G)-protected full-length poly(A) tail is reverse transcribed using the universal anti-sense primer oligo(dC9T6) and amplified by PCR with a gene-specific sense primer. After sequencing the resulting RT–PCR product the length of the poly(A) tail was unequivocally deduced from the number of adenosine residues between the oligo(G) stretch and the sequence upstream of the poly(A) tail. The efficiency and specificity of the newly developed assay was demonstrated by analysing the poly(A) tail length of the hepatitis A virus (HAV) RNA. We show here that the poly(A) tail of HAV RNA rescued after transfection of in vitro transcripts was elongated in the course of HAV replication.
INTRODUCTION
The poly(A) tail at the 3′ end of mRNA seems to have protective function against exoribonuclease degradation and is involved in initiation of translation. Stability and translatability of mRNA has been directly correlated with the length of the poly(A) tail that is added to the primary transcript in the nucleus (1). Replication of picornaviruses, a family of plus-strand RNA viruses, takes place in the cytoplasm and their 3′ terminal poly(A) tail is genome-encoded as suggested by the detection of a poly(U) tract at the 5′ end of the minus-strand replication intermediate (2–4). Upon infection of plus-strand RNA viruses, their genomes function in two ways: initially, the RNA serves as template for translation yielding RNA replication factors and subsequently for minus-strand RNA synthesis, which proceeds in the opposite direction. Recently, it was shown that picornavirus translation is strongly stimulated by their poly(A) tail (5). Moreover, the crucial importance of the poly(A) tail in the replication of picornaviruses was deduced from the observation that in vitro RNA transcripts with a short poly(A) tail had a reduced specific infectivity (3,4,6–8). In order to further understand the role of the 3′ poly(A) tail in the regulation of both translation and replication of plus-strand RNA viruses, it is of crucial interest to follow changes in the tail length during the course of viral replication.
Recently, various methods based on reverse transcription polymerase chain reaction (RT–PCR) amplification have been employed to assess the polyadenylation state of mRNA [PAT, poly(A) test assay, reviewed in 9]. Since in these assays oligo(dT) adaptor primers are used which can anneal at any position within the poly(A) tail, the products of RT might not represent the complete poly(A) tail. Here, we describe a new PCR-based oligo(G)-tailing method in which the 3′ end of the mRNA is immediately preserved from degradation by the enzymatic addition of an oligo(G) tail. With this step a poly(A)–oligo(G) junction is generated which serves as specific target for the amplification of the 3′ end of the viral genome with the universal reverse primer oligo(dC9T6) and a gene-specific forward primer. The universal antisense primer also ensures that only RNA molecules terminating with adenosine residues are amplified. The subsequent sequencing of the RT–PCR product allowed the accurate poly(A) tail length quantification. Using this method, the poly(A) tail length of hepatitis A virus (HAV) RNA rescued after transfection of in vitro transcripts with a defined numbers of adenosine residues was determined by sequencing.
MATERIALS AND METHODS
Virus and cells
HAV strain 18f was propagated in a monkey kidney (BSC-1) or a human hepatoma cell line (HuhT7). For virus infection, confluent cells were inoculated with an HAV-containing cell extract for 3 h at 37°C. The HAV-infected cells were incubated for 2 weeks before virus production was monitored by an HAV-specific enzyme-linked immunosorbent assay (ELISA) and RT–PCR (10,11).
Oligonucleotides
The following oligonucleotides were used in this study. (i) 5′-CCCCCCCCCTTTTTT-3′ [oligo(dC9T6)] was the universal reverse primer #1 for RT–PCR. It specifically anneals to the poly(A)–oligo(G) junction near the 3′ end of the RNA. (ii) 5′-ACTTGGCATGACAGCTACTTCTGCTGA-3′ (primer #2) representing nucleotides 7076–7102 of HAV strain 18f (GenBank accession no. M59808) was used as forward primer in the PCR. (iii) 5′-TTGTTCAGTCCTGTTTGGAG-3′ (primer #3) representing nucleotides 7321–7340 of HAV strain 18f was used for sequencing the PCR product.
RNA preparation and transfection
Total cellular RNA (20–40 µg) including poly(A)-containing RNA (0.2–0.4 µg) was isolated from 75 cm2 flasks of infected and uninfected cells using the RNeasy and Oligotex kits, respectively (Qiagen). The quality and quantity of isolated RNA was monitored by denaturing 1.5% agarose gel electrophoresis and spectrophotometry. The poly(A)-containing RNA was used directly for polyguanylation. The preparation of in vitro transcripts 3′RNA117 and 3′RNA524, derived from the 3′ end of HAV, has been described previously (12).
Full-length HAV in vitro transcripts were prepared from the AatII-linearised cDNAs of constructs pT7-18f-A0, pT7-18f-A14, pT7-18f-A20 and pT7-18f-A26, resulting in RNAs rHAV-A0, rHAV-A14, rHAV-A20 and rHAV-A26, respectively (Kusov,Y.Y., Shatizishvili,G. and Gauss-Müller,U., manuscript in preparation). In addition to the variable length of the poly(A) tail, the full-length HAV in vitro transcripts contained 344 vector-derived nucleotides downstream of the poly(A) tail which did not interfere with genome replication, as was observed for HAV (data not shown) and other viral transcripts (13). For RNA transfection, 2 µg of run-off transcript was added to 1 ml of OptiMEM containing 6 µl of DMRIE-C (Life Technology), mixed and immediately transferred to ∼0.5 × 106 cells grown to 80–90% confluency in a 10 cm2 plate. After incubation for 4 h at 37°C, the transfection mixture was replaced by medium supplemented with 10% fetal calf serum and G418 and incubation was continued for 16 days. Extracts of transfected cells were used to infect more cells in a second passage, which were harvested 14 days after infection. RNA-transfected and HAV-infected cells were lysed with PBS containing 0.05% Tween-20 and analysed by ELISA. Total RNA was isolated with RNeasy and applied to standard RT–PCR as described (11).
Guanylation of mRNA
The poly(A)-containing HAV RNA transcripts 3′RNA117 and 3′RNA524 and mRNA extracted from infected and uninfected cells were polyguanylated or polyadenylated using yeast poly(A) polymerase (PAP) as recommended by the manufacturer (Amersham Pharmacia Biotech) with the following modifications. To abolish the higher ordered secondary structure at the 3′ end of HAV RNA (12), RNA samples (5–10 ng) were heated at 65°C for 5 min and immediately placed on ice. They were incubated for 2 h at 37°C with 1520 U of PAP and 0.6 mM GTP or ATP in a 25 µl reaction mixture containing 20 mM Tris–HCl, pH 7.0, 50 mM KCl, 0.7 mM MnCl2, 0.2 mM EDTA, 100 µg/ml BSA and 10% glycerol. To complete the reaction, a further 760 U of PAP were added and incubation was continued for 2 h. To monitor the 3′ elongation of the RNA, the RNA electrophoretic mobility was analysed on 7 or 15% polyacrylamide gels containing 8 M urea. The number of guanosine residues added to the RNA transcripts was calculated by comparison with marker RNA molecules.
RT–PCR
The C.therm (Carboxydothermus hydrogenoformans) polymerase one-step RT–PCR kit (Roche Diagnostics, Mannheim) was applied using hot start conditions (72°C, 30 s). The first strand cDNA synthesis (RT, 60°C for 1 h) was primed by oligo(dC9T6) (6 µM). As specificity control, oligo(dT12–18) (Life Technologies) was used. The RT reaction was stopped by denaturation at 95°C (2 min) and the cDNA product was used for PCR amplification with the HAV-specific forward primer (primer #2, 0.3 µM) and the following program (45 cycles): 1 min at 94°C, 1 min at 52°C followed by 10 s of a temperature gradient from 53 to 59°C and 1 min at 60°C. Finally, product extension was completed by incubation for 7 min at 72°C. The PCR product of 0.43 kb was gel-purified (Gel extraction kit, Qiagen) after agarose electrophoresis and either used directly for sequencing or for cloning into vector pCR2.1-TOPO-TA as recommended by the manufacturer (Invitrogen). At least three individual clones were isolated and their cDNA was sequenced.
Custom service nucleotide sequencing
Primer #3 was used for sequencing of the PCR product. For sequencing of the cloned PCR products, plasmids of multiple clones were analysed using plasmid-specific primers. Sequencing was performed by AGOWA (Berlin, Germany).
RESULTS AND DISCUSSION
General outline of the method
To follow alterations in the poly(A) tail length of viral RNA during the replicative cycle, a method was developed which allows the exact determination of the number of adenosine residues of poly(A)-containing RNA. As an essential step (step 1) of the procedure (Fig. 1), oligo(dT)-purified mRNA is elongated at its 3′ end with PAP and GTP, thus creating an additional homopolymeric tail of guanosine residues. The newly created junction poly(A)–poly(G) serves as target to generate a cDNA copy by RT (Fig. 1, step 2a) with the antisense primer #1 oligo(dC9T6). The subsequent PCR specifically amplifies the tagged cDNA using a message-specific forward primer #2 (Fig. 1, step 2b). The resulting PCR product can be sequenced either directly or after cloning into a suitable vector (Fig. 1, step 3). In an initial approach, we established the method on in vitro transcripts representing the 3′ end of the HAV genome.
Figure 1.
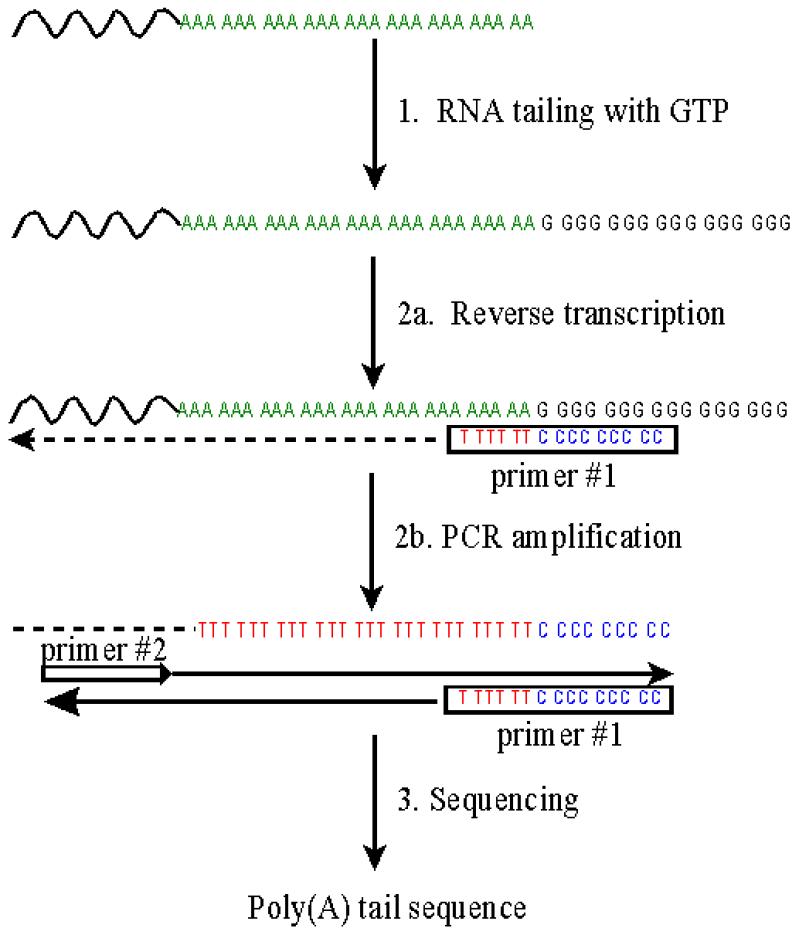
Schematic presentation of the G-tailing method. In step 1, purified mRNA (including viral RNA) is polyguanylated using PAP and GTP. In step 2a, the guanylated RNA is reverse transcribed using oligo(dC9T6) as universal primer #1. In step 2b, the RT product is amplified by PCR with oligo(dC9T6) and the HAV-specific primer #2. In step 3, the PCR product is sequenced with either primer #3 or vector-specific primers.
Addition of a GMP-homopolymer tract to RNA
Previously it has been shown in a comparative study with radiolabelled ribo-NTPs that nucleotide addition by yeast PAP to a 15mer oligo(A) RNA was more efficient with GTP than with UTP or CTP (14). Therefore, GTP in combination with yeast PAP was used here for 3′ tailing. To monitor the 3′ addition of unlabelled guanosine residues, two short HAV RNA transcripts, 3′RNA117 and 3′RNA524 encoding the 3′ end of the viral genome including a stretch of 26 adenosine residues, were used in the PAP reaction with GTP. To compare the efficiency of adenosine and guanosine addition, the reaction was performed with GTP and ATP. Elongation using ATP as substrate resulted in a pronounced shift in electrophoretic mobility demonstrating the addition up to 1000 adenosine residues to 3′RNA117 (Fig. 2A, lanes 1 and 2) and 3′RNA524 (Fig. 2A, lanes 4 and 5). The 3′ addition of GTP was less efficient which was apparent by a small shift in the electrophoretic mobility of 3′RNA117 (Fig. 2A, compare lanes 1 and 3) and 3′RNA524 (Fig. 2A, lanes 4 and 6, and B, lanes 1 and 2). Based on these data, we calculated that approximately 14 guanosine residues were added to the 3′ end of both RNAs. With the same efficiency, full-length HAV RNA in vitro transcribed from cDNA (7.5 kb length) was elongated by either GTP or ATP (see below).
Figure 2.
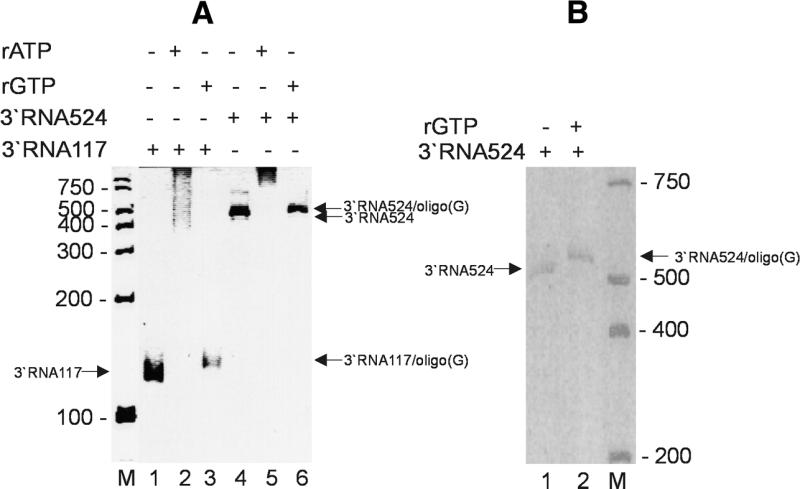
Elongation of subgenomic viral in vitro transcripts with PAP and ATP or GTP. HAV 3′RNA117 [(A) lane 1] and 3′RNA524 [(A) lane 4 and (B) lane 1] were elongated by PAP using ATP [(A) lanes 2 and 5] or GTP [(A) lanes 3 and 6 and (B) lane 2] as substrates. A scheme of combination is indicated on top. The electrophoretic mobility of the RNAs was monitored on a 7% (A) or 15% (B) denaturing polyacrylamide gel. The mobilities of tailed and parental RNAs are indicated with arrows; marker RNAs of known length are indicated on lanes ‘M’.
RT and PCR
To simplify subsequent steps, amplification of guanylated RNA was performed in a one-step RT–PCR using the universal antisense primer #1 and a HAV-specific sense primer (primer #2). Since the 3′ end of the HAV genome likely contains stable secondary structures (12), RT was performed at elevated temperature (60°C) with the thermostable polymerase of C.therm. In order to reach a sufficiently high annealing temperature, we used oligo(dC9T6) as reverse primer which hybridised to nine guanosine and six adenosine residues of the elongated RNA template. The specificity of the antisense primer and the RT reaction conditions was tested first by comparing the amplification products of the RT–PCR with either oligo(dC9T6) or oligo(dT12–18) as antisense primers. Under the RT–PCR conditions chosen, only the reverse primer oligo(dC9T6) in combination with the HAV-specific forward primer resulted in the expected amplification product of 0.43 kb with guanylated 3′RNA524 as the template (Fig. 3A, lane 2). Due to the stringency of the RT–PCR condition, reaction with oligo(dT12–18) as reverse primer did not yield the expected product (Fig. 3A, lanes 3, 6 and 9). mRNA without a GMP-homopolymer tract could not be amplified under the experimental conditions (Fig. 3A, lanes 1, 4 and 7). When the complete HAV run-off RNA transcript was used as template instead of the 3′ terminal subgenomic HAV RNA, again only the guanylated form, but not the parental transcript, generated the expected amplification product (data not shown). Taken together, the data demonstrate that the RT–PCR conditions used allowed to specifically amplify the oligo(G)-tailed 3′ end of the HAV genome. Based on these results, amplification of viral RNA, extracted from infected cells, was assessed in a similar manner. As shown in Figure 3A (lanes 4–6), the 3′ oligo(G)-elongated viral RNA was specifically amplified only when oligo(dC9T6) (Fig. 3A, lane 5) was used as reverse primer but not oligo(dT12–18) (Fig. 3A, lane 6). Neither viral RNA without the GMP-homopolymer tract (Fig. 3A, lane 4) nor RNA of uninfected cells (Fig. 3A, lanes 7–9) were amplified under the RT–PCR conditions used, demonstrating that the amplification conditions were specific for the detection of viral RNA.
Figure 3.

Specific amplification of guanylated subgenomic and genomic HAV RNA. (A) According to the scheme on top, subgenomic in vitro transcripts (lanes 1–3), RNA of HAV-infected cells (lanes 4–6) and mock-infected cells (lanes 7–9) were subjected to the PAP reaction with GTP and subsequently to the HAV-specific amplification. RT–PCR was performed either with oligo(dT12–18) or oligo(dC9T6) as antisense primers. The sense primer for PCR was primer #2 in all cases. (B) RT–PCR products derived from cell extracts containing HAV RNA rescued after transfection of transcripts rHAV-A0, rHAV-A14, rHAV-A20 and rHAV-A26. The PCR product of 432 bp is marked by arrows. DNA standards are indicated on the left (M).
Determination of the poly(A) tail length by sequencing of the PCR product
To initially test how precisely the number of adenosine residues can be determined, the sequencing reaction was performed on a PCR product derived from an in vitro transcript, which encoded 26 adenosine residues. DNA sequencing of the PCR product directly or after cloning into a vector demonstrated microheterogeneity in the poly(A) tail of the RNA template with a variance of up to ±3 residues which might be caused by slippage of the polymerases involved in the procedure (T7 RNA polymerase, reverse transcriptase and thermostable DNA polymerase). The slippage by thermostable DNA polymerase was ruled out after repeated amplifying and sequencing of PCR products containing 60 adenosine residues (data not shown). To determine the poly(A) tail length of viral RNA, the method was applied to RNA extracted from HAV-infected cells. After purification on an oligo(dT) matrix, polyguanylation, and subsequent RT–PCR using oligo(dC9T6) and primer #2 for amplification, sequencing of the cloned PCR product showed that the poly(A) tail length of viral RNA molecules varied from 41 to 60 adenosine residues. A similar number of adenosine residues was determined in an earlier report where conventional non-PCR procedures [isotope-labelling, RNAse A and T1 digestion, and oligo(dT) cellulose-hybridisation] were used (15). Since in the method described here, the 3′ end of the mRNA is protected against exonuclease degradation by the addition of an oligo(G) tail, it is reasonable to assume that the variation in the poly(A) tail length of viral RNA reflects the intrinsic heterogeneity which might originate from various functional forms of the viral RNA.
In some aspects, the procedure described here is similar to an assay in which the authentic 3′ end of the hepatitis C virus (HCV) RNA was preserved by ligating it to a phosphorylated oligoRNA (16). Fixation of the 3′ end of the HAV poly(A) tail was also important in our experimental approach and was achieved by the enzymatic addition of an oligo(G) tract. PAP-mediated elongation of the poly(A) tail resulting in an oligo(G) homopolymer tail is highly efficient and less time-consuming as compared to the addition of an oligoRNA which has to be 5′-phosphorylated prior to ligation to the 3′ end of the RNA (16). Furthermore, the addition of a homopolymeric guanosine tail to an existing poly(A) tail creates a nucleotide sequence which can be used as a universal target for priming the cDNA synthesis of any mRNA with oligo(dC9T6). Although not experimentally tested, we propose that the method described here is also suitable to analyse changes in the poly(A) tail length of specific mRNAs occurring during various biological processes (17).
3′ Addition of adenosine residues during HAV replication
In viral reverse genetics, cDNA or RNA transfection has proved to be an indispensable tool to directly analyse the role of genomic regions in viral RNA replication (18). To apply our method to a reverse genetic system, the 3′ end of viral RNA rescued after transfection of in vitro transcripts were characterised. Synthetic viral RNA transcripts derived from cDNA clones containing a defined number of adenosine residues (A0, A14, A20 and A26) followed by vector-derived sequences were transfected into mammalian cells, where they initiated an infectious cycle which was usually detectable starting 10 days after transfection. As determined by ELISA and standard RT–PCR (11) (data not shown), viral replication was apparent when transcripts with 14 or more adenosine residues at their 3′ end were transfected (Table 1). Accordingly, the 3′ end of the viral genome rescued after transfection of transcripts rHAV-A14, rHAV-A20 and rHAV-A26 (Fig. 3B, lanes 2–4), but not rHAV-A0 (Fig. 3B, lane 1), could be amplified. In contrast to poly(A)-terminating RNA molecules which were rescued during HAV genome replication (see ref. 7 for a model of picornavirus replication), input viral transcripts containing vector-derived sequences at their 3′ end were not detected with the new method. As shown in Table 1, the cloned PCR products obtained after specific amplification of the 3′ end of the poly(A)-containing RNA encoded poly(A) tails of 24–60 residues. Except in one case, all clones tested encoded a poly(A) tail which was longer than that of the transfected nucleic acid, providing direct evidence that during HAV genome replication, the poly(A) tail is elongated. Elongation of the poly(A) tail after transfection of rHAV-A26 was less efficient as compared to the other RNAs tested. This might be due to mutations in other parts of the genome that reduced the overall replication efficiency of this viral strain. Although it is not known which enzymatic activity (viral or host) catalyses poly(A) elongation of viral RNA, our results and data of others indicate that the polyadenylation state of viral RNA is regulated (3,4). This regulation might be important for the multiple functions of viral RNA during the replicative cycle, namely as template for translation and transcription, and finally packaging into viral capsids (3,4,17). How a particular polyadenylation state of viral RNA correlates with any of these RNA functions will be addressed in further studies.
Table 1. Poly(A) tail length of viral RNA obtained after transfection of in vitro transcripts.
| HAV in vitro transcript
used for transfectiona |
Number of adenosine residues in the poly(A)
tail of rescued viral RNAb |
| rHAV-A0 |
ndc |
| rHAV-A14 |
24 – 29e |
| rHAV-A20 |
54 – 60e |
| rHAV-A26 | 26 – 37e,f |
aThe name of the RNA indicates the number of adenosine residues in the cDNA.
bNumber of adenosine residues of multiple clones derived from PCR products.
cNot detected (HAV-specific ELISA and RT–PCR was negative).
eCell extracts were ELISA and RT–PCR positive.
fDue to second site mutations, rHAV-A26 replicates less efficiently and possibly poly(A) elongation is retarded.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr S. Lemon for Huh-T7 cells and HAV strain 18f, and Drs D. Reinhardt and P. Müller for critically reading the manuscript. This work was supported by the Deutsche Forschungsgemeinschaft (DFG, SFB 367, project B7). G.S. was supported by the Dr Arthur Pfungst-Stiftung, Frankfurt/Main. G.D. was supported by the Deutscher Akademische Austauschdienst (DAAD).
References
- 1.Beelman C.A. and Parker,R. (1995) Degradation of mRNA in eukaryotes. Cell, 81, 179–183. [DOI] [PubMed] [Google Scholar]
- 2.Dorsch-Hasler K., Yogo,Y. and Wimmer,E. (1975) Replication of picornaviruses. I. Evidence from in vitro RNA synthesis that poly(A) of the poliovirus genome is genetically coded. J. Virol., 16, 1512–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spector D.H. and Baltimore,D. (1975) Polyadenylic acid on poliovirus RNA IV. Poly(U) in replicative intermediate and double-stranded RNA. Virology, 67, 498–505. [DOI] [PubMed] [Google Scholar]
- 4.Spector D.H., Villa-Komaroff,L. and Baltimore,D. (1975) Studies on the function of polyadenylic acid on poliovirus RNA. Cell, 6, 41–44. [DOI] [PubMed] [Google Scholar]
- 5.Bergamini G., Preiss,T. and Hentze,M.W. (2000) Picornavirus IRESes and the poly(A) tail jointly promote cap-independent translation in a mammalian cell-free system. RNA, 6, 1781–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sarnow P. (1989) Role of 3′-end sequences in infectivity of poliovirus transcripts made in vitro. J. Virol., 63, 467–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wimmer E., Hellen,C.U.T. and Cao,X. (1993) Genetics of poliovirus. Annu. Rev. Genet., 27, 353–436. [DOI] [PubMed] [Google Scholar]
- 8.Herold J. and Andino,R. (2001). Poliovirus RNA replication requires genome circularization through a protein–protein bridge. Mol. Cell., 7, 581–591. [DOI] [PMC free article] [PubMed]
- 9.Salles F.J., Richards,W.G. and Strickland,S. (1999) Assaying the polyadenylation state of mRNAs. Methods, 17, 38–45. [DOI] [PubMed] [Google Scholar]
- 10.Kusov Y. and Gauss-Müller,V. (1999) Improving proteolytic cleavage at the 3A/3B site of the hepatitis A virus polyprotein impairs processing and particle formation, and the impairment can be complemented in trans by 3AB and 3ABC. J. Virol., 73, 9867–9878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schalasta G., Engels,V. and Lindemann,L. (1995) A rapid and simple PCR assay for detection of hepatitis A virus RNA in stool specimens. Klin. Lab., 41, 233–238. [Google Scholar]
- 12.Kusov Y., Weitz,M., Dollenmeier,G., Gauss-Müller,V. and Siegl,G. (1996) RNA–protein interactions at the 3′ end of the hepatitis A virus RNA. J. Virol., 70, 1890–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eggen R., Verver,J., Wellink,J., De Jong,A., Goldbach,R. and van Kammen,A. (1996) Improvements of the infectivity of in vitro transcripts from cloned cowpea mosaic virus cDNA: impact of terminal nucleotide sequences. Virology, 173, 447–455. [DOI] [PubMed] [Google Scholar]
- 14.Martin G. and Keller,W. (1998) Tailing and 3′-end labeling of RNA with yeast poly(A) polymerase and various nucleotides. RNA, 4, 226–230. [PMC free article] [PubMed] [Google Scholar]
- 15.Siegl G., Frosner,G.G., Gauss-Muller,V., Tratschin,J.D. and Deinhardt,F. (1981) The physicochemical properties of infectious hepatitis A virions. J. Gen. Virol., 57, 331–341. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka T., Kato,N., Cho,M.J., Sugiyama,K. and Shimotohno,K. (1996) Structure of the 3′ terminus of the hepatitis C virus genome. J. Virol., 70, 3307–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jackson R.J. and Standart,N. (1990) Do the poly(A) tail and 3′ untranslated region control mRNA translation? Cell, 62, 15–24. [DOI] [PubMed] [Google Scholar]
- 18.van Gennip H.G., van Rijn,P.A., Widjojoatmodjo,M.N. and Moormann,R.J. (1999) Recovery of infectious classical swine fever virus (CSFV) from full-length genomic cDNA clones by a swine kidney cell line expressing bacteriophage T7 RNA polymerase. J. Virol. Methods, 78, 117–128. [DOI] [PubMed] [Google Scholar]