

Abstract
Adenocarcinoma accounts for ~40% of lung cancer, equating to ~88,500 new patients in 2015, most of who will succumb to this disease, thus, the public health burden is evident. Unfortunately, few early biomarkers as well as effective therapies exist, hence the need for novel targets in lung cancer treatment. We previously identified epiregulin (Ereg), an EGF-like ligand, as a biomarker in several mouse lung cancer models. In the present investigation we used a primary two-stage initiation/promotion model to test our hypothesis that Ereg deficiency would reduce lung tumor promotion in mice. We used 3-methylcholanthrene (initiator) or oil vehicle followed by multiple weekly exposures to butylated hydroxytoluene (BHT; promoter) in mice lacking Ereg (Ereg−/−) and wildtype controls (BALB/ByJ; Ereg+/+) and examined multiple time points and endpoints (bronchoalveolar lavage analysis, tumor analysis, mRNA expression, ELISA, wound assay) during tumor promotion. At the early time points (4 and 12 wk), we observed significantly reduced amounts of inflammation (macrophages, PMNs) in the Ereg−/− mice compared to controls (Ereg+/+). At 20 weeks, tumor multiplicity was also significantly decreased in the Ereg−/− mice vs controls (Ereg+/+). IL10 expression, an anti-inflammatory mediator, and downstream signaling events (Stat3) were significantly increased in the Ereg−/− mice in response to BHT, supporting both reduced inflammation and tumorigenesis. Lastly, wound healing was significantly increased with recombinant Ereg in both human and mouse lung epithelial cell lines. These results indicate that Ereg has proliferative potential and may be utilized as an early cancer biomarker as well as a novel potential therapeutic target.
Keywords: epiregulin, tumor promotion, lung, inflammation, BHT
Introduction
Lung adenocarcinoma (ADC), a non-small cell lung carcinoma, is the primary sub-type of all lung cancers, both nationally as well as internationally [1]. Five yr. survival rates for those patients diagnosed with NSCLC (mainly ADC and squamous cell carcinoma) largely depend on stage of diagnosis. Stage 1 patients have ~45% survival rates, stage 2 ~30%, and stage 3 and higher <14% survival at 5 yrs, however most patients are not diagnosed until the advanced stages [1]. Thus additional pathways to identify early biomarkers as well as targets for therapies are critically needed for NSCLC. Tumor promotion is a critical stage in multistage lung carcinogenesis (i.e. initiation, promotion, progression) [2–4]. Initiation of cancer progenitor cells by carcinogens (genotoxic mutations) is followed by promotion where initiated cells are induced to promote further proliferation and clonal expansion that involves several factors such as inflammation, gap junction inhibition, and proliferation [2–4]. Promotion is the only reversible stage of carcinogenesis thus identification of biomarkers during promotion could provide useful diagnostic targets, however, currently few exist.
A well characterized primary lung cancer model for tumor promotion is the 2-stage model, where a low dose of the initiator (3-methylcholanthrene, MCA) is followed by a promoter (butylated hydroxytoluene, BHT) to induce tumors in those previously initiated lungs [5]. Inflammation, namely innate immune cells such as macrophages and neutrophils, is correlated to promotion in this model and deficiency in these cell types significantly reduces tumor promotion [6,7]. However, it is the combination of events including inflammation, increased proliferation, and reduced gap junctional intercellular communication that likely drive promotion in this model [6,8,9]. We previously identified a growth factor upregulated in several mouse lung cancer models (epiregulin, Ereg) [10] and herein demonstrate a role for Ereg during tumor promotion.
Ereg is a 46 amino-acid hormone peptide that resides on mouse chromosome 5 and human chromosome 4 and is nestled near two other EGFR ligands (amphiregulin, betacellulin) [11]. Ereg has receptor promiscuity and can signal through several epidermal growth factor receptors, EGFR (ERBB1) and ERBB4, as well as all possible heterodimeric receptor complexes [12–14]. Mice deficient in Ereg demonstrated that this growth factor is essential for protection from intestinal damage, but not intestinal tumors [15]. EREG protein expression is also elevated in advanced NSCLC biopsy specimens [16] and in some lung cancer cell lines (e.g. HCC827, A549) [17–21]. In addition, EREG gene expression is upregulated in inflammatory mammary cancer, an aggressive subtype of mammary carcinomas [22] and in human colon cancer [23]. Basally, Ereg has extremely low expression or none at all in most tissues, including lung, in both human and mouse [19,20]. However, EREG is observed in leukocytes (e.g. macrophages) and epithelial cells during certain diseases involving inflammation, such as cancer. For example, Ereg can promote the proliferation of adult mouse liver progenitor cells to encourage regeneration, is significantly elevated in serum from patients with acute liver failure [24], and preferentially induces wound healing above that observed with epidermal growth factor (EGF) in normal human keratinocytes [25]. In HCC827, a human lung ADC cell line, EREG was required for both cell invasion as well as cell survival [16]. Thus, evidence suggests that EREG has important roles in proliferation of some epithelial cell types and cell invasion and survival of lung tumor cells. In the MCA/BHT-induced carcinogenesis model of lung cancer, Ereg was significantly upregulated compared to MCA or oil treatment alone both at an early time point (4 wks; whole lung) and in the tumors compared to non-tumor tissue or control in BALB/cJ mice with a mutant toll-like receptor 4 [10]. Similar changes were observed in Ereg expression in urethane-induced carcinogenesis in the same background strain at both 12 (whole lung tissue) and 22 wks (tumor versus saline treated or non-tumor tissue) [26]. Thus, while Ereg expression was significantly upregulated in early and late stages of lung cancer development, a clear gap remains in our understanding of the role of the EREG growth factor in BHT-induced tumor promotion and the precursor events (i.e. inflammation) leading to tumor development.
Our overall hypothesis for these studies was that epiregulin deficiency will result in reduced lung tumor promotion in mice
We demonstrate herein that both inflammatory cells and tumors are reduced in mice lacking Ereg and that these mice have altered signaling pathways as a result of their deficiency (such as interleukin 10, Il10). In addition, using an in vitro model (human bronchial epithelial cell line, BEAS2B and an ADC progenitor cell line, C10 cells), we observed increased wound healing in the presence of recombinant Ereg, supporting a role for Ereg in proliferation.
Materials and Methods
Animals
Five to seven week old male mice were used for all studies. B6.129S6-Eregtm1Dwt/Mmnc (#016151) were purchased from the Mutant Mouse Regional Resource Center (MMRRC UNC Chapel Hill, NC) on a C57BL/6J background as described previously [15]. At the University of Colorado Denver animal facility, mice were then backcrossed onto a BALB/ByJ background (Jackson Laboratories, Sacramento, CA) until they were 99% BALB/ByJ (BALB-Eregtm1/UCD; Ereg−/−), assessed by Jackson Laboratories Genetic Analysis Lab, and then bred for experiments. Male, age-matched wild-type controls (BALB/ByJ, Ereg+/+, Jackson Laboratories) were used for these studies after 1 week acclimation. Mice were fed irradiated mouse chow (Harlan) and water ad libitum and housed in ventilated shoebox cages with humidity and temperature control. All animal procedures were conducted in facilities accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care and approved by the University of Colorado Denver Institutional Animal Care and Use Committee and followed the Helsinki convention for the use and care of animals.
The genotyping protocol used for these Ereg−/− mice is published at the MMRRC UNC at http://www.med.unc.edu/mmrrc/resources/genotyping-protocols/mmrrc-16151/view. Briefly, three primers were used Ereg-S3, Ereg-AS1, lacZ-AS2 to produce a 253 bp product if mutant Ereg (Ereg−/−) and 156 bp if wildtype Ereg (Ereg+/+). The primer sequences are located at the MMRRC site listed above. Supplementary Fig. S1 represents an example of the genotyping.
Experimental Design
Two protocols were used for these studies (please see Bauer et al, 2009 for schematic).
Protocol 1
Early events. Briefly, this protocol was used for assessment of early promotion events using 4 weekly i.p. injections of BHT (Sigma, St. Louis) starting at 150 mg/kg body weight during the first week, and followed by 200 mg/kg for the next 3 wks, all based on previous studies [10,27]. Mice were euthanized with Fatal Plus (120 mg/kg; MWI, Boise, ID) at day 3 following protocol 1, which was previously demonstrated to be a peak time point for inflammation in this model [10,27]. Bronchoalveolar lavage [28] was then performed (1/2 lung; amount determined by body weight) using Hanks Balanced Salt Solution (HBSS) to determine the types of immune cells present via cell differentials as well as determine lung hyperpermeability, reflective of lung injury, by total protein analysis (BioRad, Hercules, CA), all following previous studies [10,26,27,29,30]. The BAL macrophages were then used for phagocytosis assays. The left lung lobe was subsequently snap frozen and used for molecular analysis (RNA and ELISAs).
Protocol 2
Two-stage initiation/promotion model. Briefly, this well established protocol was used to determine differences in tumor promotion between the two strains of mice. MCA (initiator) or corn oil vehicle was ip. injected (15 μg/g body weight) on week 1 followed by 6 weekly ip. injections of BHT (150 mg/kg wk 1; 200 mg/kg for weeks 2–6) or corn oil, described in Bauer et al., 2009. Six versus four injections for Protocol 1 are used to maximize promotional efficiency since the number of tumors that arise are proportional to the number of injections [31]. Two time points were assessed. Twelve weeks after MCA, mice were euthanized for lavage analysis as described in protocol 1. Twenty weeks following MCA administration, the mice were euthanized as described above, lungs fixed in Tellyesniczky’s fixative [32], and tumors enumerated using a Olympus SZX7 dissecting microscope (Olympus, Center Valley, PA). Tumors were also sized using a digital caliper (Mitutoyo, Japan), area determined (= 3/4πr2), and morphology (Olympus BX43) determined by light microscopy using H and E sections.
For immunohistochemistry, the lung sections were stained for Ereg with a specific anti-mouse epiregulin antibody (C9, Santa Cruz) and the mouse on mouse (M.O.M.) kit (Vector Laboratories). Immunodetection of Ereg was evaluated in lungs from MCA/Oil and MCA/BHT BALB mice using peroxidase biotin-streptavidin immunohistochemistry as previously described [29,33,34].
RNA isolation, cDNA synthesis, and quantitative PCR (qRT-PCR)
Whole lung tissue was used to isolate RNA using Macherey-Nagel Nucleospin RNA II kit (Clontech Laboratories, Mountain View, CA) following their kit specifications. cDNA was prepared using oligo-dT, followed by PCR amplification in 50 μl reaction volumes [10]. qRT-PCR was then done using the cDNA stock and gene specific primers (intron-spanning) with KAPA SYBR FAST mastermix (Kapa Biosystems; Boston, MA) on a Mastercycler EP Realplex4 qRT-PCR cycler (Eppendorf; Hauppauge, NY) [9]. Primers sequences for Ereg, Mcp-1, and 18S are found in [9,10]; Il10 forward primer (5′-GCT CTT ACT GAC TGG CAT GAG-3′) and reverse primer (5′-CGC AGC TCT AGG AGC ATG TG-3′). Ct values for genes of interest were normalized to 18S [10], and calculations of fold induction performed [35], similar to previous studies [9,10,26]. Semi-quantitative PCR was used to validate an absence of Ereg expression (654 bp product) in the Ereg−/− mice using previously designed primers (Ereg-S3 and Ereg-AS4; [15] ). β-actin was used as the normalization gene for the semi-quantitative confirmation; the primers were from Lee et al. 2004 (548 bp product).
Phagocytosis assay
BAL was collected, and 50,000 cells/well (~95% macrophages) plated with macrophage complete media [Dulbecco’s Modified Eagle Medium (DMEM; Life Technologies, Grand Island NY), 50 ml fetal bovine serum (FBS; Atlanta Biological, Atlanta GA), 5 ml L-Glutamine (Life Technologies) and 5 ml Penicillin Streptomycin); incomplete media contains the same components without serum] and incubated at 37°C for 30 mins-1 h to allow macrophages to attach. The media was replaced with fresh media and the plate incubated overnight at 37°C. Zymosan bioparticles (Life Technologies) were added to each well (18 hr after plating) with macrophage incomplete media and incubated at 37°C for 1 hr. The macrophages were washed 3 times with PBS followed by staining with 0.4% trypan blue for 1 min. The cells were washed 4 times with PBS, re-suspended in macrophage complete media then stained with Hoechst’s Dye (Life Technologies) for 7 min. Macrophages were washed 3 times with PBS and fixed with 4% Formaldehyde (Electron Microscopy Sciences, Hatfield PA) for 20 min. The formaldehyde was removed, 200 μl PBS added to each well, and the plate stored at 4°C until imaging on a Perkin Elmer Operetta with guidance from the High Throughput and High Content Screening Core Facility (UCD). Harmony Software version 3.5 was then used for analyzing and imaging the nucleus with Hoechst staining and phase-contrast compared to the cells that had internal Zymosan particles. Phagocytic index = total Zymosan ingestion/total macrophages × 100. Cytochalasin D (10 μM) was used to inhibit phagocytosis.
Nuclear protein isolation and transcription factor activity assays
Nuclear protein was prepared from the lung tissue in both strains using the Nuclear Extract Kit (Active Motif, Carlsbad, CA) and following the manufacturer’s protocol. Stat3, phospho-c-Jun, and phospho-p65 NFκB activity levels were determined using the specific TransAm Transcription Factor ELISA kits for Stat3, AP-1, NFκB following manufacturer’s instructions. We used 12 μg of nuclear protein per sample for these studies.
Enzyme-linked immunosorbent assay (ELISA)
Mouse keratinocyte chemoattractant (Kc) duo-set ELISA (R and D Systems, Minneapolis, MN) was used following the manufacturer’s specifications. BAL fluid was tested for these studies and a Tecan Infinite M200 PRO Microplate Reader with Magellan 7.0 Software (Morrisville, NC) used to read the ELISA at 450 nm.
Wound healing assay
C10 cells, a non-tumorigenic alveolar type II cell pneumocyte cell line derived from BALB lung [36] and human BEAS2B, a normal bronchial epithelial cell line [37], were grown in 12-well dishes to confluence using complete media (CMRL media for C10 cells and RPMI media for BEAS2B, Invitrogen), 10% Fetal Bovine Serum (Atlanta Biological, Atlanta, GA),1% L-Glutamine; 1% Penicillin streptomycin). Following confluency, media was replaced with serum-free media for 24 hr followed by treatment with several concentrations of recombinant Ereg (rEreg; R and D systems) or recombinant epidermal growth factor (Egf, 2 nM), and a wound made in the center of the cells using a 200 μl pipette tip [38]. The plate was imaged at 0 and 24 hr using the Nikon Eclipse Ti inverted research microscope (Nikon Corporation, Tokyo, Japan) to determine any changes. Areas were determined using NIH Image J Software. Proliferation was also assessed for confirmation of the wound assay in the BEAS2B cels using a 96-well plate and CellTiter96 proliferation assays (Promega) under the same conditions described above using 2 nM rEreg, rEGF, and 10% FBS as a positive control. We followed manufacturer’s protocol for the proliferation assay.
Statistical analysis
Statistics was performed using a 2-way ANOVA (factors: strain and treatment) and comparisons of means used Student-Newman-Keuls (SNK) a posteriori test) for all experiments except tumor promotion (Kruskal-Wallis One Way Analysis of Variance on Ranks). Data presented are all mean ± SEM. Sigma Plot (12.3) software was used for all graphs and statistics. For all analyses, p<0.05 was considered statistically significant.
Results
Ereg expression in Ereg+/+ mice in response to BHT
To confirm the importance of Ereg in this model, we first examined Ereg mRNA expression. As observed previously in BALB/cJ versus these BALB/ByJ mice, Ereg mRNA expression in BALB/ByJ (Ereg+/+) mice was significantly elevated compared to oil controls following protocol 1 and was validated using both semi-quantitative (Fig. 1A, lane 2, BHT treatment) and quantitative PCR (Fig 1B). Ereg expression was absent in the Ereg deficient mice (Fig 1A lanes 3 and 4, regardless of BHT treatment). Another EGFR ligand, amphiregulin, did not differ with treatment or strain (data not shown).
Figure 1. Ereg expression in response to BHT.

(A) Semi-quantitative PCR for Ereg (654 bp product, arrow) and β-actin (548 bp product) expression in Ereg+/+ mice exposed to oil control (O) and 4 weekly doses of BHT (B, protocol 1) compared to Ereg−/− mice (oil, O; BHT, B). (B) Quantitative RT-PCR for Ereg expression in response to BHT in the Ereg+/+ mice compared to oil control. Mean fold-change ± SEM presented, n = 3 per treatment, repeated three times. *, p<0.05 for BHT compared to oil controls. The 500 bp band is noted with an arrowhead on both gels from the 100 bp ladder.
Comparison of BALF inflammatory parameters at an early time point in Ereg+/+ and Ereg−/− mice
Three days following the final BHT injection (protocol 1), lavage was performed to identify the types of immune cells present in the lung. Significant increases in all inflammatory cell types were observed in both strains with BHT treatment compared to oil controls (see Table 1). However, macrophages in the BHT-treated Ereg+/+ mice were significantly elevated above those seen in Ereg−/− mice (Fig. 2A). Mcp-1 mRNA expression, a chemokine involved in macrophage recruitment, was also significantly elevated in the Ereg+/+ mice compared to Ereg−/− mice (Fig 2B). A distinct trend is present in the BHT-treated Ereg+/+ mice in Mcp-1 levels in BALF and tissue lysates that reflect the mRNA expression (data not shown). However, phagocytosis of the BALF macrophages did not differ between strains (Fig. 2C) suggesting similar functionality of this type of macrophage. Additionally, BALF total protein levels, indicative of lung hyperpermeability, and lymphocyte numbers, were significantly elevated in both strains in response to BHT, however no strain differences were observed. Supplementary Figure S2 demonstrates a common observation in the BHT model, with significant weight loss in response to BHT compared to oil. Weight loss is an indication of injury [27]. No significant strain differences were observed in these mice with respect to weight loss.
Table 1.
Bronchoalveolar lavage fluid analysis from Ereg+/+ and Ereg−/− mice 3 days following 4 weekly doses of BHT.
| Ereg+/+ | Ereg−/− | |||
|---|---|---|---|---|
| Phenotype* | Oil | BHT | Oil | BHT |
| Macrophages | 6.7 ± 0.8 | 20.9 ± 1.9‡§ | 5.5 ± 0.6 | 12.5 ± 1.3‡ |
| Lymphocytes | 0.1 ± 0.0 | 0.8 ± 0.4‡ | 0.0 ± 0.0 | 0.5 ± 0.1 |
| PMNs | 0.1 ± 0.1 | 1.4 ± 0.5 | 0.0 ± 0.0 | 1.6 ± 0.9 |
| Epithelial | 0.2 ± 0.0 | 0.4 ± 0.1 | 0.3 ± 0.0 | 0.2 ± 0.1 |
| Total BAL Protein† | 92.6 ± 10.9 | 463.0 ± 41.5‡ | 116.1 ± 20.4 | 474.9 ± 61.3‡ |
All cell types are (× 104).
protein concentration in μg/ml.
P < 0.05 compared to oil controls;
P < 0.05 compared to Ereg−/− mice.
Figure 2. BHT-induced inflammation is significantly reduced 4 weeks following BHT in Ereg−/− mice compared to controls (Ereg+/+ mice).

(A) Bronchoalveolar lavage [28] analysis revealed macrophage infiltration in the lungs following BHT treatment (protocol 1). Mean ± SEM presented, n = 5 mice per treatment, repeated three times. (B) Ccl2 (Mcp-1) mRNA expression in whole lung tissue from Ereg+/+ mice compared to Ereg−/− mice. Mean fold change ± SEM presented, n = 3 per treatment, repeated three times. (C) Phagocytic index in BAL macrophages from Ereg+/+ mice compared to Ereg−/− mice. Phagocytic (zymosan) index was calculated by number of positive cells/total number of macrophages X 100. Cyt. D = cytochalasin D, inhibitor of phagocytosis. *, p<0.05 for BHT compared to oil controls; +, p<0.05 for Ereg+/+ mice compared to Ereg−/− mice.
At 12 weeks following MCA, the inflammatory parameters changed compared to that observed at 4 wks. Macrophage elevations were again observed in both strains above the oil and MCA/oil control groups and remained significantly increased in the Ereg+/+ mice compared to the Ereg−/− mice (Fig. 3A). However, at this 12 wk time point, PMNs and lymphocytes were also significantly elevated in the Ereg+/+ mice above that observed in the Ereg−/− mice (Fig. 3B and Table 2), suggesting involvement of these immune cells in tumor promotion. Kc, a PMN chemoattractant, was also significantly elevated in the BALF from these mice, supporting the increased PMN numbers observed (Fig 3C).
Figure 3. BHT-induced inflammation is significantly reduced 12 weeks following MCA/BHT in Ereg−/− mice compared to controls (Ereg+/+ mice).

(A) BAL macrophage infiltration into the lungs in both Ereg+/+ and Ereg−/− mice 12 weeks following the MCA or oil injection (protocol 2), oil controls (oil), or MCA/oil alone (MCA/oil). Mean ± SEM presented, n = 3–5 mice per treatment, repeated twice. (B) BAL PMN infiltration into the lungs of both strains. Mean ± SEM presented, n = 3–5 mice per treatment, repeated twice. (C) Kc (Cxcl1) content in the BAL fluid from both Ereg+/+ and Ereg−/− mice 12 weeks following the MCA or oil injection (Protocol 2). Mean ± SEM presented, n = 3–5 mice per treatment, repeated twice. *, p<0.05 for BHT compared to oil controls; +, p<0.05 for Ereg+/+ mice compared to Ereg−/− mice.
Table 2.
BALF analysis from Ereg+/+ and Ereg−/− mice 12 weeks following oil, MCA, or MCA/BHT treatment.
| Ereg+/+ | Ereg−/− | |||||
|---|---|---|---|---|---|---|
| Phenotype* | Oil | MCA/Oil | MCA/BHT | Oil | MCA/Oil | MCA/BHT |
| Macrophages | 9.1 ± 2.1 | 10.6 ± 0.9 | 19.7 ± 1.3‡§ | 9.7 ± 1.7 | 4.5 ± 1.1 | 15.0 ± 1.1‡ |
| Lymphocytes | 0.1 ± 0.1 | 0.0 ± 0.0 | 0.5 ± 0.1‡§ | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.1 ± 0.0‡ |
| PMNs | 0.1 ± 0.0 | 0.8 ± 0.2 | 1.8 ± 0.4‡§ | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.4 ± 0.1‡ |
| Epithelial | 0.1 ± 0.1 | 0.1 ± 0.1 | 0.3 ± 0.1 | 0.0 ± 0.0 | 0.2 ± 0.2 | 0.3 ± 0.1 |
| Total BAL protein† | 88.8 ± 6.9 | 97.7 ± 6.4 | 160.2 ± 10.5‡ | 107.2 ± 17.6 | 101.0 ± 19.5 | 150.0 ± 12.6‡ |
All cell types are (× 104).
protein concentration in μg/ml.
P < 0.05 compared to oil controls;
P < 0.05 compared to Ereg−/− mice.
Inflammatory signaling in the absence of Ereg
We examined several cytokines for strain and treatment differences at the early time point, however only a few changed in the absence of Ereg. As mentioned earlier, Mcp-1 was elevated in Ereg+/+ mice to a greater extent than the Ereg−/− mice. The opposite was observed with IL10, an anti-inflammatory cytokine. Il10 expression was significantly increased in the Ereg−/− mice, compared to Ereg+/+ mice (Fig. 4A). The protein activity of a known transcription factor downstream of IL10, STAT3, was also significantly increased in the Ereg−/− mice compared to wildtype controls (Fig. 4B). Several other inflammatory mediators were influenced by BHT but not Ereg, namely IL6, inducible nitric oxide synthase [39], and arginase 1 (Supplementary Fig. S3). Additional transcription factors were tested for strain differences; phospho-c-Jun and NFκB p65 subunit were elevated with BHT treatment but did not differ with strain (data not shown).
Figure 4. Il10 expression and downstream signaling was significantly elevated in response to BHT in the Ereg deficient mice compared to controls.

(A) QRT-PCR for Il10 mRNA expression in whole lung in the Ereg+/+ and Ereg−/− mice following BHT (protocol 1) or oil controls. Mean fold change ± SEM presented, n = 3 per treatment, repeated three times. (B) Stat3 transcription factor binding activity, indicated by measurement of A450 (TransAM Stat3 kit), in response to BHT or oil controls between strains. Mean ± SEM presented; n = 4. Repeated twice. *, p<0.05 for BHT compared to oil controls; +, p<0.05 for Ereg+/+ mice compared to Ereg−/− mice.
Tumor promotion studies
We used the MCA/BHT model (protocol 2) to determine any differences in tumor promotion 20 wks following MCA administration, a well-established time point for promotion in this model [5]. Tumor multiplicity, or the number of tumors per mouse, was significantly elevated in the Ereg+/+ mice compared to the Ereg−/− mice (p <0.003; Fig. 5) with a 10.5-fold increase in the Ereg+/+ MCA/BHT treated mice over MCA/oil treatment compared to a 4.3-fold increase in the Ereg−/− mice. There were no tumors in any of the oil only controls for either strain. No significant strain differences were seen in tumor size (Fig. 5) or tumor morphology. However, tumor size analysis revealed that tumors between 0.15–5 mm2 were 10% lower in number in Ereg−/− mice than Ereg+/+ mice compared to tumors <0.15 mm2 which were 10% higher in the Ereg−/− compared to the wildtype mice.
Figure 5. Tumor promotion was significantly reduced in Ereg−/− mice compared to Ereg+/+ mice in response to a 2-stage lung carcinogenesis model.

(A) Tumors were enumerated in each strain following oil, MCA/oil, or MCA/BHT treatment (protocol 2). Mean ± SEM presented for tumor multiplicity or the number of tumors per mouse; Ereg+/+ mice (MCA/oil, n = 9; MCA/BHT n=19); Ereg−/− mice (MCA/oil n=5; MCA/BHT n=15). No tumors were observed in either strain with oil alone (n=3 per strain). *, p<0.05 for BHT compared to oil controls; +, p<0.05 for Ereg+/+ mice compared to Ereg−/− mice. (B) Tumor sizes evaluated using a digital caliper did not differ between strains in response to MCA/BHT. Ereg+/+ mice n = 130 tumors; Ereg−/− mice n = 35 tumors.
We then used Ereg immunostaining to determine the cell types expressing Ereg in the Ereg+/+ MCA/oil and Ereg+/+ MCA/BHT treated lungs. Ereg expression was observed in the bronchiolar regions and alveolar type II cells as well as in the macrophages (Fig. 6A, B) in the control MCA/oil treated lungs. Following MCA/BHT treatment, Ereg expression increased in these same cell types and was observed in some, but not all tumors (>50%) to varying degrees, with some intense staining observed (Fig. 6C–F).
Figure 6. Localization of Ereg in the Ereg+/+ mice by immunostaining.
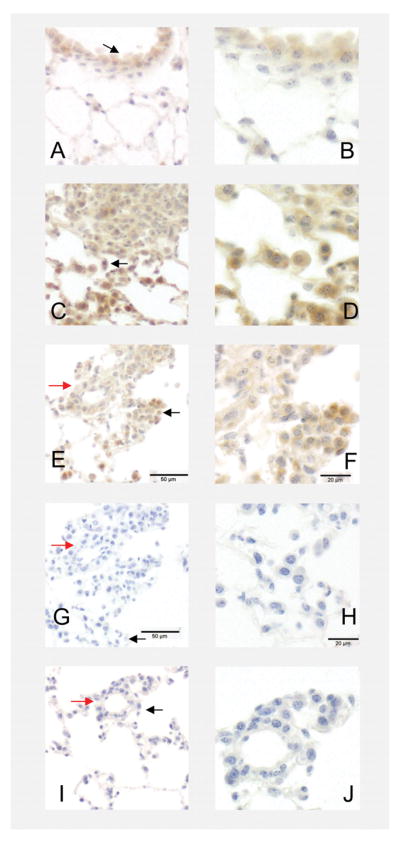
(A) Ereg localization in Ereg+/+ mice treated with MCA/oil (20 wk time point) in the bronchiolar cell types (likely Club cells) and alveolar regions (ie. type II cells) (200× magnification). (B) Same region as in A at 400× magnification. Black arrow on 200× indicates the region. (C). MCA/BHT-treated Ereg+/+ mice at the 20 wk timepoint demonstrating positive staining in the tumor cells as well as macrophages and surrounding microenvironment (eg. type II cells) (200× magnification). (D) Same region as in C but at 400× magnification. Black arrow on 200× indicates the region. (E) Another MCA/BHT-treated Ereg+/+ mouse at 200× magnification. Red arrow indicates the same regions on the G and I negative controls from same mouse, different sections. (F) Same region as in E but at 400× magnification. Black arrow on 200× indicates the region. (G) Negative control using only primary antibody, but no secondary antibody of same mouse as in E, 200x. (H) Same region as in G but at 400× magnification. Black arrow on 200× indicates the region. (I) Negative control using only secondary antibody, but no primary antibody of same mouse as in E, 200x. (J) Same region as in I but at 400× magnification. Black arrow on 200× indicates the region. 200× = 50 μm; 400× = 20 μm.
Wound healing assay using murine and human lung epithelial cells
A dose response was performed in the mouse type II cell line (C10 cells; Fig. 7A) using 0, 0.02, 0.2, 2.0, and 5 ng of rEreg in the wound assay and recombinant mouse epithelial growth factor (rEGF) as a comparison. For a species comparison, we used human bronchial epithelial cells (BEAS2B; Fig. 7B) and performed a similar dose response with rEreg (0–2 nM) and rEGF. We observed significant increases in wound healing at 0.2, 2.0 and 5.0 nM rEreg 24 hrs following treatment with the growth factor (Fig 7A) in C10 cells and at the 2.0 and 5.0 nM dose 24 hrs following treatment for the BEAS2B cells (Fig. 7B). rEreg was similar in wound healing responses to rEGF (Fig. 7). To confirm the response in the BEAS2B cells, a proliferation assay demonstrated significant increases at the 2 nM dose for both rEreg and rEgf in comparison to the positive control (10% FBS). Both growth factors were as potent as the FBS (Fig. 7C). The BEAS2B cells appear to be more sensitive to Ereg than C10 cells.
Figure 7. Ereg induces wound healing in lung epithelial cells.

(A) Murine C10 cells were exposed to several concentrations of recombinant Ereg (0–5.0 nM) following 24 hr serum deprivation and wound assays performed for a 24 hr time point. Mean ± SEM presented; n = 3 per treatment group repeated three timesrEgf (2 nM) was used in both cell lines for another growth factor comparison. *, p<0.05 for 0.2, 2 and 5 nM rEreg and rEGF compared to control (0); +, p<0.05 for rEGF compared to all doses except 5 nM. (B) Human BEAS2B cells were exposed to rEREG (0–2 nM) following 24 hr serum deprivation and wound assays performed as described above. Mean ± SEM presented; n = 3 per treatment group repeated three times. *, p<0.05 for 2 nM rEreg and rEGF compared to control (0); +, p<0.05 for rEGF compared to all doses except 2 and 5 nM. (C) BEAS2B cells were serum deprived for 24 hr followed by treatment with rEreg (2 nM), rEgf (2 nM), or 10% FBS as a positive control for 24 hr and compared to serum free media. Cell density was determined using the CellTiter96 proliferation assay (MTS). Mean ± SEM presented; n=4 per treatment group repeated twice. All experiments were graphed as fold-increase over control (0% serum, no treatment).
Discussion
In the present novel studies, we demonstrate that a lack of the growth factor Ereg results in significantly reduced lung tumor development in a primary chemically-induced tumor promotion model compared to wildtype controls (Ereg+/+ mice). Pulmonary inflammation was also reduced (primarily macrophages and PMNs) at several earlier time points, suggesting involvement of Ereg in immune responses, although baseline inflammation did not differ between strains. Chemokines with known innate immune cell chemoattraction, Mcp-1 (Ccl2) and Kc (Cxcl1), were also reduced in the Ereg−/− mice compared to Ereg+/+ mice further supporting the observed changes in macrophage and PMN numbers. However, phagocytosis of these pulmonary macrophages did not differ between strains, similar to peritoneal macrophages from Ereg−/− mice [40]. Further evaluation of cytokine expression determined that the anti-inflammatory Il10 and its downstream transcription factor (Stat3) were both significantly elevated in the Ereg deficient mice compared to wildtype in response to BHT (protocol 1). Comparable results were also determined in pilot studies with Il10−/− mice (BALB/cJ background) in response to BHT (protocol 1) where significantly increased lung hyperpermeability and slightly increased numbers of PMNs were observed compared to wildtype mice, with little change in macrophage numbers (unpublished data, Bauer and Kleeberger). Several other EREG independent inflammatory mediators were also significantly increased in response to BHT with no strain differences (Il6, Arg1, and iNos). Additionally, EGFR signaling did not change with treatment or strain at the times examined, nor did the activity of MAP kinases (phospho-ERK1/2 and phosho-p38) or transcription factors (c-jun and NFκB) (data not shown). However, future investigations will focus on additional time points for improved characterization of this pathway. Lastly, rEreg induced significant wound healing in an ADC progenitor cell line (C10 cells) and in human BEAS2B cells that further supports a role for Ereg in proliferative responses in both species, as well as tumor development in mice.
We and others have observed changes in chemokine/cytokine content in several models of lung cancer [26,30,41,42]. When macrophages are depleted in either the 2-stage MCA/BHT or urethane-induced models, there is a significant reduction in tumor development [6,41]. The same is true for PMNs; depletion of PMNs in the MCA/BHT model resulted in a significant reduction in tumor development [43]. Additionally, in a COPD-like model of promotion using nontypeable Haemophilus influenza as the promoter in a K-ras overexpressing mouse (CCSPCre/LSL-K-rasG12D), both the macrophages and PMNs are critical innate immune cells involved in tumor development [39,44]. These chemokines are both altered in response to Ereg depletion suggesting that Ereg is involved, at the least in part, in eliciting these downstream responses. Since Ereg is expressed in macrophages and epithelial cells, these events are likely either epithelial cell or macrophage-dependent mechanism(s). Kc is one of three chemokines that is in the same human IL8 cluster on chromosome 5, thus is considered an IL8-like chemokine in mice [45]. Kc, is an angiogenic factor and can recruit myeloid-derived suppressor cells (MDSCs), among other functions [46,47]. Blockage of the major receptor for Kc (Cxcr2) decreased lung tumorigenesis in the COPD-like promotion model described above [44]. Further, Kc was also elevated at more advanced stages of urethane-induced tumorigenesis [26]. In humans, elevated levels of two known neutrophil chemoattractants (IL8, LTB4) in breath condensate of NSCLC patients and IL8 gene expression in the lung of suspected cancer patients suggests these may be useful diagnostic markers for lung cancer [48,49]. Additionally, a polymorphism in human myeloperoxidase that rendered it less active was significantly associated with protection against smoking-induced lung cancer [50]. Thus, inhibition of Ereg reduced the types of immune cells which appear to be key players in the lung microenvironment during tumor development. Other studies in our lab have indicated a role for myeloid-derived suppressor cells (MDSC)s among other cell types in a different model, thus future studies will address the role of Ereg in the regulation of these other cell types and their specific phenotypes (e.g. M1 vs. M2) [51,52].
The timing for the different stages of tumorigenesis is also important to consider. During the promotion stage of lung cancer, the chemokines and cytokines are influencing immune cells differently than at later stages. For example, Kc is involved in angiogenesis, but during promotion, this function is not a major factor. However the recruitment of additional cell types, such as MDSCs early in promotion, may impact the growth of the future tumors. Mcp-1 is also elevated early and late. Ccr2 deficient mice, the receptor for Mcp-1, did not result in reduced tumorigenesis, suggesting that in the early stages of tumor development, this chemokine is not critical [41].
IL10 signaling can also have different roles if observed early versus late in lung tumorigenesis. Pulmonary IL10 is typically considered an anti-inflammatory Th2 cytokine based on observed reductions in both macrophages and dendritic cell function [53]. Some of these immune functions include reduced pro-inflammatory cytokine production, antigen presentation, and suppression of endotoxin [53]. Urethane-induced lung carcinogenesis was significantly increased in heterozygous female Il10−/+ mice [54]. Thus, if the IL10 is removed from the beginning, an increase in tumor development is observed. However, one study demonstrated that if tumor associated macrophage [55] IL10 expression was elevated in NSCLC patients, overall prognosis was significantly worse [56]. One explanation for these diverging effects is that the IL10 expressing TAMs can suppress the cytotoxic immune response favoring tumor escape [57], though this is at an advanced stage in development. Hence, timing is clearly important in all of these models and IL10 as well as the other chemokines potentially involved are dependent on the stage of tumor progression. The parallel increase observed in Stat3 activity in Ereg−/− mice provides additional evidence that Ereg is normally suppressing IL10 signaling and involved in IL10 regulation. STAT3 is a transcription factor that is downstream of many cytokines, including IL10, IL6, and G-CSF [58]. In our model, Il6 mRNA expression is elevated in response to BHT but not significantly different between strains (although there is a potential reduction of Il6 in the Ereg mice; see Supplementary Fig. S3).
The growth potential for Ereg was demonstrated using rEreg, which elicited a wound healing response in lung epithelial cells from two species indicating involvement in growth and migration of the epithelial cells. These responses in lung are similar to those observed previously in liver and keratinocytes [24,25] as well as a similar growth factor (Egf) [25]. Our model investigated early cancer development and thus did not focus on invasion or survival of tumor cells. However, in NSCLC biopsy specimens, EREG protein expression correlated with nodal metastasis and inversely with survival, further emphasizing the importance of this growth factor both in cancer development and progression [16]. Common mutations in lung cancer (Kras and EGFR) were also correlated to EREG mRNA overexpression in NSCLC cell lines compared to those with only wild-type alleles [59]. In NSCLC specimens, EREG mRNA was expressed principally in KRAS mutant ADC as well as those with pleural involvement, lymphatic involvement, or invasion into the vasculature [59]. Lastly, EREG is considered an independent prognostic marker for ADC patients, only increased when in the presence of KRAS mutations [59]. MCA initiates G→D Kras mutations in codon 12 in BALB/ByJ mice [60] in the MCA/BHT model. Therefore, it is plausible to hypothesize that in this promotion model, Kras is one of the factors driving the expression of Ereg, which is further influencing lung inflammation and proliferation of epithelial cells.
Therapeutically, tyrosine kinase inhibitors specific for EGFR, such as erlotinib or gefitinib, are effective in patients with EGFR mutations, however most tumors eventually become resistant through additional mutations in the receptor [61,62]. A mouse model created to mimic the human EGFR mutations [63] was used in a recent EGFR vaccine study. The EGFR vaccine reduced EGFR-driven carcinogenesis in mice by 76% [64]. Additionally, peptide vaccine studies in NSCLC patients have been done using EGF as the antigen, with less success, although more studies are needed for both safety and efficacy [65]. As far as therapeutic utility, since the MCA/BHT model is a chemically-induced, Kras-driven model, we suggest using this model for the initial chemoprevention studies with targeted EREG inhibition in the future or in the Kras overexpressing mice (eg. CCSPCre/LSL–K-rasG12D mice; [66]) to better mimic those NSCLC patients with Kras mutations, which accounts for ~25% of those patients in Western countries [67]. Additionally, it would be interesting to determine if the EGFR vaccine or EGFR specific inhibitors would further decrease the number of tumors in the Ereg−/− compared to wildtype mice.
Collectively, these studies provide the basis for future directions into the mechanism regulating EREG with many questions unresolved, such as the phenotypes of immune cells involved in this mechanism (i.e. macrophages (M1, M2, or a more plastic phenotype; MDSCs, etc), the receptors and downstream signaling involved (eg. KRAS, EGFR, HER4), the role of EREG in other types of lung cancer (e.g. squamous cell carcinoma, SCLC), any associations with EREG compared to stage of lung cancer (tumor, metastasis, nodal involvement; TMN status), and the role of EREG in other lung diseases such as chronic obstructive pulmonary disease (COPD) and pulmonary fibrosis, both linked to lung cancer development. Lastly, the impact of EREG on tumor development may lead to future therapeutic interventions using small peptide inhibitors or vaccines against EREG.
Supplementary Material
Ereg homozygous negative (Ereg−/−), heterozygous, and homozygous positive (Ereg+/+) mice were genotyped, as described. Ereg−/− mice (lanes 1–5) have a 253 bp product and Ereg+/+ (lanes 7, 10, 11) a 156 bp product. Heterozygotes have both bands (lanes 8, 9). The ladder shows the marker for the 250 bp band.
Body weight changes from wk 1 to 2 (2 on graph), wk 2 to 3 (3 on graph) and from wk 1 to wk 4 (Final) were determined as a typical measure of health for this model. Significant reductions in body weight were observed in both strains in response to BHT (protocol 1) from week 1 to week 2 when alveolar type 1 cells necrose and alveolar type II cells undergo hyperplasia, a well-established event with acute BHT [68]. Weight gains were then observed, as expected. Mean ± SEM presented; n = 3, repeated three times. *, p<0.05 for BHT compared to oil controls.
QRT-PCR was performed on whole lung and interleukin 6 (Il6), inducible nitric oxide synthase [39], and arginase 1 (Arg1) mRNA expression determined. All three were significantly elevated in response to BHT (protocol 1) compared to oil, however no strains differences were observed. Mean ± SEM presented; n = 3, repeated three times. *, p<0.05 for BHT compared to oil controls.
Acknowledgments
Grant sponsor: This research was funded by the American Cancer Society (RSG-10-162-01-LIB, AKB). The American Cancer Society had no involvement in the design or in conducting this research.
We would like to thank Dr. Jonathan Shannahan and Ross Osgood for critically reviewing the manuscript.
Abbreviations
- ADC
adenocarcinoma
- ARG1
arginase 1
- BAL
bronchoalveolar lavage
- BHT
butylated hydroxytoluene
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- EREG
epiregulin
- IL6
interleukin 6
- IL10
interleukin 10
- iNOS
inducible nitric oxide synthase
- KC (Cxcl1)
keratinocyte chemoattractant
- MCA
3-methylcholanthrene
- MCP-1 (Ccl2)
monocyte chemoattractant protein 1
- MDSC
myeloid-derived suppressor cell
- NSCLC
non-small cell lung carcinoma
- QRT-PCR
quantitative RT-PCR
- STAT3
signal transducer and activator of transcription 3
Footnotes
Authors Contributions
Conception and design: A.K. Bauer. Acquisition of data: C.M. Alexander, K. Velmurugan, K. Xiong, J. Xiong, R. Brooks. Analysis and interpretation of data: K. Velmurugan and A.K. Bauer. Manuscript preparation: A.K. Bauer, C.M. Alexander, and K. Velmurugan.
References
- 1.American Cancer Society; Society AC, editor. Non-small cell lung cancer survival rates by stage. Atlanta, GA: American Cancer Society; 2015. [Google Scholar]
- 2.Bauer AK, Malkinson AM, Kleeberger SR. Susceptibility to neoplastic and non-neoplastic pulmonary diseases in mice: genetic similarities. Am J Physiol Lung Cell Mol Physiol. 2004;287:L685–703. doi: 10.1152/ajplung.00223.2003. [DOI] [PubMed] [Google Scholar]
- 3.Pitot HC. Multistage carcinogenesis--genetic and epigenetic mechanisms in relation to cancer prevention. Cancer Detect Prev. 1993;17:567–573. [PubMed] [Google Scholar]
- 4.Slaga TJ. Overview of tumor promotion in animals. Environ Health Perspect. 1983;50:3–14. doi: 10.1289/ehp.83503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malkinson AM, Koski KM, Evans WA, Festing MF. Butylated hydroxytoluene exposure is necessary to induce lung tumors in BALB mice treated with 3-methylcholanthrene. Cancer Res. 1997;57:2832–2834. [PubMed] [Google Scholar]
- 6.Bauer AK, Dwyer-Nield LD, Keil K, Koski K, Malkinson AM. Butylated hydroxytoluene (BHT) induction of pulmonary inflammation: a role in tumor promotion. Exp Lung Res. 2001;27:197–216. doi: 10.1080/019021401300053948. [DOI] [PubMed] [Google Scholar]
- 7.Vikis HG, Gelman AE, Franklin A, et al. Neutrophils are required for 3-methylcholanthrene-initiated, butylated hydroxytoluene-promoted lung carcinogenesis. Mol Carcinog. 2012;51:993–1002. doi: 10.1002/mc.20870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bauer AK, Dwyer-Nield LD, Hankin JA, Murphy RC, Malkinson AM. The lung tumor promoter, butylated hydroxytoluene (BHT), causes chronic inflammation in promotion-sensitive BALB/cByJ mice but not in promotion-resistant CXB4 mice. Toxicology. 2001;169:1–15. doi: 10.1016/s0300-483x(01)00475-9. [DOI] [PubMed] [Google Scholar]
- 9.Hill T, 3rd, Osgood RS, Velmurugan K, Alexander CM, Upham BL, Bauer AK. Bronchoalveolar Lavage Fluid Utilized Ex Vivo to Validate In Vivo Findings: Inhibition of Gap Junction Activity in Lung Tumor Promotion is Toll-Like Receptor 4-Dependent. J Mol Biomark Diagn. 2013:5. doi: 10.4172/2155-9929.1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bauer AK, Fostel J, Degraff LM, et al. Transcriptomic analysis of pathways regulated by toll-like receptor 4 in a murine model of chronic pulmonary inflammation and carcinogenesis. Mol Cancer. 2009;8:107. doi: 10.1186/1476-4598-8-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riese DJ, 2nd, Cullum RL. Epiregulin: roles in normal physiology and cancer. Semin Cell Dev Biol. 2014;28:49–56. doi: 10.1016/j.semcdb.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komurasaki T, Toyoda H, Uchida D, Morimoto S. Epiregulin binds to epidermal growth factor receptor and ErbB-4 and induces tyrosine phosphorylation of epidermal growth factor receptor, ErbB-2, ErbB-3 and ErbB-4. Oncogene. 1997;15:2841–2848. doi: 10.1038/sj.onc.1201458. [DOI] [PubMed] [Google Scholar]
- 13.Riese DJ, 2nd, Komurasaki T, Plowman GD, Stern DF. Activation of ErbB4 by the bifunctional epidermal growth factor family hormone epiregulin is regulated by ErbB2. J Biol Chem. 1998;273:11288–11294. doi: 10.1074/jbc.273.18.11288. [DOI] [PubMed] [Google Scholar]
- 14.Shelly M, Pinkas-Kramarski R, Guarino BC, et al. Epiregulin is a potent pan-ErbB ligand that preferentially activates heterodimeric receptor complexes. J Biol Chem. 1998;273:10496–10505. doi: 10.1074/jbc.273.17.10496. [DOI] [PubMed] [Google Scholar]
- 15.Lee D, Pearsall RS, Das S, Dey SK, Godfrey VL, Threadgill DW. Epiregulin is not essential for development of intestinal tumors but is required for protection from intestinal damage. Mol Cell Biol. 2004;24:8907–8916. doi: 10.1128/MCB.24.20.8907-8916.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Iwanaga K, Choi KC, et al. Intratumoral epiregulin is a marker of advanced disease in non-small cell lung cancer patients and confers invasive properties on EGFR-mutant cells. Cancer Prev Res (Phila Pa) 2008;1:201–207. doi: 10.1158/1940-6207.CAPR-08-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baba I, Shirasawa S, Iwamoto R, et al. Involvement of deregulated epiregulin expression in tumorigenesis in vivo through activated Ki-Ras signaling pathway in human colon cancer cells. Cancer Res. 2000;60:6886–6889. [PubMed] [Google Scholar]
- 18.Kojima K, Musch MW, Ropeleski MJ, Boone DL, Ma A, Chang EB. Escherichia coli LPS induces heat shock protein 25 in intestinal epithelial cells through MAP kinase activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G645–652. doi: 10.1152/ajpgi.00080.2003. [DOI] [PubMed] [Google Scholar]
- 19.Toyoda H, Komurasaki T, Ikeda Y, Yoshimoto M, Morimoto S. Molecular cloning of mouse epiregulin, a novel epidermal growth factor-related protein, expressed in the early stage of development. FEBS Lett. 1995;377:403–407. doi: 10.1016/0014-5793(95)01403-9. [DOI] [PubMed] [Google Scholar]
- 20.Toyoda H, Komurasaki T, Uchida D, Morimoto S. Distribution of mRNA for human epiregulin, a differentially expressed member of the epidermal growth factor family. Biochem J. 1997;326(Pt 1):69–75. doi: 10.1042/bj3260069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toyoda H, Komurasaki T, Uchida D, et al. Epiregulin. A novel epidermal growth factor with mitogenic activity for rat primary hepatocytes. J Biol Chem. 1995;270:7495–7500. doi: 10.1074/jbc.270.13.7495. [DOI] [PubMed] [Google Scholar]
- 22.Bieche I, Lerebours F, Tozlu S, Espie M, Marty M, Lidereau R. Molecular profiling of inflammatory breast cancer: identification of a poor-prognosis gene expression signature. Clin Cancer Res. 2004;10:6789–6795. doi: 10.1158/1078-0432.CCR-04-0306. [DOI] [PubMed] [Google Scholar]
- 23.Nishimura T, Andoh A, Inatomi O, et al. Amphiregulin and epiregulin expression in neoplastic and inflammatory lesions in the colon. Oncol Rep. 2008;19:105–110. [PubMed] [Google Scholar]
- 24.Tomita K, Haga H, Mizuno K, et al. Epiregulin promotes the emergence and proliferation of adult liver progenitor cells. Am J Physiol Gastrointest Liver Physiol. 2014;307:G50–57. doi: 10.1152/ajpgi.00434.2013. [DOI] [PubMed] [Google Scholar]
- 25.Draper BK, Komurasaki T, Davidson MK, Nanney LB. Epiregulin is more potent than EGF or TGFalpha in promoting in vitro wound closure due to enhanced ERK/MAPK activation. J Cell Biochem. 2003;89:1126–1137. doi: 10.1002/jcb.10584. [DOI] [PubMed] [Google Scholar]
- 26.Bauer AK, Cho HY, Miller-Degraff L, et al. Targeted deletion of Nrf2 reduces urethane-induced lung tumor development in mice. PLoS One. 2011;6:e26590. doi: 10.1371/journal.pone.0026590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bauer AK, Dixon D, DeGraff LM, et al. Toll-like receptor 4 in butylated hydroxytoluene-induced mouse pulmonary inflammation and tumorigenesis. J Natl Cancer Inst. 2005;97:1778–1781. doi: 10.1093/jnci/dji403. [DOI] [PubMed] [Google Scholar]
- 28.Ait Yahia S, Azzaoui I, Everaere L, et al. CCL17 production by dendritic cells is required for NOD1-mediated exacerbation of allergic asthma. Am J Respir Crit Care Med. 2014;189:899–908. doi: 10.1164/rccm.201310-1827OC. [DOI] [PubMed] [Google Scholar]
- 29.Bauer AK, Rondini EA, Hummel KA, et al. Identification of candidate genes downstream of TLR4 signaling after ozone exposure in mice: a role for heat-shock protein 70. Environ Health Perspect. 2011;119:1091–1097. doi: 10.1289/ehp.1003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rondini EA, Walters DM, Bauer AK. Vanadium pentoxide induces pulmonary inflammation and tumor promotion in a strain-dependent manner. Part Fibre Toxicol. 2010;7:9. doi: 10.1186/1743-8977-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown LM, Malkinson AM, Rannels DE, Rannels SR. Compensatory lung growth after partial pneumonectomy enhances lung tumorigenesis induced by 3-methylcholanthrene. Cancer Res. 1999;59:5089–5092. [PubMed] [Google Scholar]
- 32.Obermueller-Jevic UC, Espiritu I, Corbacho AM, Cross CE, Witschi H. Lung tumor development in mice exposed to tobacco smoke and fed beta-carotene diets. Toxicol Sci. 2002;69:23–29. doi: 10.1093/toxsci/69.1.23. [DOI] [PubMed] [Google Scholar]
- 33.Cho HY, Kleeberger SR. Genetic mechanisms of susceptibility to oxidative lung injury in mice. Free Radic Biol Med. 2007;42:433–445. doi: 10.1016/j.freeradbiomed.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 34.Cho HY, Reddy SP, Debiase A, Yamamoto M, Kleeberger SR. Gene expression profiling of NRF2-mediated protection against oxidative injury. Free Radic Biol Med. 2005;38:325–343. doi: 10.1016/j.freeradbiomed.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 35.Bookout AL, Cummins CL, Mangelsdorf DJ, Pesola JM, Kramer MF. Current Protocols in Molecular Biology. John Wiley & Sons, Inc; 2001. High-Throughput Real-Time Quantitative Reverse Transcription PCR. [DOI] [PubMed] [Google Scholar]
- 36.Malkinson AM, Dwyer-Nield LD, Rice PL, Dinsdale D. Mouse lung epithelial cell lines--tools for the study of differentiation and the neoplastic phenotype. Toxicology. 1997;123:53–100. doi: 10.1016/s0300-483x(97)00108-x. [DOI] [PubMed] [Google Scholar]
- 37.Reddel RR, Malan-Shibley L, Gerwin BI, Metcalf RA, Harris CC. Tumorigenicity of human mesothelial cell line transfected with EJ-ras oncogene. J Natl Cancer Inst. 1989;81:945–948. doi: 10.1093/jnci/81.12.945. [DOI] [PubMed] [Google Scholar]
- 38.Justus CR, Leffler N, Ruiz-Echevarria M, Yang LV. In vitro cell migration and invasion assays. J Vis Exp. 2014 doi: 10.3791/51046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karabela SP, Psallidas I, Sherrill TP, et al. Opposing effects of bortezomib-induced nuclear factor-kappaB inhibition on chemical lung carcinogenesis. Carcinogenesis. 2012;33:859–867. doi: 10.1093/carcin/bgs024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shirasawa S, Sugiyama S, Baba I, et al. Dermatitis due to epiregulin deficiency and a critical role of epiregulin in immune-related responses of keratinocyte and macrophage. Proc Natl Acad Sci U S A. 2004;101:13921–13926. doi: 10.1073/pnas.0404217101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fritz JM, Tennis MA, Orlicky DJ, et al. Depletion of tumor-associated macrophages slows the growth of chemically induced mouse lung adenocarcinomas. Front Immunol. 2014;5:587. doi: 10.3389/fimmu.2014.00587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moghaddam SJ, Li H, Cho SN, et al. Promotion of lung carcinogenesis by chronic obstructive pulmonary disease-like airway inflammation in a K-ras-induced mouse model. Am J Respir Cell Mol Biol. 2009;40:443–453. doi: 10.1165/rcmb.2008-0198OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Zhang Z, Yan Y, et al. A chemically induced model for squamous cell carcinoma of the lung in mice: histopathology and strain susceptibility. Cancer Res. 2004;64:1647–1654. doi: 10.1158/0008-5472.can-03-3273. [DOI] [PubMed] [Google Scholar]
- 44.Gong L, Cumpian AM, Caetano MS, et al. Promoting effect of neutrophils on lung tumorigenesis is mediated by CXCR2 and neutrophil elastase. Mol Cancer. 2013;12:154. doi: 10.1186/1476-4598-12-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- 46.Acharyya S, Oskarsson T, Vanharanta S, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012;150:165–178. doi: 10.1016/j.cell.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rivas-Fuentes S, Salgado-Aguayo A, Pertuz Belloso S, Gorocica Rosete P, Alvarado-Vasquez N, Aquino-Jarquin G. Role of Chemokines in Non-Small Cell Lung Cancer: Angiogenesis and Inflammation. J Cancer. 2015;6:938–952. doi: 10.7150/jca.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carpagnano GE, Palladino GP, Lacedonia D, Koutelou A, Orlando S, Foschino-Barbaro MP. Neutrophilic airways inflammation in lung cancer: the role of exhaled LTB-4 and IL-8. BMC Cancer. 2011;11:226. doi: 10.1186/1471-2407-11-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spira A, Beane JE, Shah V, et al. Airway epithelial gene expression in the diagnostic evaluation of smokers with suspect lung cancer. Nat Med. 2007;13:361–366. doi: 10.1038/nm1556. [DOI] [PubMed] [Google Scholar]
- 50.Dally H, Gassner K, Jager B, et al. Myeloperoxidase (MPO) genotype and lung cancer histologic types: the MPO -463 A allele is associated with reduced risk for small cell lung cancer in smokers. Int J Cancer. 2002;102:530–535. doi: 10.1002/ijc.10756. [DOI] [PubMed] [Google Scholar]
- 51.Redente EF, Keith RC, Janssen W, et al. Tumor necrosis factor-alpha accelerates the resolution of established pulmonary fibrosis in mice by targeting profibrotic lung macrophages. Am J Respir Cell Mol Biol. 2014;50:825–837. doi: 10.1165/rcmb.2013-0386OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Redente EF, Orlicky DJ, Bouchard RJ, Malkinson AM. Tumor signaling to the bone marrow changes the phenotype of monocytes and pulmonary macrophages during urethane-induced primary lung tumorigenesis in A/J mice. Am J Pathol. 2007;170:693–708. doi: 10.2353/ajpath.2007.060566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O’Garra A, Barrat FJ, Castro AG, Vicari A, Hawrylowicz C. Strategies for use of IL-10 or its antagonists in human disease. Immunol Rev. 2008;223:114–131. doi: 10.1111/j.1600-065X.2008.00635.x. [DOI] [PubMed] [Google Scholar]
- 54.Bernert H, Sekikawa K, Radcliffe RA, Iraqi F, You M, Malkinson AM. Tnfa and Il-10 deficiencies have contrasting effects on lung tumor susceptibility: gender-dependent modulation of IL-10 haploinsufficiency. Mol Carcinog. 2003;38:117–123. doi: 10.1002/mc.10151. [DOI] [PubMed] [Google Scholar]
- 55.Tam KW, Zhang W, Soh J, et al. CDKN2A/p16 inactivation mechanisms and their relationship to smoke exposure and molecular features in non-small-cell lung cancer. J Thorac Oncol. 2013;8:1378–1388. doi: 10.1097/JTO.0b013e3182a46c0c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zeni E, Mazzetti L, Miotto D, et al. Macrophage expression of interleukin-10 is a prognostic factor in nonsmall cell lung cancer. Eur Respir J. 2007;30:627–632. doi: 10.1183/09031936.00129306. [DOI] [PubMed] [Google Scholar]
- 57.Santoni M, Massari F, Amantini C, et al. Emerging role of tumor-associated macrophages as therapeutic targets in patients with metastatic renal cell carcinoma. Cancer Immunol Immunother. 2013;62:1757–1768. doi: 10.1007/s00262-013-1487-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jarnicki A, Putoczki T, Ernst M. Stat3: linking inflammation to epithelial cancer - more than a “gut” feeling? Cell Div. 2010;5:14. doi: 10.1186/1747-1028-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sunaga N, Kaira K, Imai H, et al. Oncogenic KRAS-induced epiregulin overexpression contributes to aggressive phenotype and is a promising therapeutic target in non-small-cell lung cancer. Oncogene. 2013;32:4034–4042. doi: 10.1038/onc.2012.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fritz JM, Dwyer-Nield LD, Russell BM, Malkinson AM. The Kras mutational spectra of chemically induced lung tumors in different inbred mice mimics the spectra of KRAS mutations in adenocarcinomas in smokers versus nonsmokers. J Thorac Oncol. 2010;5:254–257. doi: 10.1097/JTO.0b013e3181c8ce04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 62.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006;20:1496–1510. doi: 10.1101/gad.1417406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ebben JD, Lubet RA, Gad E, Disis ML, You M. Epidermal growth factor receptor derived peptide vaccination to prevent lung adenocarcinoma formation: An in vivo study in a murine model of EGFR mutant lung cancer. Mol Carcinog. 2015 doi: 10.1002/mc.22405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rodriguez PC, Rodriguez G, Gonzalez G, Lage A. Clinical development and perspectives of CIMAvax EGF, Cuban vaccine for non-small-cell lung cancer therapy. MEDICC Rev. 2010;12:17–23. doi: 10.37757/MR2010.V12.N1.4. [DOI] [PubMed] [Google Scholar]
- 66.Johnson L, Mercer K, Greenbaum D, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 67.Riely GJ, Kris MG, Rosenbaum D, et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res. 2008;14:5731–5734. doi: 10.1158/1078-0432.CCR-08-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Witschi H, Malkinson AM, Thompson JA. Metabolism and pulmonary toxicity of butylated hydroxytoluene (BHT) PharmacolTher. 1989;42:89–113. doi: 10.1016/0163-7258(89)90023-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Ereg homozygous negative (Ereg−/−), heterozygous, and homozygous positive (Ereg+/+) mice were genotyped, as described. Ereg−/− mice (lanes 1–5) have a 253 bp product and Ereg+/+ (lanes 7, 10, 11) a 156 bp product. Heterozygotes have both bands (lanes 8, 9). The ladder shows the marker for the 250 bp band.
Body weight changes from wk 1 to 2 (2 on graph), wk 2 to 3 (3 on graph) and from wk 1 to wk 4 (Final) were determined as a typical measure of health for this model. Significant reductions in body weight were observed in both strains in response to BHT (protocol 1) from week 1 to week 2 when alveolar type 1 cells necrose and alveolar type II cells undergo hyperplasia, a well-established event with acute BHT [68]. Weight gains were then observed, as expected. Mean ± SEM presented; n = 3, repeated three times. *, p<0.05 for BHT compared to oil controls.
QRT-PCR was performed on whole lung and interleukin 6 (Il6), inducible nitric oxide synthase [39], and arginase 1 (Arg1) mRNA expression determined. All three were significantly elevated in response to BHT (protocol 1) compared to oil, however no strains differences were observed. Mean ± SEM presented; n = 3, repeated three times. *, p<0.05 for BHT compared to oil controls.