

Abstract
The classical examination of histology slides from a mouse model of breast cancer has been extended in this study to incorporate modern multiphoton excitation and photon-counting techniques. The advantage of such approaches is quantification of potential diagnostic parameters from the fluorescence emission signal, whereby the traditional descriptive staging process is complemented by measurements of fluorescence intensity, lifetime, and spectra. We explored whether the clinical “gold standard” of eosin and hematoxylin stained histology slides would provide optical biomarker signatures of diagnostic value. Alternatively, we examined unstained slides for changes in intensity and/or fluorescence lifetime of relevant endogenous fluorophores. Although eosin provided a strong emission signal and had distinct spectra and lifetime, we found that it was not useful as a fluorescent biological marker, particularly when combined with hematoxylin. Instead, we found that the properties of the fluorescence from the endogenous fluorophores NADH and FAD were indicative of the pathological state of the tissue. Comparing regions of carcinoma in situ to adjacent histologically normal regions, we found that tumor cells produced higher intensity and had a longer fluorescence lifetime. By imaging at 780 nm and 890 nm excitation, we were able to differentiate the fluorescence of FAD from NADH by separating the emission spectra. The shift to a longer lifetime in tumor cells was independent of the free or bound state of FAD and NADH, and of the excitation wavelength. Most forms of cancer have altered metabolism and redox ratios; here we present a method that has potential for early detection of these changes, which are preserved in fixed tissue samples such as classic histopathology slides.
Keywords: Fluorescence lifetime, FAD, NADH, Multiphoton microscopy, Carcinoma in situ, Breast cancer
Introduction
The “gold” standard for the diagnosis of tumor progression is the generation and analysis of histopathology slides, most commonly stained with hematoxylin and eosin (H&E). The rationale for this is the high level of contrast that can be observed with brightfield light microscopy and the ability to identify morphological changes that indicate tumor onset and progression. Expert diagnosis of these slides by trained pathologists can provide a high degree of accuracy for tumor staging and the prediction of patient outcome. Nevertheless, it has been a longstanding challenge to cull further information from these tissue samples, with the goal of providing quantitative data that could aid pathologists in their characterization and diagnosis [1, 2]. The addition of reproducible quantitative parameters or trends in histopathology slides could greatly improve the accuracy of qualitative (pattern recognition-based) examination of standard histopathology slides and provide objective biomarkers for the disease progression. Histopathology slides stained with H&E have historically been the source of investigation for this objective, however, all cells contain endogenous fluorophores such that examination of unstained slides is another possible route to fulfill this goal.
Two key metabolic cofactors, NAD(P)H and FAD, are autofluorescent and together can be used to determine the redox state of the cell [3]. The first step of glucose metabolism, glycolysis, is the conversion of glucose to pyruvate resulting in the conversion of two molecules of NAD+ to NADH. Subsequently during the citric acid cycle, four NADH molecules are generated and one FAD molecule is reduced to FADH2, generating reducing equivalents for oxidative phosphorylation. The final, most efficient process in ATP production is the electron transport chain whereby 10 NADH and two FADH2 molecules are oxidized to generate 36 ATP molecules. In any given cell type or developmental stage, the relative use and efficiency of these chemical pathways is regulated, and will determine the levels of the fluorophores, NADH and FAD found within the cell.
Many tumor cells have an elevated rate of aerobic glycolysis compared to their normal counterparts, a phenomenon known as the Warburg effect [4, 5]. The cause of this is not fully understood, but may be due to the transient development of hypoxia in the nascent tumor mass, resulting in a permanent switch toward metabolic programs that favor glycolysis in lieu of oxidative phosphorylation. Moreover, during aberrantly increased glycolysis, tumor cells may obtain as much as 50% of their ATP from the conversion of glucose to lactic acid [6]. Because of the high demand for energy in the form of ATP in highly proliferative tumor cells, the increased rate of glycolysis puts these cells under oxidative stress, marked by the conversion of NAD+ to NADH [7]. Additionally, tumor cells have elevated levels of reactive oxygen species (ROS), and because of the use of NADH as a cofactor by antioxidants in scavenging such molecules, this amplified pathway may also affect the ratio of oxidizing to reducing equivalents in glucose metabolism, known as the redox state of these cells. Furthermore, increased glycolysis results in elevated levels of hypoxia inducible factor (HIF-1a), which can induce angiogenesis [8]. The subsequent growth of a vascularized tumor beyond 1–2 mm in diameter may promote the development of malignancy and metastasis [9]. Hence, detecting changes in the metabolic state of tumor cells, such as the redox state (and thus the concentration of NADH and FAD), at any given time during tumor progression could have diagnostic potential. Indeed, other researchers have found that changes in the redox state of tumor cells occur during early stages of prostate carcinoma, and further change when tumors progress to invasion and metastasis [10]. It is for these reasons that methodologies that can accurately measure redox levels during specific disease stages possess potential diagnostic value.
All cells contain numerous endogenous fluorophores, but due to their relatively high quantum yield and concentration within the cell, NAD(P)H and FAD provide the strongest signal [11]. The single photon absorption of these endogenous fluorophores span the UV and visible spectrum and, importantly, emit with overlapping spectra making detection of individual species difficult without implementation of specific filtering technologies, spectral separation, or specialized analysis methods such as spectral unmixing algorithms [12]. However, when utilizing multiphoton laser-scanning microscopy (MPLSM) [13] with two different excitation wavelengths (both fluorophores can be excited at 780 nm, but only FAD is excited at 890 nm), this issue can be overcome to a large extent [11, 14, 15], particularly when combined with spectral separation of the emission signal [16, 17]. In addition, photon-counting techniques can be used to potentially detect changes in the fluorescence properties of NADH and FAD other than excitation, and exploit these properties as a way of differentiating changes in the biology of cells [16, 18–20]. For example, the duration that a fluorophore spends in the excited state, termed its fluorescence lifetime can be imaged (FLIM: Fluorescence Lifetime Imaging Microscopy). This approach can be further expanded to consider the emission spectrum and obtain the spectral lifetime (SLIM) [16]. Through these imaging techniques, one can potentially dissect out not only the relative expression levels of NADH and FAD in normal and tumor epithelium, but the proportions of each that are free and protein-bound within the cell.
To date, multiphoton FLIM has been successfully used to detect changes in autofluorescence as a function of epithelial cell density [18]; to differentiate normal and high-grade precancers [21]; and to distinguish primary from invasive mammary carcinoma cells [22] based on the metabolic biomarkers. Hence, the purpose of this study is to determine if multiphoton fluorescence lifetime analysis of normal and transformed mammary epithelium is quantitatively distinguishable in H&E stained histology sections, and whether fixed but unstained sections provide a diagnostic optical biomarker in the form of altered NADH and FAD fluorescence properties.
Materials and Methods
Mouse Model of Human Breast Cancer
The University of Wisconsin Animal Use and Care Committee approved this study. Polyomavirus middle-T mice (PyVT) mice [23, 24] under the control of the mammary specific MMTV promoter were used to generate primary tumors. This breast tumor model correlates well with many features of human breast cancer, progresses from hyperplasia to adenoma to carcinoma in a manner resembling human ductal carcinoma in situ, and is dependably invasive and metastatic. Mice were examined for palpable tumors starting at 8 weeks of age and sacrificed at 16 weeks or when the tumor burden became excessive. Selected mammary tumors were fixed in 10% formalin, paraffin-embedded, and sectioned. Following sectioning the tissue slices were de-paraffined and either remained unstained, or were stained with eosin-only or both hematoxylin and eosin using standard techniques. Sections were mounted using Crystal mount and a coverslip was applied.
Collagen Gel Culture and Fixation
MCF10A breast epithelial cells were cultured in 3D collagen gels (3 mg/ml, rat tail type I collagen) as described previously [25] in glass bottomed dishes. The day after pouring the gels, the well of the dish was rimmed to float the gel, and cultured for a total of 6–10 days before imaging. Immediately after which the gel was transferred with forceps to a beaker containing 10% formalin for 10 min before being transferred again to a mold containing partially polymerized 2% agar (which contains 1% formalin). More agar was then poured on top to encase the gel and allowed to harden. This mold was put in a histology cassette and placed in cold 70% ethanol until slide preparation. After trimming away some of the excess agar, the block was embedded in paraffin so that cutting could be performed. Multiple slices were cut and mounted per slide from multiple cutting angles and cells were identified in serial sections from de-paraffined, unstained slides. Images from six gels and subsequent tissue blocks were acquired and analyzed.
Multiphoton FLIM and SLIM
For all imaging, a custom multiphoton workstation at the University of Wisconsin Laboratory for Optical and Computational Instrumentation (LOCI, www.loci.wisc.edu) was utilized [16, 17]. The system has been recently described in detail [16–18] and allows generation of multiphoton excitation intensity images in conjunction with FLIM and SLIM. In short, the system is built around an inverted microscope (TE 2000, Nikon, Melville, NY) using illumination from a Ti:Sapphire mode-locking laser (tuning range of ~700–1000 nm, Coherent Mira, Coherent, Santa Clara, CA) pumped by an 8 W solid-state laser (Coherent Verdi) to generate pulse widths of approximately 120 fs at a repetition rate of 76 MHz. The system has multiple detectors including a 16 channel combined spectral lifetime detector (utilizes a Hamamatsu PML-16 PMT), detection range 350–720 nm, and a H7422P GaAsP photon-counting PMT (Hamamatsu) for intensity and lifetime imaging. Fluorescence lifetimes were acquired with an electronic system for recording fast light signals by time correlated single photon-counting (SPC-830, Becker and Hickl). The instrument response function (IRF) of the optical system was measured using second harmonic generated signal from a β-BaB2O4 crystal [17]. The measured full width at half maximum of the IRF was determined to be 190 ± 2 ps. It should be pointed out that this represents the IRF of the overall system where the components that mainly contribute to temporal degradation are the transient time spread of the detector, optical dispersion at mirrors and through lenses, and light scattering at diaphragms. A Nikon CFI Plan S Fluor 40× oil immersion lens (NA = 1.3) was used for all imaging. Both FLIM and SLIM data were collected for 60 s and the pixel frame size for the multiphoton intensity images is 1024 × 1024, while FLIM images are 256 × 256, and SLIM images are 128 × 128. Acquisition was performed with WiscScan (LOCI, www.loci.wisc.edu) a LOCI developed software acquisition package that can control both the MPLSM and lifetime collection.
Analysis of Lifetime and Spectral Lifetime Data
Fluorescence lifetime data was imported into SPCImage (Becker and Hickl, v.2.9.1) where a Levenberg–Marquardt routine for non-linear fitting was used to fit the fluorescence decay curve collected for each pixel in the 256 × 256 pixel image to a model multiexponential decay function of the form:
where I(t) is the fluorescence intensity at time t after the excitation pulse, n is the total number of decay components in the exponential sum, and c is a constant pertaining to the level of background light noise. The variables τi and ai are the fluorescence lifetime and fractional contribution of the ith emitting species, respectively. Corrections were made for the noise using a calibration of the instrument response function [17]. Herein, the data was fit best with a two exponential function, as assessed by the minimized Chi-square value generated during the fit. The use of Chi-squared or some other function to evaluate the goodness of fit is mandatory so that one can do the analysis in an unbiased fashion. Data with a Chi-squared value of greater than 1.3 was deemed to be of a poor fit and was eliminated from the results (the H&E data being an exception). Analysis included either the entire image field, or for the carcinoma in situ experiments, a region of interest (ROI) was drawn around specific cells or groups of cells. A threshold for analysis was applied based on photon-counts in order to eliminate background fluorescence and the data was binned to ensure that >1000 photons/pixel were analyzed to maintain high quality of the fit. We used the peak value from the corresponding histogram as the value for individual lifetime components, but one must always realize that when analysis is done in this matter that a spectrum of values is generated. Thus, our data was collected using time correlated single photon-counting techniques where the data was analyzed on a pixel by pixel analysis approach as previously reported [14, 18, 22].
SLIM data was analyzed using both SPCImage and a custom LOCI program (SLIM Plotter) as described previously [26]. After importing the data, a 3D plot was generated where time, spectra, and photon-counts were plotted in x–y–z. The emission spectra data was exported, corrected for differing sensitivities and efficiencies for each of the 16 channels in the detector, and then plotted as a line graph in SigmaPlot (SPS Science). Intensity data was analyzed in Image J (NIH, Wayne Rasband, http://rsb.info.nih.gov/ij/). As control experiments, we attempted to measure emitted photon-counts produced by the slide mounting media (CrystalMount) and hematoxylin alone but were unable to collect any photons above background levels (data not shown).
Statistical Analysis
Statistical analysis was performed using SigmaPlot (SPS Science) to perform unpaired, two-tailed Students t-tests, where P ≤ 0.05 was used as a cut-off for significance.
Results
Multiphoton Microscopy of Hematoxylin and Eosin Stained Slides
As a first step for examining the utility of fluorescence microscopy to provide relevant information from histology slides, we set out to identify and characterize the fluorescent species present in H&E stained samples so that biological interpretations of this data can be separated from the fluorescent properties (if any) of the staining dyes themselves. In order to characterize the fluorescence emission from H&E stained slides, we performed multiphoton imaging of formalin-fixed 16-week-old mouse tumors (polyoma middle-T, PyVT), sectioned and stained with various combinations of the two dyes (i.e., eosin alone, hematoxylin alone, or both H&E). Standard light microscopy examination of week 16 tumors revealed that the tumors contained mostly densely packed epithelial cells and stroma without any normal ductal structure. With multiphoton imaging at 890 nm, the fluorescence intensity of eosin-only and hematoxylin and eosin (H&E) stained slides was robust (Fig. 1). Since eosin is essentially a brominated form of fluoroscein (a well-described fluorescent dye), this was not a surprising result. Interestingly, however, the presence of hematoxylin changed the staining pattern of eosin in comparison to eosin alone. In eosin-only stained slides (Fig. 1a), where this eosinophillic dye will bind multiple targets, it was observed that the cytoplasm, extracellular matrix (ECM), and oftentimes the nucleus were each stained to varying degrees. However, because of the well reported non-fluorescence of the nuclear dye hematoxylin [27, 28], and the fact that it was applied to slides first in H&E preparations, hematoxylin staining resulted in regions of limited fluorescence at the nuclei of cells (Fig. 1b). This had the effect of diminishing the total emission per cell, thus the laser power and neutral density filter were adjusted to better visualize cells, making the ECM appear brighter than in the eosin-only stained slides, when in fact they were similar in intensity.
Fig. 1.
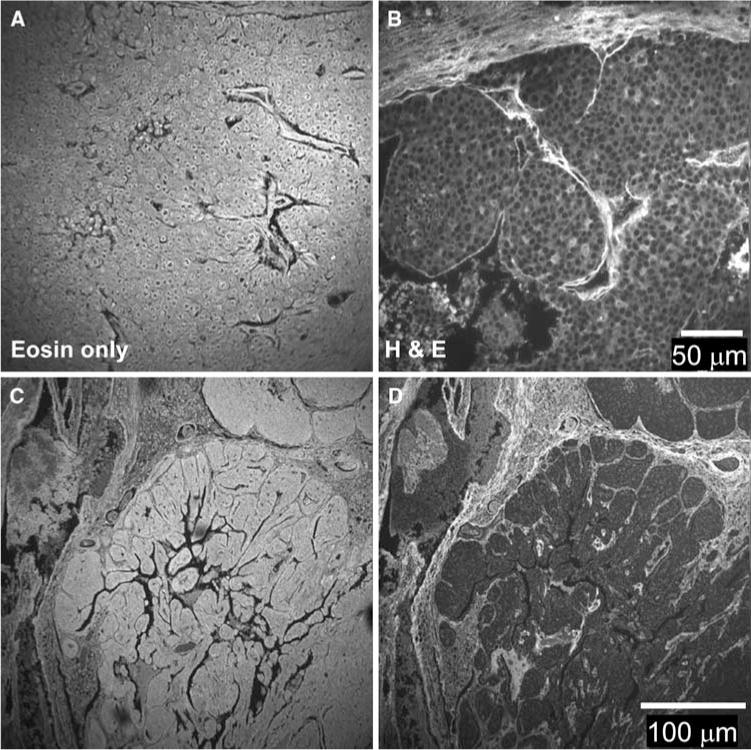
Fluorescence intensity of stained mouse breast tumor samples. a Paraffin-embedded tissue sections were stained with eosin. The fluorescence intensity showed staining in the cytoplasm of cells as well as the surrounding stroma. b Paraffin-embedded tissue sections were stained with H&E. The staining pattern of fluorescence intensity staining pattern was changed such that the intensity of eosin staining in cells was diminished but bright staining of the stroma is still visible. c, d Serial sections stained with eosin-only and H&E, respectively, are shown at lower magnification than A&B. Note the alteration in the staining pattern due to hematoxylin application
When serial sections were compared (Fig. 1c, d), with the first section stained with eosin alone and the next section stained with both H&E, we observed these effects in more detail. Because the nucleus to cytoplasm ratio is relatively high, total emission from cells is decreased in the H&E slides compared to eosin-only. In addition, we cannot rule out the possibility that hematoxylin is competing with eosin for cytoplasmic epitopes. Overall, these data suggest that while eosin-only stained slides have abundant fluorophore over the entire surface, H&E stained slides are marked by their high contrast between cells that are relatively weak in emission intensity when compared to the strong fluorescence from the ECM.
Fluorescence Lifetime Imaging of Hematoxylin and Eosin Stained Slides
In order to obtain more information about the fluorescent species found in H&E stained slides, we performed fluorescence lifetime imaging microscopy (FLIM). As described in detail in the methods section, we found that the lifetime data was fit best with a two-term exponential function (goodness of fit was assessed by the Chi-squared value generated during curve fitting using SPCImage software). Each time constant (τ1, τ2, etc.) may be representative of each of several fluorophores, or each time constant may be produced by a single fluorophore residing in multiple conditions that alter the excited state lifetime, conditions which are governed by the biology of the cell such as changes in intracellular pH, binding to molecular partners, or localization to membranes.
The fluorescence lifetimes, τ1 and τ2, of eosin stained slides was determined (Fig. 2). The τ2 color map was representative, and is shown to illustrate that the value of τ2 was reasonably uniform across the sample (Fig. 2a, b) (much like intensity data) where the cytoplasm and nuclei as well as stromal components all had similar lifetime values of ~1100 ps. In H&E stained slides the value for τ2 was significantly lengthened (P < 0.03, Fig. 2c, d), due to the addition of a non-fluorescent dye that is not known to directly interact with eosin. However, upon closer inspection it was observed that the lifetime of collagen, which is colored blue/green in the lifetime image, was actually the same in the range 1000–1500 ps as it was in eosin stained slides. Selecting smaller regions of interest only around the collagen-rich areas confirmed that eosin-stained collagen did not have an altered lifetime in H&E stained slides, which is analogous to the intensity data (Fig. 1) in which eosin-stained collagen fluorescence was not changed by the addition of hematoxylin. Therefore, the data suggests that the basis for the change in the lifetime of the entire image was due to a change in the fluorescence lifetime within stained cells.
Fig. 2.
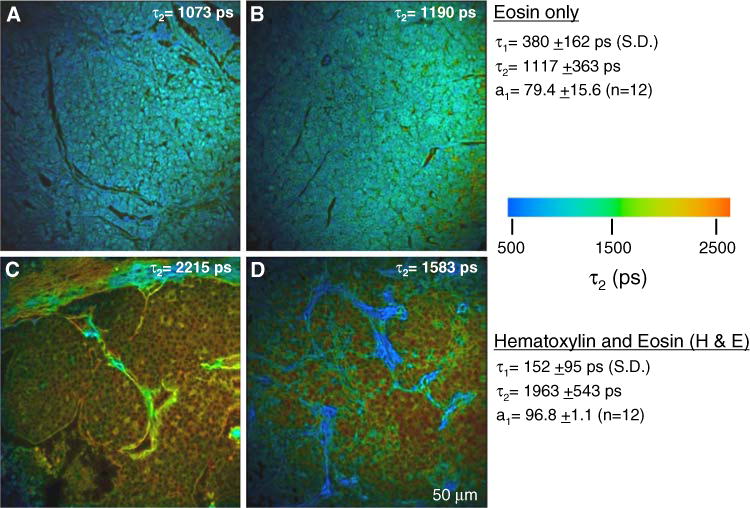
Fluorescence lifetime of stained mouse breast tumor samples. a, b In slides stained with eosin only, the fluorescence lifetime is similar throughout the field of view. Regions that encompassed cells and ECM are shown. c, d This value for eosin fluorescence lifetime was preserved in the extracellular matrix regions of H&E stained slides, where hematoxylin does not compete for staining. However, the lifetime of eosin is significantly lengthened in cells compared to eosin only. For each condition, two exemplar images out of 12 are shown. Population averages for the lifetime values are given on the right and the τ2 value for each individual panel shown is indicated
Desmoplasia, the increased deposition of collagen in the stroma, was extensive in PyVT mammary tumors. Thus, a complicating issue was the relative amount of collagen present in the image, which can bias mean lifetime values in direct proportion to its level of expression, since eosin-stained collagen had a different lifetime than H&E stained cells. Therefore, to analyze specifically the lifetime of cells, we analyzed only the fluorescence emission from within a user-defined ROI that did not include collagen. Using this approach, the change in lifetime within cells was once again due to hematoxylin, which may be blocking eosin staining or at the very least interfering with eosin fluorescence. The net result is that the lifetime of eosin is lengthened (Fig. 2c, d) when hematoxylin is present. This is highlighted in the lifetime image by cells that appear reddish-orange in color and having a τ2 value of ≥2000 ps. Alternatively, due to lessened eosin staining, the underlying endogenous autofluorescence may now represent a greater fraction of emitted fluorescence, which would result in a longer lifetime (see data below). Furthermore, the fit of the lifetime data for the H&E samples was poor, as the Chi-squared values for cells under these conditions were very high, oftentimes well beyond the defined threshold of 1.3. Hence, the complication that hematoxylin contributes in the form of negatively stained nuclei may lead to artificially calculating longer lifetimes even when data is fit on a pixel-by-pixel basis in SPC-Image. It is for this reason that one should be cautious when analyzing lifetime data from H&E stained slides.
Imaging the Endogenous Fluorescence of Unstained Slides
We found surprisingly that the endogenous fluorescence of cells was well preserved in formalin fixed, de-paraffinized, unstained tissue sections prepared from archived paraffin-embedded blocks. In Fig. 3a, at 890 nm laser excitation, and with the neutral density filtering needed for eosin imaging removed, endogenous fluorescence from thin tissue slices was observed. The intensity was uniform, and nuclei were not stained. Based on the two-photon absorption cross-section [11], the source of fluorescence at 890 nm excitation was FAD, flavin adenine dinucleotide, which is a metabolite known to be autofluorescent. The other intrinsic candidate, NADH, would only be weakly or non-fluorescent at this excitation wavelength.
Fig. 3.
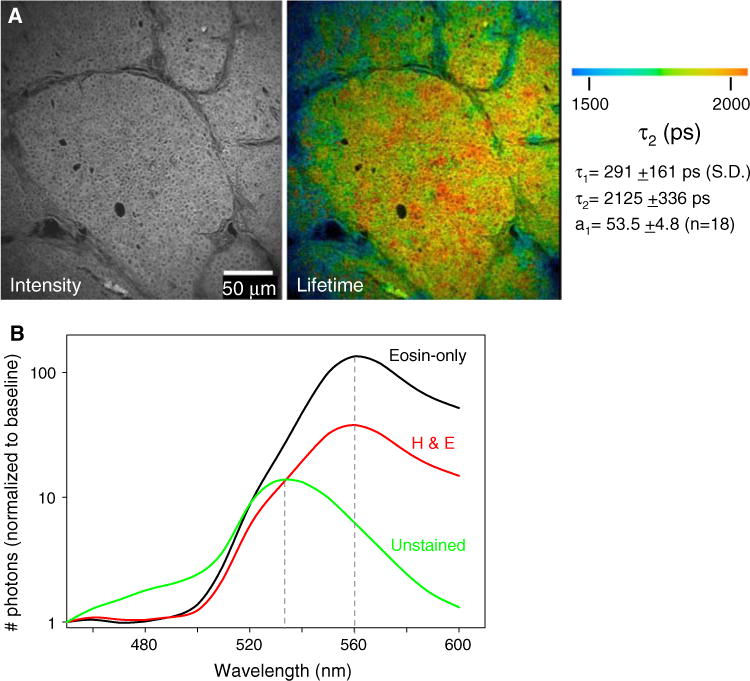
FLIM imaging of endogenous fluorophores in unstained tissue sections. a Fluorescence intensity and lifetime images of unstained tissue sections at 890 nm excitation. At this wavelength, detectable fluorescence emission intensity from FAD was generated. FAD had a distinct lifetime, and because collagen itself is autofluorescent it was observed in both intensity (left) and lifetime images (right). Lifetime values measured for the entire image field are given on the right. b Emission spectra for the variously stained slides as measured with a 16-channel spectral lifetime detector reveals a lack of change in the value of the peak emission between eosin stained slides that do or do not contain hematoxylin. Unstained slides had a shifted emission peak, because the fluorophore is endogenous FAD, not eosin. The spectra shown are from only one image for each condition, but were of the same peak emission in all samples tested
Since collagen is autofluorescent, it, too, was represented, which allows an analysis of the tissue architecture and the relationship of the cells to the ECM, even in these unstained slides. The autofluorescence of collagen is generally attributed to hydroxylysyl pyridinoline and lysyl pyridinoline crosslinkers [29, 30], and has been noted in multiphoton fluorescence images by others [31, 32]. We believe that this collagen signal was not due to second harmonic generation, which would be produced at 445 nm, since it persists when imaged with a 464 nm long-pass filter. When we measured the lifetime of cell-free collagen ROIs the value for τ2 ranges between 1500 and 2000 ps, which is also inconsistent with this being due to SHG.
The lifetime calculated across the entire image field (mostly tumor epithelium regions), was fit best with two exponentials, with values of τ1 = 291 ps and τ2 = 2125 ps, respectively (Fig. 3a, χ2 ≤ 1.3). Furthermore, the shape of the fluorescence decay curve was different than that of eosin or H&E stained slides, as the fractional representation of the first decay component was much lower in unstained slides (a1 = 53.5 vs. >80). These lifetime values are comparable to those observed in intact whole mouse tumors [22] and hamster cheek pouch [21] imaged for endogenous fluorophores on the same microscope.
In order to learn more about the sources of fluorescence, similar locations (serial sections) on both stained and unstained slides were imaged using combined SLIM microscopy [16, 17]. Separation of the emitted light spectrum was attained through the use of a diffraction grating to separate photons into a 16-channel photon-counting detector such that each bin represents 10 nm. When eosin was present, the emission peak was 560 nm; this was true whether or not hematoxylin was present (Fig. 3b), and this was confirmed in vitro by taking SLIM measurements of solutions of eosin in PBS (data not shown). In unstained slides the measured emission peak of 530 nm matched that in the literature for FAD fluorescence when measured in vitro [33, 34]. The peak photon-counts in unstained slides were much lower, and thus gave a lower emission intensity than observed for the exogenous fluorophore eosin. Overall, the greater accuracy and consistency in measuring the properties of autofluorescence in unstained tissue slides indicated that this is a preferable alternative to eosin stained slides.
To confirm that the source of fluorescence generated by tissue mounted to slides was inherent to, and preserved in, the tissue, we measured the fluorescence intensity, lifetime, and emission spectra of cells before and after slide processing. A human breast epithelial cell line, MCF10A cells, was used because these parameters have been measured in 2D cultures of these cells previously [16, 18] and the cells were shown to respond to metabolic perturbations through altered fluorescence. Cells were seeded into 3 mg/ml 3D collagen gels, floated, and grown for a period of 6–10 days before live cell imaging. Under these conditions, the cells formed acini, which is a structure analogous to breast ducts in vivo. These acini had fluorescent properties that were statistically similar (P > 0.05) to acini imaged in the same collagen gels that had been subsequently fixed, paraffin-embedded, mounted on slides and re-imaged (see Supplemental Fig. 1). Therefore, we found that the fundamental properties of endogenous fluorescence were preserved during fixation, paraffin-embedding, and subsequent slide preparation.
The Normal and Tumor Epithelium Comprising Carcinoma In Situ have Differing Fluorescence Lifetimes
The mouse tumors used in this study contained regions representing different stages of disease progression including carcinoma in situ (CIS). This stage is of high clinical relevance because it is similar to human ductal carcinoma in situ (DCIS) and is defined by a region of an otherwise normal duct containing hyperproliferating epithelium that is still within the basement membrane of the duct (Fig. 4a). Thus at this early stage of tumor progression, there is histologically normal epithelium contiguous with tumor neoplasia. In time, the hyperproliferating cells will fill the duct. Because of the fact that one can see both normal and tumor epithelium in the same image, and because of its relevance to understanding early breast disease progression, CIS is a useful region to seek endogenous optical biomarkers of cancer.
Fig. 4.
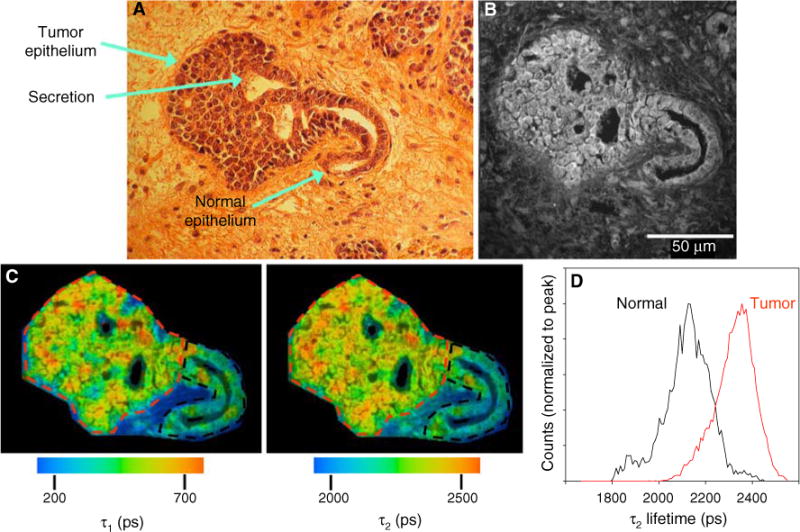
The fluorescent properties of normal and tumor epithelium differ. a Digital camera image of H&E stained mouse breast tumor. At this stage of tumor progression, which shows a carcinoma in situ, the distinction between normal epithelium and tumor is evident. Secretion of milk proteins into the remaining duct was preserved in the fixation procedure and, interestingly, was observed to be fluorescent. b Fluorescence intensity image of the sequential unstained slide at 890 nm excitation. c Color maps of the τ1 (left) and τ2 (right) components of the fluorescence lifetime, which illustrate the relatively longer lifetime values in tumor cells when compared to normal epithelium. d Histogram analysis measuring the range of lifetime values of the two ROIs drawn in (c) reveals the shift to longer lifetimes in tumor (red lines) cells compared to normal epithelium (color online)
The fluorescence intensity of CIS regions in unstained sections showed that the tumor epithelium was on average 21.6 ± 5.0% brighter than the normal epithelium (n = 13, Fig. 4b), suggesting that the tumor epithelium contained more of the fluorophore, which at an excitation wavelength of 890 nm is FAD. Because two cofactors of metabolism, FAD and NADH, are autofluorescent, we can measure changes in metabolism with fluorescence. It is well-known that tumors have radically altered metabolic pathways, which we postulate could lead to the accumulation of both fluorophores; NADH changes may be due to increased glycolysis, and FAD may be due to a drop in its reduction to FADH2 when compared to normal cells.
Continuing the analysis of normal versus tumor epithelium, we measured the fluorescence lifetime in areas of CIS. ROIs were traced around the tumor (Fig. 4c, red) and normal (Fig. 4c, black) epithelium such that the data could be analyzed separately. Both the τ1 and τ2 color maps and the histogram of τ2 lifetime values normalized to the peak illustrate a 229 ps shift to a longer lifetime in tumor cells (Fig. 4c, d). A lengthening of both τ1 and τ2 components in tumor cells was observed in 12 out of 13 DCIS regions from three different mice and is summarized in Table 1. Parallel results were obtained when these experiments were repeated at 780 nm excitation, where both NADH and FAD fluoresce (Table 1).
Table 1.
Fluorescence lifetime values for normal versus tumor epithelium
|
τ1 (ps)
|
τ2 (ps)
|
a1
|
||||
|---|---|---|---|---|---|---|
| Normal | Tumor | Normal | Tumor | Normal | Tumor | |
| 890 nm excitation (average shift) [n = 13] |
400 (68) * |
468 | 2269 (194) * |
2464 | 50 (1) |
51 |
| 780 nm excitation (average shift) [n = 10] |
456 (90) * |
546 | 2360 (178) |
2538 | 47 (2) |
49 |
Measured using two different excitation wavelengths, 890 nm and 780 nm, the two component fit of the lifetime decay showed that there is always a longer lifetime value in tumor epithelium versus normal. This was true whether or not the fluorophore, NADH or FAD, was in the free or bound state, since both τ1 and τ2 components were longer in duration and there was no change in the fractional contribution (a1) from each decay component. The values in parentheses denote the average difference between normal and tumor measurements. The asterisk denotes that the tumor value is significant compared to the normal value, P < 0.05 for a Students t-test
The Longer Lifetime in Tumor Cells is Independent of the Excitation and Emission Wavelengths
It is known that the lifetime of a fluorophore is independent of fluorophore concentration, laser power, and other imaging variations [35]. On the other hand the fluorescence lifetime is influenced by a number of factors such as pH, oxygen levels, and refractive index [36]. In particular, regions of high refractive index promote the exit of a fluorophore from the excited state and therefore shorten the lifetime [37, 38]. Examining the same CIS region as in Fig. 4 (Fig. 5a), we acquired SLIM (spectral lifetime) images, such that the total emission was separated into 16 discrete 10 nm wide channels where the lifetime was measured independently for each of these emission bins. The intensity in shorter wavelength emission channels was minimal at λex = 890 nm but significant for λex = 780 nm (Fig. 5b). This was likely due to the presence of NADH excitation and emission at λex = 780 nm, which was absent at λex = 890 nm. We examined our data based on the emission peaks previously published for FAD (530 nm, channel 9, [33, 34]) and NADH (465 nm, channel 3, [33, 39, 40]) and found that spectral channel 3, the lower wavelength emission channel, always had a shorter lifetime value than spectral channel 9, irrespective of whether normal or tumor epithelium was analyzed. The 3D plot of the SLIM data (Fig. 5c) shows this, where the time, emission wavelength, and photon-counts (x–y–z) are plotted. Here, the lifetime is clearly longer in channel 9 for each tissue type and excitation. Furthermore, the greater representation of low wavelength emission at 780 nm excitation (lower plots) in channel 3 was observed in the form of increased height and brighter cyan coloring in this channel when compared to 890 nm excitation (upper plots). However, the shift to longer lifetimes in tumor cells was difficult to resolve in these 3D plots. We found that the best way to observe the shift to longer lifetimes in tumor epithelium on a channel-by-channel basis was to look at the normalized τ2 histograms for each of these conditions. Figure 5d shows the two basic observations, that tumor epithelium always had a longer lifetime and that the longer wavelength/peak emission channel had a longer lifetime than its shorter emission wavelength counterpart.
Fig. 5.
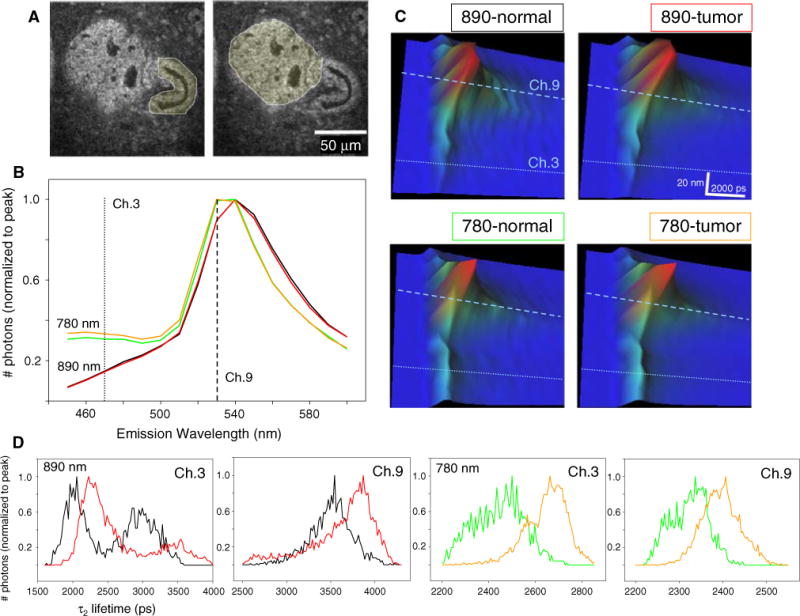
Correlation between fluorescence lifetime and emission wavelength. a Intensity images of a carcinoma in situ region where the regions of interest traced to separate normal from tumor analysis is shown. b Emission spectra normalized to peak photon-counts from 890 nm excitation (black line for normal, red line for tumor) and 780 nm excitation (green line for normal, orange line for tumor) show that the presence of higher photon-counts in channel 3 is due to excitation of NADH at shorter wavelengths and not due to differences between normal epithelium (black, green lines) versus tumor (red, orange lines). (c) 3D plot where the lifetime, emission spectra, and photon-counts are plotted in x–y–z, respectively. The longer lifetime in channel 9 is observed at each excitation wavelength and tissue type analyzed. The greater representation of emission in channel 3 when excited at 780 nm can be seen as brighter cyan coloring indicating higher amplitude in the z-axis. d Histograms of τ2 values normalized to peak photon-counts of normal (black line at 890 nm, green line at 780 nm excitation) versus tumor (red line at 890 nm, orange line at 780 nm excitation) regions. Note that there is always a shift to a longer lifetime in tumor tissue. Furthermore the lifetime value is correlated with the emission wavelength, where the shorter emission photons (channel 3) have shorter lifetimes than those found at the peak emission (channel 9) (color online)
Discussion
It has been a longstanding goal of microscopists and pathologists to diagnose and stage histopathology slides as early and accurately as possible. The rationale for fluorescence imaging of H&E stained slides is practical, however, difficulties arise due to the generalized bright staining by eosin as well as complications induced by hematoxylin. Here we demonstrate that mammary tumors can be discerned from normal epithelium by FLIM of unstained histopathology slides. Thus our data suggests that a combinatorial approach of fluorescence imaging of unstained slides along with subsequent brightfield diagnosis of concordant stained slides by a trained histopathologist can lead to a more advanced diagnosis of disease states. The gold standard process of H&E can remain intact, while our data suggests that MPSLM imaging of the tissue before it is stained would provide a useful diagnostic complement to the information gained from H&E staining. Our data indicates that it would be possible to assess unlabeled, fixed tissue with SLIM and then proceed with normal H&E methods.
The two primary endogenous fluorophores that can be exploited in unstained tissue are NAD(P)H and FAD. This is fortuitous, as the level of these molecules in cells provide information regarding the metabolic condition of the cell. In particular, the reduction:oxidation potential, also known as the redox ratio is altered in cancer [41, 42]. On the surface, this is a consequence of altered metabolism, described in part as the Warburg effect. However, NADH and FAD are enzyme cofactors in reactions beyond simple metabolism, the rates of which would change the redox ratio. Because of the endogenous fluorescence of FAD and NADH, it is possible to create redox images using the formula [FAD]/([FAD] + [NADH]) where the pixel intensity from images taken at 890 and 780 nm are used for the numerator and denominator, respectively. Although creating an image of the redox ratio in this manner may be attractive, several caveats complicate this approach. As has been pointed out, at excitation wavelengths below 880 nm, the resulting fluorescence is likely from multiple fluorophores. Standardization [15] or spectral unmixing [12] techniques need to be employed before attempting to calculate redox states using the imaging techniques used here. Furthermore, alterations in redox potential at one stage of cancer progression can be different than, or even the exact opposite of, those of another stage. Moreover, the tumor is capable of adapting to oxidative stress. For example, in breast and prostate cancer progression, tumor cells alleviate this stress by increasing fatty acid synthesis pathways that oxidize excess NADH [43]. As a result of this adaptation, the redox ratio can appear to be normal. Thus, it is always important for the researcher to consider at what stage they are imaging, and what adaptations may have occurred.
In addition to investigating intensity, the lifetime of the excited state of fluorescence contains further information about the state of the fluorophore. For example, the two components of the fluorescence lifetime decay of FAD are widely attributed to the protein-bound (short component, τ1) and free forms (long component, τ2) of FAD, respectively [44]. For NADH the opposite is true, where τ1 corresponds to the free form and τ2 to the bound form [45]. One potential cause for a change in the lifetime of the bound component is a switch in the protein to which the fluorophore is bound. For both NADH and FAD, there are hundreds of potential binding partners; thus, a comprehensive examination of this is not currently feasible.
It was notable, however, that in our experimental system both the long and short components were shifted to longer values at both excitation wavelengths. Because the unbound components are also altered, these changes cannot be solely due to changes in just the binding partner of the bound component, and instead a more general property of the fluorescence of NADH and FAD must affect the lifetime values. Because most fluorophores are expressed in environments of differing refractive index, which can significantly alter the lifetime [37, 38], it is our hypothesis that an alteration in the relative expression of the fluorophore in one refractive index location over another can shift the measured lifetime value. Although we have not measured the refractive index of these cells ourselves, van Manen et al. [37] found that GFP-Rac found in the cytosol (refractive index, η = 1.38) had a 400 ps longer τ2 lifetime than the same fluorophore expressed in the plasma membrane (η = 1.46). Thus, such a seemingly small change in refractive index alters the lifetime to a degree comparable with our results. The hypothesis is that fluorophores in membranes are quenched by the local molecular environment, and thus have a shorter lifetime. Alternatively, the shift in both long and short lifetime components might represent accumulation of NADH and FAD in the cytoplasm of tumor cells. This can occur because tumor cells have an increased rate of glycolysis, an NADH-generating process that occurs in the cytoplasm, and also have a decreased rate at which FAD is reduced in the citric acid cycle and fatty acid oxidation in the mitochondria. Moreover, it is thought that tumor cells also generate and secrete acids (H+, lactic, and others, [46]) and that a lower pH shortens the fluorescence lifetime [47–49]. In addition, Vishwasrao et al. [40] have detailed how a change in intracellular viscosity due to osmotic swelling of mitochondria in hypoxic cells (where oxygen perturbations themselves can change fluorescence intensity and lifetime) was linked to a change in the lifetime of free NADH.
The correlation we observe between short emission wavelengths having shorter lifetimes has been observed for other fluorophores and by other investigators [50, 51]. Excited fluorophores must decay to the lowest energy excited singlet state (S10) before emission, but there are multiple ground states to which it can emit, which would then result in differing emission wavelengths. Conditions that support a rapid relaxation of the fluorophore to the lowest excited singlet state (short lifetime) also support emission to the lowest possible ground state S00 (short wavelength). Another viewpoint on this topic is that fluorophores undergo spectral relaxation [51], where energy from the excited state is transferred to the solvent at a certain rate. Some fluorophores relax very little and thus have short lifetimes, whereas others relax for longer times and have longer emission wavelengths due to greater energy depletion. In this view, emission can potentially occur from any energy level during relaxation, yielding a correlated spectrum of both emission wavelength and lifetime. In our hands, endogenous fluorophores displayed a vast range of emission and lifetime values for each of several various measured conditions, suggesting that cells have inherent traits that cause this variability.
Other aspects need to be considered when analyzing the photon-count measurements in fluorescence lifetime microscopy. For one, not only do NADH and FAD have different quantum yields (quantum yield is defined as the ratio of emitted photons to the number absorbed) but the yield is altered when the coenzyme is free or bound. For example, the quantum yield of NADH is increased fourfold when bound to protein [35]. Furthermore, the excitation wavelength we used to image NADH (780 nm) was longer than the peak absorbance wavelength for this fluorophore, diminishing the relative contribution of NADH fluorescence at this wavelength compared to FAD. For NADH the absorbance at 780 nm is <0.01, whereas for FAD the absorbance is 0.04 at both 890 and 780 nm [33]. Thus, the seemingly small representation of NADH in emission spectra (Fig. 5b) could be accounted for by low absorbance at 780 nm excitation.
Several researchers have imaged the lifetime of endogenous fluorophores in the context of breast cancer tumors [18, 19, 22], skin sarcomas [52, 53], oral carcinoma [54], brain gliomas [55], and in slides [19]. The results have not been consistent across groups, but may be explained by differences in methodologies. For instance, study variables include multiple fluorophores due to the excitation wavelength, or because regions containing both cells and collagen were included in the analysis, rather than drawing regions of interest to isolate only cells or ECM for analysis. In the multiphoton imaging studies that best correlate with our use of unstained slides here, our values for the lifetime of FAD and NADH match very well with those reported in cell lines [18, 54]. In addition, in control experiments we find that the endogenous fluorophores are preserved following fixation, paraffin-embedding, and slide preparation compared to fresh, live samples. In further support of this finding, the tumor samples imaged here as histopathology slides were prepared from the same mouse tumor model that we used to image endogenous fluorescence from fresh ex vivo tumors [22]. Importantly, similar lifetime values were obtained in the fixed samples here as to those fresh tumor samples, further demonstrating the preservation of intrinsic fluorophores during fixation and slide preparation.
In summary, our data demonstrate that optical techniques are useful adjuvants to detect pathological changes associated with cancer. A challenge to pathologists is to understand which tumors are likely to progress, especially in the case of very early carcinomas such as carcinoma in situ. Here we present a way to detect the metabolic changes associated with cancer at an early stage, which has enormous potential as a diagnostic tool.
Supplementary Material
Acknowledgments
This work was supported by a Mary Kay Ash Charitable Foundation award (#080-07), a NIH NCI CA 076537 award to P.J.K., and a DOD award (W81XWH-04-1-042) to P.P.P. Histopathology slides were prepared by Jane Weeks at McArdle labs at the University of Wisconsin. We thank the members of our laboratory for discussions and comments on this manuscript.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s12013-009-9046-7) contains supplementary material, which is available to authorized users.
Contributor Information
Matthew W. Conklin, Department of Pharmacology, University of Wisconsin, 1525 Linden Dr., Madison, WI 53706, USALaboratory for Optical and Computational Instrumentation (LOCI), Laboratory for Molecular Biology, University of Wisconsin, 1525 Linden Dr., Madison, WI 53706, USAPaul P. Carbone Comprehensive Cancer Center, University of Wisconsin, 1525 Linden Dr., Madison, WI 53706, USA
Paolo P. Provenzano, Department of Pharmacology, University of Wisconsin, 1525 Linden Dr., Madison, WI 53706, USALaboratory for Optical and Computational Instrumentation (LOCI), Laboratory for Molecular Biology, University of Wisconsin, 1525 Linden Dr., Madison, WI 53706, USAPaul P. Carbone Comprehensive Cancer Center, University of Wisconsin, 1525 Linden Dr., Madison, WI 53706, USA
Kevin W. Eliceiri, Laboratory for Optical and Computational Instrumentation (LOCI), Laboratory for Molecular Biology, University of Wisconsin, 1525 Linden Dr., Madison, WI 53706, USA
Ruth Sullivan, Laboratory for Optical and Computational Instrumentation (LOCI), Laboratory for Molecular Biology, University of Wisconsin, 1525 Linden Dr., Madison, WI 53706, USAPaul P. Carbone Comprehensive Cancer Center, University of Wisconsin, 1525 Linden Dr., Madison, WI 53706, USA.
Patricia J. Keely, Department of Pharmacology, University of Wisconsin, 1525 Linden Dr., Madison, WI 53706, USALaboratory for Optical and Computational Instrumentation (LOCI), Laboratory for Molecular Biology, University of Wisconsin, 1525 Linden Dr., Madison, WI 53706, USAPaul P. Carbone Comprehensive Cancer Center, University of Wisconsin, 1525 Linden Dr., Madison, WI 53706, USA
References
- 1.Eliceiri KW, Fan CH, Lyons GE, White JG. Analysis of histology specimens using lifetime multiphoton microscopy. Journal of Biomedical Optics. 2003;8:376–380. doi: 10.1117/1.1584053. [DOI] [PubMed] [Google Scholar]
- 2.Saikia B, Gupta K, Saikia UN. The modern histopathologist: In the changing face of time. Diagnostic Pathology. 2008;3:25. doi: 10.1186/1746-1596-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chance B, Cohen P, Jobsis F, Schoener B. Intracellular oxidation–reduction states in vivo. Science. 1962;137:499–508. doi: 10.1126/science.137.3529.499. [DOI] [PubMed] [Google Scholar]
- 4.Warburg O, Dickens F, Kaiser Wilhelm-Institut für Biologie, B . The metabolism of tumours: Investigations from the Kaiser-Wilhelm Institute for Biology. Berlin-Dahlem. London: Constable; 1930. [Google Scholar]
- 5.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nature Reviews Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 6.Pedersen PL. Warburg, me and Hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers’ most common phenotypes, the “Warburg Effect”, i.e., elevated glycolysis in the presence of oxygen. Journal of Bioenergetics and Biomembranes. 2007;39:211–222. doi: 10.1007/s10863-007-9094-x. [DOI] [PubMed] [Google Scholar]
- 7.Lehninger AL, Nelson DL, Cox MM. Principles of biochemistry. New York: Worth Publishers; 1993. pp. 542–594. Chapter 18. [Google Scholar]
- 8.Gillies RJ, Gatenby RA. Adaptive landscapes and emergent phenotypes: Why do cancers have high glycolysis? Journal of Bioenergetics and Biomembranes. 2007;39:251–257. doi: 10.1007/s10863-007-9085-y. [DOI] [PubMed] [Google Scholar]
- 9.Naumov GN, Akslen LA, Folkman J. Role of angiogenesis in human tumor dormancy: Animal models of the angiogenic switch. Cell Cycle. 2006;5:1779–1787. doi: 10.4161/cc.5.16.3018. [DOI] [PubMed] [Google Scholar]
- 10.Yossepowitch O, Pinchuk I, Gur U, Neumann A, Lichtenberg D, Baniel J. Advanced but not localized prostate cancer is associated with increased oxidative stress. Journal of Urology. 2007;178:1238–1243. doi: 10.1016/j.juro.2007.05.145. discussion 1243–1234. [DOI] [PubMed] [Google Scholar]
- 11.Zipfel WR, Williams RM, Christie R, Nikitin AY, Hyman BT, Webb WW. Live tissue intrinsic emission microscopy using multiphoton-excited native fluorescence and second harmonic generation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:7075–7080. doi: 10.1073/pnas.0832308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chorvat D, Jr, Chorvatova A. Spectrally resolved time-correlated single photon counting: A novel approach for characterization of endogenous fluorescence in isolated cardiac myocytes. European Biophysics Journal. 2006;36:73–83. doi: 10.1007/s00249-006-0104-4. [DOI] [PubMed] [Google Scholar]
- 13.Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 14.Skala MC, Squirrell JM, Vrotsos KM, Eickhoff JC, Gendron-Fitzpatrick A, Eliceiri KW, et al. Multi-photon microscopy of endogenous fluorescence differentiates normal, precancerous, and cancerous squamous epithelial tissues. Cancer Research. 2005;65:1180–1186. doi: 10.1158/0008-5472.CAN-04-3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirkpatrick ND, Brewer MA, Utzinger U. Endogenous optical biomarkers of ovarian cancer evaluated with multiphoton microscopy. Cancer Epidemiology, Biomarkers and Prevention. 2007;16:2048–2057. doi: 10.1158/1055-9965.EPI-07-0009. [DOI] [PubMed] [Google Scholar]
- 16.Yan L, Rueden CT, White JG, Eliceiri KW. Applications of combined spectral lifetime microscopy for biology. Biotechniques. 2006;41:249, 251, 253. doi: 10.2144/000112251. passim. [DOI] [PubMed] [Google Scholar]
- 17.Bird DK, Eliceiri KW, Fan CH, White JG. Simultaneous two-photon spectral and lifetime fluorescence microscopy. Applied Optics. 2004;43:5173–5182. doi: 10.1364/ao.43.005173. [DOI] [PubMed] [Google Scholar]
- 18.Bird DK, Yan L, Vrotsos KM, Eliceiri KW, Vaughan EM, Keely PJ, et al. Metabolic mapping of MCF10A human breast cells via multiphoton fluorescence lifetime imaging of the coenzyme NADH. Cancer Research. 2005;65:8766–8773. doi: 10.1158/0008-5472.CAN-04-3922. [DOI] [PubMed] [Google Scholar]
- 19.Tadrous PJ, Siegel J, French PM, Shousha S, Lalani el N, Stamp GW. Fluorescence lifetime imaging of unstained tissues: Early results in human breast cancer. Journal of Pathology. 2003;199:309–317. doi: 10.1002/path.1286. [DOI] [PubMed] [Google Scholar]
- 20.Esposito A, Gerritsen HC, Oggier T, Lustenberger F, Wouters FS. Innovating lifetime microscopy: A compact and simple tool for life sciences, screening, and diagnostics. Journal of Biomedical Optics. 2006;11:34016. doi: 10.1117/1.2208999. [DOI] [PubMed] [Google Scholar]
- 21.Skala MC, Riching KM, Bird DK, Gendron-Fitzpatrick A, Eickhoff J, Eliceiri KW, et al. In vivo multiphoton fluorescence lifetime imaging of protein-bound and free nicotinamide adenine dinucleotide in normal and precancerous epithelia. Journal of Biomedical Optics. 2007;12:024014. doi: 10.1117/1.2717503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, et al. Collagen density promotes mammary tumor initiation and progression. BMC Medicine. 2008;6:11. doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin EY, Jones JG, Li P, Zhu L, Whitney KD, Muller WJ, et al. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. American Journal of Pathology. 2003;163:2113–2126. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: A transgenic mouse model for metastatic disease. Molecular and Cellular Biology. 1992;12:954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wozniak MA, Desai R, Solski PA, Der CJ, Keely PJ. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. Journal of Cell Biology. 2003;163:583–595. doi: 10.1083/jcb.200305010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Provenzano PP, Rueden CT, Trier SM, Yan L, Ponik SM, Inman DR, et al. Nonlinear optical imaging and spectral-lifetime computational analysis of endogenous and exogenous fluorophores in breast cancer. Journal of Biomedical Optics. 2008;13:031220. doi: 10.1117/1.2940365. [DOI] [PubMed] [Google Scholar]
- 27.Apgar JM, Juarranz A, Espada J, Villanueva A, Canete M, Stockert JC. Fluorescence microscopy of rat embryo sections stained with haematoxylin–eosin and Masson’s tri-chrome method. Journal of Microscopy. 1998;191:20–27. doi: 10.1046/j.1365-2818.1998.00348.x. [DOI] [PubMed] [Google Scholar]
- 28.Espada J, Valverde P, Stockert JC. Selective fluorescence of eosinophilic structures in grasshopper and mammalian testis stained with haematoxylin–eosin. Histochemistry. 1993;99:385–390. doi: 10.1007/BF00717051. [DOI] [PubMed] [Google Scholar]
- 29.Fujimoto D. Isolation and characterization of a fluorescent material in bovine achilles tendon collagen. Biochemical and Biophysical Research Communications. 1977;76:1124–1129. doi: 10.1016/0006-291x(77)90972-x. [DOI] [PubMed] [Google Scholar]
- 30.Eyre DR, Paz MA, Gallop PM. Cross-linking in collagen and elastin. Annual Review of Biochemistry. 1984;53:717–748. doi: 10.1146/annurev.bi.53.070184.003441. [DOI] [PubMed] [Google Scholar]
- 31.Kirkpatrick ND, Hoying JB, Botting SK, Weiss JA, Utzinger U. In vitro model for endogenous optical signatures of collagen. Journal of Biomedical Optics. 2006;11:054021. doi: 10.1117/1.2360516. [DOI] [PubMed] [Google Scholar]
- 32.Lilledahl MB, Haugen OA, de Lange Davies C, Svaasand LO. Characterization of vulnerable plaques by multiphoton microscopy. Journal of Biomedical Optics. 2007;12:044005. doi: 10.1117/1.2772652. [DOI] [PubMed] [Google Scholar]
- 33.Huang S, Heikal AA, Webb WW. Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophysical Journal. 2002;82:2811–2825. doi: 10.1016/S0006-3495(02)75621-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu Y, Qu JY. Autofluorescence spectroscopy of epithelial tissues. Journal of Biomedical Optics. 2006;11:054023. doi: 10.1117/1.2362741. [DOI] [PubMed] [Google Scholar]
- 35.Lakowicz JR. Principles of fluorescence spectroscopy. 3rd. New York, Berlin: Springer; 2006. p. 954. [Google Scholar]
- 36.Suhling K, French PM, Phillips D. Time-resolved fluorescence microscopy. Photochemical & Photobiological Sciences. 2005;4:13–22. doi: 10.1039/b412924p. [DOI] [PubMed] [Google Scholar]
- 37.van Manen HJ, Verkuijlen P, Wittendorp P, Subramaniam V, van den Berg TK, Roos D, et al. Refractive index sensing of green fluorescent proteins in living cells using fluorescence lifetime imaging microscopy. Biophysical Journal. 2008;94:L67–L69. doi: 10.1529/biophysj.107.127837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suhling K, Siegel J, Phillips D, French PM, Leveque-Fort S, Webb SE, et al. Imaging the environment of green fluorescent protein. Biophysical Journal. 2002;83:3589–3595. doi: 10.1016/S0006-3495(02)75359-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Villette S, Pigaglio-Deshayes S, Vever-Bizet C, Validire P, Bourg-Heckly G. Ultraviolet-induced autofluorescence characterization of normal and tumoral esophageal epithelium cells with quantitation of NAD(P)H. Photochemical & Photobiological Sciences. 2006;5:483–492. doi: 10.1039/b514801d. [DOI] [PubMed] [Google Scholar]
- 40.Vishwasrao HD, Heikal AA, Kasischke KA, Webb WW. Conformational dependence of intracellular NADH on metabolic state revealed by associated fluorescence anisotropy. Journal of Biological Chemistry. 2005;280:25119–25126. doi: 10.1074/jbc.M502475200. [DOI] [PubMed] [Google Scholar]
- 41.Chaiswing L, Bourdeau-Heller JM, Zhong W, Oberley TD. Characterization of redox state of two human prostate carcinoma cell lines with different degrees of aggressiveness. Free Radical Biology and Medicine. 2007;43:202–215. doi: 10.1016/j.freeradbiomed.2007.03.031. [DOI] [PubMed] [Google Scholar]
- 42.Chaiswing L, Zhong W, Cullen JJ, Oberley LW, Oberley TD. Extracellular redox state regulates features associated with prostate cancer cell invasion. Cancer Research. 2008;68:5820–5826. doi: 10.1158/0008-5472.CAN-08-0162. [DOI] [PubMed] [Google Scholar]
- 43.Hochachka PW, Rupert JL, Goldenberg L, Gleave M, Kozlowski P. Going malignant: the hypoxia-cancer connection in the prostate. Bioessays. 2002;24:749–757. doi: 10.1002/bies.10131. [DOI] [PubMed] [Google Scholar]
- 44.Tanaka F, Tamai N, Yamazaki I, Nakashima N, Yoshihara K. Temperature-induced changes in the coenzyme environment of D-amino acid oxidase revealed by the multiple decays of FAD fluorescence. Biophysical Journal. 1989;56:901–909. doi: 10.1016/S0006-3495(89)82736-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lakowicz JR, Szmacinski H, Nowaczyk K, Johnson ML. Fluorescence lifetime imaging of free and protein-bound NADH. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:1271–1275. doi: 10.1073/pnas.89.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gatenby RA, Gawlinski ET, Gmitro AF, Kaylor B, Gillies RJ. Acid-mediated tumor invasion: A multidisciplinary study. Cancer Research. 2006;66:5216–5223. doi: 10.1158/0008-5472.CAN-05-4193. [DOI] [PubMed] [Google Scholar]
- 47.Nakabayashi T, Wang HP, Kinjo M, Ohta N. Application of fluorescence lifetime imaging of enhanced green fluorescent protein to intracellular pH measurements. Photochemical & Photobiological Sciences. 2008;7:668–670. doi: 10.1039/b800391b. [DOI] [PubMed] [Google Scholar]
- 48.Gannot I, Ron I, Hekmat F, Chernomordik V, Gandjbakhche A. Functional optical detection based on pH dependent fluorescence lifetime. Lasers in Surgery and Medicine. 2004;35:342–348. doi: 10.1002/lsm.20101. [DOI] [PubMed] [Google Scholar]
- 49.Hille C, Berg M, Bressel L, Munzke D, Primus P, Lohmannsroben HG, et al. Time-domain fluorescence lifetime imaging for intracellular pH sensing in living tissues. Analytical and Bioanalytical Chemistry. 2008;391:1871–1879. doi: 10.1007/s00216-008-2147-0. [DOI] [PubMed] [Google Scholar]
- 50.Lakowicz JR, Gratton E, Cherek H, Maliwal BP, Laczko G. Determination of time-resolved fluorescence emission spectra and anisotropies of a fluorophore–protein complex using frequency-domain phase-modulation fluorometry. Journal of Biological Chemistry. 1984;259:10967–10972. [PubMed] [Google Scholar]
- 51.Lakowicz JR. On spectral relaxation in proteins. Photochemistry and Photobiology. 2000;72:421–437. doi: 10.1562/0031-8655(2000)072<0421:OSRIP>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.De Beule PA, Dunsby C, Galletly NP, Stamp GW, Chu AC, Anand U, et al. A hyperspectral fluorescence lifetime probe for skin cancer diagnosis. Review of Scientific Instruments. 2007;78:123101. doi: 10.1063/1.2818785. [DOI] [PubMed] [Google Scholar]
- 53.Galletly NP, McGinty J, Dunsby C, Teixeira F, Requejo-Isidro J, Munro I, et al. Fluorescence lifetime imaging distinguishes basal cell carcinoma from surrounding uninvolved skin. British Journal of Dermatology. 2008;159:152–161. doi: 10.1111/j.1365-2133.2008.08577.x. [DOI] [PubMed] [Google Scholar]
- 54.Skala MC, Riching KM, Gendron-Fitzpatrick A, Eickhoff J, Eliceiri KW, White JG, et al. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19494–19499. doi: 10.1073/pnas.0708425104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leppert J, Krajewski J, Kantelhardt SR, Schlaffer S, Petkus N, Reusche E, et al. Multiphoton excitation of autofluorescence for microscopy of glioma tissue. Neurosurgery. 2006;58:759–767. doi: 10.1227/01.NEU.0000204885.45644.22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.