

Abstract
Human apolipoprotein A-I (apoA-I) is the most abundant protein in high-density lipoprotein, an anti-atherogenic lipid-protein complex responsible for reverse cholesterol transport. The protein is composed of an N-terminal helix bundle domain, and a small C-terminal (CT) domain. To facilitate study of CT-apoA-I, a novel strategy was employed to produce this small domain in a bacterial expression system. A protein construct was designed of insect apolipophorin III (apoLp-III) and residues 179–243 of apoA-I, with a unique a methionine residue positioned between the two proteins and an N-terminal His-tag to facilitate purification. The chimera was expressed in E. coli, purified by Ni-affinity chromatography, and cleaved by cyanogen bromide. SDS-PAGE revealed the presence of three proteins with masses of 7 kDa (CT-apoA-I), 18 kDa (apoLp-III), and a minor 26 kDa band of uncleaved chimera. The digest was reloaded on the Ni-affinity column to bind apoLp-III and uncleaved chimera, while CT-apoA-I was washed from the column and collected. Alternatively, CT-apoA-I was isolated from the digest by reversed-phase HPLC. CT-apoA-I was α-helical, highly effective in solubilizing phospholipid vesicles and disaggregating LPS micelles. However, CT-apoA-I was less active compared to full-length apoA-I in protecting lipolyzed low density lipoproteins from aggregating, and disrupting phosphatidylglycerol bilayer vesicles. Thus the novel expression system produced mg quantities of functional CT-apoA-I, facilitating structural and functional studies of this critical domain of apoA-I.
Keywords: apolipoprotein A-I, chimera, fusion protein, C-terminal domain, apolipophorin III, lipid binding, lipoprotein
Graphical abstract

1. Introduction
Human apolipoprotein A-I (apoA-I) is the main protein associated with high-density lipoprotein (HDL) (1–3). ApoA-I exhibits remarkable structural flexibility existing in a lipid free form or bound to HDL of various sizes, which require structural adaptations accommodating changes in lipoprotein size and shape (4). The protein is a major player in heart disease and has been well documented for its roles in reverse cholesterol transport, inflammation, and innate immunity (5). ApoA-I is a 243 residue protein with residues 1–178 folded into a helix bundle forming the N-terminal (NT) domain (6, 7). This domain contains several amino acid residues responsible for lecithin cholesterol acyl transferase activity (2). The C-terminal (CT) tail, residues 179–243, is referred to as the CT domain, and is relatively unstructured in its monomeric form but can adopt helical structure upon self-association or lipid binding (8). When the CT domain was removed from apoA-I, this resulted in significant changes in apoA-I function. The remaining NT helix bundle was less effective in solubilizing phospholipid vesicles, and binding to macrophages was impaired resulting in decreased cholesterol and phospholipid efflux (9). Moreover, other CT deletion variants showed reduced capability of ABCA-I driven lipid efflux in J774 macrophages and preference for smaller HDL, and increased plasma clearance (10, 11). It has been reported that the CT domain of apoA-I initiates contact with the lipid surface, bringing the NT domain in close proximity to lipids. A conformational change then provides access of hydrophobic residues of the NT domain for a stable binding interaction with the lipid environment to form HDL (1, 12).
To gain more insight in the properties of the CT domain of apoA-I, it would be beneficial to have access to sufficient quantities of this protein domain. De novo peptide synthesis has been employed to produce CT-apoA-I (8), but this can be expensive, especially due to the large number of amino acid residues. Furthermore, purchase of the peptide may have to be done successively, and while changes can be made in the amino acid sequence upon synthesis, this can become rather costly. Normally, over-expression in Escherichia coli is the preferred method for producing small proteins, which also permits changes at the amino acid level by site-directed mutagenesis. While full-length apoA-I and the NT helix bundle can be expressed in large quantities in E. coli (13–16), the CT-domain may be too small for efficient recombinant overexpression. Recently, we engineered a chimeric protein made of insect apolipophorin III (apoLp-III) and CT-apoA-I, which was produced in large quantities in E. coli (17). This apolipoprotein construct provided an opportunity to isolate CT-apoA-I, by expression and purification as a full-length chimeric protein followed by removal of apoLp-III. To facilitate this, a unique methionine residue was introduced between apoLp-III and CT-apoA-I. This allowed for cyanogen bromide cleavage producing apoLp-III and CT-apoA-I. This would have been much more difficult to accomplish in full-length apoA-I as it includes four methionine residues in its NT domain. Since the cyanogen bromide reaction may not be complete, the cleavage reaction produces many fragments, making isolation of CT-apoA-I challenging. In the present report we demonstrated that purification of CT-apoA-I from the cyanogen bromide digest of the chimera yielded a high purity protein preparation with sufficient quantities required for functional studies of this critical part of apoA-I.
2. Materials and Methods
2.1 Chimeric Protein, site-directed mutagenesis, protein expression and purification
The chimeric construct was custom made by Eurofins MWG Operon as described previously and inserted into the pET-20b(+) vector (17). Asparagine 189 was replaced by methionine using the QuikChange-II site directed mutagenesis kit (Qiagen). To generate the substitution, the following primers were used: 5′-GGTTCAGAAACCGGCGATGGAAGCGCTGAAAGAAAACG-3′ (forward) and 5′-CGTTTTCTTTCAGCGCTTCCATCGCCGGTTTCTGAACC-3′ (reverse). The mutation was confirmed by DNA sequencing (Genewiz). The chimeric protein and apoA-I were overexpressed in E. coli BL21 (DE3) pLysS cells (Agilent Technologies). Overnight cultures in 2xYT broth with 50 μg/mL ampicillin and chloramphenicol were used to seed 2 L cultures. Cells were grown at 37°C until an optical density of 0.6 at 600 nm was reached, and protein over-expression was induced with 0.5 mM isopropyl-β-D-1-thiogalactopyranoside (Gold Biotechnology). Cells were grown for another 4 h and harvested by centrifugation at 8,000 g at 4°C. Cells were resuspended in phosphate buffered saline (PBS; 150 mM NaCl, 10 mM NaH2PO4, 10 mM Na2HPO4, pH 7.4) and lysed using a digital sonicator (Branson) at 30% amplitude for five intervals of 30 s. Lysed cells were centrifuged 2× at 20,000 g for 30 min to remove cell debris. The supernatant was mixed with an equal volume of sample loading buffer (2× PBS, 6 M guanidine-HCl, pH 7.4) and loaded onto 5 mL HiTrap™ chelating columns (GE Healthcare). The column was washed with 20 mL of 40 mM imidazole, 3 M guanidine-HCl in PBS. Proteins were eluted using 20 mL elution buffer (500 mM imidazole in PBS). The eluate was dialyzed in 4 L of 10 mM ammonium bicarbonate buffer with 1 mM EDTA, pH 7.8, and three buffer changes were made over 48 h. The samples were then freeze-dried (Labconco) stored at −20 °C until use. For structural or functional analysis, dried recombinant proteins were resuspended in 6 M guanidine HCl, and refolded by dialysis for 48 h in the appropriate buffer, depending on the analysis. Protein concentrations were obtained using the bicinchoninic acid assay (Thermo Fisher Scientific) or absorbance at 280 nm. Protein purification and purity was assessed using polyacrylamide gel electrophoresis (PAGE). Protein samples (15 μg) were incubated at 70° for 10 min after adding lithium dodecyl sulfate sample loading buffer (Thermo Fisher Scientific). Samples were separated on NuPAGE 10% Bis-Tris gels at 200 V for 35 min in 2-(N-morpholino)ethanesulfonic acid, sodium dodecyl sulfate (SDS) running buffer (Thermo Fisher Scientific). Gels were stained with 0.5 % (m/v) naphthol blue-black, in 10 % glacial acetic acid and 45 % methanol (v/v).
2.2 Chemical cleavage, peptide isolation, and circular dichroism
The chimera was cleaved with cyanogen bromide (Sigma-Aldrich) using a 1:100 molar ratio of methionine to cyanogen bromide. Twenty-six mg of dry protein was dissolved in 0.5 mL of 70% formic acid, and added to 0.5 mL of 200 mM cyanogen bromide solution in 70% formic acid. The reaction was carried out for 24 h at 25 °C, after which the sample was diluted five-fold with Milli-Q water, frozen at −80 °C and freeze-dried for one day. The lyophilized digest was dissolved in 2 mL of 50 mM ammonium bicarbonate buffer and freeze-dried.
Nickel-affinity chromatograph was used to isolate the CT-apoA-I from the cyanogen digest. The lyophilized digest was dissolved in 1 mL PBS and loaded onto a 1 mL HiTrap™ chelating column (GE Healthcare). The column was washed with 1x PBS, 40 mM imidazole, pH 7.4, releasing CT-apoA-I. ApoLp-III and uncleaved chimera were eluted with 500 mM imidazole in PBS. Column fractions were dialyzed in 2 L of 10 mM ammonium bicarbonate buffer and freeze-dried; the purity of CT-apoA-I was verified by SDS-PAGE. Alternatively, CT-apoA-I was purified from the cyanogen bromide digest with reversed-phase HPLC. Approximately 6 mg of digest was dissolved in 1 mL of 20 mM sodium phosphate buffer (Na2HPO4, NaH2PO4, pH 7.4) and loaded on a Zorbax 300SB-C8 column (Agilent Technologies). Proteins were eluted using a linear gradient of water and acetonitrile in 0.05 % trifluoroacetic acid (0.5% acetonitrile increase per min). The flow rate was set at 1 mL/min and elution was monitored at 210 nm.
CT-apoA-I was analyzed for secondary structure by circular dichroism in a Jasco 810 polarimeter. The ellipticity of the protein samples were determined from 185 to 260 nm at a concentration of 0.2 mg/mL in 20 mM sodium phosphate buffer, pH 7.4 (50 nm/min scan rate).
2.3 Phospholipid vesicle solubilization
Lipid films were prepared by dissolving 10 mg of dry lipid (Avanti Polar Lipids) in 1 mL chloroform methanol (1:3, v/v). After evaporation of solvent, the phospholipid film was further dried in a Labconco freeze-dryer for 24 h and stored at −20 °C until use. Multilamellar vesicles (MLV) were prepared from 1,2-dimyristoyl-sn-glycero-3-phosphoglycerol (DMPG) or 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) (Avanti Polar Lipids). Lipid films were rehydrated with 1 mL PBS and vortexed vigorously for 1 min. The vesicles were then extruded through 200 nm membranes at 45°C to form large unilamellar vesicles (LUV). DMPC LUV dispersions (250 μg) were solubilized by 75 μg protein. For DMPG, 250 μg MLVs were mixed with 50 μg protein. Vesicle solubilization was monitored at 325 nm in a spectrophotometer (Shimadzu UV-2401PC) at a constant temperature of 24°C.
2.4 Phosphatidylglycerol vesicle destabilization
Lipid films were prepared using 5 mg of egg phosphatidylglycerol (PG) (Avanti Polar Lipids) as described above. One mL of 100 mM calcein in Tris buffer (20 mM Tris-HCl, 150 mM NaCl, 0.5 mM EDTA) was added to the lipid film and vortexed vigorously. The vesicle suspension was extruded at 45°C using 100 nm membranes to encapsulate calcein in small unilamellar vesicles (SUV). Gel filtration chromatography (Sephadex G-75 resin, GE Healthcare) was used to separate vesicles from free calcein. Concentration of phospholipids was calculated according to the AMES phosphate assay (18). Calcein release of the vesicle suspension was measured in a fluorometer (LS 55B Perkin Elmer), with excitation and emission wavelengths set at 490 and 520 nm, respectively (19). Lipid to protein molar ratios of 1000:1, 100:1 and 25:1 were used for CT-apoA-I and 1000:1 and 100:1 for apoA-I. Protein was added after 2 min of equilibration, and at 10 min Triton X-100 (0.1%) was added to release the remaining calcein from the vesicles.
2.5 Lipopolysaccharide binding interaction
Binding interaction between the apolipoproteins and lipopolysaccharides (LPS) was measured using non-denaturing 4–20% Tris-Glycine PAGE, as described previously (20). Twenty μg of LPS (O55:B55, Sigma-Aldrich) was incubated with 0, 1, 3, or 8 μg of protein for 1 h at 37 °C and electrophoresed for 2 h. LPS was visualized with Pro-Q Emerald 300 LPS gel stain kit (Thermo Fisher Scientific). Alternatively, 40 μg LPS was incubated with 40 μg protein, analyzed by native-PAGE and proteins were visualized by naphthol blue-black.
2.6 Lipoprotein binding
Lipoprotein aggregation was measured using modified human low density lipoproteins (LDL) (21). Fifty μg of protein was mixed with an equal amount of LDL protein in a total volume of 200 μL reaction buffer (50 mM Tris-HCl, 150 mM NaCl, 2 mM CaCl2, pH 7.4) in a 96-well plate (Greiner Bio-One). The reaction was started by addition of 200 mU phospholipase-C (Sigma-Aldrich) to the LDL apolipoprotein mixture, and the absorbance at 340 nm was recorded at 5 min intervals in a Thermo Varioskan plate reader for 2 h at 37 °C.
3. Results
3.1 Recombinant expression of chimeric apoLp-III/apoA-I (179–243), cleavage, and isolation
ApoLp-III is a well-characterized invertebrate exchangeable apolipoprotein. Similar to human apolipoproteins, apoLp-III is made of amphipathic α-helices (22). The helix bundle is different from the common 4-helix bundle in that it contains a 5th helix (23). The protein can be successfully expressed in large quantities in E. coli, ensuring a large protein supply needed for structural and functional analyses (24). Recently, we produced a chimeric protein made of Locusta migratoria apoLp-III and residues 179–243 from human apoA-I; this protein construct formed the basis to isolate CT-apoA-I. Site-directed mutagenesis was employed to change the last residue of apoLp-III (N164) to methionine, allowing apoLp-III removal by cyanogen bromide. Overexpression of the chimeric protein, apoLp-IIIN164M/CT-apoA-I, yielded ~20 mg purified protein per L of expression medium (Fig. 1, lane 1). The chimera was then cleaved by cyanogen bromide, and SDS-PAGE analysis showed the two expected fragments of 17.5 kDa (apoLp-III) and 7 kDa (CT-apoA-I) (Fig. 1, lane 2). A small amount of uncleaved chimeric protein was also visible, and based on the intensity of the protein bands the reaction was estimated to be ~ 90% complete. To isolate CT-apoA-I from the reaction mixture, two different strategies were employed. The first method made use of the NT His-tag of the chimera, which is also present in apoLp-III after cyanogen bromide cleavage and absent in CT-apoA-I. The mixture was reloaded onto the Ni-column to bind both His-tagged proteins, while CT-apoA-I was washed from the column and collected (Fig. 1, lane 3). The His-tagged proteins were then eluted with imidazole elution buffer (Fig. 1, lane 4). Alternatively, reversed-phase HPLC was employed to isolate CT-apoA-I. The HPLC profile using a linear gradient of water and acetonitrile displayed two prominent peaks (Fig. 2). SDS-PAGE analysis showed that the sharp peak at 100 min was CT-apoA-I (Fig 2 inset, lane 1). The peaks between 110 and 115 min contained uncleaved chimera and apoLp-III, which showed significant overlap in the HPLC profile. Thus, both isolation methods achieved complete separation of CT-apoA-I from the larger, His-tagged proteins; the purification scheme is depicted in Fig. 3. Purified CT-apoA-I was then analyzed for secondary structure. The Far UV CD scan of CT-apoA-I showed that the protein was helical (32.6%), although the amount of secondary structure was less compared to apoA-I (51.9%) (Fig. 4). To ensure functionality of isolated CT-apoA-I, lipid binding analysis was performed by examining the ability to solubilize phospholipid vesicles and interact with PG, LPS, and lipolyzed LDL.
Fig 1.

SDS-PAGE of the isolation of CT-apoA-I. Lane 1: apoLp-III (N164M)/CT-apoA-I; lane 2: apoLp-III (N164M)/CT-apoA-I digested with cyanogen bromide; lane 3: flow through fraction of Ni-affinity chromatography containing CT-apoA-I; lane 4: elution of apoLp-III and undigested chimera from the Ni-affinity column.
Fig 2.
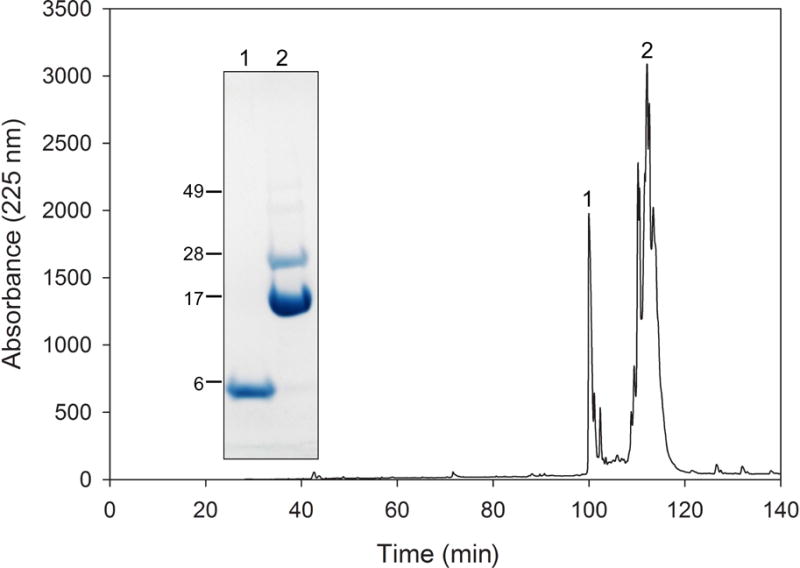
Isolation of CT-apoA-I by RP-HPLC. The apoLp-III (N164M)/CT-apoA-I cyanogen bromide digest was separated into the fragments showing two main peaks (labeled 1 and 2). Both peaks were collected and subjected to SDS-PAGE (inset), showing that CT-apoA-I eluted at 100 min, and that peak 2 (110–115 min) contained uncleaved chimera and apoLp-III.
Fig 3.

Strategy of the expression and purification of CT-apoA-I. The apoLp-III (N164M)/CT-apoA-I construct bearing an N-terminal poly-His tag was cloned into the p20b(+) expression vector, expressed in E. coli and purified by Ni-affinity chromatography. Treatment with cyanogen bromide cleaves the chimeric protein at position M178 producing two fragments. The digest was subsequently applied to Ni-affinity chromatography to isolate CT-apoA-I, which was present in the Ni-column wash fraction. CT-apoA-I can also be purified by reversed-phase HPLC.
Fig 4.

Secondary structure analysis. Far-UV spectra of CT-apoA-I (dashed line) and apoA-I (solid line) at a protein concentration of 0.2 mg/mL.
3.2 Phospholipid vesicle solubilization
The ability of CT-apoA-I to solubilize phospholipid vesicles was measured with LUVs made of pure DMPC, using 250 μg vesicles and 75 μg protein. ApoA-I transformed the vesicles into small discoidal particles, as seen by the decrease in sample turbidity measured by the absorbance at 325 nm (Fig. 5A). CT-apoA-I solubilized the vesicles at a significantly faster rate compared to apoA-I. Based on the decrease in sample turbidity, first order rate constants were 34.7 ± 0.4 × 10−3 s−1 for CT-apoA-I and 6.7 ± 0.1 × 10−3 s−1 for apoA-I. Similarly, CT-apoA-I was also better than full-length apoA-I in solubilizing DMPG vesicles (Fig. 5B). Using 250 μg vesicles and 50 μg protein, a rate constant of vesicle solubilization of 9.4 ± 0.2 × 10−3 s−1 was calculated for CT-apoA-I, and 2.7 ± 0.1 × 10−3 s−1 for apoA-I.
Fig 5.

Phospholipid vesicle solubilization. Panel A: DMPC vesicles (250 μg) were incubated with 75 μg of apoA-I (solid line), an equal amount of CT-apoA-I (dashed line), or in the absence of protein (dotted line). Absorbance at 325 nm was continuously recorded for the times indicated to monitor conversion of vesicles into small discoidal complexes. Panel B: solubilization of DMPG vesicles (250 μg phospholipid, 50 μg protein).
3.3 Binding to phosphatidylglycerol vesicles and lipopolysaccharides
ApoA-I interacts strongly with PG membranes and is able to associate with LPS (19). Both are abundant components of the membrane of gram-negative bacteria, and by measuring the binding activity, insight in potential antimicrobial properties of apoA-I can be obtained. To measure binding to PG membranes, calcein was encapsulated into SUVs made of egg PG, which contains a mixture of fatty acids, mainly palmitoleic, oleic, linolenic and arachidonic acid. The fluorescence intensity of calcein containing vesicles was initially monitored for 2 min, after which protein was added, continuously measuring the increase in fluorescence intensity caused by release of calcein from vesicles. After equilibrium was reached, detergent was added to liberate any remaining calcein trapped in the vesicles. Incubations were carried out at a 1000:1 and 100:1 molar ratio of lipid to protein, and are shown in Fig. 6. ApoA-I induced an almost complete release of calcein (95% at a 100:1 molar ratio) or 86.4% when using 10× less protein (1000:1). However, CT-apoA-I was significantly less effective, inducing 61.3 % calcein release (100:1) and 16.5 % (1000:1). However, when the amount CT-apoA-I was increased to obtain the same protein mass as was used for apoA-I (25:1 molar ratio for CT-apoA-I and a 100:1 molar ratio for apoA-I), the potency of CT-apoA-I improved, releasing 72.4% calcein from the vesicles.
Fig 6.

Binding interaction of apoA-I with zwitterionic PG. Vesicles made of egg PG containing calcein were incubated with full-length apoA-I (FL) or CT-apoA-I (CT) using a molar ratio of lipid to protein of 25:1, 100:1, and 1000:1. The solid line represents PG vesicles in the absence of protein showing no release of measurable amounts of calcein. Release of calcein was measured by the increase in fluorescence intensity upon addition of apolipoprotein at the 2 min time point. At the 10 min time point, Triton X-100 was added to determine the maximum fluorescence intensity.
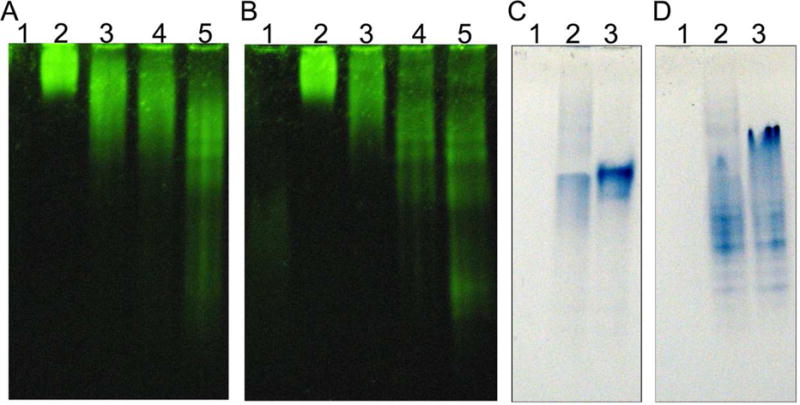
ApoA-I has been shown to disaggregate LPS micelles, which can be measured by incubating a suspension of LPS micelles with protein. LPS disaggregation can then be observed by PAGE in the absence of SDS. As shown in the native PAGE analysis of Fig. 7, LPS displayed limited electrophoretic mobility and remained at the top of the gel. However, upon addition of small quantities of CT-apoA-I (Fig. 7A) or apoA-I (Fig. 7B), the LPS micelles were broken apart into smaller particles with increased electrophoretic mobility. In addition, equal amounts of apoA-I and LPS were mixed and electrophoresed followed by protein staining of the non-denaturing gel. This showed marked shifts in the electrophoretic mobilities of CT-apoA-I (Fig. 7C) and apoA-I (Fig. 7D), indicating association of both proteins with LPS.
Fig 7.

PAGE analysis of LPS binding. Panel A (CT-apoA-I) and B (apoA-I) show LPS disaggregation. LPS (20 μg) was incubated with 0, 1, 3, or 8 μg protein (lanes 2–5). Lane 1 contains 8 μg of protein in the absence of LPS. Panel C (CT-apoA-I) and D (apoA-I) show the LPS-induced change on the electrophoretic mobility of the protein. Lane 1: 40 μg LPS, lane 2: 40 μg LPS and 40 μg protein, lane 3: 40 μg of protein.
3.4 Prevention of phospholipase-C induced aggregation of lipoproteins
To assess binding of CT-apoA-I to the lipoprotein surface, LDL was incubated with phospholipase-C, which results in the appearance of diacylglycerol on the LDL surface and causes the lipoprotein to aggregate (21). The increase in LDL diacylglycerol creates binding sites for apolipoproteins, which upon association protect LDL from aggregation. As is shown in Fig. 8, the absorbance of LDL incubated with phospholipase-C increased after 5 min, indicating formation of LDL aggregates. However, when the LDL phospholipase-C mixture was incubated simultaneously with apoA-I, the sample turbidity remained at background levels. Thus apoA-I provided full protection and no LDL aggregates were formed. CT-apoA-I was much less effective; a similar amount of protein delayed the onset of aggregation until approximately 25 min, after which the turbidity of the sample rose to values indistinguishable from phospholipase-C-treated LDL.
Fig 8.
Protection of LDL against phospholipase-induced aggregation. LDL incubated in the presence of phospholipase C rapidly aggregated as seen by the increase in sample turbidity (closed circles). ApoA-I was able to provide full protection against lipase-induced LDL aggregation as the absorbance remained at background levels (open circles), while CT-apoA-I (closed triangles) delayed the onset of aggregation by approximately 15 min after which the sample turbidity steadily increased indicating formation of LDL aggregates.
4. Discussion
The C-terminal domain of apoA-I is a critical part of the protein, and study of its properties may considerably improve our understanding of apoA-I function. The expression method using the chimeric protein apoLp-IIIN164M/CT-apoA-I produced mg quantities of functional CT-apoA-I. Insect apoLp-III proved to be an excellent carrier for CT-apoA-I, and 20 mg purified protein was routinely produced per L culture medium. A unique methionine was inserted between apoLp-III and CT-apoA-I to allow removal of the carrier protein. Efficiency of cyanogen bromide cleavage was approximately 90%, and the released CT-apoA-I could be isolated by either HPLC or a second passage on the Ni-affinity column employing the absence of a His-tag in CT-apoA-I. Both isolation procedures produced a pure protein preparation, but the affinity column procedure has the advantage of simplicity and does not require an expensive HPLC set-up. Starting with a 1 L bacterial expression culture, approximately 2 mg of pure CT-apoA-I was isolated. The availability of CT-apoA-I may help understand apoA-I domain organization and the specific functions of each domain.
The isolated apoA-I domain displayed significant helical character, with an α-helical content of ~ 33% at 0.2 mg/mL, similar to reported values of a slightly shorter CT fragment of apoA-I (8). This peptide, comprised of residues 198–243, was monomeric at a low protein concentration with a helical content of 16%. Higher protein concentrations resulted in self-association and increase in helical content to about 50% (8), similar to the CT domain used in the current study with amino acid residues 179–243. CT-apoA-I was effective in solubilizing DMPC and DMPG vesicles, being more potent then apoA-I itself, confirming that CT-apoA-I is essential in initiating vesicle binding and transformation into discoidal particles (25–27). However, the CT-apoA-I was less effective, compared to the full-length protein, in releasing calcein from PG vesicles. This suggests that the NT domain of apoA-I is required for a full response in destabilizing PG vesicles. Similarly, when the chimeric protein was designed, and the effect of CT-apoA-I addition to apoLp-III was measured, the activity of the apoLp-III chimera was much improved but remained less potent compared to apoA-I (17). Previous studies showed that when residues 190–243 were removed from apoA-I, this resulted in a decreased ability to release calcein from PG vesicles (19). Since we observed a similar result for CT-apoA-I, this indicates that both apoA-I domains need to be present for an optimal response when added to the negatively charged phospholipid vesicles.
CT-apoA-I was not capable in preventing aggregation of phospholipase-C-treated LDL. The delay in the onset of aggregation was shifted from 10 min in the absence of apolipoprotein to 20 min in the presence of CT-apoA-I, but then LDL rapidly started to aggregate. In contrast, full-length apoA-I was able to provide complete protection against aggregation. This shows that the CT-domain of apoA-I alone is insufficient to prevent aggregation, and the presence of the NT domain is required for complete protection, similar as observed for PG membrane destabilization. This result also resembles previous analysis of truncation variants of apoLp-III which were less effective in protecting lipolyzed LDL from aggregation, but were significantly better in solubilizing DMPC vesicles (28). Similar to CT-apoA-I, these truncation variants displayed reduced helical content, which may have promoted vesicle solubilization through increase in α-helical content upon lipid association. CT-apoA-I was effective in disaggregating LPS micelles, which supports a previous study in which apoA-I fragments generated by cyanogen bromide were analyzed for LPS binding, identifying a CT fragment spanning residues 149–243 with the highest LPS binding activity (29).
Recombinant apoLp-III has been an excellent protein to express, yielding large amounts of recombinant protein, either using a vector in which the protein is released into the media assisted by a pelB leader sequence, or by use of a His-tag thioredoxin fusion protein to facilitate purification from cell extracts (24, 30–32). The simple protein fold of apoLp-III, an up-and-down bundle of five amphipathic helices, is fully reversible (33–35). This helix bundle arrangement may have contributed to the high yield of the apoLp-III/CT-apoA-I chimeric protein. The apoLp-III helix bundle may also be used to express other peptides that are otherwise difficult to express in bacteria. In earlier studies, methionine has been used successfully to produce an apoLp-III deletion variant and a helix fragment by cyanogen bromide digestion. This was done by employing the single methionine already present in Manduca sexta apoLp-III (36) or by introduction of a methionine in the L. migratoria helix bundle (23). Nevertheless, alternative strategies to remove the carrier protein, such as inclusion of a target sequence for enzyme-mediated proteolysis, can be easily implemented as custom made nucleotide synthesis, which include codon optimization, has become relatively inexpensive. For example, a TEV or thrombin cleavage site can be introduced in between apoLp-III and the target peptide to facilitate isolation of the peptide of interest (37).
Highlights.
A novel method to produce the CT domain of apoA-I is presented
ApoLp-III was used a carrier protein, and was removed by cyanogen bromide cleavage
CT-apoA-I was helical and functional
CT-apoA-I was effective in solubilizing vesicles and LPS disaggregation
CT-apoA-I binding to phosphatidylglycerol and low density lipoprotein was impaired
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number T34GM008074 and GM089564. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We are very grateful to Dr. Jason Schwans, Department of Chemistry and Biochemistry CSU Long Beach, for his assistance with the HPLC analysis.
Abbreviations
- apoA-I
apolipoprotein A-I
- apoLp-III
apolipophorin III
- CT
C-terminal
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphocholine
- DMPG
1,2-dimyristoyl-sn-glycero-3-phosphoglycerol
- HDL
high density lipoprotein
- LPS
lipopolysaccharides
- LDL
low density lipoprotein
- LUV
large unilamellar vesicles
- MLV
multilamellar vesicles
- NT
N-terminal
- PAGE
polyacrylamide gel electrophoresis
- PBS
phosphate buffered saline
- PG
phosphatidylglycerol
- SDS
sodium dodecyl sulfate
- SUV
small unilamellar vesicles
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Saito H, Lund-Katz S, Phillips MC. Contributions of domain structure and lipid interaction to the functionality of exchangeable human apolipoproteins. Prog Lipid Res. 2004;43:350–380. doi: 10.1016/j.plipres.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Mei X, Atkinson D. Lipid-free Apolipoprotein A-I Structure: Insights into HDL Formation and Atherosclerosis Development. Arch Med Res. 2015;46:351–360. doi: 10.1016/j.arcmed.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phillips MC. Molecular mechanisms of cellular cholesterol efflux. J Biol Chem. 2014;289:24020–24209. doi: 10.1074/jbc.R114.583658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang R R, Silva RA, Jerome WG, Kontush A, Chapman MJ, Curtiss LK, Hodges TJ, Davidson WS. Apolipoprotein A-I structural organization in high-density lipoproteins isolated from human plasma. Nat Struct Mol Biol. 2011;18:416–422. doi: 10.1038/nsmb.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sorci-Thomas MG, Thomas MJ. High density lipoprotein biogenesis, cholesterol efflux, and immune cell function. Arterioscler Thromb Vasc Biol. 2012;32:2561–2565. doi: 10.1161/ATVBAHA.112.300135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davidson WS, Hazlett T, Mantulin WW, Jonas A. The role of apolipoprotein AI domains in lipid binding. Proc Natl Acad Sci U S A. 1996;93:13605–13610. doi: 10.1073/pnas.93.24.13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chetty PS, Mayne L, Lund-Katz S, Stranz D, Englander SW, Phillips MC. Helical structure and stability in human apolipoprotein A-I by hydrogen exchange and mass spectrometry. Proc Natl Acad Sci USA. 2009;106:19005–19010. doi: 10.1073/pnas.0909708106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu HL, Atkinson D. Conformation and lipid binding of a C-terminal (198–243) peptide of human apolipoprotein A-I. Biochemistry. 2007;46:1624–1634. doi: 10.1021/bi061721z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burgess JW, Frank PG, Franklin V, Liang P, McManus DC, Desforges M, Rassart E, Marcel YL. Deletion of the C-terminal domain of apolipoprotein A-I impairs cell surface binding and lipid efflux in macrophage. Biochemistry. 1999;38:14524–14533. doi: 10.1021/bi990930z. [DOI] [PubMed] [Google Scholar]
- 10.Favari E, Bernini F, Tarugi P, Guido F, Calabersi L. The C-terminal domain of apolipoprotein A-I is involved in ABCA1-driven phospholipid and cholesterol efflux. Biochem Biophys Res Commun. 2002;299:801–805. doi: 10.1016/s0006-291x(02)02745-6. [DOI] [PubMed] [Google Scholar]
- 11.Schmidt HH, Remaley AT, Stonik JA, Ronan R, Wellmann A, Thomas F, Zech LA, Brewer HB Jr, Hoeg JM. Carboxyl-terminal domain truncation alters apolipoprotein A-I in vivo catabolism. J Biol Chem. 1995;270:5469–5475. doi: 10.1074/jbc.270.10.5469. [DOI] [PubMed] [Google Scholar]
- 12.Oda MN, Forte TM, Ryan RO, Voss JC. The C-terminal domain of apolipoprotein A-I contains a lipid-sensitive conformational trigger. Nat Struct Biol. 2003;10:455–460. doi: 10.1038/nsb931. [DOI] [PubMed] [Google Scholar]
- 13.Bergeron J, Frank PG, Emmanuel F, Latta M, Zhao Y, Sparks DL, Rassart E, Denèfle P, Marcel YL. Characterization of human apolipoprotein A-I expressed in. Escherichia coli Biochim Biophys Acta. 1997;1344:139–152. doi: 10.1016/s0005-2760(96)00136-1. [DOI] [PubMed] [Google Scholar]
- 14.Rogers DP, Roberts LM, Lebowitz J, Engler JA, Brouillette CG. Structural analysis of apolipoprotein A-I: effects of amino- and carboxy-terminal deletions on the lipid-free structure. Biochemistry. 1998;37:945–955. doi: 10.1021/bi9713512. [DOI] [PubMed] [Google Scholar]
- 15.Panagotopulos SE, Witting SR, Horace EM, Maiorano JN, Davidson SW. Bacterial expression and characterization of mature apolipoprotein A-I. Protein Expr Purif. 2002;25:353–361. doi: 10.1016/s1046-5928(02)00020-7. [DOI] [PubMed] [Google Scholar]
- 16.Ryan RO, Forte TM, Oda MN. Optimized bacterial expression of human apolipoprotein A-I. Protein Expr Purif. 2003;27:98–103. doi: 10.1016/s1046-5928(02)00568-5. [DOI] [PubMed] [Google Scholar]
- 17.Horn JVC, Ellena RA, Tran JJ, Beck WHJ, Narayanaswami V, Weers PMM. Transfer of C-terminal residues of human apolipoprotein A-I to insect apolipophorin III creates a two-domain chimeric protein with enhanced lipid binding activity. Biochim Biophys Acta. 2017 doi: 10.1016/j.bbamem.2017.04.017. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ames BN. Assay of inorganic phosphate, total phosphate and phosphatases. Methods Enzymol. 1966;8:115–118. [Google Scholar]
- 19.Beck WHJ, Adams CP, Biglang-Awa IM, Patel AB, Vincent H, Haas-Stapleton EJ, Weers PMM. Apolipoprotein A-I binding to anionic vesicles and lipopolysaccharides: role for lysine residues in antimicrobial properties. Biochim Biophys Acta. 2013;1828:1503–1510. doi: 10.1016/j.bbamem.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oztug M, Martinon D, Weers PMM. Characterization of the apoLp-III/LPS complex: insight into the mode of binding interaction. Biochemistry. 2012;51:6220–6227. doi: 10.1021/bi300619a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu H, Scraba DG, Ryan RO. Prevention of phospholipase-C induced aggregation of low density lipoprotein by amphipathic apolipoproteins. FEBS Lett. 1993;316:27–33. doi: 10.1016/0014-5793(93)81730-n. [DOI] [PubMed] [Google Scholar]
- 22.Narayanaswami V, Kiss RS, Weers PMM. The helix bundle: a reversible lipid binding motif. Comp Biochem Physiol A Mol Integr Physiol. 2010;155:123–133. doi: 10.1016/j.cbpa.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dwivedi P, Rodriguez J, Ibe NU, Weers PMM. Deletion of the N- or C-terminal helix of apolipophorin III to create a four-helix bundle protein. Biochemistry. 2016;55:3607–3615. doi: 10.1021/acs.biochem.6b00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weers PMM, Wang J, Van der Horst DJ, Kay CM, Sykes BD, Ryan RO. Recombinant locust apolipophorin III: characterization and NMR spectroscopy. Biochim Biophys Acta. 1998;1393:99–107. doi: 10.1016/s0005-2760(98)00063-0. [DOI] [PubMed] [Google Scholar]
- 25.Laccotripe M, Makrides SC, Jonas A, Zannis VI. The carboxyl-terminal hydrophobic residues of apolipoprotein A-I affects its rate of phospholipid binding and its association with high density lipoprotein. J Biol Chem. 1997;272:17511–17522. doi: 10.1074/jbc.272.28.17511. [DOI] [PubMed] [Google Scholar]
- 26.Lyssenko NN, Hata M, Dhanasekaran P, Nickel M, Nguyen D, Chetty PS, Saito H, Lund-Katz S, Phillips MC. Influence of C-terminal α-helix hydrophobicity and aromatic amino acid content on apolipoprotein A-I functionality. Biochim Biophys Acta. 2012;1821:456–463. doi: 10.1016/j.bbalip.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagao K, Hata M, Tanaka K, Takechi Y, Nguyen D, Dhanasekaran P, Lund-Katz S, Phillips MC, Saito H. The roles of C-terminal helices of human apolipoprotein A-I in formation of high-density lipoprotein particles. Biochim Biophys Acta. 2014;1841:80–87. doi: 10.1016/j.bbalip.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dettloff M, Niere M, Ryan RO, Luty R, Kay CM, Wiesner A, Weers PMM. Differential lipid binding of truncation mutants of Galleria mellonella apolipophorin III. Biochemistry. 2002;41:9688–9695. doi: 10.1021/bi0200108. [DOI] [PubMed] [Google Scholar]
- 29.Henning MF, Herlax V, Bakas L. Contribution of the C-terminal end of apolipoprotein AI to neutralization of lipopolysaccharide endotoxic effect. Innate Immun. 2011;17:327–337. doi: 10.1177/1753425910370709. [DOI] [PubMed] [Google Scholar]
- 30.Ryan RO, Schieve D, Wientzek M, Narayanaswami V, Oikawa K, Kay CM, Agellon LB. Bacterial expression and site-directed mutagenesis of a functional recombinant apolipoprotein. J Lipid Res. 1995;36:1066–1072. [PubMed] [Google Scholar]
- 31.Soulages JL, Pennington J, Bendavid O, Wells MA. Role of glycosylation in the lipid-binding activity of the exchangeable apolipoprotein, apolipophorin-III. Biochem Biophys Res Commun. 1998;243:372–376. doi: 10.1006/bbrc.1998.8099. [DOI] [PubMed] [Google Scholar]
- 32.Niere M, Meisslitzer C, Dettloff M, Weise C, Ziegler M, Wiesner A. Insect immune activation by recombinant Galleria mellonella apolipophorin III. Biochim Biophys Acta. 1999;1433:16–26. doi: 10.1016/s0167-4838(99)00148-x. [DOI] [PubMed] [Google Scholar]
- 33.Breiter DR, Kanost MR, Benning MM, Wesenberg G, Law JH, Wells MA, Rayment I, Holden HM. Molecular structure of an apolipoprotein determined at 2.5-Å resolution. Biochemistry. 1991;30:603–608. doi: 10.1021/bi00217a002. [DOI] [PubMed] [Google Scholar]
- 34.Wang J, Sykes BD, Ryan RO. Structural basis for the conformational adaptability of apolipophorin III, a helix-bundle exchangeable apolipoprotein. Proc Natl Acad Sci USA. 2002;99:1188–1193. doi: 10.1073/pnas.032565999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weers PMM, Kay CM, Oikawa K, Wientzek M, Van der Horst DJ, Ryan RO. Factors affecting the stability and conformation of Locusta migratoria apolipophorin III. Biochemistry. 1994;33:3617–3624. doi: 10.1021/bi00178a019. [DOI] [PubMed] [Google Scholar]
- 36.Narayanaswami V, Kay CM, Oikawa K, Ryan RO. Structural and binding characteristics of the carboxyl terminal fragment of apolipophorin III from Manduca sexta. Biochemistry. 1994;33:13312–13320. doi: 10.1021/bi00249a018. [DOI] [PubMed] [Google Scholar]
- 37.Waugh DS. An overview of enzymatic reagents for the removal of affinity tags. Protein Expr Purif. 2011;80:283–293. doi: 10.1016/j.pep.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]