

Abstract
Oxidative [1,2]-Brook rearrangements via hypervalent silicon intermediates induced by photoredox-catalyzed single-electron transfer have been achieved, permitting the formation of reactive radical species that can engage in alkylations and arylations.
Brook rearrangements comprise the transfer of a silyl group from a carbon to an oxygen atom with concomitant migration of charge resulting in a carbanion from the initiating alkoxide species.1 This rearrangement, initially introduced by Brook et al.,2 has been well studied1b,3 and is now known to proceed reversibly via a hypervalent silicon (Scheme 1a).4
Scheme 1.
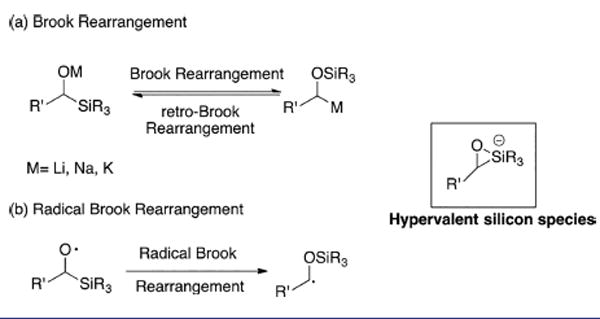
Brook and Radical Brook Rearrangement
Brook rearrangements have been employed extensively in our and other Laboratories in conjunction with a multicomponent coupling tactic we now term Anion Relay Chemistry (ARC).5 Although negative charge interchange via migration of a silyl group is now well-known, Brook rearrangements via single-electron transfer (SET) have not been extensively explored (Scheme 1b). Moreover, such rearrangements have not been employed synthetically,6 presumably due to the difficulty associated with the formation of an alkoxy radical. Organosilicon compounds, especially alkylsilanes, are generally known to be quite resistant to oxidants given their high oxidation potential (for example, Eox = +1.68 V vs SCE7 for I in Scheme 2). Recently however a mild catalytic photoredox system has been introduced to employ hypervalent organosilicates as alkyl radical precurors.8 In these reports, pentacoordinate silicates9 are prepared and demonstrated to have relatively low oxidation potentials (for example, Eox = +0.61 V8b vs SCE for II in Scheme 2). As illustrated in Scheme 1a, the Brook rearrangement builds a direct linkage between alkylsilanes and hypervalent silicates. We therefore hypothesized that it might be possible to access a pentacoordinate alkylsilicate in situ via a Brook rearrangement, followed by achieving the oxidative cleavage of a strong C–Si bond via single-electron transfer employing photoredox catalysts.10 This chemistry might provide an alternative to the direct generation of an α-hydroxy radical. To the best of our knowledge, such a reaction sequence is unprecedented.
Scheme 2.
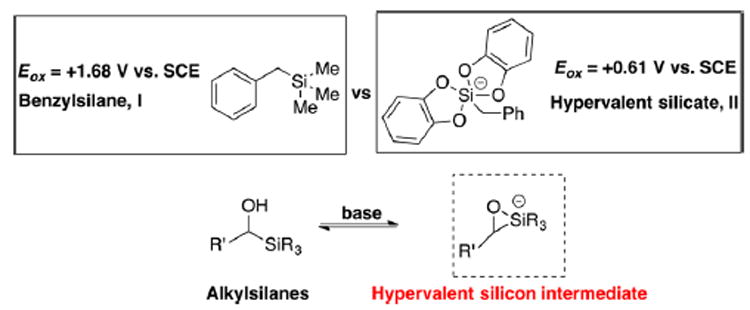
Comparison of Hypervalent Silicon Species
To achieve an oxidative [1,2]-Brook rearrangement, two criteria would be required: (i) the reduction potential of the excited-state photoredox catalysts must be sufficiently high to cleave a strong Si–C bond; and (ii) selective cleavage of the C–Si bond to form the alkyl radical must be achieved when the silicon is tetraalkyl-substituted (i.e., TMS, TBS, or TIPS). Thus, we report here the first visible-light-induced SET tactic to achieve an oxidative [1,2]-Brook rearrangement. Both photoredox-catalyzed alkylations and arylations have been achieved.
Based on the above hypothesis, we propose the reaction sequence in Scheme 3. First, deprotonation of an aryl-(trialkylsilyl)methanol to set up an equilibrium between the alcohol and the hypervalent silicon (i.e., A) in analogy to the well-known anionic Brook rearrangement. Intermediate A could then serve either as a radical precursor of the α-benzyl silyl ether radical (i.e., B), capable of alkylation with an electron-deficient olefin (catalytic cycle in Scheme 3a), or arylation with an electron-deficient aromatic compound (catalytic cycle in Scheme 3b).11 For alkylation (Scheme 3a), oxidation of intermediate A by the excited state of an Ir photocatalyst must occur. The resulting open-shell intermediate B could then engage in a conjugate addition with an electron-deficient olefin to form an α-carbonyl radical C. Electron transfer from the reduced photocatalyst would then generate an enolate anion, which in turn would undergo protonation to provide the alkylation product D, with the photocatalyst returning to the ground state. For arylation (Scheme 3b), a photoactivated Ir photocatalyst would first undergo SET with an electron-deficient arene to furnish the corresponding arene radical anion E, with generation of the Ir(IV) photocatalyst. We hypothesize that the Ir(IV) species would then be able to oxidize the hypervalent silicon intermediate A to form radical B. A bond-forming step could then occur via radical union between intermediates B and E, followed by elimination of cyanide to yield the arylated product (i.e., F).
Scheme 3.
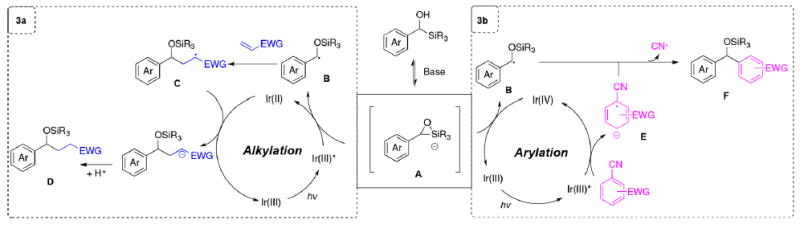
Proposed Reaction Route
To begin, we selected (tert-butyldimethylsilyl)phenylmethanol (1b) as a model substrate and methyl vinyl ketone (2a) as the radical acceptor. After extensive screening, we found that employing KOPiv as the base in dichloroethane (DCE) led to alkylation product 3b in 38% yield employing 3 mol % of Ir[dF(CF3)ppy]2(dtbpy)PF6 as the photocatalyst (entry 1 in Table 1). Other photocatalysts led to poor results (entries 2–3).
Table 1.
Optimization of the Alkylation Reaction a

| |||||
|---|---|---|---|---|---|
| entry | photocatalyst | equiv. of 2a | base | solvent | yieldb |
| 1 | Ir[dF(CF3)ppy]2(dtbpy)PF6 | 2 | KOPiv | DCE | 38% |
| 2 | Ru(bpy)3PF6 | 2 | KOPiv | DCE | 19% |
| 3 | Ru(bpz)3PF6 | 2 | KOPiv | DCE | trace |
| 4 | Ir[dF(CF3)ppy]2(dtbpy)PF6 | 2 | KOPiv | Acetone | trace |
| 5 | Ir[dF(CF3)ppy]2(dtbpy)PF6 | 2 | KOPiv | CH3CN | < 10% |
| 6 | Ir[dF(CF3)ppy]2(dtbpy)PF6 | 2 | NaOH | DCE | trace |
| 7 | Ir[dF(CF3)ppy]2(dtbpy)PF6 | 2 | K2CO3 | DCE | 31% |
| 8 | Ir[dF(CF3)ppy]2(dtbpy)PF6 | 2 | CsOAc | DCE | 36% |
| 9 | Ir[dF(CF3)ppy]2(dtbpy)PF6 | 2 | KOPiv+CsOAcc | DCE | 83% (78%) |
| 10d | Ir[dF(CF3)ppy]2(dtbpy)PF6 | 1 | KOPiv+CsOAcc | DCE | 77% (72%) |
Reactions were run with 1b (0.1 mmol, 1 equiv), 2a, photocatalyst (3 mol %), base (1.0 equiv), in 1 mL of solvent.
1H NMR yields. Isolated yield is given in parentheses.
1.0 equiv of KOPiv and 1.0 equiv of CsOAc.
0.5 mmol scale of 1b in 0.2 mL of DCE for 4 days.
The specific choice of base and solvent significantly influenced the yield. For example, polar solvents such as acetone and CH3CN led to no product (entries 4–5). Stronger bases such as NaOH (entry 6) led to the formation of (benzyloxy)trimethylsilane, which is the byproduct of a base-mediated Brook rearrangement. A weaker base such as K2CO3 (entry 7) led to low conversion. The best combination of bases proved to be 1:1 equiv of KOPiv and CsOAc and 1:2 equiv of 1b and 2a; the isolated yield was then 78% (entry 9). To our delight, the loading of radical acceptor 2a could be further decreased to 1 equiv with a 72% isolated yield when the reaction was run in high concentration (2.5 M) on 0.5 mmol scale (entry 10).
We next evaluated the scope of the alkylation protocol (Scheme 4). Initially, aryl(trialkylsilyl)-methanols were explored (1a–1e). Groups such as TMS, TES, TBS, and TIPS delivered over 70% of 3a–3e, indicating that steric hindrance on the silicon atom does not influence the reaction. Generally, TES and TBS substrates proceeded in higher yield than the corresponding TMS congeners. This result is attributed to the higher stability of the TES- and TBS-carbon bonds. However, changing the silyl group to the dimethylphenylsilyl (DMPS) witnessed a modest decrease in yield (3d, 56% yield). The electron-withdrawing effect of the phenyl group on the Si atom appears to play a negative role in this photoredox reaction. Next, different functional groups on the aromatic ring attached to the carbinol were explored. Electron-donating groups were well tolerated leading in high yield to products 3f–3k. Halogenated substrates were also well tolerated leading to 3l–3n. However, in contrast with fluoro, the stronger electron-withdrawing group CF3 in 3o led to diminished reactivity. Sterically hindered systems such as ortho-methyl substituted aryl(trimethylsilyl)methanol also reacted well to provide a 67% yield of 3j. Equally pleasingly, substrates possessing heterocyclic substituents proved to be viable substrates leading to 3p–3q in moderate yields. We reason that the decrease in yield of 3q is mainly due to facile decomposition of the corresponding substrate 1q. Finally, the alkylation could be scaled up for gram-scale synthesis employing a high concentration (1.0 M) of 1f using 2 mol % of Ir photocatalyst (Scheme 4b).
Scheme 4. Arylsilylmethanol Substrate Scopea.
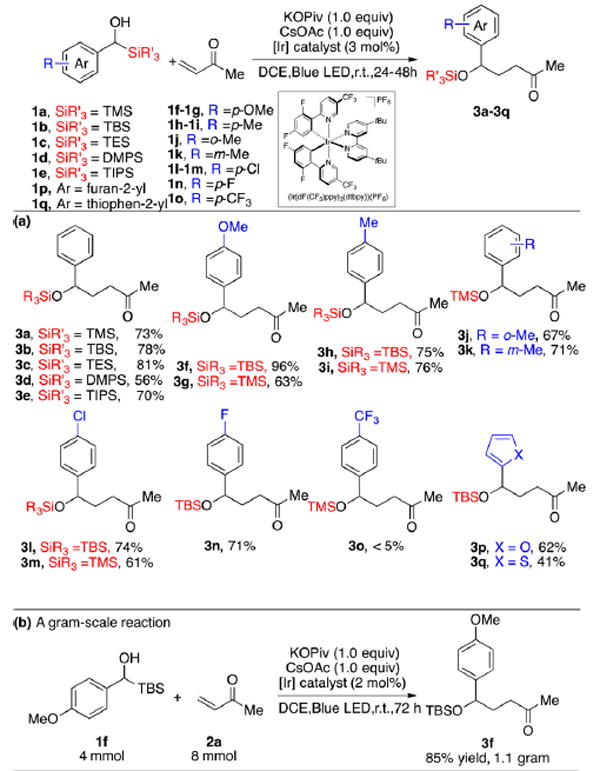
aReactions were run with 1a–q (1.0 equiv), methylvinylketone (2.0 equiv), photocatalyst (3 mol %), KOPiv (1.0 equiv), CsOAc (1.0 equiv) in 2 mL of DCE on 0.2 mmol scale.
We next explored diverse radical acceptors testing both alkyl and aryl enones (Scheme 5, 2b–2k). The presence of bulky alkyl groups did not influence the reactivity; good to high yields were obtained (4b–4c). An alkyl enone with a remote OTBS group (2d) was also well tolerated, thus providing a scaffold amenable to synthetic applications. Aryl enones normally gave better results than alkyl enones. Different functional groups on the aromatic ring were also explored (2e–2i). Neither electronic nor steric effects reduced the reactivity. However, a 1- and 2-substituted enone (2k and 2j) revealed that steric and electronic effects on the enone can strongly influence the yield. For example, with a methyl group as in 2j, the yield was significantly reduced, but with an electron-withdrawing phenyl group as in 2k, an excellent yield was obtained. However, simple acrylates provided only a trace amount of the alkylation product. We reasoned that this outcome is likely due to the difficulty associated with reducing α-carbonyl radicals in simple acrylates.12 However, when a phenyl group was introduced at the α-position, alkylation product 4l was produced in 86% yield.
Scheme 5. Radical Acceptor Scope a.
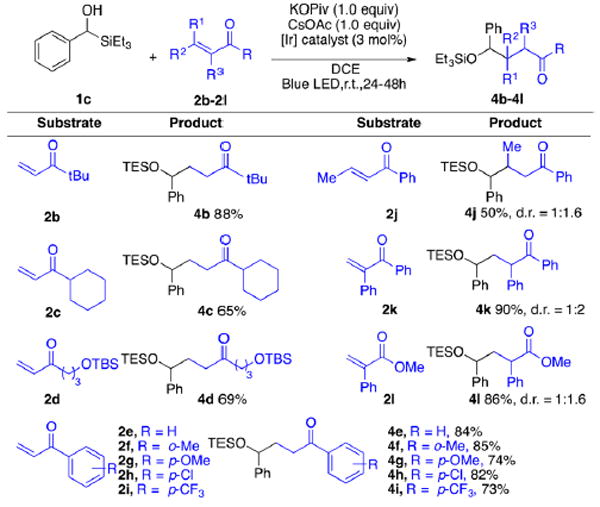
aReactions were run with 1c (1.0 equiv), 2b–2l (2.0 equiv), KOPiv (1.0 equiv), CsOAc (1.0 equiv), photocatalyst (3 mol %) in 2 mL of DCE on 0.2 mmol scale.
With the alkylation protocol established, we next explored whether a SET pathway could be employed to achieve arylation (catalytic cycle in Scheme 3b). We reasoned that radical B could enter the catalytic cycle of a photoredox-catalyzed radical–radical union protocol.11 Thus, a formal arylation would be achieved, which in turn would increase the synthetic utility of this chemistry.
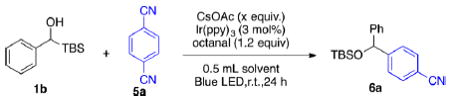
To this end, we selected (tert-butyldimethylsilyl)phenyl-methanol (1b) as a model substrate and terephthalonitrile (5a) as the radical acceptor. Octanal was used as an additive to sequester the cyanide anion formed in the reaction process.11 Initially, dichloroethane was employed as solvent with 1 equiv of CsOAc as the base. We were pleased to find that a small amount of desired product could be detected (entry 1, Table 2). Again, solvent selection had a significant effect on reaction performance. Polar solvents such as DMF and DMA led to the best results (entries 2–3). Based on the persistent radical effect, transient radical precursor 1b self-terminates to cause a buildup of excess persistent radicals, thus accelerating the cross-reaction.13 Therefore, increasing the loading of the transient radical precursor 1b provided an effective way to enhance conversion (entry 4).11,14 When we adjusted the amount of the base, we were surprised to find that a substoichiometric amount of base furnished a similar yield (entry 5). Finally, 83% isolated yield of 6a was achieved with 3 equiv of 1b (entry 6).
Table 2.
Optimization of thes Arylation Reactiona
| ||||
|---|---|---|---|---|
| entry | equivalent of 1b | solvent | equiv. of CsOAc | yieldb |
| 1 | 1 | DCE | 1 | trace |
| 2 | 1 | DMF | 1 | 58% |
| 3 | 1 | DMA | 1 | 61% |
| 4 | 2 | DMA | 1 | 68% |
| 5 | 2 | DMA | 0.5 | 67% |
| 6 | 3 | DMA | 1 | 87%(83%) |
Reactions were run with 1b, terephthalonitrile (0.1 mmol, 1 equiv), photocatalyst (3 mol %), base in 0.5 mL of solvent.
Yields were determined by 1H NMR analysis, and isolated yield is given in parentheses.
With these conditions in hand, we examined the structural diversity of both coupling partners. As shown in Scheme 6, both electron-donating and -withdrawing groups were well tolerated on the transient radical precursor leading to high yields (6a–6e, 66–85% yield). Furan and thiophene aromatic systems also proceeded in moderate to high yield (6f–6g). Changing the TBS group to a TES group also led to similar reactions, although a lower yield was observed for 6h due to the desilylation. Turning to arene coupling partners, substituted terephthalonitriles, and cyanopyridines readily react with the benzylic radicals to yield similar products (7a–7c). Also of interest, benzonitriles substituted with sulfones and esters undergo this reaction, although lower yields were observed (7d–7e).
Scheme 6. Substrate Scope for the Arylation Reaction a.
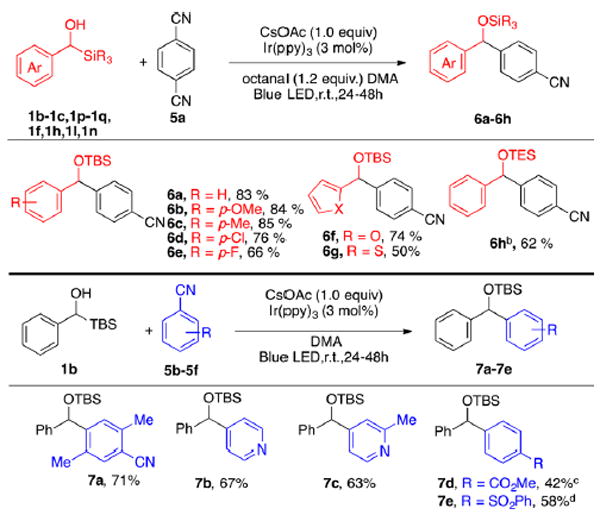
aReactions were run with arylsilylmethanol (3.0 equiv), aryl cyanide (1.0 equiv), Ir(ppy)3 (3 mol %), CsOAc (1.0 equiv), octanal (1.2 equiv) in 1 mL of DMA on 0.2 mmol scale. b2 equiv of 1c. cCsOAc (1.0 equiv) and KOPiv (1.0 equiv). dCsOAc (0.5 equiv)
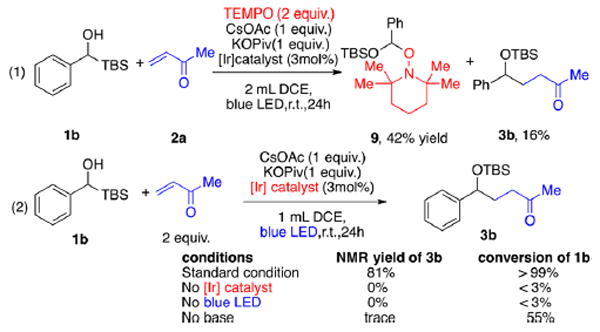
To gain mechanistic insights of the photochemical/oxidation Brook rearrangement, we conducted several control experiments. We first carried out a TEMPO radical quenching experiment with the (tert-butyldimethylsilyl)phenylmethanol as the substrate; a 42% yield of the TEMPO adduct 9 was isolated, with the yield of 3b decreasing significantly to 16% (Scheme 7, eq 1). Also, control experiments revealed the requirement for base, light, and photocatalyst (eq 2). Without light or a photocatalyst, almost all starting material remained. Also, if a base is not added to the reaction, only a trace amount of 3b could be detected with the conversion of 1b at 55% due to over-oxidation to form benzaldehyde.
Scheme 7.
Control Experiments
Notwithstanding the above results, consistent with the proposed photoredox-catalyzed mechanism in Scheme 3, there is the possibility of proton-coupled electron transfer (PCET)15 activation of the initial stronger O–H bond16 of the silyl alcohol, followed by a radical Brook rearrangement to generate the benzylic radical. Kinetic studies however revealed that although the reaction rate decreases as the basicity of the anion decreases, employing a higher oxidizing photocatalyst does not compensate for the lower rate of the weaker base. Moreover, cyclic voltammetry revealed no oxidation potential for 1b within the tested range (0–2.00 V vs SCE in MeCN). Yet, in the presence of CsOAc, an oxidation potential was observed ( vs SCE in MeCN), indicating the necessity of the base. Finally, when employing weaker bases such as CsOBz, the oxidation potential remained the same (see Supporting Information). These results indicate the PCET model is less likely.
In summary, we have designed and validated an oxidative [1,2]-Brook rearrangement involving visible-light-induced SET exploiting the oxidation of a hypervalent silicon species. The resulting alkyl radical was found to engage both in conjugate additions to achieve formal alkylation and in radical coupling reactions to achieve arylation. Studies to extend Brook rearrangements involving visible-light-induced SET continue in our laboratory.
Supplementary Material
Acknowledgments
Financial support was provided by the NIH through Grants CA-19033 and GM-29028. We thank Yusen Qiao for collecting cyclic voltammetry data. We also thank Prof. Tehshik P. Yoon for discussions about mechanism.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b05165.
Experimental procedures, NMR spectra for obtained compounds, and mechanistic studies (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Brook AG. Acc Chem Res. 1974;7:77.Moser WH. Tetrahedron. 2001;57:2065. and references therein.
- 2.(a) Brook AG. J Am Chem Soc. 1958;80:1886. [Google Scholar]; (b) Brook AG, Warner CM, McGriskin ME. J Am Chem Soc. 1959;81:981. [Google Scholar]; (c) Brook AG, Schwartz NV. J Am Chem Soc. 1960;82:2435. [Google Scholar]; (d) Brook AG, Iachia B. J Am Chem Soc. 1961;83:827. [Google Scholar]
- 3.(a) Linderman RJ, Ghannam A. J Am Chem Soc. 1990;112:2392. [Google Scholar]; (b) Jankowski P, Raubo P, Wicha J. Synlett. 1994;1994:985. [Google Scholar]; (c) Fleming I, Roberts RS, Smith SC. J Chem Soc Perkin Trans. 1998;1:1215. [Google Scholar]
- 4.(a) Jiang X, Bailey WF. Organometallics. 1995;14:5704. [Google Scholar]; (b) Kawashima T, Naganuma K, Okazaki R. Organometallics. 1998;17:367. [Google Scholar]; (c) Naganuma K, Kawashima T, Okazaki R. Chem Lett. 1999;28:1139. [Google Scholar]; (d) Speier JL. J Am Chem Soc. 1952;74:1003. [Google Scholar]; (e) West R, Lowe R, Stewart HF, Wright A. J Am Chem Soc. 1971;93:282. [Google Scholar]
- 5.For reviews: Smith AB, III, Adams CM. Acc Chem Res. 2004;37:365. doi: 10.1021/ar030245r.Smith AB, III, Wuest WM. Chem Commun. 2008;45:5883. doi: 10.1039/b810394a..
- 6.Paredes MD, Alonso R. J Org Chem. 2000;65:2292. doi: 10.1021/jo9912855. and references therein.
- 7.Maruyama T, Mizuno Y, Shimizu I, Suga S, Yoshida J. J Am Chem Soc. 2007;129:1902. doi: 10.1021/ja068589a. [DOI] [PubMed] [Google Scholar]
- 8.(a) Matsuoka D, Nishigaichi Y. Chem Lett. 2014;43:559. [Google Scholar]; (b) Corcé V, Chamoreau L-M, Derat E, Goddard J-P, Ollivier C, Fensterbank L. Angew Chem Int Ed. 2015;54:11414. doi: 10.1002/anie.201504963. [DOI] [PubMed] [Google Scholar]; (c) Jouffroy M, Primer DN, Molander GA. J Am Chem Soc. 2016;138:475. doi: 10.1021/jacs.5b10963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chuit C, Corriu RJP, Reye C, Young JC. Chem Rev. 1993;93:1371. and references therein.
- 10.For reviews: Hopkinson MN, Sahoo B, Li J-L, Glorius F. Chem - Eur J. 2014;20:3874. doi: 10.1002/chem.201304823. Romero NA, Nicewicz DA. Chem Rev. 2016;116:10075. doi: 10.1021/acs.chemrev.6b00057. Skubi KL, Blum TR, Yoon TP. Chem Rev. 2016;116:10035. doi: 10.1021/acs.chemrev.6b00018.Shaw MH, Twilton J, MacMillan DWC. J Org Chem. 2016;81:6898. doi: 10.1021/acs.joc.6b01449. and references therein.
- 11.Qvortrup K, Rankic DA, MacMillan DWC. J Am Chem Soc. 2014;136:626. doi: 10.1021/ja411596q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bortolamei N, Isse AA, Gennaro A. Electrochim Acta. 2010;55:8312. [Google Scholar]
- 13.Fischer H. Chem Rev. 2001;101:3581. doi: 10.1021/cr990124y. [DOI] [PubMed] [Google Scholar]
- 14.For examples: Hager D, MacMillan DWC. J Am Chem Soc. 2014;136:16986. doi: 10.1021/ja5102695.Cuthbertson JD, MacMillan DWC. Nature. 2015;519:74. doi: 10.1038/nature14255..
- 15.Gentry EC, Knowles RR. Acc Chem Res. 2016;49:1546. doi: 10.1021/acs.accounts.6b00272. and references therein.
- 16.There is only one report about PCET activation of O–H bond via photoredox catalysis: Yayla HG, Wang H, Tarantino KT, Orbe HS, Knowles RR. J Am Chem Soc. 2016;138:10794. doi: 10.1021/jacs.6b06517..
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.