

Abstract
The fact that the heritable neurodegenerative disorder Huntington’s Disease (HD) is autosomal dominant means that there is one wild type and one mutant allele in most HD patients. The CAG repeat expansion in the exon 1 of the protein huntingtin (HTTex1) that causes the disease, leads to the formation of HTT fibrils in vitro and vivo. An important question for understanding the molecular mechanism of HD is which role wild type HTT plays for the formation, propagation, and structure of these HTT fibrils. Here we report that fibrils of mutant HTTex1 are able to seed the aggregation of wild type HTTex1 into amyloid fibrils which in turn can seed the fibril formation of mutant HTTex1. Solid-state NMR and EPR data showed that wild type HTTex1 fibrils closely resemble the structure of mutant fibrils, with small differences indicating a less extended fibril core. These data suggest that wild type fibrils can faithfully perpetuate the structure of mutant fibrils in HD. However, wild type HTTex1 monomers have a much higher equilibrium solubility compared to mutant HTTex1 and only a small fraction incorporates into fibrils.
Huntington’s Disease (HD) is a fatal neurodegenerative disease that is caused by a CAG repeat expansion in the exon-1 of the protein huntingtin (HTT). HTT exon-1 (HTTex1) is translated as an alternative splice variant 1 and was found to be predominant in the fibril deposits of post-mortem HD brains.2 HD occurs if the CAG repeat, encoding for a poly glutamine (polyQ) domain, that usually ranges from 9–35 repeats, is expanded above a threshold of about 36.3,4 HD is inherited in an autosomal dominant fashion and the age of onset is inversely correlated with the length of the expanded CAG repeat, that can get as long as 120 repeats.4 The length of the CAG repeat and thereby the polyQ domain also correlates with the ability of HTTex1 model peptides to form fibrils in vitro.5
The mechanism of how the CAG repeat expansion causes HD is an active field of research and protein misfolding has long been considered to be one of the factors contributing to pathogenesis. Thus, a detailed molecular understanding of HTT misfolding is of potential diagnostic and therapeutic relevance. A number of misfolded species have been identified and it has been proposed that cytotoxic and non-toxic fibrils as well as toxic protofibrils and oligomeric forms of HTT exist.6–8 The structure of fibrils formed by synthetic polyQ containing peptides and more recently HTTex1 with expanded polyQ domains has been studied using electron paramagnetic resonance (EPR), solid-state nuclear magnetic resonance (solid-state NMR), electron microscopy (EM), and other biophysical techniques.5,9–15 These studies show that the N-terminal residues preceding the polyQ domain (N17) form an α-helix of intermediate dynamics in the fibrils state.10,11 The polyQ domain in the center of HTTex1 forms the static core of the amyloid fibril. An EPR study on HTTex1Q46 showed that the Gln residues were not in an in-register parallel β conformation unlike many other amyloid fibrils.9 Solid-state NMR studies showed that this polyQ domain has Gln in two distinct conformations that occur in a 1:1 ratio. One of the Gln types has a chemical shift typical for a β-sheet conformation, the other an unusual chemical shift that was hypothesized to be the result of an specific side-chain conformation.10–12,14 Although the two Gln types are found throughout the polyQ domain and are close in space, they are sequentially clustered.12,13 There are several models for the structure of the polyQ fibril core.13,14,16–18 The C-terminal Pro-rich domain of HTTex1 is in a mixed polyproline II helical and random coil conformation forming dynamic bristles at the outside of the fibril similar to a bottlebrush.9,12,19
HD is not only an autosomal dominant disease where one expanded allele is sufficient for pathogenesis, but, curiously, the Q-length of the second allele does not even appear to have a pronounced impact on disease onset.4 This finding might be somewhat surprising as two alleles with expanded Q-length might lead to more pronounced misfolding than a single allele. However, one scenario in which the impact of the allele with shorter polyQ length is negligible involves cross-seeding. In such a case the mutant allele with the longer Q-length could readily misfold and consequently act as a template that will promote the misfolding of the protein with shorter Q-length. Cross seeding has been shown for small, polyQ containing peptides,10 and for HTTex1 fusion proteins in vitro and cell culture.20 However, whether the intrinsic structure of the seed is retained in the less aggregation-prone HTTex1 with shorter Q-lengths remains unclear. A faithful propagation of seed structure may not be a given for HTTex1 where fibril formation requires the concerted formation of contacts between helical N17 regions and β-sheet containing polyQ regions and it is unclear whether all of these contacts can be maintained in the case of proteins with different Q-lengths. To learn more about the interaction of HTTex1 with different Q-lengths, in particular to get structural information of wild type HTTex1 in the presence of preformed mutant HTTex1 fibrils, we asked the following specific questions: Can wild type HTTex1 be effectively seeded with fibrils formed from mutant HTTex1 fibrils with expanded Q length and if so, can the wild type fibrils, in turn be used to seed mutant fibril growth? If wild type HTTex1 can be seeded, how does its structure compare to fibrils formed by mutant HTTex1? In the following we will address these questions using a combined thioflavin T (Tht) fluorescence kinetics, EM, EPR, and solid-state NMR approach.
Materials and Methods
Protein expression and purification
Plasmids that express HTTex1Q25 and HTTex1Q46 were a gift from Pamela Bjorkman’s lab. Cysteine mutations were introduced by site-directed mutagenesis into these vectors (pET32a-HDx25Q), which expresses HTTex1 with an N-terminal thioredoxin tag and a C-terminal His tag. For HTTex1Q25, single cysteines mutations were introduced at the following positions: 11, 15, 18, 21, and 42. All HTTex1Q46 mutants used in this study were described previously.9
The expression of HTTex1 was started by transforming BL21 (DE3) E.coli cells with the pET32a-HDx25Q or Q46 plasmid and plated onto LB agar with 100 mg/ml ampicillin (Amp). A single colony was inoculated into 20 ml LB medium and grown for about 3 to 4 hours at 37°C. This starter culture was then split and expanded to 2 x 500 ml of LB/Amp and grown to an optical density at 600 nm (OD600) of 0.6 to 0.8. The expression was then induced by adding 1 mM final concentration of Isopropyl 1-thio-β-d-galactopyranoside and the cells were incubated overnight at 18°C. Finally, cell pellets were collected by centrifugation and stored at −80°C until further use. The expression of uniformly labeled 13C, 15N HTTex1 was done as described previously.12
Purification was carried out as described previously12 with few minor modifications: Briefly, 1 × CelLytic™ B cell lysis reagent (Sigma-Aldrich) was replaced with 1% Triton X-100. Nickel-nitrilotriacetic acid-agarose beads were replaced with His60 resin (Clontech). The nickel based His60 column was washed with 50 mM imidazole instead of 20 mM.
For spin-label experiment Htt contain a single cysteine mutant was expressed and purified as described above except for the following modifications: All buffers used prior to the elution from His60 resin contained 1mM dithiothreitol (DTT). Prior to elution and spin labeling, residual DTT was removed with 100 ml of additional column wash containing no DTT. Protein was eluted with 25 ml elution buffer (20 mM Tris, 300 mM NaCl, 250 mM Imidazole pH 7.4). The concentration of eluted protein was estimated via the absorbance at 280 nm using an extinction coefficient ε=14105 M−1cm−1. Consequently, the MTSL (1-oxyl-2,2,5,5 tetramethyl-Δ3-pyrroline-3-methylmethanethiosulfonate) spin label was added to the protein in a 3 to 5 fold molar excess at room temperature and incubated for an hour. Unbound spin label was removed by diluting the sample 10 fold with 20 mM Tris, pH 7.4 and loading it onto a HiTrap Q XL anion exchange chromatography column (GE Healthcare) on an AKTA FPLC system (Amersham Pharmacia Biotech). The protein was eluted using a NaCl gradient.
For the preparation of HTTex1Q46 seeds, fusion protein was diluted with 20 mM Tris, 150 mM NaCl to between 20 to 25 μM. The thioredoxin fusion tag was removed and fibril growth initiated by adding 1 unit EKMax (Invitrogen) per 280 μg of protein and incubating the solution at 4°C for several days.12 Fibril seeds were prepared by sonicating the so formed fibrils as described previously.9 Seed morphology was confirmed by electron microscopy (EM) as described below. The concentrations of sonicated fibrils were determined via a BCA assay (Thermo Fisher Scientific).
Fibrils for NMR and EPR studies were formed by bringing HTTex1 fusion proteins to a concentration of 25 μM (632 μg/ml) in 20 mM Tris, 150 mM NaCl and adding HTTex1Q46 fibril seeds in a 5% molar ratio. Fibril formation was then started by adding 1 unit of EKMax per 280 μg of protein. The mixture was incubated without agitation at 4°C for 2 weeks to complete fibril formation.
Electron microscopy
Electron microscopy grids (150 mesh copper) were prepared by absorbing 10 μl of sample on them for 5 to 10 minutes. The grids were negatively stained by adding 10 μl of 1% uranyl acetate solution for 2 min and then dried at room temperature for 1 hour. All samples were photographed with a Gatan digital camera using a JEOL JEM-1400 electron microscope (JEOL, Peabody, MA) at 100 kV.
Fibril forming kinetics
The ThT kinetics assay was done in an Eppendorf AF2200 plate reader using falcon 96 well (clear flat bottom) plates. Samples of 200 μl total volume contained 25 μM protein in 20 mM Tris, 150 mM NaCl pH 7.4, 25 μM ThT, and 0.02% (v/v) sodium azide. For seeded reactions, fibril seeds were added in 0.2% to 5% molar ratio. To initiate fibril growth, EKMax (1 unit/280 μg of protein) was added. Fluorescence was measured at 25°C every 30 minutes for 20 hours. Each reading was preceded by 2 s for orbital agitation. The following settings were used: excitation at 440 nm with a bandwidth of 20 nm and emission at 484 nm with a bandwidth of 25 nm, the gain was 25, and 25 acquisitions were integrated over of 25 μs. All samples were repeated at least three times.
EPR spectroscopy
Fibrils were pelleted by ultracentrifugation, resuspended in 10 μl of 20 mM Tris, 150 mM NaCl, and loaded into quartz capillaries (0.6-mm inner diameter × 0.84-mm outer diameter, VitroCom, Mt. Lakes, NJ). EPR spectra were recorded on an X-band Bruker EMX spectrometer (Bruker Biospin Corporation) at ambient temperature. The scan width was 150 gauss using a HS cavity at an incident microwave power of 12.60 milliwatts. All spectra were normalized by double integration.
The amount of fibril formation was estimated as follows: a 25 μM solution of spin labeled HTTex1Q25 or HTTex1Q46 fusion protein was mixed with HTTex1Q46 fibril seeds at a 5% molar ratio. One unit of EKMax per 280 μg of protein was added to initiate fibril growth and 20 μl of sample were then loaded into quartz capillaries and a EPR spectrum was recorded immediately after the sample was prepared and 4.5 days after the preparation using the parameters described above.
Solid-state NMR spectroscopy
All NMR spectra were acquired on an Agilent DD2 600 MHz solid-state NMR spectrometer using a T3 1.6 mm probe operating at 25 kHz magic angle spinning (MAS) and 0°C set temperature. A recycle delay of 3 s was used in all spectra. Proton and 13C hard radio frequency (rf) pulses had amplitudes of 200 kHz and 100 kHz, respectively. 1H-13C cross polarizations (CPs) were done using the Hartman-Hahn condition at 60 kHz and 85 kHz on 13C and 1H, respectively with a 10% amplitude ramp and 1 ms contact time. Two pulse phase modulation (TPPM)21 of 120 kHz was applied during the direct and indirect dimensions for 1H decoupling.
One dimensional spectra were recorded with a spectral width of 50 kHz, 650 complex points and 256 and 128 acquisitions for the HTTex1Q25 and HTTex1Q46 sample, respectively. The 13C-13C DARR (dipolar assisted rotational resonance) spectra22 were recorded using a 25 kHz 1H recoupling field during the 50 ms of mixing time and spectral widths of 50 kHz in both dimensions. For each of the 400 indirect increments 24 and 16 acquisitions were recorded for the HTTex1Q25 and HTTex1Q46 sample, respectively. All spectra were processed using Lorentz to Gauss transform window functions. Chemical shifts were referenced externally to DSS (4,4-dimethyl-4-silapentane-1-sulfonic acid) using adamantane.23
Results
Seeding of HTTex1Q25 fibrils
To determine if recombinant, wild type HTTex1Q25 is able to form fibrils in the presence of seeds prepared from HTTex1Q46, we mixed N-terminally thioredoxin tagged HTTex1 fibrils with 5% of preformed and sonicated HTTex1Q46 seeds and initiated aggregation by removing the thioredoxin using the enzyme EKMax. Starting the aggregation with the cleavage of the thioredoxin solubility tag assures seed-free, monomeric HTTex1 starting conditions that are difficult to achieve by e.g. disaggregating HTTex1 without fusion tag. After 24 h, fibril formation was monitored via negatively stained EM images. As can be seen from Figure 1, both HTTex1Q46 and HTTex1Q25 formed amyloid fibrils in the presence of HTTex1Q46 seeds. Although both fibrils have a similar diameter (~15 nm) and show a comparable number of single fibrils and fibril bundles, the HTTex1Q25 fibrils are generally shorter compared to the HTTex1Q46 fibrils. We measured the length of 40 well separated HTTex1Q46 and HTTex1Q25 fibrils each and determined an average length of 345 ±83 nm and 152 ±41 nm, respectively. Additional EM images of these HTTex1Q46 and Q25 fibrils can be seen in Figure S1.
Figure 1.

HTTex1Q25 forms fibrils in the presence of HTTex1Q46 seeds. Electron micrographs of negatively stained fibrils formed by HTTex1Q25 and HTTex1Q46 in the presence of 5% HTTex1Q46 fibril seeds grown for 24h at 4°C. HTTex1Q25 fibrils are shorter than the comparable HTTex1Q46 fibrils.
Could this difference in fibril length be a result of slower growth of the HTTex1Q25 fibrils in the presence of seeds as compared to HTTex1Q46? To answer this question, we measured fibril forming kinetics via a ThT fluorescence assay. In this assay 25 μM HTTex1Q25 and HTTex1Q46, were aggregated in the presence of 0.2%, 1%, and 5% of preformed HTTex1Q46 seeds and the ThT fluorescence was monitored over 20 h at 25°C. All experiments were run at least in triplicate and the aggregation was started by adding 1 unit EKMax (Invitrogen) per 280 μg of protein. HTTex1Q46 without EKMax, and HTTex1Q25 and HTTex1Q46 with EKMax but in the absence of seeds served as controls. As can be seen from the normalized Tht fluorescence curve in Figure 2a and the average half time of these kinetics in Figure 2c, both HTTex1Q25 and HTTex1Q46 formed Tht positive aggregates in the presence of seeds. The Tht kinetics were faster with increasing concentration of seeds for HTTex1Q46. For HTTex1Q25 significant Tht fluorescence increase could only be observed when adding 5% of seeds and the maximal absolute Tht fluorescence intensity of the HTTex1Q25 kinetic curves was about 4 times smaller compared to HTTex1Q46. We did not observe an increase in Tht fluorescence for HTTex1Q25 in the absence of seeds. To test if the so formed HTTex1Q25 fibrils were in turn able to seed monomeric HTTex1Q46, we repeated the experiments by adding 0.2%, 1%, and 5% HTTex1Q25 seeds (obtained by seeding with Httex1Q46) to HTTex1Q46. As can be seen from Figure 2b and c, HTTex1Q25 seeds significantly sped up the Tht kinetics of HTTex1Q46 whereas HTTex1Q46 in the absence of seeds led to an increase in Tht fluorescence after a lag phase of about 3 h. The Tht fluorescence curves not only depend on the fibril forming kinetics but also on how efficiently the fusion tag is cleaved by EKMax. To get an estimate of the efficiency of the EKMax reaction, we monitored the decrease in fusion protein and increase in free thioredoxin and HTTex1 using SDS-PAGE. As can be seen from Figure S2, most of the fusion protein was cleaved after 2.5 h. These data indicate that the cleavage reaction is less important for low seed concentration or no seeds but might be rate limiting at high seed concentration.
Figure 2.

a) HTTex1Q46 seeds accelerate the fibrillization of HTTex1Q46 and HTTex1Q25. Tht fluorescence assay of 25 μM HTTex1Q46 and HTTex1Q25 in the presence of 0.2%, 1%, and 5% HTTex1Q46 fibril seeds. Protein aggregation is initiated by the addition of 1 unit/280 μg EKMax. HTTex1Q46 and HTTex1Q25 without seeds and HTTex1Q46 without seeds and without EKMax served as controls. Only 5% of seeds led to significant Tht fluorescence for HTTex1Q25. Fluorescence intensities of curves showing a significant increase were baseline corrected and normalized to maximum intensity. Controls that did not show an increase in fluorescence were normalized according to HTTex1Q46 without seeds b) HTTex1Q25 seeds can seed the fibrillization of HTTex1Q46. Tht fluorescence assay of 25 μM HTTex1Q46 in the presence of 0.2%, 1%, and 5% of HTTex1Q25 fibril seeds. HTTex1Q46 without seeds served as control. c) Average of the Tht kinetics half times calculated from 3 repeats of the Tht time courses shown in a) and b).
Tht fluorescence intensity is generally a poor quantitative measure of fibril formation when comparing different proteins or constructs. Therefore, we asked if the smaller absolute Tht fluorescence intensity measured for HTTex1Q25 is the consequence of a smaller Tht fluorescence of the HTTex1Q25 fibril compared to HTTex1Q46 or if it indicates incomplete aggregation. To address this question, we monitored the amount of aggregation via the EPR signal decay of HTTex1Q25 and Q46 that both had been MTSL spin labeled at residue 35 (35R1) within the polyQ domain. We chose residue 35 since it is right inside the polyQ domain of HTTex1Q25 and Q46 and thereby a good site to monitor fibril formation. The narrow EPR linewidth of monomeric HTTex1 with its the highly dynamic and fast-tumbling polyQ domain results in a large line intensity. The EPR linewidth broadens when the protein forms fibrillar aggregates leading to a decay in intensity to about 10%. As can be seen from Figure 3, the EPR signal of seeded HTTex1Q46 after 4.5 days of incubation has broadened significantly and does only show a very small narrow component comparable to the EPR spectrum at the beginning of the experiment. The EPR spectral changes of HTTex1Q25, however, are very different and most of the signal is still compatible to monomeric protein after 4.5 days of incubation. To calculate the remaining monomer concentration, we measured the sharp component of monomeric HTTex1 by subtracting scaled EPR spectra of monomeric HTTex1 from the spectra taken after 4.5 days (See Figure S3). Starting from a monomer concentration of 25 μM there were 0.94 μM and 16.25 μM remaining of HTTex1Q46 and HTTex1Q25, respectively. The free energy difference ΔG between the protein in solution and incorporated into the fibril can be calculated as from the equilibrium monomer concentration Ceq as follows ΔG=−RT ln(1/Ceq).24,25 Consequently we calculated free energy differences of −33.83 kJ/mol and −26.88 kJ/mol for HTTex1Q46 and HTTex1Q25, respectively. Taken together our Tht and EPR data indicate that HTTex1Q46 aggregation is completed within a few hours. Likewise the aggregation of HTTex1Q25 reaches an equilibrium after a few hours when measured via Tht fluorescence kinetics. However, the large amount of HTTex1Q25 monomer we detected in solution several days after seeding shows that the ΔG of fibril formation is larger for HTTex1Q25 compared to HTTex1Q46.
Figure 3.

The aggregation of HTTex1Q25 is incomplete 4.5 days after seeding. The normalized EPR intensities of HTTex1Q25 and HTTex1Q46 spin labeled at residue 35 (i.e. within the polyQ domain) are shown immediately after adding 1 unit/280 μg EKMax in the presence of 5% preformed HTTex1Q46 seeds and after 4.5 days. The change in intensity reflects the amount of monomer remaining in solution (see Figure S3). Where the HTTex1Q46 signal descended to about a tenth of its original intensity and a significantly broadened line indicated complete aggregation, the HTTex1Q25 signal showed only a modest decline in intensity and no change in lineshape indicating that the majority of the protein stayed monomeric even after 4.5 days of aggregation.
Structural characterization of HTTex1Q25 fibrils
How does the size, location, and structure of the HTTex1Q25 fibril core compared to HTTex1Q46? To answer this question, we first recorded 1D CP MAS of solid-state NMR spectra of seeded HTTex1Q25 fibrils and compared them to spectra of non-seeded HTTex1Q46. As can be seen from Figure 4, the resonance lines of these two spectra overlap very well. The intensity of the two spectra was normalized according to their Pro peaks. In this case the intensity of the Gln Cα peak at ~55 ppm of the HTTex1Q25 fibrils is about half the corresponding intensity of the HTTex1Q46 fibrils and the other aliphatic peaks with Gln contributions are reduced as well.
Figure 4.

The intensity of the NMR spectra reflects the smaller size of the fibril core. 1D 13C CP MAS spectra of HTTex1Q46 fibrils and HTTex1Q25 fibrils seeded with HTTex1Q46, recorded at 25 kHz MAS, 0°C and normalized according to the intensity of the proline peaks. Consequently, the intensity of the Gln Cα lines of the HTTex1Q25 fibrils is half of the corresponding line in the HTTex1Q46 fibrils showing that roughly the same percentage of Gln are incorporated into the fibrils formed by both proteins.
To further test how the local atomic structure of HTTex1Q25 fibrils compares to HTTex1Q46 fibrils, we recorded the 2D 13C-13C DARR spectrum shown in Figure 5. Despite small variations in signal intensity, these HTTex1Q25 fibril spectra are virtually identical to those of HTTex1Q46 fibrils, published previously.12 Both spectra are dominated by the two Gln spin systems termed Gln A and B that we showed to be separated in sequence, but within 7Å of each other.12 The difference in chemical shift between Gln A and B is attributed to a difference in side chain conformation.13,14 The relatively weaker, third Gln type (Gln C), that we showed to be more dynamic than Gln A and B12 is also present in both HTTex1 fibril spectra. Besides Gln there are two types of Pro observed in both spectra. The chemical shifts of the more intense Pro A spins system are compatible with a polyproline II helical conformation and the chemical shifts of Pro B indicate a random coil conformation.12 Both types of Pro were detected for both the HTTex1Q25 and HTTex1Q46 fibril spectra at comparable intensity. The only significant difference is that the Glu Cγ-Cδ cross peak visible in the HTTex1Q46 spectra is absent in the HTTex1Q25 spectra.
Figure 5.

The structure of the static amyloid core of HTTex1Q46 fibrils and HTTex1Q25 fibrils seeded with HTTex1Q46 is very similar. 2D 13C-13C DARR spectra recorded at 25 kHz MAS, 0°C, with a 50 ms mixing time. Amino acid type assignments are indicated. Both spectra give essentially the same cross peak pattern indicating that the structural motif of the amyloid core formed by the two proteins does not depend on the length of the polyQ region. The spectrum of HTTex1Q46 was previously published as Figure 2 by Isas et al.12
CW EPR spectroscopy has previously been used to investigate the structural features of spin labeled HTTex1Q46 fibrils.9 The EPR spectra of these fibrils were predominantly governed by local side chain mobility as well as spin-spin interactions when labeled sites were within 20 Å of each other. To compare the structure of the fibrils grown here to those of HTTex1Q46, we labeled analogous sites in HTTex1Q25 fibrils and compared their EPR spectra to those of HTTex1Q46 fibrils. Toward this end, two sites each were chosen in the N17 (11R1 and 15R1) and the polyQ region (18R1 and 21R1). In addition, residues 42R1, 45R1, and 90R1 (correspond to 63R1, 66R1, and 111R1 in HTTex1Q46) were labeled at the end of the polyQ and inside the Pro-rich domain. As shown in Figure 6, the EPR spectra for all of the pairs are very similar in nature. For example, the spectra for 11R1 clearly exhibit two components of different mobility in HTTex1Q46 (blue traces) and HTTex1Q25 (red traces). One of these components is of low mobility indicating a buried structure while the other component indicates a less restricted motion. In agreement with the prior EPR study,9 we also observe some broadening beyond the typical ~70 Gauss width of fully immobilized spectral components. This broadening is due to spin-spin interactions and indicates spatial proximity between N17 regions from different proteins.9 Interestingly, this broadening is slightly more pronounced in HTTex1Q25 fibrils. In the case of 15R1, the overall line shapes (i.e. mobilities) were also comparable for the two different fibrils, while slightly stronger spin-spin coupling was visible in HTTex1Q25 fibrils. Also, the spectra for the labeling sites in the polyQ region (18R1 and 21R1) have similar line shapes, consistent with similar, strong immobilization in both fibril types. As in the case of the N-terminal sites, spectra from HTTex1Q25 fibrils show more spin-spin interaction than the corresponding spectra from HTTex1Q46 fibrils. This increased spin-spin interaction is further supported by the significantly narrower EPR spectrum of 10% MTSL labeled HTTex1Q25 21R1 that was expected to have no significant spin coupling. Figure S4 shows the 1st integral of all spectra shown in Figure 6 and a version normalized to the same number of spins as another way of illustrating the broader lines observed for HTTex1Q25. To quantify this difference in linewidth, we deconvoluted the spectra of HTTex1Q25 and HTTex1Q46 21R1 into an uncoupled component (corresponding to the 10% MTSL labeled spectrum of Figure 3) and a coupled component that was determined from the difference spectrum of HTTex1Q25 and HTTex1Q46 21R1 (see Figure S5). Where the spectrum of HTTex1Q25 21R1 is almost entirely coupled (~94%), HTTex1Q46 21R1 has a sizable (~33%) uncoupled component, indicating that not every spin in these fibrils is involved in spin-spin coupling. Towards the C-terminus the difference in EPR spectra between HTTex1Q25 and HTTex1Q46 becomes smaller (42R1/63R1 and 45R1/66R1) until there is no difference at residue 90R1/111R1. Collectively, the EPR data are consistent with the NMR data which suggested that the overall structural environment is highly similar in HTTex1Q25 and HTTex1Q46 fibrils. The only difference in the EPR spectra between the HTTex1Q25 and HTTex1Q46 fibrils is that for some sites the spin-spin interactions are frequent in HTTex1Q25. As discussed below, this is likely due to a structurally similar, but a less extended polyQ core region in HTTex1Q25 fibrils, which allows more intermolecular spin-spin interaction.
Figure 6.
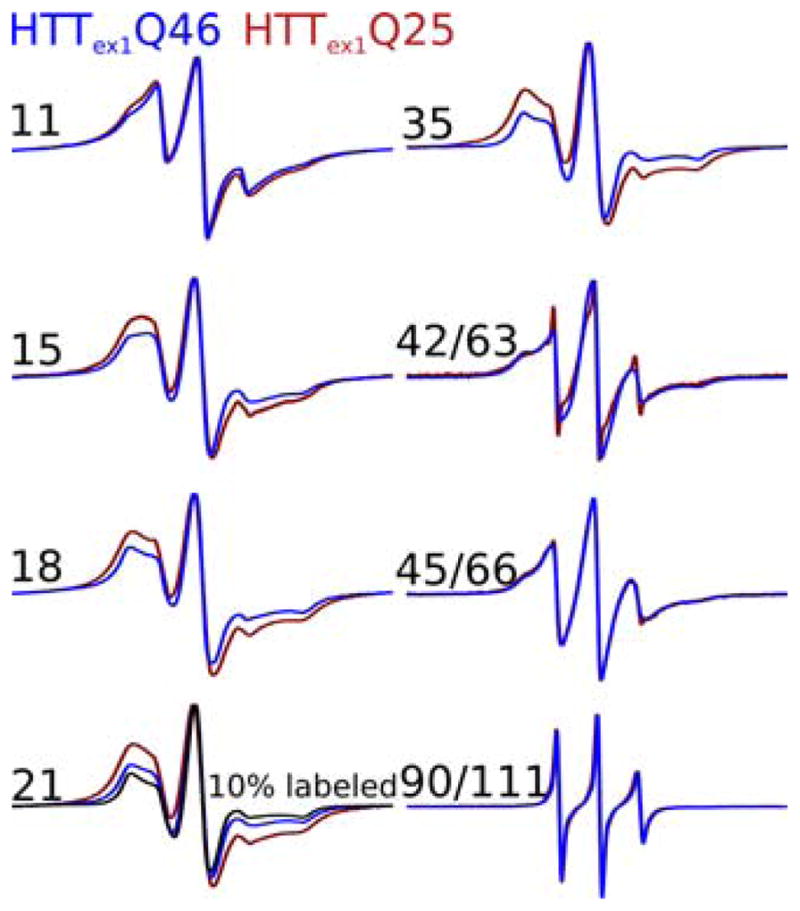
Overlay of EPR spectra of 100% MTSL labeled HTTex1Q25 (red) compared to HTTex1Q46 (blue). For residue 21 the spectrum of 10% MTSL labeled HTTex1Q25 is shown in black. Where the spectra have very similar line shapes indicating an overall similar structure and dynamics, the HTTex1Q25 spectra are generally broader compared to HTTex1Q46, except for the C-terminus. This line broadening indicates more frequent spin-spin interactions in HTTex1Q25 compared to HTTex1Q46. The EPR spectra of HTTex1Q46 were previously published by Bugg et al. as part of Figure 3.9
Discussion
Using a combination of methods including ThT, EM, EPR and solid-state NMR, we find that HTTex1 of different Q-lengths can efficiently cross-seed each other. Under the present conditions, we detect negligible amounts of misfolding for HTTex1Q25 in the absence of HTTex1Q46 seeds. In the presence of such seeds; however, HTTex1Q25 misfolds within a few hours (as seen in Figure 2a), a time frame similar to that of HTTex1Q46 fibril formation.
Our solid-state NMR and EPR data indicate that related structures are being formed in both fibril types. The remarkable part about this finding is that the helical N17 region as well as the β-sheet polyQ regions retain their structural arrangements. It should be pointed out that the dimensions of the two secondary structure elements are rather different. While a helix is on the order of about 10 Å in diameter, the spacing between strands is about 4.8 Å. Although the precise structural details remain unknown, these findings indicate a remarkable plasticity of polyQ aggregation that allow different Q-lengths to misfold in analogous manners while still accommodating the different rises of the N-terminal helix and the β-strands. Moreover, the fact that N17 structural arrangement is retained is consistent with the notion that the N17 is an additional driver of misfolding that has significant contributions to the overall stability of fibrils.5,10,11 However, this structural plasticity of polyQ aggregates can only go so far and increasingly short polyQ domains approaching zero Gln certainly cannot be seeded with preformed fibrils.
Although our NMR and EPR data indicate that the structure of HTTex1Q25 and HTTex1Q46 fibrils are very similar, the two structures cannot be identical. After all, they differ by 21 Gln residues. One of the subtle differences that the EPR experiments revealed is the more pronounced presence of spin-spin coupling present in HTTex1Q25. The enhanced spin-spin interaction in the HTTex1Q25 fibrils may not be entirely surprising since fewer glutamines will likely result in a less extended fibril core thereby facilitating spin-spin interactions between same residues in adjacent monomers. Such contacts could be caused by enhanced proximity along the fibril axis or between individual filaments. The fact that our EM images show generally shorter HTTex1Q25 fibrils that have same thickness compared to HTTex1Q46, and that the overall structural motif of the polyQ domain of HTTex1Q25 is the same compared to HTTex1Q46 but with smaller relative signal intensity, supports this conclusion. Regardless of the precise origin, it needs to be emphasized that the enhanced spin-spin interactions for sites in the polyQ region of HTTex1Q25 are not caused by exchange narrowing. Exchange narrowing requires close spatial proximity of more than two spin labels and is a characteristic feature of sites in fibrils with parallel, in register structure.26 Thus, even in the less extended HTTex1Q25, we find no evidence for a parallel, in-register structure for any of the labeling sites. Our solid-state NMR chemical shift data, being more sensitive to short range order, show that there is no significant change in conformation of the Gln inside the polyQ domain, even though the Gln signal is, as expected, weaker compared to the HTTex1Q46 fibrils simply because there is less Gln in the polyQ domain. This consistent structural environment of the Gln residues confirms that wild type polyQ domains can form fibrils similar to pathologically expanded polyQ and that the structural motif and short-range order of the polyQ fibril core is independent of the polyQ length.
Recently, Hoop and co-workers proposed a polyQ fibril model in which every monomer forms a β-strand–turn–β-strand motif with all strands contributing to the same β-sheet and intra- and intermolecular hydrogen bonds parallel to the fibril axis.13 The authors assumed one turn per monomer and elongated β-strands with increasing polyQ length, which would result in an extension of the fibril core perpendicular to the fibril axis. A similar polyQ length dependent extension would follow from the model proposed by Schneider and coworkers.14 This model proposes a similar antiparallel β-sheet with the difference that the hydrogen bonds parallel to the fibril axis are exclusively intermolecular and the individual monomers form, depending on polyQ length, one or multiple β-strands in separate sheets that are connected by small loops.
Both models are compatible with the structural plasticity of the polyQ domain of HTTex1 reported in this paper and accommodate different polyQ lengths while not being in an in-register parallel β-sheet conformation. However, our observation of more frequent EPR spin-spin coupling in HTTex1Q25 compared to HTTex1Q46 is not compatible with the model by Schneider and coworkers since it predicts every other monomer to be in-register in parallel and therefore regular 9 Å spin-spin distances that should be independent of the polyQ length. The more frequent spin-spin coupling is compatible with the model proposed by Hoop and co-workers, which proposes stochastic monomer interfaces, especially if additional spin-couplings between individual filaments are taken into account.
The model in Figure 7 summarizes the interpretation of our results. Where HTTex1Q46 forms fibrils in the presence and absence of seeds, HTTex1Q25 only forms significant amounts of fibrils in the presence of preformed fibrils seeds. Nevertheless, HTTex1Q25 fibrils can be used to seed HTTex1Q46. Although our NMR data show that the HTTex1Q25 fibril core is similar in overall structure compared to HTTex1Q46, the EPR data suggest that it is less extended.
Figure 7.
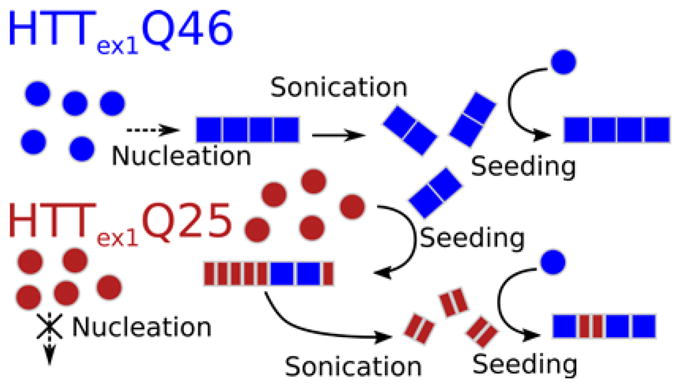
Model of the fibril formation of HTTex1. HTTex1Q46 spontaneously forms amyloid fibrils in the absence of the thioredoxin solubility tag. Seeds of HTTex1Q46 can be produced via sonication and speed up fibril formation of HTTex1Q46 considerably. HTTex1Q25 does not nucleate well from monomeric solutions but forms readily fibrils in the presence of HTTex1Q46 seeds. The HTTex1Q25 fibril core is hypothesized to be less extended compared to HTTex1Q46 resulting in shorter overall fibrils and increased spin-spin coupling in the EPR spectra.
The present results show that significant misfolding occurs for seeded HTTex1Q25 and the time course of this formation is comparable for HTTex1Q46 and HTTex1Q25. However, despite these similarities there are also some differences. The EPR spectra show that a larger amount of protein remains in solution at the end of misfolding indicating that the equilibrium solubility of HTTex1Q25 is higher than that of HTTex1Q46 and consequently the free energy difference ΔG between the protein in solution and incorporated into the fibril is smaller for HTTex1Q25 compared to HTTex1Q46. The trend that additional number of Q residues provide additional driving force for misfolding is consistent with prior studies.5 Inasmuch as the time course of formation (on-rate) seems to be similar in the case the different Q-lengths, it would seem likely that the reduced thermodynamic stability is reflected in an enhanced off rate of HTTex1Q25 from the fibrils. However, these thermodynamic parameters still have to be determined experimentally. The shorter fibrils seen in the EM images could be the result of this reduced thermodynamic stability, or, alternatively, due to HTTex1Q25’s inability to sustain fibrillization over a large number of monomers or its less extended fibril core. None of these effects exclude each other and the shorter fibril length might well be a result a combination of these effects.
In addition to providing new structural and mechanistic insights into HTTex1Q25 misfolding, the present data also provide an insight into co-aggregation of HTT proteins of different Q length in vivo. Cellular work has previously shown that it is possible to seed the formation of puncta using polyQ proteins.20,27 Furthermore, Serpionov and co-workers showed that HTTQ25 aggregation can be seeded and in turn seed the aggregation of other Q/N-rich proteins in yeast.28 Our data show that for HTTex1 this is a direct seeding effect in which the seed structure is almost entirely reproduced by the new fibril that is formed. The HTTex1Q46 fibril type used as a seed here has previously been reported to be a toxic fibril type. Thus, the ability of the normally non-toxic and not aggregation prone HTTex1Q25 to incorporate into potentially toxic fibril types (albeit at a slightly reduced rate) could further contribute to toxicity. Whether this seeding-dependent structural conversion of proteins with low Q-length contributes to the pathogenesis in vivo and whether it is part of the reason why homozygosity for longer Q-lengths does not lead to earlier disease onset than heterozygosity needs to be determined.
Supplementary Material
Acknowledgments
Funding Information
We would like to acknowledge support from the National Institutes of Health NIGMS Award R01GM110521, NINDS Award R01NS084345, and the University of Southern California.
Abbreviations
- HD
Huntington’s Disease
- HTT
huntingtin
- HTTex1
huntingtin exon-1
- EPR
electron paramagnetic resonance
- NMR
nuclear magnetic resonance
- EM
electron microscopy
- DTT
dithiothreitol
- MTSL
1-oxyl-2,2,5,5 tetramethyl-Δ3-pyrroline-3-methylmethanethiosulfonate
- Amp
ampicillin
- ThT
thioflavin T
- MAS
magic angle spinning
- rf
radio frequency
- CP
cross polarization
- TPPM
two pulse phase modulation
- DARR
dipolar assisted rotational resonance
- DSS
4,4-dimethyl-4-silapentane-1-sulfonic acid
Footnotes
Figure S1 showing additional EM images of negatively stained HTTex1Q25 and HTTex1Q46 fibrils; Figure S2 SDS page illustrating the EKMax digestion of thioredoxin tagged HTTex1 Q25 and Q46; Figure S3 measurement of equilibrium monomer concentration from the EPR spectra of Figure 3; Figure S4 EPR spectra of Figure 6 plotted as 1st integral and normalized by the same number of spins; Figure S5 deconvolution of the HTTex1Q25 and HTTex1Q46 21R1 spectra into coupling and non-coupling component.
References
- 1.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 2.Sathasivam K, Neueder A, Gipson TA, Landles C, Benjamin AC, Bondulich MK, Smith DL, Faull RLM, Roos RAC, Howland D, Detloff PJ, Housman DE, Bates GP. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci U S A. 2013;110:2366–2370. doi: 10.1073/pnas.1221891110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet. 2005;6:743–755. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- 4.Lee JM, Ramos EM, Lee JH, Gillis T, Mysore JS, Hayden MR, Warby SC, Morrison P, Nance M, Ross CA, Margolis RL, Squitieri F, Orobello S, Di Donato S, Gomez-Tortosa E, Ayuso C, Suchowersky O, Trent RJA, McCusker E, Novelletto A, Frontali M, Jones R, Ashizawa T, Frank S, Saint-Hilaire MH, Hersch SM, Rosas HD, Lucente D, Harrison MB, Zanko A, Abramson RK, Marder K, Sequeiros J, Paulsen JS, Landwehrmeyer GB, Myers RH, MacDonald ME, Gusella JF PREDICT-HD study of the Huntington Study Group (HSG), REGISTRY study of the European Huntington’s Disease Network, HD-MAPS Study Group, COHORT study of the HSG. CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology. 2012;78:690–695. doi: 10.1212/WNL.0b013e318249f683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jayaraman M, Kodali R, Sahoo B, Thakur AK, Mayasundari A, Mishra R, Peterson CB, Wetzel R. Slow amyloid nucleation via α-helix-rich oligomeric intermediates in short polyglutamine-containing huntingtin fragments. J Mol Biol. 2012;415:881–899. doi: 10.1016/j.jmb.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoffner G, Djian P. Monomeric, oligomeric and polymeric proteins in huntington disease and other diseases of polyglutamine expansion. Brain Sci. 2014;4:91–122. doi: 10.3390/brainsci4010091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nekooki-Machida Y, Kurosawa M, Nukina N, Ito K, Oda T, Tanaka M. Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc Natl Acad Sci U S A. 2009;106:9679–9684. doi: 10.1073/pnas.0812083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pieri L, Madiona K, Bousset L, Melki R. Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys J. 2012;102:2894–2905. doi: 10.1016/j.bpj.2012.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bugg CW, Isas JM, Fischer T, Patterson PH, Langen R. Structural features and domain organization of huntingtin fibrils. J Biol Chem. 2012;287:31739–31746. doi: 10.1074/jbc.M112.353839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sivanandam VN, Jayaraman M, Hoop CL, Kodali R, Wetzel R, van der Wel PCA. The aggregation-enhancing huntingtin N-terminus is helical in amyloid fibrils. J Am Chem Soc. 2011;133:4558–4566. doi: 10.1021/ja110715f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoop CL, Lin HK, Kar K, Hou Z, Poirier MA, Wetzel R, van der Wel PCA. Polyglutamine amyloid core boundaries and flanking domain dynamics in huntingtin fragment fibrils determined by solid-state nuclear magnetic resonance. Biochemistry. 2014;53:6653–6666. doi: 10.1021/bi501010q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Isas JM, Langen R, Siemer AB. Solid-State Nuclear Magnetic Resonance on the Static and Dynamic Domains of Huntingtin Exon-1 Fibrils. Biochemistry. 2015;54:3942–3949. doi: 10.1021/acs.biochem.5b00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoop CL, Lin HK, Kar K, Magyarfalvi G, Lamley JM, Boatz JC, Mandal A, Lewandowski JR, Wetzel R, van der Wel PCA. Huntingtin exon 1 fibrils feature an interdigitated β-hairpin-based polyglutamine core. Proc Natl Acad Sci U S A. 2016;113:1546–1551. doi: 10.1073/pnas.1521933113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider R, Schumacher MC, Mueller H, Nand D, Klaukien V, Heise H, Riedel D, Wolf G, Behrmann E, Raunser S, Seidel R, Engelhard M, Baldus M. Structural characterization of polyglutamine fibrils by solid-state NMR spectroscopy. J Mol Biol. 2011;412:121–136. doi: 10.1016/j.jmb.2011.06.045. [DOI] [PubMed] [Google Scholar]
- 15.Kar K, Baker MA, Lengyel GA, Hoop CL, Kodali R, Byeon IJ, Horne WS, van der Wel PCA, Wetzel R. Backbone Engineering within a Latent β-Hairpin Structure to Design Inhibitors of Polyglutamine Amyloid Formation. J Mol Biol. 2017;429:308–323. doi: 10.1016/j.jmb.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perutz MF, Finch JT, Berriman J, Lesk A. Amyloid fibers are water-filled nanotubes. Proc Natl Acad Sci U S A. 2002;99:5591–5595. doi: 10.1073/pnas.042681399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dougan L, Li J, Badilla CL, Berne BJ, Fernandez JM. Single homopolypeptide chains collapse into mechanically rigid conformations. Proc Natl Acad Sci U S A. 2009;106:12605–12610. doi: 10.1073/pnas.0900678106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sikorski P, Atkins E. New model for crystalline polyglutamine assemblies and their connection with amyloid fibrils. Biomacromolecules. 2005;6:425–432. doi: 10.1021/bm0494388. [DOI] [PubMed] [Google Scholar]
- 19.Lin HK, Boatz JC, Krabbendam IE, Kodali R, Hou Z, Wetzel R, Dolga AM, Poirier MA, van der Wel PCA. Fibril polymorphism affects immobilized non-amyloid flanking domains of huntingtin exon1 rather than its polyglutamine core. Nat Commun. 2017;8:15462. doi: 10.1038/ncomms15462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Busch A, Engemann S, Lurz R, Okazawa H, Lehrach H, Wanker EE. Mutant huntingtin promotes the fibrillogenesis of wild-type huntingtin: a potential mechanism for loss of huntingtin function in Huntington’s disease. J Biol Chem. 2003;278:41452–41461. doi: 10.1074/jbc.M303354200. [DOI] [PubMed] [Google Scholar]
- 21.Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. Heteronuclear decoupling in rotating solids. J Chem Phys. 1995;103:6951–6958. [Google Scholar]
- 22.Takegoshi K, Nakamura S, Terao T. 13C–1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem Phys Lett. 2001;344:631–637. [Google Scholar]
- 23.Harris RK, Becker ED, De Menezes SMC, Granger P, Hoffman RE, Zilm KW. Further Conventions for NMR Shielding and Chemical Shifts (IUPAC Recommendations 2008) Magn Reson Chem. 2008;46:582–598. doi: 10.1002/mrc.2225. [DOI] [PubMed] [Google Scholar]
- 24.O’Nuallain B, Shivaprasad S, Kheterpal I, Wetzel R. Thermodynamics of A beta(1-40) amyloid fibril elongation. Biochemistry. 2005;44:12709–12718. doi: 10.1021/bi050927h. [DOI] [PubMed] [Google Scholar]
- 25.Williams AD, Shivaprasad S, Wetzel R. Alanine scanning mutagenesis of Abeta(1-40) amyloid fibril stability. J Mol Biol. 2006;357:1283–1294. doi: 10.1016/j.jmb.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 26.Margittai M, Langen R. Fibrils with parallel in-register structure constitute a major class of amyloid fibrils: molecular insights from electron paramagnetic resonance spectroscopy. Q Rev Biophys. 2008;41:265–297. doi: 10.1017/S0033583508004733. [DOI] [PubMed] [Google Scholar]
- 27.Tan Z, Dai W, van Erp TGM, Overman J, Demuro A, Digman MA, Hatami A, Albay R, Sontag EM, Potkin KT, Ling S, Macciardi F, Bunney WE, Long JD, Paulsen JS, Ringman JM, Parker I, Glabe C, Thompson LM, Chiu W, Potkin SG. Huntington’s disease cerebrospinal fluid seeds aggregation of mutant huntingtin. Mol Psychiatry. 2015;20:1286–1293. doi: 10.1038/mp.2015.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Serpionov GV, Alexandrov AI, Antonenko YN, Ter-Avanesyan MD. A protein polymerization cascade mediates toxicity of non-pathological human huntingtin in yeast. Sci Rep. 2015;5:18407. doi: 10.1038/srep18407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.