

Abstract
Naturally produced halogenated compounds are ubiquitous across all domains of life where they perform a multitude of biological functions and adopt a diversity of chemical structures. Accordingly, a diverse collection of enzyme catalysts to install and remove halogens from organic scaffolds has evolved in nature. Accounting for the different chemical properties of the four halogen atoms (fluorine, chlorine, bromine, and iodine) and the diversity and chemical reactivity of their organic substrates, enzymes performing biosynthetic and degradative halogenation chemistry utilize numerous mechanistic strategies involving oxidation, reduction, and substitution. Biosynthetic halogenation reactions range from simple aromatic substitutions to stereoselective C-H functionalizations on remote carbon centers and can initiate the formation of simple to complex ring structures. Dehalogenating enzymes, on the other hand, are best known for removing halogen atoms from man-made organohalogens, yet also function naturally, albeit rarely, in metabolic pathways. This review details the scope and mechanism of nature’s halogenation and dehalogenation enzymatic strategies, highlights gaps in our understanding, and posits where new advances in the field might arise in the near future.
TOC Image

1. Introduction
Halogens are biologically available in the environment as halides (F−, Cl−, Br−, and I−). A remarkable diversity of halogenated organic natural products are synthesized in nature ranging from single-carbon halogenated methanes, simple halogenated aromatic phenols, more complex terpenoids, polyketides, oligopeptides, and alkaloids, all the way to some of the most complex and synthetically challenging to access molecules such as palau’amine.1,2 The majority of organohalogen natural products are brominated or chlorinated with over 1,000 examples each. Fewer examples of iodinated compounds, and even rarer examples of fluorinated compounds, currently exist.2 While chlorination is represented in both marine and terrestrial environments, bromination is almost exclusively a feature of marine natural products.2,3 Remarkably, despite the fact that brominated natural products outnumber their chlorinated counterparts, chloride occurs in seawater at a concentration nearly three orders of magnitude greater than that of bromide at 500 mM to 0.9 mM. Biological fluorination is exceedingly rare despite the great natural abundance of fluorine, which is likely due to the extreme electronegativity of fluorine and the high enthalpic cost associated with fluoride desolvation.4,5 On the other hand, the scarcity of organoiodine natural products (~180 reported),6 that still outnumber their organofluorine counterparts by over tenfold, is explained by the extremely low natural availability of iodide.4,7,8
For natural products that are halogenated, halogenation can be of critical importance to their bioactivities. This includes both, activities of halogenated natural products in their native physiological context, as well as bioactivities that make halogenated natural products attractive pharmaceuticals. For example, in a natural physiological context, activity of the thyroid hormones triiodothyronine (1, Figure 1) and prohormone, thyroxine (2) is critically dependent on the number of iodine atoms appended to the bicyclic core.9 As a therapeutic, the antibiotic vancomycin (3) exhibits a marked drop in bioactivity following the removal of one or both of the chlorine atoms decorating its glycopeptide core.10 Halogenation plays a direct role in the mechanism of action of the chlorinated anticancer candidate salinosporamide A (4), which forms a covalent adduct with the active site threonine residue of the 20S proteasome facilitated by a chlorine as a leaving group.11,12 In addition, halogen identity also affects the potency of halogenated molecules. For example, replacing the two chlorines of balhimycin (5), a differently glycosylated variant of 3, to bromines profoundly alters its antimicrobial profile.13 Another well-known example of a clinically used halogenated natural product is the antibiotic chloramphenicol (6).14 Furthermore, fluorination, while exceedingly rare in biology, is a well-proven strategy for increasing the efficacy of pharmaceuticals.7,15 Halogenation strategies involved in the biosynthesis of the abovementioned natural products, among several others, will be discussed as a part of this review.
Figure 1.

Examples of halogenated natural products relevant to human health and disease.
In addition to naturally produced halogenated molecules, the environment also bears a heavy load of halogenated anthropogenic molecules that are often recalcitrant to degradation and are toxic to humans and wildlife. Indeed, all molecules on the list of persistent organic pollutants regulated by the Stockholm convention, are polyhalogenated. As a result, degradation and bioremediation of organohalogen pollutants has brought renewed interest to studies that have suggested a native role for microbial dehalogenation as part of a biogeochemical halogen cycle in marine and terrestrial environments.16,17 A large number of biocatalysts capable of removing halogens from anthropogenic compounds through processes coupled to respiration or carbon metabolism have been described. However, in contrast to the abundance of reported halogenated natural products, the biochemical transformations leading to their natural formation and degradation remain largely unexplored. While the link between biological halogenation and dehalogenation may be tenuous, the authors of this review hope that the inclusion of a brief overview of dehalogenation will stimulate the investigation of the cognate process of dehalogenation from the fresh perspective of natural product biosynthesis.
1.1 Historical perspective on natural product halogenation
The first natural product halogenating biocatalyst reported nearly fifty years ago was a hydrogen peroxide (H2O2) and heme-dependent chloroperoxidase (CPO) secreted from the terrestrial fungus Caldariomyces fumago, and implicated in the biosynthesis of halogenated cyclopentanediol fungal halometabolite caldariomycin (7, Figure 2A).18–20 CPO has since been thoroughly studied and even marketed as an electrophilic halogenating reagent with broad substrate specificity.21 The structure of the enzyme has been determined (Figure 2A).22 The operative mechanism, as has been reviewed extensively elsewhere,23–26 is similar to heme-dependent hydroxylases and oxygenases. Briefly, a cysteine thiolate ligated heme Fe(III)-porphyrin is activated by hydrogen peroxide to generate the oxo-Fe(IV) species. In a departure from P450 hydroxylases, nucleophilic addition of a halide would generate the catalytic Fe(III)-hypohalite species. Halogenation can conceivably proceed via two routes: the CPO can bind the substrate proximal to the Fe(III)-hypohalite leading to the formal delivery of the electrophilic halenium ion to an electron rich substrate with, or without, an intermediary transfer of the halenium to an active site residue side chain, a scheme reminiscent of the halogenation strategy employed by flavin-dependent halogenases (vide infra). A second route, that has been favored in literature involves the release of hypohalite by the enzyme to non-selectively halogenates electron-rich substrate centers. This proposed route is also suggested for vanadium-dependent haloperoxidases (V-HPOs) as discussed in Section 3. Control exercised by the enzyme over its catalytic cycle seems to be rather relaxed as the CPO catalyzes a host of other reactions in addition to halogenation, including oxidation, sulfoxidation, epoxidation, and hydroxylation. Whether the physiological role of CPO indeed involves the production of 7, or whether halogenation is a side activity demonstrated by this fairly catalytically promiscuous enzyme are still open questions. As reviewed by Drennan and coworkers, the halide and substrate binding sites for heme-dependent haloperoxidases have not been identified.24
Figure 2. Heme-dependent haloperoxidases in biological halogenation reactions.

(A) Structure (PDB: 2CPO) and mechanism of CPO that participates in the biosynthesis of 7. For the sake of simplicity, throughout the review, crystal structures of only the monomeric units of multimeric enzymes are shown. (B) Halogenation of 8 leads to loss in absorption at 277 nm. The MCD-assay has been widely used to monitor formation of freely diffusible hypohalite by halogenases. (C) Diiodination of two tyrosyl side chains of the thyroglobulin protein, followed by oxidative coupling, β-elimination, rearomatization, and proteolytic cleavage leads to the production of 2. Note that halogenation and bi-radical coupling is postulated to be affected within the same enzyme active site.
The hypothetical release of hypohalite by haloperoxidases can be efficiently monitored spectrophotometrically by utilizing monochlorodimedone (8, MCD, Figure 2B) that exhibits a loss in absorption at 290 nm following electrophilic halogenation.18 The MCD-assay relies on the ability of a candidate enzyme to oxidize a halide substrate (X− = Cl−, Br−, and I−) to form a diffusible hypohalite species, and hence inherently selects for promiscuous oxidative halogenation enzyme catalysts.27 Utilizing the MCD-assay, numerous haloperoxidases have been discovered, including V-HPOs found in seaweeds some twenty years after the initial report of the first CPO.28
The notion that a freely diffusible hypohalite is released by heme-dependent haloperoxidases as the effective halogenating agent without subsequent control over substrate halogenation is perhaps challenged by the human thyroid epithelial heme-dependent iodoperoxidase that selectively diiodinates two tyrosine side chains (Tyr5 and Tyr130) ortho to the phenoxyl harbored within the 1000-residue thyroglobulin protein. Subsequent oxidative bi-radical coupling of the diiodinated tyrosyl side chains by the same peroxidase, followed by scavenging of the two amino acids from thyroglobulin by dedicated proteases releases 2 that is subsequently deiodinated to its biologically active form 1. The enzymatic deiodination of 2 to furnish 1 is covered in a later section in this review (Section 6).
The paucity of alternative biological halogenation enzymes and strategies discovered over the decades that followed the discovery of haloperoxidases led to the initial assumption that haloperoxidases were the primary halogenating catalysts in the biosynthesis of halogenated natural products.29 In other words, absence of evidence became evidence for absence. Indeed, at the time of the discovery of the first CPO, this argument was helped by the fact that biological halogenation was largely considered to be an accident of biology as only thirty halogenated natural products had been described.30 However, with the subsequent discovery of thousands of bioactive natural products bearing halogens attached to both electron rich and aliphatic scaffolds in regiospecific configurations, it became clear that halogenated natural products had well defined roles in biology and ecology, and that they were biosynthesized by dedicated enzymatic processes.1,2 Moreover, the discovery of fluorinated natural products, despite the inability of any known haloperoxidase to accept fluoride as a substrate, further challenged the notion that these enzymes were the sole strategy for biological halogenation.7 Hence, the broad structural diversity of organohalogen natural products signaled a wealth of substrate-specific biocatalysts utilizing alternative halogenation strategies beyond promiscuous haloperoxidases.27 One of the main obstructions to the discovery of new halogenation biocatalysts has been the inability to link enzymes with their natural substrates. However, progress in DNA sequencing and computational assembly of genomes, particularly bacterial genomes, over the last two decades have led to a renaissance in the discovery of natural product biosynthetic pathways and tailoring enzymes.31 This is also reflected in the current inventory of halogenating enzymes that is heavily dominated by bacterial natural product halogenases.
The marine environment has been a prolific source of halogenated natural products.32,33 A cornucopia of halogenated marine natural products has been discovered, aided in part by the distinctive isotopic signature associated with chlorine and bromine atoms.34 Several instances in which marine natural products and the mechanisms for their production have led to advancement in halogenation biochemistry will be highlighted throughout this review.
1.2 Strategies for natural product halogenation
Halogenases exploit three oxidation states of halogen atoms, and can thus be divided into three broad groups. The first group that will be discussed in this review are halogenases that oxidize the halide (X−) to halenium (X+),35 formally envisaged as hypohalite (XO−) in aqueous media. The halenium ion in turn catalyzes electrophilic substitution reactions on electron-rich substrates, the majority of which are aromatic. The second class of enzymes catalyzes 1e− oxidations of the halide to the X• radical and can thus catalyze halogenation on unactivated aliphatic carbon centers, a transformation that is difficult to realize in synthetic organic chemistry. Both the above classes of oxidative enzymes unsurprisingly cannot utilize fluoride due to its electronegativity. Fluoride incorporation, among other halides, falls under the domain of the third class of halogenases that utilize halides as nucleophiles to catalyze SN2 substitutions on electron deficient carbon centers. Hence, it is evident that the wide inventory of halogenating catalysts has evolved to serve a variety of substrates. All three classes of halogenases are discussed with examples presented for their participation in natural product biosynthesis. The review also emphasizes the progression of the structure-mechanism studies for halogenases and the remaining mechanistic questions for these fascinating catalysts. Structural and biochemical characterization of natural product halogenases, combined with biomolecular, chemical, and process engineering promises to transcend these biological catalysts to laboratory and industrial small molecule synthetic schemes. Recent engineering and development efforts with halogenases are also presented. The review covers literature as current to 2016 with a focus on natural product biosynthesis.
2. Flavin-dependent halogenases
Perhaps the most extensively characterized class of halogenating enzymes is the flavin adenine dinucleotide (FAD) dependent halogenases (FDHs). FDHs are ubiquitous across all domains of life. FDHs catalyze the 2e− oxidation of the halide to the hypohalite, and thus, for the most part, the activity of FDHs is restricted to aromatic substitution reactions. The discussion regarding FDHs, in this review, is divided to reflect the diversity of their substrates and their participation in natural product biosynthetic schemes. Despite the diversity of substrates and the wide repertoire of natural product biosynthetic reactions they catalyze, FDHs follow a conserved mechanism involving halide oxidation followed by regiospecific delivery of the halenium to aromatic substrates.
2.1 Structure and mechanism
Mechanistically and structurally, FDHs resemble FAD-dependent oxygenases (Figure 3). Multiple routes of the redox biochemistry associated with the flavin-cofactor in natural product biosynthetic schemes have been reviewed recently.40,41 Over the last decade, a consensus mechanistic proposal for FDHs has emerged based primarily on the biochemical characterization and crystal structures of L-tryptophan FDHs. Briefly, a 2e− transfer from NAD(P)H to an oxidized flavin cofactor (Flox), catalyzed by a flavin-reductase, generates the reduced flavin cofactor (Flred, Figure 4). The flavin-reductase may, or may not, be encoded with the FDH in the natural product biosynthetic gene cluster. To date, FDHs have not been shown to make specific contacts with the flavin-reductase and the activity of numerous FDHs has been reconstituted using the E. coli flavin-reductases SsuE42 and Fre43 as reaction partners. Flred diffuses to the FDH active site where it reacts with molecular oxygen (O2) to generate the Fl-C4a-OOH transient species. Until this stage, the reaction scheme for FDHs and FAD-dependent oxygenases is indistinguishable. FAD-dependent oxygenases position the aromatic substrate such that the distal oxygen of the Fl-C4a-OOH, which can also be thought of as the electrophilic ‘OH+’ equivalent, is directly transferred to hydroxylate electron-rich aromatic substrates, thereby leaving the Fl-C4a-OH species to resolve via the loss of water to regenerate Flox. FDHs use the cosubstrate halide anions (X−, X = Cl, Br, I) to attack the distal Fl-C4a-OOH oxygen atom, leading to the scission of the labile oxygen-oxygen single bond to generate the electrophilic hypohalite and the Fl-C4a-OH. The hypohalite is thus the effective halenium delivering species. Each of the three flavin-cofactor states, Fl-C4a-OOH, Fl-C4a-OH, and Flox, have been spectroscopically detected in FDHs.44 The kinetics for the formation of these species has been shown to be unaffected by the addition of the substrate, suggesting that substrate engagement either occurs after the flavin redox chemistry, or in a pocket distant to the cofactor. These findings are in concert with the substrate binding site as identified first in the co-crystal structure of the tryptophan-7-chlorinase PrnA,36 and corroborated since with the co-crystal structures of tryptophan-7-chlorinase RebH,45,46 and tryptophan-5-chlorinase PyrH.47 In each of these crystal structures, the substrate binds in a pocket distant to the site of generation of the hypohalite. The hypohalite is not freely diffusible from the enzyme active site, but is rather delivered regiospecifically to the aromatic substrate via the formation of a haloamine species at the side chain primary amine of a highly conserved lysine residue, the mutation of which renders the FDH catalytically inactive (Figure 4).45 Whether the hypohalite is delivered via a covalent haloamine intermediate (as shown in Figure 4), or a tightly coordinated hypohalous acid,48 is presently not clear. Furthermore, the dynamics via which the hypohalite reaches the lysine side chain, while not halogenating the two sterically proximal and highly conserved tryptophan side chains is also not fully understood. While a halide binding site has been identified at the re face of the cofactor isoalloxazine ring, whether this a productive site for halide binding has been debated in the literature.24,40
Figure 3. Structural similarity of FDHs with flavin-dependent oxygenases.

(A) Overlay of crystal structures of FDH PrnA (PDB: 2AQJ) (in blue) and kynurenine-3-monooxygenase (PDB: 5FN0) (in magenta). The structures were overlaid so as to align the cofactor FAD isoalloxazine rings. FAD is shown in stick-ball representation. Chloride ions bound to PrnA is shown as an orange sphere. L-Trp (bound to 2AQJ) and kynurenine-3-monooxygenasae inhibitor (GSK180, bound to 5FN0) are shown as spheres with carbon atoms colored yellow. (B) Zoomed in view of the active sites, with the PrnA catalytic lysine (vide infra) shown in stick-ball representation with carbon atoms colored yellow. (C) Crystal structure of FDH Bmp2 (PDB: 5BVA) (in green) overlaid with 4-hydroxybenzoate hydroxylase (PDB: 1YKJ) (in cyan). 4-hydroxybenzoate (PHBA, bound to 1YKJ) is shown as spheres with carbon atoms colored yellow. (D) Zoomed in view of the active sites, with the Bmp2 catalytic lysine (vide infra) shown in stick-ball representation with carbon atoms colored yellow.
Figure 4. Reaction mechanism for FDHs.
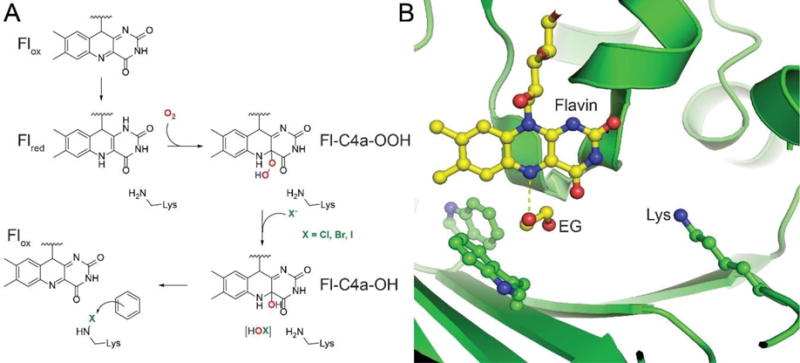
(A) The reaction scheme for FDHs and flavin-dependent oxygenases is identical until the formation of Fl-C4a-OOH. FDHs resolve this intermediate by the displacement of the distal oxygen atom by the halide anion, from where the hypohalite is captured by the catalytic lysine side chain by the formation of a haloamine intermediate, finally affecting an electrophilic aromatic substitution on an electron-rich substrate. (B) The relative positioning of the flavin cofactor isoalloxazine ring and the FDH catalytic lysine side chain as identified in the recently described crystal structure of the FDH Bmp2 (PDB: 5BVA). In the Bmp2 crystal structure, an ethylene glycol (EG) molecule was found in the vicinity of the cofactor proximal to the two tryptophan amino acids that are conserved in all FDH sequences. The dashed line represents the distance of 2.7 Å between the isoalloxazine N5 and one of the EG oxygen atoms. The EG binding site likely represents the region where Flred engages molecular oxygen, finally leading to the formation of the hypohalite that is then transferred to the lysine side chain.
The above discussion is restricted to two-component FDHs, which, just like two-component flavin-dependent oxygenases, require the flavin-reductase as a reaction partner to transfer two electrons from NAD(P)H to Flox and generate Flred. However, flavin-dependent oxygenases also occur as ‘stand-alone’ single-component systems that have a distinct NAD(P)H binding domain and can catalyze the reduction of Flox to Flred, and the subsequent substrate oxygenation all within the same polypeptide. To date, only one single-component FDH has been reported with the discovery of Bmp5 from epiphytic marine bacteria of the genera Pseudoalteromonas and Marinomonas.49,50 Bmp5 catalyzes the bromination of 4-hydroxybenzoate to 3-bromo-4-hydroxybenzoate, followed by decarboxylative-bromination to yield the marine natural product 2,4-dibromophenol (9, Figure 5A). This two-step reaction scheme for Bmp5 combines the activities of two previously reported single-component flavin-dependent oxygenases: 4-hydroxybenzoate hydroxylase51 that yields 3,4-dihydroxybenzoate as the product (Figure 5B), and 4-hydroxybenzoate decarboxylative hydroxylase52 that yields hydroquinone as the product (Figure 5C). Discrete single-component flavin-dependent hydroxylases that hydroxylate distinct positons of the phenoxyl ring derived from 4-hydroxybenzoate are also represented in microbial production of ubiquinone.53 Just as for the single-component flavin-dependent oxygenases, Bmp5 does not require the presence of a flavin reductase for in vitro activity.
Figure 5. Resemblance of bromination activity of single-component halogenase Bmp5 to single-component monooxygenases.

(A) The two-step conversion of 4-hydroxybenzoate to 9 catalyzed by Bmp5. (B) Hydroxylation and (C) decarboxylative-hydroxylation of 4-hydroxybenzoate catalyzed by single-component monooxygenases. Aryl-rings bearing the halogen adducts are highlighted in blue in this section.
2.2 Small molecule halogenases
To date, the inventory of FDHs acting on small molecule substrates is populated, for the most part, by bacterial FDHs chlorinating the amino acid L-tryptophan. The tryptophan-7-chlorinase PrnA, encoded within the biosynthetic gene cluster54,55 for the production of Pseudomonas derived antifungal natural product pyrrolnitrin (10), was the first FDH for which the in vitro biochemical activity was reconstituted using cell free extracts (Figure 6).56 The activity of PrnA was subsequently demonstrated using purified enzyme, and the dependence of FDH activity on a flavin-reductase was identified.57 PrnA has been crystallized in various catalytic states,36 and together with the crystallographic and kinetic studies with the indolocarbazole antitumor natural product rebeccamycin (11) tryptophan-7-chlorinase RebH,44,45,58 forms the basis for much of our understanding about FDH mechanistic enzymology. The celebrated discovery, in vitro enzymatic activity reconstitution, and the structural characterization of L-tryptophan halogenases has been reviewed extensively.23,24,26,41,59–61 Indolocarbazoles can be converted to indolotryptolines,62 and tryptophan halogenases such as AbeH, BorH, and ClaH have been detected in the biosynthetic gene clusters for indolotryptoline natural products BE-54017 (12),63 borregomycin (13),64 and cladoniamide (14),65 respectively (Figure 6). Apart from indolocarbazoles and indolotryptolines, the tryptophan-5-chlorinase PyrH that participates in the biosynthesis of pyrroindomycin B (15) has been biochemically and structurally characterized, together with the biochemical66 and crystallographic67 characterization of the tryptohan-6-chlorinase SttH that is involved in an as yet unidentified biochemical pathway in Streptomyces toxytricini. Another tryptophan-6-chlorinase, ThdH, that participates in the biosynthesis of the Streptomyces derived plant growth regulating hormone theinodolin (16) has been characterized,68 while the contribution of an unnamed tryptophan-6-chlorinase AORI_5336 has been implicated in the biosynthesis of plant growth-regulating compound LYXLF2 (17) from the actinobacterium Amycolatopsis orientalis that also produces 3.69
Figure 6. Regiospecific tryptophan halogenases and their participation in natural product biosynthesis.

The halogenated indole rings are shaded blue. Note that the chlorination on the pyrrole ring of 10 is catalyzed not by PrnA but by PrnC, a second FDH encoded with the pyrrolnitrin biosynthetic gene cluster.
With a repertoire of enzymes that catalyze the regioselective chlorination of tryptophan at three different positions, efforts started to understand the structural bases for this regioselectivity and to engineer tryptophan FDHs as tailored catalysts using mutagenesis and activity screening assays. The reader is directed to several excellent recent reviews dedicated to this topic.26,70–72 Thus, these efforts are not covered in detail here. Combined with efforts directed towards improving the thermostability and organic solvent tolerance of FDHs,73,74 enzyme immobilization to affect gram-scale product transformations,75 and the development of high throughput assays to screen through mutant libraries76 hold promise to push FDHs towards industrially viable catalysts.
Perhaps more relevant from a natural product biosynthesis perspective, a second engineering approach asked the question whether tryptophan FDHs could be used in vivo to generate novel natural products, or intermediate structures to be used in synthetic derivatization via cross-coupling schemes. Regiospecifically halogenating aryl rings can be challenging via purely synthetic means, especially for natural products where other electron rich aryl rings might also be present. Tryptophan FDHs have been successfully used to address these challenges. The Pseudomonas FDH PrnA was conjugated into the genome of the natural product pacidamycin producer Streptomyces coeruleorubidus that led to the production of a halogenated derivative of pacidamycin that was chlorinated only on the indole, and not on the phenyl ring.77 Chloropacidamycin was then used as the substrate in cross-coupling reactions with boronic acids to generate a variety of derivatives. In another approach, tryptophan halogenases were expressed in the medicinal plant Catharanthus roseus to drive the production of chlorinated and brominated derivatives of highly complex plant alkaloids78 that were then amenable to Suzuki-Miyaura cross-coupling reactions with boronic acid substrates.79 A recent report describing the use of regioselective tryptophan halogenases followed by Boc-protection of the main chain amine open the possibility of incorporating halogenated tryptophan intermediates in peptide synthesis and subsequent downstream cross-coupling reactions.80
All FDHs discussed above derive from bacterial sources. This observation is not reflective of the lack of halogenative potential among other domains of life, but rather the bias in DNA sequencing efforts. For example, filter feeding benthic marine invertebrates such as sponges and marine algae are prolific producers of halogenated natural products. However, to date, only a solitary halogenating enzyme has been identified from a marine sponge within a genetic purview of halogenated natural product biosynthesis.81 This bias is slowly starting to shift. FDHs have been found in halogenated fungal natural product gene clusters (Figure 7), such as the Hsp90 inhibitor radicicol (18) chlorinase RadH from Chaetomium chiversi,82 also called as Rdc2 from a different producer Pochonia chlamydosporia.83 In a series of in vitro assays, the fungal FDH Rdc2 could be paired with the E. coli flavin-reductase Fre, and in a substrate promiscuous fashion, chlorinate a series of lactones on the electron rich resorcylic ring.84,85 Substrate promiscuity was also reported for the halogenation of the mushroom toxin melleolide F (19) at the resorcylic acid ring by each of the five different FDHs identified in the mushroom cDNA.86
Figure 7.

Structures of eukaryotic halogenated natural products biosynthesized by the action of FDHs.
Identification of the physiological substrates and the precise timing for halogenation in natural product biosynthetic schemes can be challenging, and the abovementioned substrate tolerance hints towards the possibility that the physiological substrates for these FDHs are yet to be identified. Noteworthy here is the observation that tryptophan halogenases also demonstrate broad substrate tolerance, albeit with reduced catalytic efficiencies, towards a wide swathe of electron rich substrates.87–89 Advances in the identification of the physiological substrate for a fungal FDH was provided by the recent study examining the biosynthesis of chaetoviridin A (20) from Chaetomiun globosum via the caz biosynthetic gene cluster.90 The postulated small molecule substrate for the FDH CazI accumulates in the culture extracts when the cazI gene is deleted, and the chlorination of this accumulated small molecule was experimentally demonstrated by the FDH CazI.91 However, halogenation occurring en route modular polyketide elongation on a substrate tethered to a carrier protein (CP) cannot be entirely discounted based on these findings, as also applicable for the halogenation of the resorcylic ring of the Streotomycete natural product venemycin.92 The applicability of using the small molecule substrate to experimentally demonstrate the dichlorinating activity of amoeba Dictyostelium discoideum FDH ChlA93 can be called into question as well, as halogenation on the two electron rich positions can be easily envisaged while the polyketide chain is still bound to the upstream carrier protein to generate the Dictyostelium differentiation inducing factor-1 (DIF-1, 21).94
2.3 Halogenases for substrates acylated to carrier proteins
CPs are ubiquitous small proteins that undergo a post-translation condensation of coenzyme A (CoA) derived phosphopantetheine to a highly conserved serine side chain hydroxyl. By virtue of the phosphopantetheine terminal thiol, CPs act as molecular shuttles for carboxylic acids that are transiently acylated to it by the formation of a thioester bond (Figure 8A). CP-mediated modifications are ubiquitous in natural product biosynthesis, particularly as it relates to modular extension of peptide and polyketide chains by non-ribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs), respectively.
Figure 8. Halogenation of acyl-S-CP substrates.

(A) Post-translational acylation of apo-CP with CoA-derived phosphopantetheine generates holo-ACP that then undergoes thioesterification with L-proline and oxidation of the prolyl heterocycle to generate pyrrolyl-S-CP. The action of six different pyrrolyl-S-CP FDHs with corresponding natural product structures is shown. Note that the fourth bromine atom in 24 is also installed by Bmp2, while the dibromophenol moiety of 29 is generated by aforementioned single-component FDH Bmp5. Note that 30 is not derived from the action of Bmp2 and Bmp5. (B) Representative L-tyrosine derived halogenated natural products. Chlorination of β-hydroxytyrosine by SgcC3 is shown. The timing of halogenation for other natural products shown here has not been experimentally determined.
The first report of a FDH acting on a CP-tethered substrate described the dichlorination of the pyrrole ring en route the production of the pseudomonad antifungal natural product pyoluteorin (22, Figure 8A).97 PltA did not chlorinate pyrrole-2-carboxylic acid, thus demonstrating the absolute requirement of the substrate to be acylated to a CP. Subsequently, the activity of the FDH SgcC3 was demonstrated to catalyze the chlorination of the CP-acylated β-tyrosine intermediate in the biosynthetic scheme for the Streptomyces globisporus enediyne antitumor antibiotic C-1027 (23, Figure 8A).96 Just like PltA, enzyme SgcC3 only accepted substrates acylated to the CP, though the substrate specificity was relaxed to include both (R)- and (S)-enantiomers of β-tyrosine indicating flexibility in the substrate recruitment site.
Since these reports, the detection of FDHs that work on CP-tethered substrates has blossomed to include the tetrabromopyrrole (24), marinopyrrole A (25), chlorizidine (26), pyrrolomycin (27), and hormaomycin (28) pyrrolyl-S-CP halogenases Bmp2,38,40,49 Mpy16,98 Clz5,99 Pyr29,100 and HrmQ,101 respectively, though the activities for only Bmp2 and Mpy16 have been reconstituted in vitro. Derived from 24, Bmp2 also participates in the biosynthesis of pentabromopseudilin (29).49 In the biosynthesis of each of the abovementioned natural products, the CP carries L-proline through the oxidation of the prolyl heterocycle to pyrrole, followed by halogenation of the pyrrole residue by a FDH. This short but highly conserved biosynthetic route, first elucidated for the biosynthesis of 22,95,97,102 has served as a beacon for genome mining and discovery of the biosynthetic gene clusters for bacterial natural products bearing a halopyrrole moieties, one that is likely to extend further to illuminate the biosynthetic routes for the exceptionally abundant and pharmacologically interesting marine pyrrole aminoimidazole alkaloids.103 Furthermore, the abovementioned FDHs can be used to modify other pyrrole containing natural product biosynthetic pathways to generate halogenated derivatives, as was reported for the engineering of the clorobiocin pathway by the introduction of HrmQ.104 Note that the pyrrole-3-chlorinase PrnC participating in the biosynthesis of 10 is not postulated to follow the CP-mediated route,55 while the physiological substrate for the pentachloropsuedilin (30) FDH HalB is yet to be deciphered.105 Additional tyrosyl-S-CP FDHs have also been discovered, such as the marine proteobacterial brominase AltN participating in the biosynthesis of bromoalterochromides, such as dibromoalterochromide A (31).106,107 The myxobacterial chlorinase CndH, that likely chlorinates the tyrosyl ring in chondrochlorens has been identified with the crystal structure of the enzyme determined as well.108,109 However, the physiological substrate and the timing of halogenation remains to be elucidated. The cyanobacterial FDH AerJ that participates in the biosynthesis halogenated congeners of aeruginosins, such as aeruginosin 101 (32), has been postulated to act on a tyrosyl-S-CP substrate110,111 and will be discussed in a later section together with FDHs that encode halogenation of other cyanobacterial products such as cryptophycin 1 (33), cyanopeptolin 954 (34), and anabaenopeptilide 90-B (35, Figure 8B).
While numerous crystal structures of acyl-S-CP utilizing FDHs have been reported (PltA,112 Mpy16,38 Bmp2,38 CndH108), key mechanistic questions remain unresolved. Primary among these is the catalytic base that abstracts the proton to rearomatize the product during the electrophilic aromatic substitution by the halenium. Based on the structure of PrnA,36 a conserved glutamate residue side chain carboxylate was proposed to serve this role (PrnA-Glu346). While alternate roles for PrnA-Glu346 have been proposed in coordinating hypohalous acid in conjunction with PrnA-Lys79,48 no glutamate or aspartate residues are located in analogous positions in the PltA, Myp16, Bmp2, or CndH crystal structures (Figure 9). In these pyrrolyl-S-CP halogenases, a phenylalanine residue replaces the PrnA glutamate. In PrnA, a Glu→Gln mutation of the proposed catalytic base leads to reduction, rather than abolishment of enzymatic activity, while a Glu→Asp mutation led to complete loss of enzymatic activity without changing the enzyme structure or the substrate binding site.36,48 Whether acyl-S-CP FDHs, that in fact possess retarded turnover numbers, pay this kinetic penalty remains to be experimentally elucidated. Considering that the active site is deeply buried within the enzyme core, an earlier proposal that the catalytic base can be donated by the CP108 seems rather unlikely without invoking large scale conformational changes in the halogenase structure.
Figure 9. Proposed CP binding site in acyl-S-CP halogenases.

Active site of L-tryptophan chlorinase PrnA and pyrrolyl-S-CP halogenases Mpy16 and Bmp2 demonstrating conserved positioning of the FAD cofactor and the postulated haloamine bearing Lys side chain. However, the catalytic base in PrnA- Glu346, is replaced with Phe323 and Phe300 in Mpy16 and Bmp2, respectively. The product, 7-chlorotryptophan is also shown in the PrnA structure to identify the substrate recruitment site.
It should be noted that no crystal structures of acyl-S-CP FDHs have been reported in complex with their CP-tethered substrates. While the requisite positioning of the CP relative to the FDH has not been experimentally determined, the recently reported crystal structures of PltA,112 Mpy16,38 and Bmp238 allow us to advance a postulate. In each of the three crystal structures, a positively charged region on the surface of the enzyme (in blue, Figure 10) can be identified in a concave cavity above the FAD binding site. Electrostatic recruitment of acidic CPs to positively charged surfaces on enzymes is well documented.113,114 Furthermore, in the crystal structure of Mpy16, starting from this concave cavity, a chain of water molecules can be identified that likely represent the path traversed by the phosphopantetheine arm of the acyl-S-CP. This water filled cavity terminates at the reaction site that is defined by the catalytic haloamine bearing lysine side chain (Mpy16-Lys72) and is positioned analogously to the tryptophan binding site in the reported PrnA co-crystal structure (Figure 3).36
Figure 10. Proposed CP binding site in acyl-S-CP halogenases.

Semi-transparent electrostatic surface representation of pyrrolyl-S-CP halogenases PltA (PDB: 5DBJ), Bmp2 (PDB: 5BVA) and Mpy16 (PDB: 5BUK) showing conserved positioning of a positively charged (in blue) concave cavity in the vicinity of the FAD cofactor (in stick-ball representation, carbon atoms colored yellow). In Mpy16, a chain of water molecules (in red) starting at the positively charged cavity traverses the FAD binding site terminating at the halogenation active site defined by the catalytic lysine side chain.
The acyl-S-CP FDHs, such as AltN, PltA, Mpy16, Clz5, and Bmp2, among others, catalyze multiple halogenations upon their CP-loaded aromatic substrates. Recently, the remarkable tetrahalogenating activity of the marine bacterial FDH Bmp2 was reported.38 Bmp2 brominates all four carbon atoms of the CP-tethered pyrrole ring setting up a thioesterase mediated hydrolytic release of a 2,3,4,5-tetrabromo-pyrrole-2-carboxylic acid that undergoes a non-catalytic decarboxylation to yield the coral settlement chemical cue 24 (Figure 8A).115 Multiple halogenations, thought of as relaxation in the regiospecificity for halogenation by acyl-S-CP FDHs, is in stark contrast to small molecule halogenases such as PrnA,36 PyrH,116 RebH,58 and SttH67 that display remarkable regiocontrol in halogenating tryptophan.47 While it is not clear why acyl-S-CP halogenases have lost the regiocontrol exercised by tryptophan FDHs, engineering the Bmp2 enzyme via structure guided site directed mutagenesis has revealed that halogenation regiocontrol in both classes of FDHs is encoded within the halogenase substrate binding active site.38,47,67,117 In light of the above discussion, an unresolved mechanistic question presents itself. Are multiple halogenation reactions catalyzed by acyl-S-CP FDHs processive, in that, does the acyl-S-CP substrate remain bound to the enzyme during successive halogenation events, or distributive, so that the acyl-S-CP substrate is released by the enzyme and has to find the enzyme active site again for the subsequent halogenation? Such a question has also been asked for other natural product biosynthetic enzymes such as the lanthipeptide dehydratases and cyclases that catalyze multiple reactions on spatially distinct sites on a substrate peptide.118–121 Regeneration of the acyl-S-CP FDH between subsequent halogenations would minimally require the following events to occur: (i) loss of FAD, (ii) diffusion of FADH2 back into the FDH active site, (iii) reengagement of O2, and (iv) binding of the halide anion. The abovementioned hypothesis for the site for CP binding proximal to the FAD binding site precludes that FAD and FADH2 can be exchanged while the CP is still bound, though a different site for exchange of FAD/FADH2 cannot be ruled out. Moreover, reduction of O2 at FAD-C4a, followed by the oxidation of the halide and generation of the postulated lysine side chain haloamine can be impeded by the phosphopantetheine arm of the acyl-S-CP substrate that would sterically hinder these processes to occur. Hence, it is likely that acyl-S-CP FDHs reside as ‘cocked guns’ with the reduction of O2, oxidation of the halide, and generation of the haloamine occurring prior to substrate engagement. In an event of productive substrate binding, likely a thermodynamically controlled process, substrate halogenation would occur. Disengagement of the CP would then allow for FAD to be exchanged for FADH2, and the enzyme to be regenerated for the next catalytic cycle. This hypothetical order of events is in line with prior stopped flow kinetic characterization of the RebH-catalyzed chlorination of tryptophan where the presence or absence of substrate did not influence the dynamics of the FAD redox chemistry, and that the substrate chlorination occurred subsequent to the flavin redox reactions.44 Clearly, a co-crystal structure of an acyl-S-CP FDH in complex with its physiological substrate will go a long way in verifying or refuting the above hypotheses. Advances in the biosynthesis of acyl-coenzyme A thioesters,122,123 mass spectrometry based analytical techniques124 that circumvent alkaline offloading of substrates and products from acyl-S-CPs prior to the characterization, and synthesis of molecular probes to interrogate flavin-dependent enzymes125 will undoubtedly aid in answering these mechanistic conundrums that have persisted for more than a decade.
2.4 Halogenases acting en route modular elongation
Each of the acyl-S-CP FDHs discussed in the previous section, with the exception of Bmp2 and SgcC3, halogenate the initiating building block for the biosynthesis of natural products via downstream NRPS or PKS modular assembly line pathways. For example, the 5-chloropyrrolyl-S-CP product of HrmQ is incorporated into the downstream NRPS derived scaffold of 28,101 the 4,5-dichloropyrrolyl-S-CP products of PltA, Pyr29, Mpy16, and Clz5 are incorporated in respective downstream PKS pathways (Figure 8A), and the 3-bromo- or the 3,5-dibromo-p-coumaroyl-S-CP product of AltN is postulated to be elongated via iterative fatty acid biosynthetic enzymes, finally getting incorporated in a NRPS pathway (Figure 8B).107 While the tetrabromination of the pyrrole ring by Bmp2 catalyzes the decarboxylative-offloading of 24,38 chlorination of CP-loaded β-tyrosine by the SgcC3 also does not involve downstream modular NRPS or PKS elongations, rather, aryl hydroxylation,126 followed by direct transesterification127 to the enediyne core to furnish 23 (Figure 8B).128,129 However, when halogenated building blocks occur not as the first building block incorporated in the NRPS or PKS chain, several different routes via which halogenation occurs can be envisaged.
The simplest mechanism would be the accumulation of a pool of halogenated precursor amino acids or polyketide building blocks, and their eventual incorporation into the assembly line by dedicated adenylation and ketosynthase domains that preferentially use these halogenated precursors over their corresponding deshalo congeners. For NRPS derived peptides, such as the recently reported actinomycete cryptic natural product taromycin A (36) that was accessed via transformation associated recombination-based direct capture of the biosynthetic gene cluster and refactoring in a heterologous host,130 this strategy is likely operative. A single FDH, Tar14, encoded with the taromycin biosynthetic gene cluster, likely generates 6-chlorotryptophan that is then incorporated directly into the growing peptide chain by the NRPS Tar8 module-1 adenylation domain (Figure 11A). Sequence comparison revealed the closest structural homolog of Tar14 to be the recently reported tryptophan-6-chlorinase SttH,67 followed by the tryptophan halogenases PrnA, RebH, and PyrH. 6-Chlorotryptophan generated by Tar14 is envisaged to be also converted into 4-chlorokynurenine, that is in turn incorporated by the NRPS Tar10 module-13 adenylation domain. Rationalizing based on the meta-positioning of the chlorine relative to the ortho- and para- directing aniline in 4-chlorokynurenine amino acid,88 it is unlikely that Tar14 halogenates both tryptophan and kynurenine. Furthermore, as deschlorotaromycin congeners are not produced at detectable levels together with 36, the Tar8 module-1 and Tar10 module-13 adenylation domains are likely specific for the recognition and activation of 6-chlorotryptophan and 4-chlorokynurenine, respectively, in contrast to the corresponding adenylation domains encoded within the biosynthetic gene cluster of closely related lipopeptide antibiotic daptomycin (37) that conceivably are specific for the incorporation of tryptophan and kynurenine (Figure 11A). This chemical logic for the incorporation of halogenated building blocks in NRPS peptides is likely conserved in the biosynthesis of kutznerides,131,132 such as kutzneride E (38), actinobacterial depsipeptides bearing a 6,7-dichlorotryptophan derived amino acid building block that is putatively recognized and activated by the NRPS KtzH module-6 adenylation domain (Figure 11B).133 The two FDHs encoded with the kutzneride biosynthetic gene cluster,133 KtzQ and KtzR, bear greatest sequence homology to tryptophan halogenases SttH, PrnA, RebH, and PyrH. Whether the 1,2-epoxidation of the indole ring, followed by cyclization via the main chain nitrogen atom to generate the tricyclic pyrroloindoline motif occurs post or prior to incorporation in the NRPS peptide remains to be discerned. Chlorinated pyrroloindolines, postulated to be synthesized by as yet unidentified FDHs, also occur in the NRPS-derived chloptosin134 (39) and other structurally related peptides isolated from Streptomyces alboflavus 313 (Figure 11C).135 Similarly, while the abovementioned hypotheses for the action of the FDHs Tar14, KtzQ, and KtzR have not been experimentally verified, adenylation domains that recognize and activate halogenated precursors in NRPS assembly lines are attractive ‘plug-and-play’ elements that can be used to regiospecifically engineer natural product scaffolds to bear halogens, a modification that is likely challenging to be affected using chemical derivatization techniques.
Figure 11. Incorporation of halogenated tryptophan in NRPS-derived peptide natural products.
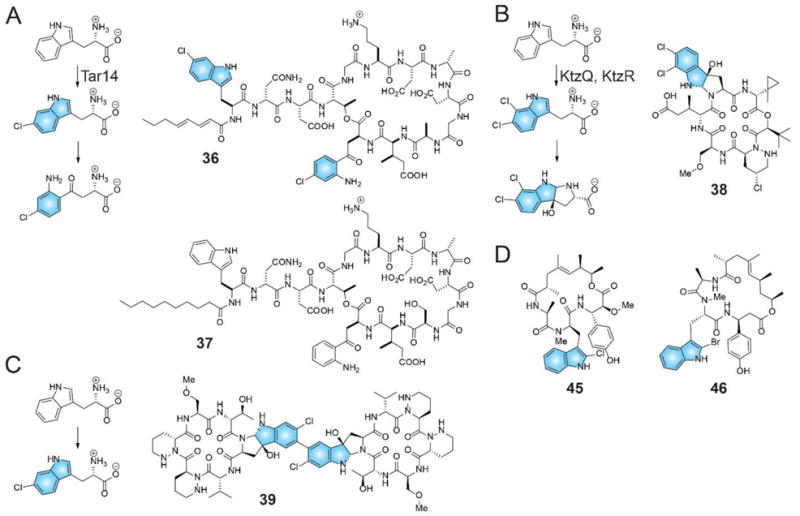
(A) 6-chlorotryptophan, generated by FDH Tar14 is converted to 4-chlorokyunerine. Both amino acids are incorporated in 36. (B) FDHs KtzQ and KtzR generate 6,7-dichlorotryptophan that is incorporated as a pyrroloindoline in the kutznerides family of natural products, such as 38. (C) The natural product 39 also bears the chlorinated pyrroloindoline motif that is generated via an as yet unidentified enzymatic route. (D) The FDH CmdE encoded in the biosynthetic gene cluster for the production of 45 bears homology to acyl-S-CP utilizing FDHs rather than Tar14, KtzQ, and KtzR (vide infra). It is thus likely that the mechanism for incorporation of the 2-chlorotryptophan moiety in 45, and in closely related jasplakindolide (46), might differ from that for 36 and 38.
Perhaps the most celebrated example of halogenation in a NRPS-derived natural product is the dichlorination for the glycopeptide antibiotic-of-last-resort, 3 (Figure 1). Though the emergence of vancomycin resistant pathogenic bacteria dilutes this proud distinction,136 it is well established that the aryl-chlorination of the two β-hydroxytyrosine building blocks by the FDH VhaA is critical to the antibiotic efficacy of 3.10,137–139 For a structurally related natural product, deletion of the FDH participating in the biosynthesis of 5, BhaA,140 from the biosynthetic gene cluster141 led to the production of the deschloro derivative of 5.142 Further gene deletion and precursor feeding experiments143 established that the module-2 and module-6 adenylation domains incorporate β-hydroxytyrosine and not the 3-chloro-β-hydroxytyrosine amino acid precursor in 5. Thus, contrary to the biosynthetic proposal for 36 and 38 presented above, it is unlikely that chlorination of the β-hydroxytyrosines in 3 and 5 occurs prior to the NRPS assembly of the respective heptapeptides. At this stage, the physiological substrate for the VhaA and BhaA FDHs, and the timing of the halogenation relative to the peptide assembly remained elusive. In the temporal vicinity of these findings, biosynthetic gene clusters encoding production of structurally analogous bacterial NRPS derived chlorinated peptides such as chloroeremomycin (40),144 complestatin (41),145 A47934 (42),146 A40926 (43),147 and teicoplanin (44)148 were reported (Figure 12). In each of these gene clusters, only a single FDH was located, as more recently also observed in environmental DNA (eDNA) derived vancomycin-like eDNA gene cluster (VEG) and teicoplanin-like eDNA gene clusters (TEG).149,150 With the assumption that halogenation in these NRPS peptides is intertwined with the NRPS peptide assembly, the rules of regiospecificity for FDHs as discussed in the preceding sections need to be further relaxed, such that the single FDH encoded within the biosynthetic gene cluster for 41, ComH, needs to perform a remarkable six halogenations on three phenyl rings.
Figure 12. Halogenated NRPS derived glycopeptide antibiotics.

A CP loaded hexa-peptide was used as a substrate for the in vitro reconstitution of the activity for the FDH VhaA. The VhaA reaction product would require further NRPS elongation by the 3,5-dihydroxyphenylglycine amino acid, oxidative coupling of the aryl rings, glycosylation, and offloading from the NRPS assembly line for maturation into 3. The order or amino acid addition during the NRPS assembly is denoted by numerals for 3 and halogenated aryl rings are shaded blue.
The longstanding biosynthetic riddle regarding the timing of halogenation in glycopeptide antibiotic biosynthesis was recently resolved with the in vitro characterization of the FDH VhaA.151 This study demonstrated, for the first time, that halogenation in biosynthesis of 3 occurs en route the NRPS assembly of the linear heptapeptide. A biomimetic CP-loaded hexapeptide substrate was dichlorinated by VhaA. Whether the physiological substrate for VhaA is the CP-loaded hexapeptide (as used in these investigations) or the heptapeptide (formed after the addition of the terminal 3,5-dihydroxyphenylglycine amino acid by NRPS module-7) remains to be determined. Nevertheless, this study demonstrated the challenge nascent in generating the substrates that are required to characterize these FDHs. Interestingly, VhaA halogenates the aryl rings at positions 2 and 6 of the substrate, while analogous phenoxyl activated aryl rings at positions 4 and 5 remain unmodified. This regiospecific decision making by VhaA can be attributed, in part, to the β-hydroxyl moieties at positions 2 and 6 of the hexapeptide in the absence of which chlorination did not occur. Halogenation also did not transpire when a CP-loaded dipeptide substrate comprising of amino acids at positions 1 and 2 (Figure 12) were used as substrate for VhaA, demonstrating that halogenation occurs on a relatively mature NRPS-derived peptide. No crystal structures of the NRPS glycopeptide FDHs are presently available. The underlying reason why the biosynthetic logic for 3 involving the incorporation of chlorinated β-hydroxytyrosines differs from that for the incorporation of chlorinated tryptophan, and its derivatives, in 36 and 38 is presently not clear.
NRPS derived cyanobacterial natural products, such as aeruginosins and cyanopeptolins, constitute both halogenated and nonhalogenated congeners (Figure 8B).152 Cyanobacteria of the genera Microcystis and Planktothrix have been shown to harbor the FDHs AerJ and McnD within aeruginosin and cyanopeptolin biosynthetic gene clusters, respectively, the presence of which corresponds to the production of halogenated congeners such as 32 and 34.110,111 While the activity of the two halogenases has not been reconstituted in vitro and physiological substrates have not been experimentally established, chemical logic suggests that the aeruginosin FDH AerJ mono- or di-chlorinates the first building block of the NRPS peptide, the L-tyrosine-derived 4-hydroxyphenyl lactate while it is acylated to the AerA CP domain. Hence, in effect, AerJ could resemble the β-tyrosine chlorinase SgcC3 in its activity (Figure 8B), though halogenation at a different stage of NRPS elongation cannot be presently ruled out. Chlorination by McnD is however localized to a L-tyrosine-derived late stage building block. Hence, as before, whether chlorination of the L-tyrosine aryl ring occurs during the NRPS assembly of 34, or whether a chlorinated tyrosine amino acid is incorporated by the associated adenylation domain cannot be rationalized based on chemical logic alone. This biosynthetic conundrum also presents itself en route to the production of chlorinated anabaenopeptilides, such as 35, by cyanobacteria of the genus Anabaena153 where the timing for halogenation by the FDH ApdC relative to the NRPS assembly has not been discerned, as well as during the production of chlorinated cryptophycins154 such as 33 by cyanobacteria of the genus Nostoc that likely involves the chlorination of the methoxytyrosine aryl ring by the FDH CrpH (Figure 8B).155 Sequence analyses of the cyanobacterial FDHs AerJ, McnD, ApdC, and CrpH reveal closest structural homologs to FDHs acting on acyl-S-CP substrates, pointing towards substrates for these enzymes to be loaded onto CPs as well. A close sequence homolog to CrpH is the myxobacterial tryptophan-2-halogenase CmdE involved in the biosynthesis of chlorinated cytotoxic NRPS derived chondramides, such as chondramide B (45, Figure 11D).156 Whether the biosynthetic scheme for the incorporation of chlorinated tryptophan residue in 45 differs from that of 36 and 38 remains to be experimentally verified. 45 shares close structural homology to the marine sponge Jaspis derived brominated jasplakinolides,157,158 such as 46 (Figure 11D), that also bear brominated tryptophan building blocks in a NRPS-derived scaffold. However, halogenation at the 2-position of tryptophan in 45 and 46 posits towards the presence of a tryptophan halogenase, such as CmdE, that differs in its regiospecificity for halogenation from all other tryptophan halogenases discussed previously. Interestingly, a shift in halogenation regiospecificity, including halogenation at the 2-position on the indole ring, has been reported for tryptophan-7-chlorinases with changes in the substrate chemical structure.89
Other notable halogenated natural products in which the timing of halogenation by FDHs has not yet been elucidated include the NRPS-derived antibiotics ramoplanin (47) and enduracidin (48) that target cell wall biosynthesis,159,160 and the unique 3-amino-5-hydroxybenzoic acid (AHBA) starter unit initiated PKS-derived chlorinated ansamacrolactams such as napthomycin A (49) and ansamitocin AP-1 (50) that inhibit RNA polymerase (Figure 13).161 Notably, the ansamitocin and napthomycin FDHs-Asm12162 and Nat1, respectively, could functionally replace each other in their respective deletion strains,138 possibly implying broad substrate tolerance. Deletion of the asm12 gene demonstrated the accumulation of mature deschloro-ansamitocin by the Actinosynnema pretiosum, implying that either halogenation could likely be late stage biosynthetic step, or that other Asm enzymes are promiscuous enough to accommodate both halogenated and deshalo intermediates in the ansamitocin biosynthetic scheme.163 A combinatorial replacement of FDHs implicated in the biosynthesis of 48 and 47, End30139 and Ram20, respectively, led to the production of novel derivatives of these antibiotics.164–166 However, unlike 3, chlorination is not essential for the bioactivity of 47.167 In a recent study using degenerate primers to screen for FDHs in mangrove actinomycetes, gene clusters for the production of 48 and ansamacrolactams were detected and novel producers of these antibiotics were identified.168 Using a similar approach but for Arctic marine actinomycetes, a homolog of the FDH Asm12 was identified, among several other FDHs that likely participate in the biosynthesis of as yet unidentified halogenated natural products.169
Figure 13. Chemical structure of chlorinated natural products 47–50.

The halogenated rings are shaded in blue.
Another intriguing knowledge gap exists in the timing of halogenation by the FDH CmlS en route the biosynthesis of 6 (Figure 1), a commonly employed antibiotic. Though the activity of CmlS has not been reconstituted, intriguingly, CmlS is the only FDH known to date that demonstrates a covalently tethered FAD cofactor to the enzyme active site,170 calling into question the hypothesis that reduced and oxidized flavin cofactors are diffusible from the FDH active site. Even though the biosynthetic gene cluster for the production of 6 has been known for more than a decade,171 the molecular details, and indeed the genetic players involved in the biosynthesis, are still being worked out.172
Though outside the purview of halogenated natural products biosynthesized via modular NRPS or PKS pathways, it is interesting to speculate on the timing of halogenation relative to the maturation of the lanthipeptide antibiotic microbisporicin (51)173,174 bearing the 5-chlorotryptophan residue that is likely synthesized by the FDH MibH encoded within the biosynthetic gene cluster (Figure 14).175 While the activity of MibH has not been reconstituted in vitro and the identity of the physiological substrate remains elusive, fidelity of peptide synthesis by the ribosome precludes that L-tryptophan will be the physiological substrate for MibH. Hence, like VhaA that accepts a peptidic late stage biosynthetic intermediate as the substrate, MibH would likely accept either the microbisporisin prepeptide as a substrate for halogenation, or a biosynthetic intermediate en route ring closures and removal of the leader peptide. Either way, MibH promises to expand the substrate scope for FDHs to now include ribosomally synthesized post-translationally modified peptides (RiPPs).176 Already, feeding of potassium bromide to the producer strain of 51 has revealed the production of brominated variants of 51 with improved antimicrobial efficacy.177
Figure 14. Chemical structure of the lanthipeptide antibiotic 51.

The halogenated indole ring is shaded in blue.
2.5 Predictive functional assignments
Can we use the inventory of FDHs presented above to glean information for as yet experimentally uncharacterized FDHs in natural product biosynthetic gene clusters? A phylogenetic analysis reveals that these FDHs have a propensity to cluster according to their substrate specificity. For instance, as shown in Figure 15, all FDHs halogenating pyrrolyl-S-CP substrates (red nodes), regardless of the source bacterial phyla, cluster together. Similarly, tryptophan (blue nodes) and CP-tethered phenoxyl FDHs (in purple) are organized in their respective clades. Notable is the absence of the myxobacterial chondramide tryptophan halogenase CmdE that instead clades with the fungal FDHs Rdc2, RadH, and CazI, together with the cyanobacterial FDH CrpH. As has been mentioned previously, whether or not CmdE catalyzes the halogenation of free tryptophan remains to be verified. If not, it begets the question whether Rdc2, RadH, and CazI catalyze halogenation of small molecule or CP-tethered substrates. It should be noted that the small molecules used for the activity reconstitution of Rdc2 and CazI bear resorcylic rings that are highly susceptible to electrophilic aromatic substitution by hypohalites. In themselves, the next nearest neighbors of Rdc2, RadH, CazI, CrpH, and CmdE are FDHs that catalyze halogenation for phenolic CP-tethered substrates. This analysis certainly has its limitations. Why CrpH clusters away from other cyanobacterial phenolic FDHs such as AerJ, McnD, and ApdC is presently not clear.
Figure 15. Neighbor-joining tree showing the relatedness of FDHs.

Phylogenetic analysis was performed using Mega. The scale bar indicates 0.2 changes per amino acid. Full length primary sequences of select FDHs that have been biochemically characterized, or identified within genetic context of natural product biosynthetic gene clusters are included as discussed in the text. The FDH Amm3 is implicated in the biosynthesis of the ammosamide alkaloids.
Another curious case is the clustering together of the FDH PrnC, implicated in the chlorination of the pyrrole ring of 10, with the FDH HalB (as yet unexplained contribution to the biosynthesis of 30) and the chlorinases Mpy10 and Mpy11 participating in the biosynthesis of 25 (green nodes). While the physiological substrate for HalB is yet to be identified, the contribution of Mpy10 and Mpy11 in the atroposelective cross-coupling of two monodeoxypyoluteorin monomers to generate 25 has been demonstrated in vivo.98 In a proposed mechanistic scheme, a cryptic halogenation at either the 3-position of the pyrrole ring, or on the pyrrole nitrogen should set up a cross-coupling reaction with the elimination of the halogen. In such a scenario, both Mpy10 and Mpy11 being present in the neighborhood of PrnC in a phylogenetic analysis makes biochemical sense. However, why does this scheme need the presence of two FDHs? Deletion of either mpy10 or mpy11 leads to loss in production of 25.98 Is one of the Mpy10/Mpy11 FDHs masquerading as a cross-coupling catalyst that also introduces atroposelectivity in the final natural product outcome? A comprehensive in vitro biochemical characterization of Mpy10 and Mpy11 enzymes is required to answer these open questions.
3. Vanadium-dependent haloperoxidases
With the majority of biogenic organohalogens being marine in origin, it was originally thought that heme-dependent haloperoxidases were responsible for catalyzing the bulk of the halogenation events. Using the MCD-assay that was employed in the initial characterization of CPO (Figure 2), the first V-HPO was isolated from the marine brown alga Ascophyllum nodosum.180 Upon its characterization, stoichiometric amounts of vanadium were identified instead of the proposed heme as the enzyme cofactor. To date, characterized V-HPOs have been primarily of marine origin. Vanadium-dependent bromoperoxidases (V-BPOs) are predominantly isolated from marine algae, while vanadium-dependent chloroperoxidases (V-CPOs) are found in fungi and marine-derived bacteria.
3.1 Structure and mechanism
V-HPOs utilize a chemical oxidant, such as hydrogen peroxide, to catalyze the oxidation of halides and, as for FDHs, are named after the most electronegative halide they are able to oxidize. Thus, V-CPOs can oxidize chloride, bromide and iodide, whereas V-BPOs only oxidize bromide and iodide. Because hydrogen peroxide lacks the thermodynamic potential to oxidize fluoride, vanadium-dependent fluoroperoxidases have not been found in nature.181 Vanadate is a required prosthetic group and is coordinated to the protein in a trigonal bipyramidal fashion through the side chain imidazole nitrogen atom of a conserved histidine residue (Figure 16). The overall negative charge of the vanadate decreases through an extensive hydrogen bonding network between vanadate’s three equatorial oxygen atoms and conserved amino acid residues in the active site.182–184 This vanadate coordination geometry is also observed for the transition-state mimics for di-metal ion-dependent phosphate and phosphonate esterases.185–188 As with the heme-dependent haloperoxidases discussed previously in the biosynthesis of 2 and 7, V-HPOs utilize hydrogen peroxide to catalyze the two-electron oxidation of halides to the corresponding reactive hypohalites. However, unlike the heme-dependent haloperoxidases, V-HPOs do not succumb to oxidative inactivation during turnover, as vanadate maintains the V(v) oxidation state throughout the catalytic cycle and is not redox active. After formation of the hypohalite intermediate, if the appropriate nucleophile is present, the reactive X+ species will react with the substrate to form a halogenated product with the overall stoichiometry being one halogenated product for every one equivalent of hydrogen peroxide consumed (Figure 16A). If the correct substrate is not present, the oxidized species can react with another equivalent of hydrogen peroxide to regenerate the halide and produce dioxygen in the singlet state.189 Whether hydrogen peroxide is the physiological halide oxidant has not been established, however, no other oxidants have yet been identified for V-HPOs and the V-HPO biochemistry has relied on using hydrogen peroxide in in vitro biochemical reactions.190 Unlike FDHs, crystal structures of V-HPOs have failed to reveal either the halide binding site, or the site at which substrates are bound by these enzymes. Moreover, the order of halide oxidation relative to substrate binding has not been revealed by kinetic studies. These shortcomings have precluded, for the most part, a rational engineering of V-CPOs and V-BPOs for tailored applications.
Figure 16. Overall mechanism and vanadate coordination in V-HPOs.

(A) Proposed catalytic scheme for generating the halogenating agent X+ in V-HPOs (adapted from Winter and Moore, 2009). In the first step of catalysis, hydrogen peroxide is coordinated to the vanadium center in a side-on manner. The peroxide bond is broken through a nucleophilic attack on the partially positive oxygen by a halide resulting in the release of the hypohalite. If the appropriate substrate (R) is present, a halogenated compound will be formed with the loss of water and regeneration of vanadate. If the appropriate substrate is not present, the hypohalite can react with hydrogen peroxide to regenerate the halide and form dioxygen in the singlet state. (B) Active site of native V-CPO from Curvularia inaequalis (PDB accession number 1IDQ). Active site residues are shown as yellow ball and sticks and vanadate is shown coordinated to the conserved His496 residue.
With respect to natural product biosynthesis, investigations involving V-HPOs have traversed two broad areas: first being the transformation of electron rich substrates to their halogenated products by marine eukaryotic V-BPOs. As illustrated below, these investigations lacked the genetic context of a dedicated natural product biosynthetic gene cluster and thus utilized electron rich substrates as biogenic precursors. The identities of the precursors were selected based on chemical logic dictating the biomimetic scheme via which the targeted natural product could be biosynthesized. The second, and more recent line of investigation, involves marine bacterial V-CPOs within confirmed genetic context of natural product biosynthetic schemes and gene clusters. The discussion below is thus divided to follow these two broad directions.
3.2 Halogenation by marine Eukarya
As has been mentioned previously, given the halide concentration in seawater, the majority of naturally occurring organohalogens, including nearly all brominated compounds, are of marine origin.193 Many of these halometabolites are produced by marine algae; with a majority of the compounds being isolated from species belonging to red macroalgae.34,189,194–198 It was hypothesized that V-BPOs could be responsible for the observed chemistry, and surveys of red, green and brown algae for bromoperoxidase activity demonstrated that red algae belonging to the genus Corallina had the highest activity.199,200 Initial characterization studies, however, with the V-BPO from C. pilulifer and its capability of halogenating anisole and prochiral aromatic compounds failed to show any regio- or stereospecific bromination selectivity.201 Because of its lack of selectivity, it was speculated that V-BPOs generated a hypobromite intermediate that was freely diffusible and would carry out bromination reactions outside the enzyme’s active site. However, several subsequent characterization studies discussed below have shown that V-BPOs can indeed halogenate a range of organic compounds in a regio- and stereospecific manner.
Halogenated sesquiterpenes are one of the largest groups of natural products isolated from marine algae, and as a consequence, it was initially hypothesized that most halogenated cyclic sesquiterpenes were biosynthesized via a halenium-induced cyclization of an acyclic terpene precursor.202,203 V-BPOs isolated from marine red algae (e.g. C. officinalis, Laurencia pacifica, and Plocamium cartilagineum) were shown to catalyze the cyclization and asymmetric bromination of the acyclic sesquiterpene (E)-(+)-nerolidol (52) to yield the α-, β-, γ-snyderols (53–55) and (+)-3β-bromo-8-epicaparrapi oxides (56–57) marine natural products (Figure 17).194 After the selective bromination of the C10-C11 olefin, the bromonium-intermediate is attacked by the internal olefin of 52 to yield the tertiary carbocation. This intermediate can then undergo one of three different elimination reactions leading to 53–55. Additionally, quenching of the reaction with the tertiary alcohol of 52 produces 56–57. Single diastereomers of 54 and 55 were produced in the enzyme reaction, whereas, in the synthetic reaction with 2,4,4,6-tetrabromocyclohexa-2,5-dienone, two diastereomers of each were formed. This study established the role of V-BPOs in the biosynthesis of brominated cyclic sesquiterpenes from marine red algae.
Figure 17.

V-BPO-catalyzed cyclization of 52 to form the brominated sesquiterpene marine natural products. Co-VBPO: C. officinalis V-BPO.
C15 acetogenins are nonterpenoid cyclic ether metabolites containing varying oxane ring systems and a conjugated enyne or bromoallene terminus.204 Eight-membered cyclic ethers are the most abundant C15 acetogenins, and Laurencia spp. are common producers of these halogenated metabolites. The eight-membered cyclic ethers can be divided into two subclasses: the lauthisan-type, which contains the metabolite laurencin (58, Figure 18), and the laurenan-type, which contains the metabolites laureatin (59) and laurallene (60).204 It was proposed that linear laurediols were a precursor to both subtypes, and that the assembly of the cyclic ether metabolites could proceed through a bromoetherification of the linear precursor. Incubation of (3E, 6R, 7R)-laurediol (61) with a crude preparation of a V-BPO from the red alga Laurencia nipponica produced the cyclic bromoether deacetyllaurencin (62, Figure 18A).205 When (3Z, 6S, 7S)-laurediol (63) was used as the substrate, the C15 acetogenin prelaureatin (64), which belongs to the laurenan class of cyclic ethers, was observed (Figure 18B).205 While yields of the brominated cyclic metabolites were low, results from the experiment established that cyclization of the acyclic laurediol precursors to the eight-membered bromoethers was initiated by a bromonium ion. The partially purified V-BPO from L. nipponica was also shown to convert (Z)-prelaureatin (65) to 59 and isolaureatin (66). (Figure 18C).206 However, unlike the lactoperoxidase that could also catalyze the bromonium-ion induced cyclization of (E)-prelaureatin to form the bromoallene-containing natural product laurallene, no turnover with the (E)-isomer was observed with the V-BPO thereby further supporting the notion that V-BPOs possess inherent substrate selectivity. Recently, results of the deacetyllaurencin study were validated using the more stable trimethylsilyl-capped (3E, 6R, 7R)-laurediol precursor and purified V-BPO from L. nipponica.207
Figure 18. V-BPO-catalyzed chemoenzymatic synthesis of brominated acetogenins.

(A) Bromolactonization of 61 to 62 using the L. nipponica V-BPO (Ln-VBPO). (B) Bromolactonization of 63 using partially purified Ln-VBPO. (C) Conversion of 65 to 59 and 66 using a partially purified Ln-VBPO.
Quorum sensing is a process used by bacteria to monitor cell density and regulate phenotypic responses such as biofilm formation.208 Many bacteria secrete small signaling molecules, such as 3-oxo-acyl homoserine lactones to signal cell density and to coordinate gene regulation within a population. In the ocean, all surfaces are susceptible to biofouling and in order to survive, marine algae must have access to sunlight and nutrients. It was proposed that for some macroalgal species, V-HPOs are present on the plant surface and produce hypohalous acids that function as anti-fouling agents.209 Studies of gene expression in the brown alga Laminaria digitata demonstrated that V-BPOs and vanadium-dependent iodoperoxidases (V-IPOs) are up-regulated upon defense elicitation.210 Interestingly, the 3-oxo-acyl homoserine lactone signaling molecules are readily susceptible to electrophilic halogenations at the C2 position.211 When incubated with the red alga Delisea pulchra, dibromination at the C2 position of 3-oxo-acyl homoserine lactone 67 was observed (Figure 19A). The resulting α,α-3-oxo-hexanoylhomoserine lactone 68 disrupts bacterial quorum sensing, suggesting that V-BPOs are localized at the alga’s surface to defend against microbial colonization.212 D. pulchra also produces a series of bromofuranone natural products that inhibit quorum sensing in select bacteria by interfering with receptor-mediated phenotypic responses.212 As the precursors for the naturally occurring bromofuranones are unknown, 4-pentynoic acid (69) was used as a substrate for biomimetic characterization experiments. The V-BPO isolated from D. pulchra was shown to catalyze the bromolactonization of 69 to form 5E-bromomethylidenetetrahydro-2-furanone (70, Figure 19B). Compared to the naturally occurring bromofuranones (Figure 19C),213,214 70 lacks a degree of unsaturation within the furanone ring. However, the enzymatically synthesized 70 was still able to inhibit quorum sensing in Agrobacterium tumefaciens. Altogether, results from these studies demonstrated that to mitigate microbial fouling on its surface, the red alga D. pulchra has evolved a two-pronged approach for disrupting bacterial quorum sensing, that is, brominative inactivation of 3-oxo-homoserine lactones and synthesis of brominated furanone inhibitors of quorum sensing. These results posit towards the ecological roles of halogenases in small molecule mediation of marine chemical ecology.
Figure 19. V-BPO-catalyzed chemoenzymatic synthesis of brominated furanones.

(A) Bromination of acylhomoserine lactone 67 to 68 was demonstrated using whole pieces of D. pulchra. (B) Bromolactonization of 69 to 70. (C) Bromofuranones isolated from D. pulchra.
3.3 Biosynthetic schemes in marine bacteria
The first V-CPOs were identified from the terrestrial fungus Curvularia inaequalis in 1987215,216 and marine fungus Embellisia didymospora in 1998.217 However, no halogenated metabolites were reported from either source. It was therefore suggested that fungal V-CPOs generate hypochlorous acid for the chlorination and degradation of lignin.218 While there are numerous chlorinated natural products, especially from the marine environment, it wasn’t until 2007 that the first V-CPO affiliated with a natural product was identified.219
Meroterpenoids are natural products of mixed polyketide-terpenoid origin and while they are commonly isolated from fungi and higher plants, only a small group of metabolites have been identified from bacteria.220–222 The biosynthetic gene cluster responsible for the synthesis of meroterpenoids belonging to the napyradiomycin family of chlorinated dihydroquinones, including the tri-chlorinated napyradiomycin A80915C (71), was identified in Streptomyces aculeolatus NRRL 18422 and the marine-derived Streptomyces sp. CNQ-525.219 Through heterologous expression, the nap biosynthetic gene cluster from S. sp. CNQ-525 was shown to be wholly responsible for the synthesis of the chlorinated meroterpenoid natural products. Three putative V-HPO genes were annotated within the nap biosynthetic gene cluster, which was consistent with the structural inspection of the chlorinated napyradiomycins in which their terpenoid fragments might undergo chloronium ion-induced cyclization biochemistry in a manner reminiscent of snyderol biosynthesis as depicted in Figure 17.
Structural and functional comparisons of V-HPOs from eukaryotic organisms have shown that seven amino acids are required for halogenating activity.217,223–225 All three nap V-HPO enzymes contain His-496 (which covalently binds to the vanadate cofactor) and five of the six residues that participate in the hydrogen bonding of vanadate: Lys-353, Arg-360, Ser-402, Gly-403, and Arg-490 (numbering from C. inaequalis). Conserved residue His-404, which is proposed to form a hydrogen bond to the apical oxygen of the cofactor, is replaced with Ser, as seen in NapH1 and NapH4, or Phe as seen in NapH3. Mutagenesis studies have shown that the V-CPO mutant H404A from C. inaequalis loses chlorinating activity225 and a similar natural exchange is observed in NapH3. Hence, NapH3 is most likely not involved in halogenation and may have a different biosynthetic function in the assembly of the napyradiomycins. NapH1 and NapH4, on the other hand, contain a hydrophilic Ser residue at this position that can still hydrogen bond with the apical oxygen atom and were therefore predicted to catalyze the chloronium-induced cyclization of the two terpene units. It is worth pointing out that based on the MCD-assay, NapH1 was predicted to be a V-BPO.226 Additionally, a critical review of the MCD-assay was written after attempts to identify the enzymes involved in the synthesis of the ambigols and tjipanazoles in the cyanobacterium Fischerella ambigua yielded false positive results. Caution should therefore be exercised when using the MCD-assay as the sole source for identifying a putative V-HPO and/or assigning its physiological halide specificity.227 However, the utility of the MCD assay in predicting halogenating potential for a haloperoxidase enzyme at an early stage in the characterization process remains unquestioned. Ultimately, the proposed role of NapH1 in the biosynthesis of the napyradiomycins was established through in vitro chemoenzymatic conversion of the meroterpenoid SF2415B1 (72) to SF2415B3 (73, Figure 20).226 Single diastereomers of 73 were observed confirming that NapH1 mediates the stereoselective chlorination-cyclization of the hemiterpene moiety in which the chloronium ion is quenched by a nucleophilic attack from the hydroxyl group on the dihydroquinone core. It is still unclear if NapH4 or NapH1 is responsible for the second oxidative cyclization event to convert 73 to 71. As the V-CPOs were identified within a biosynthetic operon, the napyradiomycin system provided a unique opportunity to characterize V-HPOs in their biosynthetic context.
Figure 20.

Chloronium-mediated cyclization of the napyradiomycin intermediate 71 to 72 catalyzed by the bacterial V-CPO NapH1. Tetrahydroxynaphthalene is the PKS-derived building block and is assembled by a type III PKS encoded within the biosynthetic gene cluster.
Recently, a new group of chlorinated meroterpenoids, merochlorins A−D (74–77), were identified from the marine-derived bacterium Streptomyces sp. CNH-189.228,229 While 74 contains a highly unusual bicycle[2.2.1]-heptanone ring system, 75 and 76 possess distinctive carbon skeletons that are unprecedented in natural products (Figure 21). A close inspection of the structures suggested that the merochlorins were possibly assembled by a V-CPO-dependent chloroetherification reaction similar to the napyradiomycins. Through genome sequencing and heterologous expression, the merochlorin biosynthetic cluster was identified and found to contain two V-CPO homologs (Mcl24 and Mcl40). Through differential expression of the biosynthetic pathway, V-CPO Mcl40 was implicated in the chloronium-induced macrocyclization of the isosesquilavandulyl terpenoid moiety to convert 77 to 76, however, characterization of this enzyme in vitro has yet to be achieved (Figure 21).228
Figure 21. Chloronium-mediated cyclization of the merochlorin meroterpenoids.

Mcl40 and Mcl24 are characterized V-CPOs from the merochlorin biosynthetic cluster, whereas Mcl23 is a characterized prenyltransferase that attaches the isosesquilavandulyl terpenoid moiety onto the tetrahydroxynapthalene-derived polyketide building block.
Subsequent in vitro studies revealed the role of the V-CPO Mcl24. Surprisingly, only four enzymes (Mcl17, 22–24) are required for the complete biosynthesis of 74 and 75 from the biosynthetic precursors malonyl-CoA, dimethylallyl pyrophosphate (DMAPP), and geranyl pyrophosphate (GPP).230 The prenyl diphosphate synthase Mcl22 was shown to catalyze the unusual “head-to-torso” coupling of DMAPP and GPP to form the newly established sesquiterpene isosesquilavandulyl pyrophosphate. The prenyltransferase Mcl23 then attaches the isosesquilavandulyl moiety to the aromatic tetrahydroxynaphthalene scaffold, previously formed through the activity of the type III polyketide synthase Mcl17, to yield the triene intermediate pre-merochlorin (78).230 The identification of 78 suggested an oxidative cyclization event must occur in order to form the polycyclic cores of 74 and 75. This was confirmed by in vitro characterization of Mcl24, wherein incubation with enzymatically prepared 78 afforded both 74 and 75. Ultimately, the entire pathway was reconstituted in a one-pot in vitro reaction.230
In an accompanying study, the novel Mcl24 transformation was further interrogated.231 Mcl24 was inactive when assayed with other simple terpene alcohols and phenolic and naphtholic substrate analogs, establishing enzyme specificity and confirming its role in a biosynthetic pathway, in contrast to the non-specific halogenation observed in many V-HPOs characterized to date. As with NapH1, Mcl24 was unable to oxidize 8 in the presence of potassium chloride, and oxidation only occurred with addition of potassium bromide underscoring the importance of exhibiting caution when conducting assays with this model substrate. The mechanism for intramolecular cyclization was hypothesized to take place via a chloronium-induced oxidative dearomatization. To further probe the enzyme mechanism, a chemical chlorination reaction was designed to induce oxidative dearomatization/cyclization of 78. The most efficient reaction conditions included N-chlorosuccinimide and diisopropylamine.231 Formation of a lysine chloramine intermediate has been observed in the FDH RebH, as previously described.45 Therefore, it is possible that V-HPO enzymes have evolved to direct regiospecific chlorination, perhaps via a conserved lysine residue in the vicinity of the vanadate cofactor. In addition, the product distribution using the biomimetic preparation of merochlorins was compared to that of the Mcl24 enzyme reaction with 78 as the starting substrate. Structural characterization of the products identified three major compounds of the synthetic reaction as minor components in the enzymatic reaction (isochloro-merochlorin B and deschloro-merochlorin A and B), while the major components merochlorin A and B in the enzymatic reaction were only minor components in the biomimetic synthesis – essentially a reversal of selectivity.231 Re-isolation of the merochlorin series from the natural producing strain confirmed the presence of these minor compounds, albeit in small amounts, demonstrating the utility of biomimetic synthesis.
Based upon both the enzymatic and synthetic studies, a hypothetical mechanism was posited that begins with selective C2 chlorination of 78. This is followed by an additional O-chlorination forming an aromatic hypochlorite intermediate, which then rearranges to a benzylic carbocation intermediate upon chloride loss. The formation of a benzylic carbocation intermediate had previously been postulated in separate studies on the total syntheses of both merochlorin A and B.232,233 Finally, differential cation-induced terpene cyclizations result in either merochlorin A or B. This study established the novel activity of the V-CPO Mcl24, in that, it not only mediates the site-selective chlorination of the tetrahydroxynaphthalene core, but sets off the cascade of oxidative dearomatization and isosesquilavandulyl cyclization events to yield the complex scaffolds of the merochlorins.
To date, bacterial NapH1 and Mcl24 remain the only V-HPOs that have been characterized within the genetic and biochemical context of natural product biosynthesis. This reflection is tempered, in no small part, by the advances in sequencing and assembly of bacterial draft genomes, and clustering of natural product biosynthetic genes in bacterial genomes that allow for heterologous expression and biochemical rationale for substrates to mature at a faster rate than for eukaryotic halogenases discussed previously. Recently, a V-BPO was identified in the sequenced genome of marine cyanobacterium Synechococcus sp. CC9311.234,235 While the physiological substrate for the V-BPO has not been identified, the genetic proximity of the V-BPO to phosphoenolpyruvate mutase enzyme lends speculation to its involvement in the biosynthesis of marine halogenated phosphonate natural products.236,237
3.4 Structure-sequence relationships
There are currently five V-HPO crystal structures available and include the V-CPO from the fungus C. inaequalis,182,238,192 V-BPO from the brown alga Ascophyllum nodosum,184 V-BPO from the red alga C. officinalis,183 V-IPO from the marine bacterium Zobellia galactanivorans,239 and the V-CPO NapH1 from S. sp. CNQ-525 (Table 1). While the proteins share very little sequence identity and differ dramatically in their reported oligomeric states, their structures are almost superimposable when overlaid around the vanadate center. This core structure consists of mainly α-helices with two four-helix bundles as the tertiary structural motif and an almost identical arrangement of amino acids surrounding the vanadate active site (Figures 16B, 22).183,192,238,239 Given the similarities in active site residues between the three sub-classes of V-HPOs, several site-directed mutagenesis studies have targeted subtle differences between the three classes in hopes of bringing to light the residue(s) responsible for halide selectivity and binding.183,201,225,238–243 As mentioned previously, no halide or substrate co-crystal structures are available for this class of halogenases. One such difference that has been targeted is Phe397 (numbering from C. inaequalis). Phe is present at this position in the fungal V-CPO and bacterial V-IPO structures, whereas V-BPOs and the V-CPO NapH1 contain His. It was proposed that while this residue does not directly interact with vanadate, the His side chain could affect the overall oxidation of the peroxo-vanadate complex by acting as a proton donor and acceptor during catalysis.201,244 Interestingly, while the bacterial V-IPO contains Phe at this position and is unable to oxidize chloride in the thymol blue assay,245 the bacterial V-CPO NapH1 contains His and was unable to oxidize chloride in the MCD-assay. However, when provided its natural substrate, 72, NapH1 catalyzed the stereoselective chlorination-cyclization of the hemiterpene moiety. With the crystal structure of the V-CPO NapH1 in hand, further co-crystallization experiments with napyradiomycin intermediates will provide further information on biosynthetic function and identify residues involved in substrate selectivity.
Table 1.
PDB accession codes for crystal structures of halogenases
| Enzyme | Natural product | Organism | PDB id (mutant enzyme) | Physiological substrate | Structurally described cofactors and ligands |
|---|---|---|---|---|---|
| FAD-dependent halogenases | |||||
| PrnA | 10 | Pseudomonas fluorescens | 2APG 2AQJ 2AR8 2ARD 4Z44 (F454K) 4Z43 (E450K) 2JKC (E346D) |
L-tryptophan | FAD, Cl− FAD, Cl−, Trp FAD, Cl−, 7-Cl-Trp FAD FAD, Cl− FAD, Cl− FAD, Cl−, Trp |
| RebH | 11 | Lechevalieria aerocolonigenes | 2OAM 2OAL 2E4G 2OA1 2O9Z 4LU6 |
L-tryptophan | Apo FAD, Cl− Trp FAD, Cl−, Trp Apo Apo |
| PyrH | 15 | Streptomyces rugosporus | 2WEU 2WET 2WES (E46Q) |
L-tryptophan | Trp FAD, Cl−, Trp FAD, Cl− |
| SttH | Unidentified | Streptomyces toxytricini | 5HY5 | L-tryptophan | FAD, Cl− |
| Bmp2 | 24 | Pseudoalteromonas PS-5 | 5BVA 2BUL (Y302S F306V A345W) |
pyrrolyl-S-CP | FAD FAD |
| Mpy16 | 25 | Streptomyces sp. CNQ-418 | 5BUK | pyrrolyl-S-CP | FAD |
| PltA | 22 | Pseudomonas protegens | 5DBJ | pyrrolyl-S-CP | FAD, Cl− |
| CndH | chondrochlorens | Chondromyces crocatus | 3E1T | Unidentified | FAD, Cl− |
| CmlS | 6 | Streptomyces venezuelae | 3I3L | Unidentified | FAD |
| Vanadium-dependent haloperoxidases | |||||
| Fungus (V-CPO) |
Unidentified | Curvularia inaequalis | 1IDQ 1IDU 1VNE (D292A) 1VNF (R360A) 1VNG (H404A) 1VNH (H496A) 1VNI 1VNS 1VNC |
Unidentified | VO4 VO4 VO4 VO4 VO4 VO4 VO4 SO4 VO4 |
| Red algae (V-BPO) |
54–57 and brominated furanones | Corallina officinalis | 1QHB | Unidentified | PO4 |
| Brown alga (V-BPO) | Unidentified | Ascophyllum nodosum | 1QI9 5AA6 (A68V V87I V103M A203V A204V L327M) |
Unidentified | PO4, I− VO4 |
| zobellia_1262 (V-IPO) | Unidentified | Zobellia galactanivorans | 4USZ 4CIT |
Unidentified | VO4 VO4 |
| NapH1 | 73 | Streptomyces sp. CNQ-525 | 3W36 3W35 |
72 | VO4 Apo |
| Red algae (V-BPO) |
54–57 and brominated furanones | Corallina pilulifera | 1UP8 | Unidentified | PO4 |
| Non-heme iron-dependent halogenases | |||||
| SyrB2 | 82 | Pseudomonas syringae | 2FCU 2FCV 2FCT |
L-threonine-S-CP | α-KG α-KG, Fe(II), Br− α-KG, Fe(II), Cl− |
| CytC3 | 83 | Streptomyces sp. | 3GJA 3GJB |
aminobutyryl-S-CP | Apo α-KG, Fe(II) |
| CurHal | 88 | Moorea producens | 3NNF 3NNL 3NNM 3NNJ |
(S)-3-hydroxy-3-methylglutaryl-CP | α-KG, Fe(II), Cl− α-KG, Fe(II), Cl− Apo Apo |
| WelO5 | 92 | Hapalosiphon welwitschii | 5IQT 5IQS 5IQU 5IQV |
12-epi-fischerindole U | α-KG, Fe (II), Cl−, 12-epi-fischerindole U α-KG, Fe(II), Cl− α-KG, Fe (II), 12-epi-fischerindole U α-KG, Fe (II), Cl−, 12-epi-fischerindole U, nitric oxide |
| SAM-dependent halogenases | |||||
| FlA | 94, 95 | Streptomyces cattleya | 1RQR 1RQP 2C2W |
SAM, F− | 5′-FDA, L-Met SAM 5-ClDA |
| SalL | 4 | Salinispora tropica | 2Q6I 2Q6K 2Q6O (Y70T) 2Q6L (Y70T G131S) |
SAM, Cl− |
5′-ClDA, L-Met Adenosine SAM, Cl− 5′-ClDA, L-Met |
| MJ1651 | Unidentified | Methanococcus jannaschii | 2F4N | SAM, H2O† | |
| PH0463 | Unidentified | Pyrococcus horikoshii | 1WU8 | SAM, H2O† | Adenosine |
| TTHA0338 | Unidentified | Thermus thermophilus | 2CW5 | SAM, H2O† | |
| AtHTMT | 97–99 | Arabidopsis thaliana | 3LCC | SAM, X− | SAH, Cl− |
SAM has been shown to be a substrate in vitro. It remains to be demonstrated if SAM is indeed the physiological substrate
Figure 22. Comparison of V-HPO structures.

(A) V-CPO from C. inaequalis (PDB: 1IDQ); (B) V-CPO from S. sp. CNQ-525 (PDB: 3W36); (C) V-BPO from A. nodosum (PDB: 1QI9); (D) V-IPO from Z. galactanivorans (PDB: 4USZ); and (E) V-BPO from Corallina officinalis (PDB: 1QHB). (Top panel): Ribbon structures with active site residues highlighted in yellow. (Bottom panel): Electrostatic surface representation showing a cross section of the cavity leading to the active site with bound vanadate or phosphate. Active site side chains, vanadate and phosphate are shown in ball and stick representation.
The high conservation among the V-HPOs around the active site suggests that they arose from a common ancestor and may share an evolutionary relationship with the acid phosphatases.191 Phylogenetic analysis of V-BPOs, V-CPOs, V-IPOs, and bacterial non-specific acid phosphatases shows that the enzymes form clades based on proposed enzymatic function (Figure 23). The three V-CPOs from the napyradiomycin biosynthetic cluster (NapH1, NapH3 and NapH4) and two V-CPOs from the merochlorin cluster (Mcl24 and Mcl40) do not clade with the fungal or other postulated bacterial-derived V-CPOs that are proposed to synthesize hypohalous acid. Instead, these enzymes form a distinct clade that is consistent with their proposed dedicated roles in natural product biosynthesis. While NapH1 and Mcl40, which have both been shown to facilitate chlorolactonization reactions, are closely related, the multitasking V-CPO Mcl24 is surprisingly more related to the non-halogenating V-CPO NapH3. This suggests that NapH3 may too catalyze unprecedented biochemical reactions and characterization studies will help elucidate this new chemistry. With genome sequencing becoming a common laboratory technique, new prokaryotic V-HPO homologs possessing new biological functions are emerging. Several promising homologs have been identified and clade with the bacterial V-CPOs involved in natural product biosynthesis. A closer investigation of these putative clusters may reveal new biological functions that go beyond the known chemistry of currently characterized V-HPOs.
Figure 23. Neighbor-joining tree showing the relatedness of V-BPOs, V-CPOs, V-IPOs and acid phosphatases identified from fungi, algae and bacteria.

Phylogenetic analysis was performed using Mega. The scale bar indicates 0.2 changes per amino acid. Sequences in which crystal structures are available are in bold. Sequence identification codes include Ci_VCPO from Curvularia inaequalis (GenBank accession number CAA59686); Rb_VCPO from Rhodopirellula baltica SH1 (CAD72609); Ed_VCPO from Embellisia didymospora (CAA72622); NapH1 VCPO from Streptomyces sp. CNQ-525 (ABS50486); NapH3 VCPO from Streptomyces sp. CNQ-525 (ABS50491); NapH4 VCPO from Streptomyces sp. CNQ-525 (ABS50492); Mcl40 VCPO from S. sp. CNH-189 (469658154); Mcl24 V-CPO from S. sp. CNH-189 (469658138); Cs_VCPO from Cellulophaga sp. MED134 (ZP_01050453); Ss1_VCPO1 from Streptomyces sp. CNQ-766 (WP_018841016); Ss2 VCPO from Streptomyces sp. CNQ-766 (WP_018836294); Ss3 VCPO from Streptomyces sp. CNQ-329(WP_027774460); Ss4_VCPO from Streptomyces sp. CNT-371 (WP_027745869); Ss5_VCPO from Streptomyces sp. CNQ-865 (WP_027769314); Ss6_VCPO from Streptomyces sp. CNT-371 (WP_027744001); Ss7_VCPO from Streptomyces sp. CNQ-509 (WP_027745869); Ss8_VCPO from Streptomyces sp. SBT349 (WP_049575625); Ss9_VCPO from Streptomyces sp. CNQ-509 (WP_047020372); Ss10_VCPO from Streptomyces sp. CNS-335 (WP_018842950); Ss11_VCPO from Streptomyces sp. MMG1121 (WP_053666624); Sac_VCPO from Saccharomonospora saliphila (WP_019819575); Am_VCPO from Actinopolyspora mortivallis (WP_019852972); An_VBPO from Ascophyllum nodosum (P81701); Co_VBPO from Corallina officinalis (AAM46061); Cp1_VBPO from Corallina pilulifera (BAA31261); Cp2_VBPO from Corallina pilulifera (BAA31262); Sys_VBPO from Synechococcus sp. CC9311 (YP_731869); Fd_VBPO from Fucus distichus (AAC35279); Ld1_VBPO from Laminaria digitata (CAD37191); Ld2_VBPO from Laminaria digitata (CAD37192); Ld1_VIPO from Laminaria digitata (CAF04025); Ld3_VIPO from Laminaria digitata (CAQ51446); Am1_VIPO from Algoriphagus marincola HL-49 (KPQ20079); Am2_VIPO from Algoriphagus marincola HL-49 (KPQ13775); Zg1_VIPO from Zobellia galactanivorans (4USZ); Zg2_VIPO from Zobellia galactanivorans (CAZ96246); Pi_ACP acid phosphatase from Prevotella intermedia, phoC (AB017537); Kp_ACP acid phosphatase from Klebsiella pneumoniae, phoC (AJ250377); St_ACP acid phosphatase from Salmonella typhimurium, phoN (X63599); Ps_ACP acid phosphatase from Providencia stuartii¸ phoN (X64820); and Eb_ACP acid phosphatase from Escherichia blattae (AB020481).
It was originally thought that V-HPOs were non-specific halogenases. However, with recent in vitro and in vivo characterization experiments, the enzymes have received increasing attention due to their ability to not only halogenate a range of organic compounds, but because they have the capability to do so in a regio- and stereospecific manner. Marine bacteria, and in particular actinomycetes, produce a wide range of halogenated secondary metabolites and represent an untapped resource for the discovery of new halogenating enzymes. Whole genome sequence in combination with phylogenetic analysis will provide a powerful platform for identifying promising candidates.
4. Non-heme iron-dependent halogenases
As shown for the heme-dependent HPOs, FDHs, and V-HPO halogenating enzymes, convergent strategies have evolved for addition of an oxidized electrophilic halogen atom to electron rich centers. However, this chemical logic cannot account for natural products that are halogenated on unactivated alkyl carbon atoms. Therefore, a separate halogenation strategy must be employed. To accomplish a reaction of this type, radical chemistry can be invoked negating the need for prior functionalization of an aliphatic substrate, and indeed, the α-ketoglutarate-dependent non-heme iron (NHFe) halogenases have evolved to utilize this strategy during halogenation. The reaction is dependent upon O2, α-ketoglutarate, and Fe(II) for halogenation of the substrate, ultimately forming succinate, CO2, and the halogenated product. The reducing power of α-ketoglutarate is harnessed through oxidative decarboxylation of this co-substrate to drive the overall oxidation reaction. Just like FDHs that borrow their underlying mechanistic logic from oxygenases and hydroxylases, NHFe halogenases are closely related to NHFe hydroxylases that catalyze analogous hydroxylations on unactivated carbon atoms. The evolution of our understanding for NHFe halogenases is however opposite of that of FDHs, in that, NHFe halogenases acting on CP-tethered substrates were discovered and characterized earlier than NHFe halogenases acting on free small substrates. But, like for FDHs, a crystallographic description of substrate engagement in the halogenase active site had to await the recent discovery of a free-standing small molecule NHFe halogenase, as will be described below.
The structural and mechanistic description of the NHFe halogenase reaction is provided in a later section (Section 4.d) in light of the evolution of the discovery and understanding of these remarkable enzymatic catalysts. Furthermore, a mechanistic switch between NHFe halogenases and NHFe hydroxylases has recently been realized with the characterization of a small molecule NHFe halogenase, a conversion that is as yet unrealized for FDHs, and will be described together with the structure-mechanism description of NHFe halogenases in Section 4.d.
Natural products synthesized by NHFe halogenases can be separated into two distinct groups based on the presence of a visible chlorine atom, or a cyclopropyl ring present in the final product, the latter of which has been termed cryptic chlorination due to the requirement of a chlorination event prior to cyclopropyl ring formation. The loss of a chloride ion facilitates ring closure, and chlorine is no longer visible in the final product.
4.1 Halogenation leading to visible chlorination
The first notion that radical chemistry may play a role in halogenation was introduced during the investigation of the biosynthesis of barbamide (79), a marine natural product containing a trichloromethyl group (Figure 24).246 Studies using various 13C- and 2H-labeled leucine substrates fed to the native producer, the marine cyanobacterium Moorea producens (formerly Lyngbya majuscula), established that no prior activation of the methyl group was necessary for the trichlorination reaction to occur.247 This observation suggested a different mechanistic route to chlorination than what had been reported previously. Subsequently, in vitro reconstitution of halogenation activity was accomplished by the sequential action of the NHFe halogenases BarB2 and BarB1 on the substrate L-leucine attached to the CP BarA.248 Additional BarB2 and BarB1 homologs (DysB2 and DysB1) were identified in cyanobacterial symbionts known to produce the hexachlorinated natural products dysidenin (80) and dysideathiazole (81),81 however, recapitulation of activity has yet to be accomplished. The first example of in vitro reconstitution of enzymatic NHFe halogenase activity was demonstrated in the context of the phytotoxin syringomycin E (82) biosynthesis in the organism Pseudomonas syringae pv. syringae B301D. This nine-residue lipopeptide harbors a 4-chloro-L-threonine residue formed through radical mediated halogenation catalyzed by the NHFe annotated SyrB2. Recombinant expression, purification, and reconstitution of SyrB2 with α-ketoglutarate and Fe(II) afforded ~0.85–0.95 mol Fe per monomer. Activity was recapitulated by incubation of SyrB2 with the tethered amino acid substrate L-threonine-S-SyrB1 to form 4-chloro-L-threonine-S-SyrB1and the reaction was found to be dependent upon O2, α-ketoglutarate, Fe, and chloride.249
Figure 24. Natural products bearing halogen atoms on non-activated aliphatic carbon centers.

Representative natural products shown here are biosynthesized via the action of NHFe halogenases as described in the text.
In the biosynthesis of dichloroaminobutyrate, also known as the actinomycete natural product armentomycin (83), the NHFe halogenase CytC3 is responsible for the formation of both 4-chloro- and 4,4-dichloroaminobutyryl-S-CytC2 from aminobutyryl-S-CytC2, whereby the thioesterase CytC4 releases 83 from the CytC2 CP.250 The chlorine substituent in the natural product hectochlorin (84) was recently confirmed to be the result of activity by the enzyme HctB that catalyzes the conversion of a CP-tethered fatty acyl moiety, hexanoyl-S-HctB, to 5,5-dichlorohexanoyl-S-HctB.251 Note that the halogenase and the CP domains are encoded within the same peptide in HctB. A similar transformation has been hypothesized in the biosynthesis for the structurally related lyngbyabellins, such as lyngbyabellin A (85) shown in Figure 24.252 Finally, as has been discussed previously, kutznerides are a series of cyclic depsipeptides that harbor multiple chlorine atoms on non-proteinogenic residues (Figure 11, representative molecule 38). One residue of particular interest harbors a 5-chloropiperizate moiety. Initial identification and genetic characterization of the biosynthetic gene cluster suggested a single NHFe halogenase, in addition to two putative FDHs.133 Subsequent analysis of the entire genome sequence revealed an additional NHFe halogenase, KthP, just upstream of the initially proposed cluster that catalyzes the conversion of (3S)-piperazyl-S-KtzC to (3S,5S)-5-chloropiperazyl-S-KtzC.253 Each of the abovementioned NHFe halogenases work on CP-tethered substrates, and as discussed for pyrrolyl-S-CP dependent FDHs, catalyze one or up to two halogenation events on their respective substrate carbon centers. How an NHFe halogenase tailors this activity is not known, but the discrimination is likely encoded within the halogenase active site itself as demonstrated for FDHs.38
4.2 Cryptic halogenation leading to cyclopropane ring formation
While addition of a halogen substituent is the outcome of a NHFe halogenase reaction, the same class of enzymes are involved in biosynthetic pathways where halogenation may not be obvious. In addition to the evident multiple chlorine atoms adorning the kutznerides (Figure 11), the biosynthetic pathway contains an additional NHFe halogenase that is responsible for the biosynthesis of the 2-(1-methylcyclopropyl)glycine residue. Characterization in vitro showed that the NHFe halogenase KtzD catalyzes formation of 4-chloro-L-isoleucine-S-KtzC from L-isoleucine-S-KtzC.254 However, subsequent cyclopropyl ring formation catalyzed by the flavoprotein KtzA, affords (1S,2R)-allocoronamic acid tethered to KtzC. This intramolecular elimination erases the previous evidence of chlorination justifying the use of “cryptic” to describe the initial chlorination event. An analogous pathway of coronamic acid (86) biosynthesis is present in the biosynthesis of the phytotoxin coronatine (87) (Figure 25A). The NHFe halogenase CmaD converts L-allo-isoleucine-S-CmaD to 4-chloro-L-allo-isoleucine-S-CmaD.255 In this instance however, subsequent cyclopropyl ring formation is catalyzed by the zinc-dependent enzyme CmaC.256 Activation via cryptic halogenation to assist the formation of the cyclopropane rings in natural products mentioned above is in line with the underlying chemical logic for N-C coupling and ring closure in marinopyrrole (25) and merochlorin (74–76) biosynthesis, respectively. However, a completely different logic of methylation of an alkene by a radical S-adenosyl-L-methionine (SAM) dependent methyltransferase and cyclopropane ring closure is followed in the biosynthesis of mycolic acids257 and the recently described natural product yatakemycin,258 while a radical modification of the valine side chain to afford a norcoronamic acid building block in NRPS-derived natural products has also been reported.259 The reader is directed to an excellent review on this topic.260
Figure 25. Halogenation assisted biosynthesis of cyclopropane rings and vinyl chlorides in natural products.

(A) Structures of 86 and 87. (B) The biosynthetic origin of both the vinyl chloride (Jam, 88 and 89) and cyclopropane group (Cur, 87) arises from the initial action of an NHFe halogenase (Hal). A conserved dehydratase domain (ECH1, 94% sequence identity between Cur/Jam) is responsible for an identical dehydration reaction, however, the pathways then deviate through differing activities of the respective decarboxylase domains (ECH2). In jamaicamide biosynthesis, this results in the vinyl chloride moiety present in both 89 and 90. In the biosynthesis of 88, the product of the ECH2 domain is further modified by a novel NADPH-dependent enoyl reductase (ER) forming the cyclopropyl moiety.
The biosyntheses of the jamaicamides and curacin in the marine cyanobacterium M. producens provides a unique example of evolutionary interplay as the pathways bear striking homology, yet result in both forms of chemistry discussed above (Figure 25B). The mixed polyketide-nonribosomal peptide natural products differ in the presence of either a cyclopropane or a vinyl chloride moiety in curacin A (88) or jamaicamides A (89) or B (90), respectively. Surprisingly, each NHFe halogenase domain is located in a multidomain protein and the halogenase domains share 92% sequence identity. A thorough biosynthetic investigation revealed the activity of the predicted NHFe halogenase (Cur Hal) involved in the biosynthesis of 88 to be chlorination at the aliphatic carbon of a (S)-3-hydroxy-3-methylglutaryl-CP ((S)-HMG-CP) substrate.261 While the activity of the jamaicamide halogenase was not investigated due to the high sequence identity to the Cur Hal, the structural diversity between the two natural products was determined to arise due to the difference in activity of the downstream decarboxylase and enoyl reductase domains, the latter of which was found to catalyze a novel NADPH-dependent intramolecular nucleophilic substitution to afford the cyclopropane ring in 88.
4.3 Halogenation of small molecules
Recently, the welwitindolinone alkaloid biosynthetic gene cluster from the cyanobacterium Hapalosiphon welwitschii was identified and characterized.262,263 Welwitindolinone alkaloids are biosynthetically related to hapalindole, ambiguine, and fischerindole alkaloids, and bear a chlorine at C-13 (Figure 26).264 Bioinformatic analysis of the cluster identified a putative NHFe halogenase (WelO5) that was subsequently characterized in vitro to catalyze the monochlorination of the aliphatic C-13 of 12-epi-fischerindole U and 12-epi-hapalindole C to form 12-epi-fischerindole G (91) and 12-epi-hapalindole E (92), respectively.265 This is the first reported example of an NHFe halogenase catalyzing the chlorination of a small molecule, specifically a substrate not tethered to a CP, expanding the scope of NHFe halogenase catalysis. An additional biosynthetic pathway from the cyanobacterium Fischerella ambigua has been characterized and shown to be responsible for the production of the structurally related ambiguines, such as ambiguine K (93) shown in Figure 26.266 An NHFe halogenase is also present in this pathway (AmbO5) that shares ~79% sequence identity with WelO5 and has recently been shown to exhibit chlorination activity on a variety of deschloroambiguines.267
Figure 26.

Alkaloids with chlorine atoms installed via the action of NHFe halogenases.
4.4 Structure and mechanism
To date, SyrB2, CytC3, Cur Hal, and WelO5 have been structurally characterized by X-ray crystallography.268–271 All exhibit an antiparallel β-sandwich, or “jelly roll”, topology characteristic of the cupin fold superfamily and similarly observed in NHFe hydroxylases (Figure 27).272,273 In addition, all structures have been obtained with Fe, chloride, and α-ketoglutarate bound in the active site. In the case of NHFe hydroxylases, a conserved 2 His, 1 carboxylate (either Asp or Glu side chains) motif occupies three positions of the octahedral coordination geometry of Fe, forming a ‘facial triad’.274 The remaining coordination positions are occupied by bidentate coordination with α-ketoglutarate and a water molecule that will ultimately become displaced by molecular oxygen during the catalytic cycle. This coordination sphere is retained in NHFe halogenases with one exception; replacement of the coordinating carboxylate residue by the halide.
Figure 27. Structural conservation between NHFe halogenases and hydroxylases.

Crystal structures of NHFe halogenases (A) SyrB2 (PBD: 2FCT), (B) CytC3 (PBD: 3GJB), (C) Cur Hal (PBD: 3NNF), (D) WelO5 (PBD: 5IQT), and (E) the NHFe hydroxylase TauD (PDB: 1GY9) are shown in cartoon representation with Fe(II) as orange spheres and α-ketoglutarate in stick-ball representation with carbon atoms colored blue. Note the conservation of the anti-parallel β-sandwich motif (in grey) that binds the Fe(II) ion and α-ketoglutarate. For the sake of simplicity, active site comparisons with bound substrates and halide ions are shown separately in Figure 28B–D.
No crystal structures of NHFe halogenases that utilize CP-tethered substrates have been obtained in complex with their substrate, perhaps due to many of the inherent experimental difficulties with such an effort that have also been encountered with FDHs. The only structural information available regarding the interaction between enzyme and a CP-tethered substrate is an NMR structure of a CP domain from the curacin biosynthetic pathway and mapping of the CP residues that modulate Cur Hal activity based on mutational analyses.276 However, the discovery of WelO5 has facilitated the attainment of a co-crystal structure with the substrate 12-epi-fischerindole U,271 and furthered the elucidation of the mechanism of halogenation/hydroxylation control, as discussed below.
NHFe halogenases exhibit significant conformational changes during the course of catalysis. Studies on the curacin halogenase (Cur Hal) in the biosynthesis of 88 revealed a conformational switch, wherein a 27-residue lid covers the active site (closed form) triggered by binding of α-ketoglutarate.270 This closed form is proposed to allow for (S)-HMG-CP substrate binding and subsequent catalysis. In the absence of α-ketoglutarate, the enzyme is in the open form and the lid region is completely disordered. In the case of WelO5, the enzyme adopts a unique open form prior to substrate binding and the active site is exposed to solvent.271 Upon substrate binding, an external helix shifts to occlude the active site from solvent, forming the mainly hydrophobic active site necessary to bind the similarly hydrophobic substrate.
NHFe halogenases so far characterized have only been implicated in chlorination of natural products. However, SyrB2 exhibits bromination ability when assayed with significantly higher concentrations of NaBr, yet the enzyme shows a 180-fold preference for chloride during incorporation.277 This observation supports previous isolation studies of bromosyringomycin E (brominated derivative of 82) production in vivo when the producing bacterium is grown in a medium with an excess of NaBr.278 HctB also exhibits bromination activity when assayed with its native substrate and high concentrations of KBr (1 M).251 Bromination activity is also present in WelO5, as pre-saturation dialysis with NaBr results in formation of (13R)-bromo-12-epi-fischerindole U, yet the enzyme prefers chlorine over bromine at an approximate ratio of 500:1.279 In no instance has an NHFe halogenase been reported to catalyze iodine or fluorine incorporation.
In addition to flexibility in regards to Cl/Br incorporation, NHFe halogenases often catalyze multiple chlorination events within biosynthetic pathways. In the biosynthesis of 79, BarB2 was shown to catalyze both the mono- and dichlorination of L-leucine-S-BarA, while BarB1 catalyzes the final chlorination to obtain (2S,4S)-5,5,5-trichloroleucine-S-BarA.248 Interestingly, both enzymes possess the capacity to di- and trichlorinate, albeit in trace amounts, confirming the necessity of both halogenases in biosynthesis. Furthermore, the previously described CytC3 and HctB can catalyze both mono- and dichlorination, while SyrB2 has shown the ability to catalyze a dichlorination when assayed in equimolar amounts with native substrate.277
NHFe halogenases possess a significant degree of promiscuity, both in the substrate scope and the reaction catalyzed. Remarkably, a single point mutation switches the activity of WelO5 from halogenation to hydroxylation.271 The mutation (G166D) provided the necessary carboxylate ligand, re-establishing the ‘facial triad’ observed in NHFe hydroxylases, and switched the activity of WelO5 to solely hydroxylation; a conversion that is as yet unrealized for FDHs. Both HctB and SyrB2 exhibit hydroxylation of their native substrates.251,280 However, the propensity for hydroxylation is negligible compared to halogenation during multiple turnovers, and attempts to convert the activity to solely hydroxylation through mutations have been unsuccessful for these enzymes.
SyrB2 can also chlorinate and hydroxylate different substrate analogs with the chemoselectivity modulated based upon the specific substrate.280 Remarkably, SyrB2 harbors nitration and azidation activity towards the non-native substrate L-2-aminobutyrate-S-SyrB1 when assayed in the presence of the corresponding N3− and NO2− anions.281 A single point mutation or a residue in the active site known to stabilize halide binding (A118G) significantly decreased the affinity for Cl−, while enhancing nitration and azidation activity. Studies on the substrate scope of NHFe halogenases acting on freestanding substrates, WelO5 and AmbO5, revealed a relaxed substrate specificity of AmbO5 towards a variety of hapalindole-type molecules.267 In addition, the substrate scope could be modulated by generating a variety of WelO5/AmbO5 chimeras, all while retaining regio- and stereoselectivity.
The mechanism of NHFe halogenases (Figure 28A) closely mirrors that of the NHFe hydroxylases.282,283 Briefly, NHFe hydroxylases catalyze hydroxylation reactions by first binding α-ketoglutarate then substrate, which displaces the coordinated water. Next, O2 binds in the vacated water position, followed by subsequent oxidative decarboxylation of α-ketoglutarate forming the reactive Fe(IV)-oxo (ferryl) species. This intermediate abstracts a hydrogen atom from the substrate to form Fe(III)-OH and the substrate radical, which subsequently combines with the hydroxyl radical from the newly formed Fe(III)-OH species (termed radical rebound) resulting in the hydroxylated product and Fe(II) with an open coordination position. NHFe halogenase X-ray crystal structures clearly lack the canonical carboxylate ligand found in NHFe hydroxylases, with the coordination position instead occupied by a halide, and would therefore utilize a halo-ferryl intermediate for oxidation (Figures 28B–D). Furthermore, after H-atom abstraction by the haloferryl intermediate, recombination with the hydroxyl radical is suppressed in favor of transfer of the halogen to the substrate radical. Indeed, analogous haloferryl species have been directly observed in the reaction mechanisms of CytC3 and SyrB2 using a combination of X-ray absorption, UV/visible absorption, and Mössbauer spectroscopy.284–286 Surprisingly, the haloferryl intermediate exhibits a half-life orders of magnitude longer than any other NHFe hydroxylase previously characterized.286
Figure 28. Reaction mechanism and active site comparisons of NHFe-dependent enzymes.

(A) Scheme of the general reaction mechanism for both NHFe halogenases and hydroxylases. Comparison of the active sites of (B) TauD, (C) Cur Hal, and (D) WelO5, with substrate-bound, where available. The conserved 2 His, 1 carboxylate coordination of Fe (orange sphere) observed in the NHFe hydroxylase TauD is analogous to a 2His, 1 halide (green sphere) arrangement, wherein the halide replaces the carboxylate in the same coordination position. The substrate is bound in an orientation that is perpendicular to the plane in which the halide resides. The side chains of the active site amino acids are shown in stick-ball representation. Taurine (panel B), 12-epi-fischerindole U (panel D), and α-ketoglutarate (panel B-D) are shown in stick-ball representation with the carbon atoms colored yellow.
Undoubtedly, one of most intriguing questions surrounding the NHFe halogenase mechanism is the ability of the enzyme to selectively control for halogenation over rebound hydroxylation. This “radical rebound” is facilitated by the proximity of the resultant Fe(III)-OH species to the newly formed substrate radical. However, generation of the haloferryl intermediate in NHFe halogenases results in five-coordinate trigonal bipyramidal geometry with the halide in an equatorial coordination position, perpendicular to the axial oxo group and the presumed point of H-atom abstraction. Therefore, the question as to how the enzyme discriminates between the two reactivities has been an active area of investigation. A significant body of research on NHFe halogenases acting on CP-tethered substrates supports the positioning of the substrate, coupled with the sluggish rate of H-atom abstraction,285,286 as the major factors dictating halogenation over hydroxylation. Kinetic and deuterium-labeling studies on the promiscuous activity of SyrB2 revealed this enzyme capable of utilizing SyrB1-tethered L-threonine, L-2-aminobutyrate, and L-norvaline as substrates to catalyze varying degrees of both chlorination and hydroxylation.280 Comparison of the kinetic parameters and identification of the precise location of chlorination/hydroxylation on the various substrates proved these enzymes position the substrate in an orientation such that the target carbon is closer to the halide than the reactive Fe(IV)-oxo species.280 This observation aligns with the sluggish H-atom abstraction rates and the extended lifetime of the haloferryl intermediate, as the enzyme positions the substrate to favor radical halogen transfer, while sacrificing the efficiency of H-atom abstraction. The concept that substrate positioning is the main determinant in halogenation is also supported by additional computational and electron paramagnetic resonance spectroscopy studies.287–290 Furthermore, geometries and molecular orbitals accessed within the catalytic trajectory may play an additional role in selectivity.291
The recent X-ray crystal structure of WelO5 with substrate bound (Figure 28D) provides invaluable insight into the catalytic mechanism, one that entails rearrangement of the reactive haloferryl species during turnover. The structure reveals that the target carbon of halogenation is actually positioned closer to the coordination position initially occupied by the water molecule and replaced by oxygen during turnover, and further from the halide,271 prior to formation of the reactive haloferryl intermediate. Therefore, to negate radical rebound, the reactive oxo species is hypothesized to rotate into the same plane as the halide at some point during formation, in the space vacated by a coordinating carbon of α-ketoglutarate lost as CO2 during conversion to succinate. This rearrangement results in a configuration in which the target carbon is now closer to the halide and further from the reactive oxo intermediate prior to H-atom abstraction. An additional outer-sphere hydrogen bond between the oxo group and a serine residue (S189) is presumed to stabilize the rotated conformation, as mutation of this residue (S189A) results in a mixture of halogenation and hydroxylation activity. Active site rearrangement has previously been observed in the case of the NHFe enzyme clavaminate synthase,292 and was also predicted to occur in NHFe halogenase catalysis through computational analyses.291 While this rearrangement takes places after oxygen binding in WelO5, it remains to be determined if it takes place prior to or after oxygen binding in NHFe halogenases acting on CP-tethered substrates. It must be noted that this rearrangement is not at odds with previous reports, but rather reinforces the conclusion that the target carbon is closer to the halide than the reactive oxo species during H-atom abstraction, and substrate positioning is critical for discrimination between the two competing activities.
NHFe halogenases have been the focus of much attention due to their unique mechanism of halogenation on unreactive substrates. A combination of kinetic and spectroscopic studies, in addition to structural validation in the form of an X-ray crystal structure with substrate bound, provides a convincing mechanistic paradigm. Further understanding of active site dynamics and outer-sphere hydrogen bonding participants will be necessary to fully ascertain how these enzymes control substrate preference, multiple halogenations, and ultimately catalyze halogenation over hydroxylation. In addition, enzymatic halogenation has long been implicated in the development of “green catalysts” for organic chemistry.71,293 With attainment of a substrate-bound structure, and the knowledge that NHFe halogenases harbor other reactivities such as nitration and azidation, this enzyme class has the potential for development into potent biocatalysts through further engineering.
5. S-Adenosyl-L-methionine-dependent halogenases
Each class of halogenases discussed previously activate the halide via oxidation, either a two-electron oxidation for FDHs or haloperoxidases (heme- and vanadate-dependent), or a single-electron oxidation (NHFe halogenases). Nucleophilic halogenases, on the other hand, utilize halogens without oxidation, as halides. Two classes of nucleophilic halogenases are known, the 5′-halo-5′-deoxyadenosine synthases and the halide methyltransferases. Both use SAM as co-substrate, catalyzing nucleophilic substitution reactions. The structure-mechanism of these two enzyme classes are described below.
5.1 5′-Halo-5′-deoxyadenosine synthases
The first crystal structure of a nucleophilic halogenase to be reported was that of the fluorinating enzyme 5′-fluoro-5′-deoxyadenosine synthase (FlA) in 2004, from the soil bacterium Streptomyces cattleya.294 FlA participates in the biosynthesis of fluoroacetate (94) and fluorothreonine (95) and is the only fluorinase characterized to date (Figure 29). In addition to S. cattleya, the enzyme has been identified in other bacteria, including the marine actinomycete Streptomyces xinghaiensis NRRL B-24674,295 the soil isolate Streptomyces sp. MA37, the hospital pathogen Nocardia brasiliensis HUJEG-1, and the soil strain Actinoplanes sp. N902-109.296 FlA catalyzes a SN2-type displacement reaction in which fluoride attacks the electrophilic C-5′ carbon of SAM to generate 5′-fluoro-5′-deoxyadenosine (5′-FlDA) with concomitant displacement of L-methionine.297
Figure 29. Reaction catalyzed by nucleophilic, SAM-dependent halogenases of the 5′-halo-5′-deoxyadenosine synthase class.

Fluorinase FlA catalyzes the first committed step in the biosynthesis of 94 and 95 in the soil bacterium Streptomyces cattleya. Analogously, chlorinase SalL catalyzes the first committed step towards chloroethylmalonyl-CoA, a precursor to 96.
While the fluorinase FlA also accepts chloride to a much lesser extent as later shown in coupled enzyme assays,298 the first physiological 5′-chloro-5′-deoxyadenosine (5′-ClDA) synthase was reported in 2008 in association with the biosynthesis of the potent proteasome inhibitor salinosporamide A (96).299,300 The chlorinase SalL has 35% identity to fluorinase FlA, yet does not accommodate fluoride as a substrate but rather chloride, bromide and iodide (in this order of preference).299 The SalL product 5′-ClDA is converted in a series of enzymatic reactions to the novel halogenated PKS substrate chloroethylmalonyl-CoA that provides the key reactive substrate to the PKS-NRPS derived 96 that is presently in clinical trials to treat multiple myeloma and other cancers (Figure 29).301,302
Comparison of the crystal structures of the two enzymes294,299 and of active site mutants299,303 (Figure 30), along with stereochemical,297 theoretical,304 and kinetic studies,299,303 helped shed light on the nucleophilic halogenating mechanism and on halide specificity. The reader is also directed to two previous reviews on this topic.24,305 The first indication of a SN2 nucleophilic substitution mechanism arose from the observation that the reaction proceeds with inversion of configuration at the C-5′ carbon.297,303 The crystal structures of both fluorinase (Figure 30A–B) and chlorinase (Figure 30C–D) also support a SN2 mechanism. For fluorinase, comparison of the substrate and product complexes aligns the C-F bond of the product 164° anti to the C5′-S bond of the substrate.294 Analogously, for a virtually inactive chlorinase active site mutant (Y70T) in which substrates SAM and chloride were trapped in the active site, a nearly 180° angle was observed between chloride and the C5′-S bond of SAM (Figure 30C–D).299
Figure 30. Crystal structures of 5′-halo-5′-deoxyadenosine synthases.

(A) Overview structure of fluorinase FlA (PDB: 1RQR) displaying monomers colored in pink, green and cyan (top view). Note that the active site lies in the interface between monomers, with three active sites per homotrimer (ligands 5′-FDA and L-met shown as spheres and colored by element). The fluorinase has been shown to be a hexamer in solution (dimer of trimers), according to gel filtration, whereas the chlorinase is a trimer in solution according to ultracentrifugation studies. (B) Fluorinase active site with bound SAM (PDB: 1RQP); chains are colored the same way as in panel A, with the ligand colored by element and hydrogen bonds with key enzyme residues shown as yellow dashes. (C) Chlorinase SalL Y70T mutant (PDB: 2Q6O) active site with bound substrates SAM (colored by element) and chloride (green sphere). (D) Chlorinase SalL Y70T mutant active site showing a view through the chloride ion. Note that the chloride makes a hydrogen bond with the amide backbone of Gly131. The water molecule shown would be displaced by the Tyr70 residue in the wild-type enzyme. (E) Overview of one monomer each of fluorinase FlA (PDB: 1RQR, pink) and chlorinase SalL G131S Y70T double mutant (PDB: 2Q6L, wheat) highlighting a 23-residue loop in FlA (magenta, position 93 to 115) which is absent in SalL (yellow, position 87 to 90) and sits just above the active site. (F) Close up view of the active sites of panel E. Overlay of the active sites of fluorinase FlA (PDB: 1RQR, pink) and chlorinase SalL G131S Y70T double mutant (PDB: 2Q6L, wheat) with bound products 5′-FDA and 5′-ClDA, respectively, highlighting the displacement of the chlorinase loop carrying Ser131 away from the product when compared to fluorinase.
In addition, a common theme that emerges is catalysis by desolvation.5 Fluoride in particular is a potent nucleophile in its desolvated state. However, it is tightly hydrated in aqueous solutions and, thus, effectively inert. In their desolvated state, the order of nucleophilicity for halides is F− > Cl− > Br− > I−.24 Crystallographic294,299,303 and QM/MM theory studies304 support complete desolvation of fluoride prior to catalysis. Fluoride makes H-bond contacts with Ser158 (to both the backbone -NH and the side chain OH, as observed in the crystal structure294) and with Thr80 (as it progresses towards the transition state and as predicted in theory studies304), which should help offset the energy of desolvation and assist with stabilization of the desolvated fluoride for catalysis, respectively. It is proposed that fluoride diffuses into the enzyme and binds first, and that subsequent SAM binding further drives dehydration of fluoride.303
The major change in the active site of chlorinase299 compared to fluorinase294 is replacement of Ser158 with a glycine (Gly131 in SalL), eliminating one H-bond with the halide and increasing the size of the halide binding pocket, which matches the halide discrimination of SalL for larger halides (Figure 30C–D). Other notable differences around the active site include Tyr70 (Thr75 in fluorinase) and Trp129 (Phe156 in fluorinase). Thus, the halide binding pocket of SalL is large enough to accommodate Cl−, Br−, and I− and the relative enzyme efficiency (kcat/Km) matches the nucleophilicity of the desolvated halides (Cl− > Br− > I−). While no halogenating activity could be detected for a G131S mutant of SalL, a Y70T mutation resulted in a 100-fold reduction in activity while affording the observation of SAM and chloride trapped in the active site. Importantly, chloride is observed interacting with water molecules in the Y70T mutant (Figure 30C). Thus, the Y70T SalL structure also supports the hypothesis that excluding water from the active site is important to activate halides for nucleophilic substitution.
The inability of SalL to catalyze fluorination may be partially explained by the serine to glycine substitution mentioned above (Ser158 in FlA and Gly131 in SalL) since the H-bond donor side chain is eliminated. Yet, an Y70T G131S mutant of SalL – i.e. a more “fluorinase-like” mutant – is still unable to catalyze fluorination. Another notable difference between the two enzymes is the presence of an additional 23-residue loop in fluorinase that sits just above the active site (Figure 30E) and appears to contribute to the size of the halide binding pocket. In fact, a structural overlay of fluorinase and SalL Y70T G131S shows a 1.4-Å displacement of the loop carrying Ser131 (SalL numbering) away from the product that results in disruption of the H-bonding network in the SalL double mutant (Figure 30F).299 Thus, the halide binding site for nucleophilic halogenases appears to have evolved to fully dehydrate halide ions and to position halides for SN2 nucleophilic substitution. Halide specificity seems to depend on the size of the halide binding pocket – which is determined by active site residues and by the presence or absence of an extra loop in the N-terminus – and on H-bond donors.
FlA and SalL belong to a large family of >200 homologous proteins referred to as DUF62 in various bacteria and archaea. Protein sequence alignments show that Gly131 (SalL numbering) is conserved in most cases, while Tyr70 is replaced by nonpolar, aliphatic residues including Val, Gly and Ile.306–308 On the basis of structure–function studies of FlA and SalL, these DUF62 proteins would most likely not act as nucleophilic halogenases due to the predicted larger size of the halide binding pocket and likely presence of water in the active site. In fact, two DUF62 proteins have been biochemically analyzed, i.e. archaeal PH0463 from Pyrococcus horikoshii and marine bacterial SARE1364 from Salinispora arenicola CNS-205.306–308 Both enzymes showed SAM hydroxide adenosyltransferase activity (Km SAM = 1.6 μM and kcat = 0.5 min−1 for SARE1364306 and Km SAM = 39 μM and kcat = 0.14 s−1 for PH0463307) but no significant halogenase activity in vitro. Only iodide was accepted by SARE1364 but with Km for iodide (20 mM, which is 40,000-fold higher than the concentration of iodide in seawater) and kcat (0.5 h−1) that do not appear biologically relevant.306 Moreover, DUF62 proteins contain a strictly conserved Asp-Arg-His triad that is absent in fluorinase and chlorinase and is proposed to be involved in water activation for SAM hydrolysis (Figure 31).306–308 It remains to be shown if this SAM hydrolase activity measured in vitro is physiologically relevant. It seems counterintuitive to hydrolyze SAM as it is a high-energy, essential metabolite, biosynthesized in an ATP-dependent reaction and widely used as the methyl donor in biological methylation reactions.309,310 Yet, biochemical studies oppose halogenase activity and the active site is tailored for acid-base catalysis using water as a nucleophile. It is also conceivable that the physiological substrate is not SAM itself but a related substrate that has yet to be identified. In any case, similarly to other halogenases that appear to have evolved from hydroxylases,25,311 5′-halo-5′-deoxyadenosine synthases may have originated from a common enzyme that utilizes hydroxide from water instead of halides as nucleophiles.
Figure 31. SAM hydroxide adenosyltransferases.

(A) Overview structure of DUF-62 PH0463 from archaeon Pyrococcus horikoshii (PDB: 1WU8) showing the same fold as 5′-halo-5′-deoxyadenosine synthases. The three monomers of the homotrimer are colored magenta, green and cyan. (B) Previously proposed mechanism for water activation involving a conserved Asp-Arg-His triad not found in 5′-halo-5′-deoxyadenosine synthases.
Halogenation often improves biological activity. For example, 96 is 500-fold more active against cancer cell lines than its deschloro analog.302,312 Fluorine, in particular, is extremely desirable in medicinal chemistry with about 20% of available drugs being fluorinated.313 However, fluorinated natural products are extremely rare with only a handful known.314 Thus, nucleophilic halogenases, and fluorinase in particular, are attractive targets for biotechnological applications. Three studies have been reported that provide proof of concept for engineering fluorometabolite biosynthesis. First, the incorporation of fluoroacetyl-CoA as a starter unit in aromatic polyketide biosynthesis in vitro was demonstrated.315 Second, the fluorinase from soil S. cattleya was engineered in the marine bacterium S. tropica to achieve the production of fluorosalinosporamide directly from fluoride ion by fermentation (via fluoroethylmalonyl-CoA as an extender unit).316,317 Thirdly, the site-selective incorporation of fluoromalonate into a simplified, mini-PKS model system was realized.318 These studies set the stage for broader applicability of nucleophilic halogenases towards engineering halometabolites of biomedical utility.71,319,320 Furthermore, both FlA and SalL have been used for the biosynthesis of SAM and its derivatives,321,322 complementing other recent approaches323,324 to generate derivative of this ubiquitous biological cofactor to enable synthetic biological efforts.
5.2 Halide methyltransferases
Biosynthesis of halomethanes, MeCl, MeBr, and MeI (97–99), has received attention due to their role in ozone depletion,325 and, more recently, due to their application as end products (e.g. solvents) and as intermediates in the synthesis of more complex organic compounds (e.g. fuels).326 SAM-dependent halide methyltransferases present in plants, algae, bacteria, and fungi have been proposed to be responsible for the presence of halomethanes in the atmosphere. Indeed, halomethane emission has been correlated with halide methyltransferase activity in e.g. agricultural plants.327
A comprehensive study analyzing 89 recombinant, putative halide methyltransferases from bacteria, fungi, plants, and archaea showed that 56% of the enzymes were active with chloride, 85% with bromide, and 69% with iodide (6% of the proteins tested showed no activity).326 A halide methyltransferase from the halophytic plant Batis maritima exhibited the highest activity on each halide and several enzymes displayed unique specificities for given halides. Moreover, the highest activity was observed for the production of 99, which was about 10-fold higher than that for 97 and 98.
Halide methyltransferases catalyze a nucleophilic substitution reaction between SAM and halide ions to form methyl halides and S-adenosylhomocysteine (SAH) (Figure 32A). A carbocation route based on the halide preference (I− > Br− > Cl−) of an enzyme from the marine alga Endocladia muricata has been proposed.328 In addition to halide ions chloride, bromide, and iodide, thiocyanate and bisulfide have been shown to act as nucleophiles in some cases, e.g. for enzymes from radish Raphanus sativus and cabbage Brassica oleracea.327,329
Figure 32. SAM-dependent halide methyltransferase.

(A) Reaction catalyzed by SAM-dependent halide methyltransferases. (B) Overview structure of SAM-dependent halide methyltransferase from A. thaliana (PDB: 3LCC) with SAH ligand shown as spheres. (C) Halide methyltransferase active site. Waters coordinating to Tyr172 are shown as red spheres; SAH is shown with carbon atoms colored yellow.
The crystal structure of a halide methyltransferase from the plant Arabidopsis thaliana331 co-crystallized with SAH has been reported (Figure 32B–C).330 A methyl group modeled at the sulfonium center to represent SAM occupies an open cavity consistent with the promiscuity of the enzyme towards larger halides and thiocyanate, with catalytic efficiency in the order NCS− > I− > Br− > Cl− (F− not accepted). The model proposed by the authors is that ordered H-bonding through a bridging water molecule to the side chain of Tyr172 helps orient the nucleophile, particularly smaller nucleophiles. Consistent with this model, a Y172F mutant shows reduced efficiency only with chloride.
In conclusion, much remains to be understood regarding halide methyltransferases, their mechanism, their physiological role and the implications of methyl halide emissions for atmospheric chemistry. Nonetheless, halide methyltransferases are promising biocatalysts for the production of halomethanes, which are of interest as final products and as intermediates in the synthesis of complex organic compounds.325,326
6. Enzymatic dehalogenation
In contrast to the halogenating enzymes described in this review with dedicated physiological roles in natural product biosynthesis schemes, the field of biological dehalogenation has largely focused on the identification of enzymes on the basis of their ability to act on anthropogenic compounds toward the end goal of bioremediation. This anthropocentric approach has succeeded in identifying numerous dehalogenases utilizing diverse strategies—reductive, oxidative, and hydrolytic, as will be discussed in this section—to dehalogenate a wide range of molecules. However, whether dehalogenases evolved as a result of recent industrial input of polyhalogenated persistent organic pollutants in the environment, or are coincident to polyhalogenated natural products that bare resemblance to man-made molecules, remains uncertain.332 What is clear is that contextual and mechanistic disconnects exist between what is known about biological halogenation versus dehalogenation, which is exacerbated by the fact that halogenation in the context of natural product synthesis appears to be a biosynthetic end-point in that halogenated carbon centers rarely undergo further modification. This is in sharp contrast to the ubiquity of halogenated starting units and intermediates in organic syntheses in which halogenation is routinely utilized as a carbon center activating strategy. Moreover, in contrast to halogenation biocatalysts described from the context of secondary metabolite biosynthesis, the vast majority of dehalogenation biocatalysts identified to date are implicated in primary metabolic processes such as respiration. This divide is likely just as much a reflection of the different approaches taken to study these enzymes as it is due to the inherent challenge in confirming the physiological nature of dehalogenation. While dehalogenation has been studied almost exclusively from the perspective of activity-guided screens, the renaissance in biological halogenation has been sparked by the ability to study halogenation in the unequivocal contexts of biosynthetic pathways. To bridge the gap between halogenation and dehalogenation in natural products, this section will begin with a description of the few dehalogenases known from physiological processes, namely, two classes of deiodinases from the thyroid hormone system, and a recently reported debrominase from the context of the biosynthesis of a microbial natural product. This section also provides an overview on progress in reductive dehalogenation from the context of halorespiration of anthropogenic compounds due to the recent spate of structure-function studies in this area, while the remaining dehalogenation strategies will be given sparer treatment in recognition of comprehensive review articles that cover these areas as highlighted below.
6.1 Natural product reductive dehalogenases
Reductive dehalogenases (RDHs) catalyze net 2e− reductive elimination reactions that result either in the cleavage of a carbon-halogen bond and formation of a carbon-hydrogen bond and a halide (α-elimination), or the breakage of two adjacent halogen substituents resulting in the release of two halides and the formation of an unsaturated product (β-elimination).16 Perhaps the best-studied class of RDHs are cobalamin (B12)-dependent enzymes for their potential applications in bioremediation. However, physiological and biotechnological studies have also revealed several additional classes of non-B12-dependent reductive dehalogenases from aerobic organisms. Indeed, despite a perpetuating notion in the literature on the rarity of biological reductive dehalogenation in aerobic organisms, the aerobic realm has actually afforded a greater diversity of enzymes catalyzing reductive dehalogenations.333–335 This section will emphasize the current mechanistic understanding of dehalogenation enzymes utilizing reductive strategies beginning with a description of the few known examples of natural product dehalogenases.
6.1.1 Reductive deiodinases from the thyroid hormone system
The earliest reported examples of RDHs for which physiological substrates are known are selenoprotein iodothyronine (thyroid hormone) deiodinases and flavoprotein iodotyrosine deiodinases that, respectively, modulate thyroid hormone activity and salvage of the scarce micronutrient iodide from iodotyrosine waste products of thyroglobulin proteolysis.336 These enzymes are also the only known examples of dehalogenases from mammals. Due to the importance of thyroid hormone as “master control of metabolism”, both classes of deiodinases play an essential role in human health and dysfunctions in their activities lead to diseases such as hypo- and hyper-thyroidism and cancers. Remarkably, although both of these enzymes act on similar substrates, they arise from distinct structural families and utilize entirely different electron donors. Recent crystal structures of the catalytic domains of these enzymes has provided preliminary insight into their reaction mechanisms and substrate binding modes.337,338 This section summarizes structure-function studies on these two distinct classes of physiological deiodinases from the thyroid system.
Thyroid hormone is mainly released from the thyroid gland as the prohormone thyroxine (2, Figure 1), and is activated and deactivated by a series of deiodinases that catalyze monodeiodination of the aromatic rings of the thyroid hormone. The first suggestion of deiodinases from thyroid physiology came about in 1955 with the discovery of 1, the outer-ring triiodinated variant of 2, and active form of the thyroid hormone. Almost twenty years later, proof of extrathyroidal production of 1 was demonstrated, and several years on in 1975, the first thyronine 5′-deiodinase was described shortly followed by two additional classes of iodothyronine deiodinases.339–341 At present, iodothyronine deiodinases are classified into one of three types (Type I–III, abbreviated Dio1–3) on the basis of their regiospecificities with respect to monodeiodination of the inner and outer rings of thyroid hormone (Figure 33A).342 Dio1 or Dio2 both catalyze outer-ring deiodination of 2 to active form 1. This can further undergo deiodination of the inner ring by Dio1 or Dio3 to afford 3,3′-T2 (100), which is thought to also be an active form of thyroid hormone. Initial deiodination of the inner ring of 2 by Dio1 or Dio3 leads to the inactive metabolite reverse T3 (rT3) that can be converted to 100 by outer-ring deiodination by either Dio1 or Dio2.343 The catalysis of deiodination is selenocysteine(Sec)-dependent and mutation of the active site Sec to cysteine (Cys) leads to a 100-fold reduction in enzyme activity.344 The favored mechanistic route for dehalogenation of 2 proceeds through the direct attack of the selenocysteine selenolate of the iodine leaving group to form a selenyl iodide species analogous to the Cob(II)alamin halide intermediary formed in the recently proposed mechanism for B12-dependent dehalogenases (Figure 33B). An alternative proposal that proceeds through formation of an organoselenium adduct via a keto-enol intermediate. However, keto-enol tautomerization is only viable for outer-ring deiodination, while individual dehalogenases are able to perform deiodination of both inner and outer rings of 2. Hence, assuming dehalogenation of the inner- and outer- rings of 2 proceed through the same mechanism, a keto-mechanism would not be viable. Due to the high degree of amino acid similarity of Dio1–3, it is likely that all three types of iodothyronine dehalogenases act by the same catalytic mechanism.343
Figure 33. Selenoprotein iodothyronine deiodinase structure-mechanism.

(A) Transformations catalyzed by Dio1–3. (B) Peroxiredoxin-like mechanism proposed for dehalogenation of 2 with residues corresponding to mouse Dio3 in panel ‘C’. Direct attack of the leaving iodine by the selenolate of Sec170 leads to formation of a selenyl iodide intermediate. The selenyl-iodine bond is broken by the addition of the thiolate from the Cys239 side chain to form an endogenous selenyl-sulfide bond in line with the mechanism of 2-Cys peroxiredoxins. The selenyl-sulfide bond is subsequently reduced by attack of the thiolate side chain of a third cysteine residue (Cys168) leading to formation of an endogenous disulfide bridge that is then reduced by a small protein thioredoxin to complete the catalytic cycle. (C) Crystal structure of the mouse Dio3cat (PDB: 4TR3) in green, and 2-Cys peroxiredoxin PtGPx5 (PDB: 2P5Q) in yellow showing the side chains of the catalytic selenocysteine and cysteine residues. Note that the selenocysteine (Sec170) in Dio3cat is mutated to a cysteine in the crystal structure.
The integral membrane nature of Dio1–3 and Sec-dependency has hindered their recombinant heterologous production and consequently, structure-function characterization of iodothyronine deiodinases. Indeed, nearly forty years separate the first in vitro characterization of a thyroid hormone deiodinase and the first reported crystal structure of the catalytic domain of mouse Dio3 (Dio3cat) (Figure 33C).337 While the mechanism of thyroid hormone deiodinases remains uncertain, the peroxiredoxin-like fold of Dio3cat has provided further insights into the reaction mechanisms of selenocysteine-dependent deiodinases. Superposition of a structure of Dio3cat on the structure of prototypical 2-Cys peroxiredoxin PtGPx5 reveals that Sec170 (by proxy of a substituted Cys) of Dio3cat overlays with the anticipated peroxidatic Cys residue (the electron donating residue in peroxiredoxins) (Figure 33C).346 Moreover, a second Cys residue involved in resolving the protein-substrate complex in peroxiredoxins is also conserved in Dio3cat (Cys239). The Dio3cat structure further reveals that the side chain thiol of Cys170 points into an elongated cleft that is proposed to be the substrate-binding site, consistent with its role as the electron donor.337 A substrate-binding model constructed by superposition of the binding residues of a thyroxine receptor co-crystal structure on the proposed binding residue of Dio3cat further shows the substrate orientated with the departing 5-iodine of 2 in proximity to the Cys170 side chain thiol as anticipated for an E1-type haloelimination leading to formation of the proposed Sec170 selenyl iodide species. In a mechanism, akin to recycling of the active site cysteine in peroxiredoxins, an endogenous cysteine thiolate would resolve the selenyl iodide bond to form an endogenous selenyl-sulfide bond (Figure 33B). A third Cys residue conserved in Dio3, but not in peroxiredoxins, that is shown to enhance the deiodinase activity, is then thought to resolve the endogenous selenyl sulfide bond, returning Sec170 to its reduced state and resulting in formation of an endogenous Cys-Cys disulfide bond. The newly formed disulfide bond would then be reduced by an exogenous small protein thioredoxin to complete the catalytic cycle (Figure 33B).337 Although the mechanism for selenoprotein deiodinases remains unresolved, recent structural advances have shed light on a peroxiredoxin-like mechanism for dehalogenation in line with the favored mechanism for reductive dehalogenation.
Continued structure-function studies of iodothyronine deiodinases promise to aid in the development of small molecule modulators of thyroid function as therapeutics in the treatment of thyroid diseases. Moreover, the fact that the polyhalogenated diphenyl core of 2 closely resembles the structure of polybrominated diphenyl ether (PBDE) anthropogenic and natural persistent organic compounds, such as from marine bacteria49 and sponges,347–349 suggests that deiodinases might also be engineered for bioremediation function, a role predominantly entertained for bacterial B12-dependent RDHs.350,351 Structural understanding of thyroid hormone deiodinases may also provide insight into the mechanisms of toxicity of endocrine-disrupting PBDE molecules. For further details on advances in selenoprotein deiodinase structure and mechanism the reader is referred to a recent review of the topic.343
The synthesis of thyroid hormone from the proteolysis of the precursor protein thyroglobulin leads to release mono- and di-iodinated L-tyrosine (101 and 102, respectively). Molecules 101 and 102 are thought to be waste products that are present in six- to seven-fold excess over the thyroid hormone itself.336 To salvage scarce iodide from organic iodine, mammals have evolved a remarkable enzyme, iodotyrosine deiodinase (IYD), to catalyze the complete reductive deiodination of 102.352 Dysfunction of IYD results in hypothyroidism, presently the leading congenital endocrine disorder affecting 1 in 3000 newborns.353 Iodotyrosine deiodinating activity in the thyroid gland was first reported in 1950s, and further investigations of IYD demonstrated its NADPH-dependent/O2-independent catalytic requirement (Figure 34A).352,354,355 Over half a century later, the first IYD gene sequence- human DEHAL1 was identified, and specific mutation-associated disease states were revealed shortly thereafter.353,356–359 Intriguingly, despite their critical role in thyroid physiology, IYDs have also recently been identified in Eukarya that lack thyroid systems as well as in bacteria.360
Figure 34. Structure-function of IYD.

(A) Net reaction catalyzed by IYD. (B) mIYD (PDB: 3GFD) co-crystal structure with the substrate 101 (left), and close-up of active site (right) showing FMN cofactor (yellow) and 101 (blue). The substrate 101 bridges active site residues of mIYD with the isoalloxazine ring of the FMN cofactor. (C) Single electron transfer mechanism proposed for deiodination of iodotyrosines beginning with keto-enol tautomerization followed by a single electron transfer from FMN hydroquinone (hq) to release iodide and form phenoxyl radical. The phenoxyl radical is then reduced by a second single electron transfer from the FMN semiquinone (sq) to afford the deiodinated tyrosine.
Like iodothyronine deiodinases, IYDs are transmembrane proteins. Unlike their thyroid hormone-acting counterparts, IYDs utilize a flavin mononucleotide (FMN) cofactor rather than a genetically encoded selenocysteine electron donor.356 In contrast to the majority of flavoenzymes characterized to date that catalyze O2-dependent oxidative chemical reactions (including halogenation as discussed earlier in this review), IYDs catalyze an O2-independent reductive reaction.40,41 Moreover, apart from a cursory report of a flavoprotein dehalogenases, IYD is the only confirmed example of a flavoprotein that directly catalyzes reductive dehalogenation.352,361 Mammalian IYDs possesses a highly conserved three-domain architecture consisting of a membrane anchor region (~20 amino acids) and intermediate domain with no homology to any known protein domains (~50 amino acids), and the C-terminal Nitro-FMN-like domain (~200 amino acids).362 Notably, IYD lacks a nicotinamide binding domain and relies on an external reductase to deliver NADPH. On the basis of sequence homology, IYD has been assigned to the Nitro-FMN superfamily of enzymes, which includes nitroreductases that catalyze the reduction of nitroaromatic compounds, flavin reductase P (FRP) which provides reduced FMN to luciferase in bacterial bioluminescence, and FMN-dependent NADH oxidase, all of which catalyze O2-dependent two-electron redox chemistry. The C-terminal domain of IYD shows highest homology to bacterial Thermus thermophilius FMN-dependent NADH oxidase.363
Following the progress in molecular understanding of IYD, the unbound as well as co-crystal structures with 101 and 102 of the catalytic domain of mouse IYD (mIYD, Figure 34B) were recently reported.338 mIYD shows closest structural homology to BluB flavin destructase which catalyzes the O2-dependent degradation of its FMN cofactor via serial single-electron transfer mechanism to form the lower axial ligand of vitamin B12.364 Upon binding of the substrate, an active site lid sequesters the substrate from solvent in mIYD. The amino acid residues forming the active site lid align with the corresponding active site lid-forming residues in BluB and are distinct from the residues forming the active site lid in other members of the nitro-FMN superfamily. Hence, based on their FMN binding mode and alignment of amino acids forming their active site lids, BluB and mIYD form their own subfamily within the nitro-FMN superfamily. In contrast to other members of the nitro-FMN superfamily, the side chain residues of the active sites of mIYD and BluB do not coordinate with the pyrimidine moiety of FMN, hinting at the alternative redox mechanism employed by these enzymes. Moreover, the co-crystal structure of mIYD with 101 and 102 revealed polar interactions between the FMN pyrimidine and amino side chain residues with the zwitterionic side of the substrate, suggesting that the substrate plays a direct role in modulating the reactivity of the FMN cofactor (Figure 34B). It was recently demonstrated that titration of mIYD with the substrate mimic iodotyrosine leads to a switch from the two-electron oxidized and reduced forms of the IYD-bound FMN cofactor to the neutral one-electron reduced semiquinone. Hence, preliminary experimental evidence favors a serial single-electron transfer mechanism.365 While no direct interaction with the substrate halogen substituents were observed, the substrate C-I bond is optimally positioned for electron transfer just above C4a in the FMN isoalloxazine ring. Consistent with lack of halogen binding, the identity of the halogen (with the exception of fluorine) of the halotyrosine substrate does not significantly affect substrate binding.365 However, halogenation does appear to play a significant role in substrate recognition, potentially due to reduction of the pKa of the phenol hydroxyl, which is suggested by the mIYD structure to coordinate the ribityl 2′-hydroxyl group of the FMN cofactor.338
While the mechanism of IYD remains speculative, its similarities to BluB and mIYD hint at a serial single-electron transfer strategy in contrast to the two-electron chemistry catalyzed by other members of the nitro-FMN superfamily. With respect to the nature of the substrate radical-generating species, earlier studies showed high affinity of IYD for substrates intended to mimic the keto form of the iodotyrosine.366 Taken together, these observations lead to a mechanistic proposal proceeding through a stabilized keto form of the iodotyrosine phenoxyl which undergoes halide elimination concomitant to a first single-electron transfer to generate the phenoxy radical, which is then reduced by a second single electron transfer by the neutral semiquinone to the deiodinated phenol (Figure 34C).365 In line with recent progress in the molecular understanding of IYDs, an improved understanding of its enzyme mechanism is surely on its way.
The structure of mIYD has had important implications for human health, providing a basis for mechanistic understanding of disease states. For example, mutations associated with hyperthyroidism in human DEHAL1 mapped to key residues involved in coordination of the FMN cofactor in mIYD.338,352 In addition to its importance to human health, the widespread distribution among diverse phyla and oxygen-insensitivity make IYDs an attractive and underappreciated contender to bioremediation applications. Indeed, it has been shown that IYDs are also capable of dechlorination and debromination of halotyrosines.367 Moreover, further investigation of halotyrosine deiodinases in the context of the synthesis and degradation of abundant halotyrosine bacterial natural products could provide an under-appreciated link to global halogen cycling, and in turn expand the substrate scope of these unusual flavoproteins. While anaerobic dehalogenation continues to be a focal point for microbial bioremediation due to the status quo of B12-dependent RDHs, IYDs open the door to aerobic strategies. Illustratively, RDH-mediated dehalogenation activity has been investigated in the anaerobic microbiome fraction of the marine sponge Aplysina aerophoba which harbors a diversity of bromotyrosine natural products.368 However, the aerobic communities where IYD-like enzymology might be present were neglected by this study.
6.1.2 Reductive dehalogenase from microbial natural product biosynthesis
Tremendous attention has been devoted to identifying microbial enzymes capable of dehalogenating anthropogenic pollutants toward the goal of bioremediation. Remarkably, the study of enzymatic dehalogenation as it relates to the formation and degradation of naturally produced halogenated molecules, as discussed in detail in the previous sections, remains largely uncharted. The bioaccumulation and recalcitrance of many anthropogenic pollutants to degradation is due to the high degree of halogenation that renders their carbon scaffolds inaccessible to oxidative catabolism.369–371 An analogous problem manifests itself in the biosynthesis of the highly brominated marine microbial natural product 29 in which dehalogenation of the intermediate 24 is required to unmask the C-2 position of the pyrrole to furnish 2,3,4-tribromopyrrole (103), which is the physiological substrate for the CYP450-Bmp7 catalyzed oxidative coupling to 9 to afford 29. (Figure 35A).38,49,372 This requisite dehalogenative transformation was recently shown to be enzymatically catalyzed by the unique thioredoxin-like reductive dehalogenase, Bmp8, encoded in the biosynthetic gene cluster responsible for the production of 29.372 Of note, Bmp8 is the first, and to date only example of a dehalogenase enzyme that performs a dedicated role in the biosynthesis of a natural product. Moreover, characterization of the catalytic role of Bmp8 enabled a complete in vitro reconstitution of all enzymatic reactions in the biosynthesis of 29, a half century after its initial report as a remarkable marine microbial natural product bearing 70% of its molecular weight as halogen atoms.373,374
Figure 35. Dehalogenation in natural products biosynthesis.

(A) The oxidative coupling reaction in the final step in the biosynthesis of 29 necessitates the dehalogenation of the highly brominated intermediate 24 to 103 catalyzed by the dehalogenase Bmp8. (B) Scheme depicting the role of the active site CXXC Cys residues in the Bmp8 reaction mechanism. The attacking Cys residue is denoted ‘CysA’, while the resolving Cys residue denoted ‘CysR’. (C) Proposed role for a Bmp8-like dehalogenase in the biosynthesis of 104.
Bmp8 was found to utilize a cofactor-independent reaction mechanism akin to the homologous thioredoxin AhpD. The primary amino acid sequence of Bmp8 possesses a signature thioredoxin motif consisting of a cysteine residue separated by two amino acid residues, followed by another cysteine residue (i.e. Cys-X-X-Cys, CXXC, Figure 35). As in the reaction mechanisms for thioredoxins, the side chain thiolates of the C- and N- terminal cysteine residues for the CXXC motif were found to act as the “attacking” electron donor that forms a thioether bond with the substrate and “resolving” nucleophile that liberates the substrate leading to the formation of an endogenous disulfide bond, respectively (Figure 35B). Indirect evidence suggests that the electron acceptor in the reaction catalyzed by Bmp8 is the C-2 bromine of 24. Attack at the C-2 bromine by the cysteinyl thiolate would result in the formation of a transient cysteinyl bromide (Figure 35B). The cysteinyl bromide is either directly thiolyzed by the side chain thiolate of the resolving active site cysteine to form an intramolecular disulfide bond (route ‘i’ in Figure 35B). In an alternate route, the cysteinyl bromide can be first hydrolyzed to form a sulfenic acid species, followed by disulfide bond formation and release of water (route ‘ii’ in Figure 35B) by the resolving cysteine. The proposed mechanism for Bmp8 is analogous to that presently favored for the peroxiredoxin-like thyroid hormone deiodinase which invokes the formation of selenyl iodide intermediate resulting from direct attack of the an active selenocysteinyl selenolate (in place of a cysteinyl thiolate) on the departing iodide, as previously discussed in Section 6.1.1.343 Notably, Bmp8 bears no sequence homology to thyroid hormone deiodinases, suggesting convergent evolution of their respective molecular mechanisms.
In line with cryptic halogenation events catalyzed by NHFe halogenases that participate in the construction of cyclopropane rings in natural product scaffolds (Section 4.2), Bmp8 too encrypts halogenation at the C-2 position of 24.38 However, in contrast to NHFE halogenase-mediated cyclopropane ring formation, dehalogenation that occurs en route the biosynthesis of 103 and 29 is explicitly catalyzed by a dedicated dehalogenase enzyme. In light of the focus of this review on natural product halogenation biochemistry, it is worth highlighting the intimate link between halogenation and dehalogenation presented by the biosynthesis of 29. As detailed in Section 2.3, with the exception of Bmp2, all pyrrolyl-S-CP FDHs described to date halogenate pyrroles in a regioselective manner. In contrast, Bmp2 halogenates all four carbon atoms of the acyl-pyrrole in a regiopromiscuous manner, setting up a non-catalytic decarboxylation reaction to liberate 24. Therefore, the conversion of 24 to 103 by Bmp8 effectively confers back-end regiocontrol on the halogenation of the pyrrole by Bmp2, providing an alternative biosynthetic strategy for cryptic halogenation in natural product biosynthesis.
As the first example of a dehalogenase in natural products biosynthesis, Bmp8 expands the repertoire natural product biosynthetic logic to include not only enzymes that add halogens, but also enzymes that specifically eliminate them to facilitate downstream transformations. One example where the biochemical logic illuminated by the characterization of Bmp8 might be applicable is toward the biosynthesis of the N-C coupled bipyrrole natural product ‘Q1’ (104), a heptachlorinated molecule ubiquitously detected in the marine food web.375,376 The fully halogenated structure of 104 consists of a tetrachlorinated pyrrole with N-1 coupling to the C-2 of a trichlorinated pyrrole (Figure 35C). In light of the discovery of Bmp8, one might envisage a pathway for the biosynthesis of 104 in which tetrachloropyrrole (105) is biosynthesized in a similar manner to its brominated counterpart 24 and enzymatically dehalogenated by a Bmp8-like dehalogenase. Subsequent oxidative coupling to another molecule of 105 can lead to the formation of 104 (Figure 35C). Lastly, an important distinction is to be made between Bmp8 and other microbial dehalogenases characterized to date, in that, the definition of Bmp8 as a dehalogenase is derived from an explicit biosynthetic context. In contrast, other microbial “dehalogenases” discussed later in this section are so-named because of their ability to dehalogenate compounds, which may or may not be their physiological substrates. In this respect, dedicated natural product biosynthetic dehalogenases tailored by evolution to act efficiently on manifold organic scaffolds could lead the way to more efficient dehalogenation biocatalysts than those discovered outside of a natural context.
6.2 Reductive dehalogenases acting on anthropogenic substrates
Over the course of last two decades, numerous bacterial RDHs have been characterized via functional proteomics or transcriptional assays, revealing a repertoire of anthropogenic substrates, which include halogenated phenols, dioxins, biphenyls, and aliphatic hydrocarbons.16 Meanwhile, many hundreds more homologous sequences await functional characterization.379 The following section describes the discovery and structure-function details for two classes of RDHs that act on anthropogenic substrates.
6.2.1 Reductive dehalogenases from halorespiring prokaryotes
Anaerobic halorespiring bacteria utilize a broad spectrum of organohalogens as terminal electron acceptors in energy-conserving and catabolic processes using metalloenzymes. With the exception of a single reported example of heme-dependent RDH, the majority of RDHs characterized from phylogenetically diverse genera of halorespiring bacteria (and archaea) are B12/iron-sulfur (Fe-S) cluster-containing RDHs and are classified as their own subclass of oxidoreductases.377–379 Cytoplasmic B12/Fe-S-containing RDHs are also found in the context of organohalogen catabolic pathways in microaerophilic bacteria.380 These RDHs contain two Fe-S clusters (usually two 4Fe-4S clusters, but sometimes one 4Fe-4S and one 3Fe-4S). Respiratory dehalogenases possess N-terminal signal sequences used for transport of the pre-folded protein to the periplasm, while catabolic dehalogenases possess an NAD(P)H binding domain that is absent in respiratory dehalogenases.380 In addition to significant sequence variation, B12-dependent RDHs are highly dissimilar at the amino acid level from the other major subfamilies of B12-dependent methyltransferases and isomerases that catalyze radical Co(III)-C chemistry.378
The sub-families of B12-dependent RDHs utilize a spectrum of organohalogens, but individual RDHs possess strict substrate specificities and regiocontrol with respect to haloelimination.378 Historically the detailed mechanistic characterization of B12-dependent RDHs has been hindered by slow growth rates of the native halorespiring microbes that harbor them, as well as challenges in their heterologous expression, and the instability of the proteins under aerobic conditions.16 Functional heterologous expression of a B12-dependent trichloroethene (106) dehalogenase PceA from Desulfitobacterium hafniense strain Y51 was only recently reported in the non-dehalogenating B12-producing bacterium Shimwellia blattae over a decade after its initial characterization.381,382
Two recent breakthrough structure-function studies of oxygen-tolerant B12-dependent dehalogenases—a canonical respiratory PceA functionally characterized nearly two decades prior, and a newly characterized catabolic 2,6-dibromophenol dehalogenase (NpRdhA)—are strongly suggestive of dehalogenation mechanism invoking unprecedented Co-halogen chemistry (Figure 36A–C).383–385 Previously, three mechanistic routes were suggested for B12-dependent RDHs beginning with the coordinated Co in its initial Co(I) state (Figure 36D). The first mechanistic proposal (‘Route I’ in Figure 34D) invokes formation of Co(III)-C bond with the organic backbone of the substrate. This mechanism is inspired by Co(III)-C chemistries catalyzed by B12-dependent methyltransferases and isomerases, and is unsupported experimentally in RDHs.378 A second route (‘Route II’ in Figure 36D) supported by limited experimental evidence is initiated by a single electron transfer from Co(I) to the organohalogen substrate leading to the formation of a transient substrate radical.386 Halide elimination is followed by single electron transfer leading to the reduced substrate, or by addition of the organic substrate to cobalt to again form an organocobalt intermediate (‘Route III’ in Figure 36D). A co-crystal structure of PceA with substrate 103 shows the dichlorinated end of 106 in proximity to the coordinated Co, with the leaving chlorine positioned axially to the Co (Figure 36C). Further, iodide was shown to form a complex with Co(II) at the position aligning with the putative chlorine leaving group, suggestive of a weak halide-binding site.383 While a co-crystal structure of NpRdhA with the substrate 3,5-dibromo-4-hydroxybenzoic acid (107) has not been reported yet, computational docking of the substrate in the enzyme active site places the departing bromine atom in proximity to the Co ion. Further, EPR analysis of NpRdhA with 107 strongly suggested the formation of a Co(II)-bromine adduct.384 Active site sterics in PceA and NpRdhA exclude the formation of an organocobalt intermediates with respective to substrates, but still accommodate the previously proposed radical mechanism (‘Route II’ in Figure 36D). However, structure-function evidence strongly favors a new paradigm of B12-mediated Co-halogen chemistry. The unusual ‘base-off’ configuration of the norpseudo-B12 and B12 cofactors of PceA and NpRdhA, respectively, further alludes to unconventional B12-mediated chemistry. Hence, the structure-function studies of PceA and NpRdhA disfavors Co-C chemistry (‘Routes I and III’ in Figure 36D), while suggesting an unanticipated alternative mechanism (‘Route IV’ in Figure 36D) proceeding through a Co(III)-halide intermediate.
Figure 36. Structure-mechanism B12-dependent RDHs.

(A) Dehalogenation reactions catalyzed by PceA (top) and NpRdhA (bottom). (B) Overall crystal structures of PceA (PDB: 4UR0) in magenta and NpRdhA (PDB: 4RAS) in green. (C) Active sites of PceA and NpRdhA showing the B12 cofactor in yellow and the Fe-S clusters. In the PceA structure, two alternate conformations of 106 are modeled above the Co, while the similar site in NpRdhA is occupied by a chloride ion shown as a grey sphere. (D) Scheme showing hypothesized mechanistic routes for dehalogenation by B12-dependent RDHs illustrated with NpRdhA substrate 107.
B12-dependent RDHs exhibit strict substrate specificities and regioselectivities with respect to haloelimination. For example, PceA selectively dehalogenates the dihalogenated end of 106, while NpRdhA exhibits substrate specificity for ortho-substituted halophenols, with a strong preference for 2,6-dihalogenated phenols.383,384 In PceA the substrate first encounters a “letterbox” shaped substrate filter that leads into a narrow hydrophobic channel that opens up into an amphiphilic substrate binding pocket situated at β-face of the B12 cofactor confirmed by co-crystallization of PceA with 106 (Figure 36C). Investigation of the brominated analog of the product cis-dichloroethene- cis-dibromoethene demonstrated a vacancy at the position of the chlorine nearest the coordinated Co ion in the co-crystal structure with 106, consistent with the regioselectivity of dehalogenation.383
Over three decades after the discovery bacterial reductive dehalogenation and two decades after the first report of the first RDH sequence, structural and mechanistic understanding of B12-dependent RDHs remains at its infancy.387–389 Further structural studies sampling from the wide sequence space of B12-dependent RDHs promise to shed light on the basis for substrate selectivity, and aid in the engineering of dehalogenases with expanded substrate scopes. In addition to anticipated differences for respiratory versus catabolic dehalogenases, the structures of PceA and NpRdhA exhibited striking similarities. Intriguingly, the B12 binding domains of both RDHs are structurally homologous to that of the human B12-trafficking chaperone CblC, suggesting, for the first time, a distant common ancestor for B12-dependent RDHs.383,384 As B12-dependent RDHs vary widely in the primary amino acid sequence homology, the extent of their structural diversity remains to be explored through further structure-function studies from within the subfamily. While initial reports have indicated the potential for reductive dehalogenation in pristine unpolluted environments, nothing is known of the physiological substrates of RDHs.389 Study of the native transformation of RDHs may further shed insight into their evolutionary origins. Finally, the reader is referred to recent reviews on the B12-dependent RDHs for a more extensive treatment of the topic.16,379
6.2.2 Glutathione S-transferase-like reductive dehalogenases
An additional well-studied class of reductive dehalogenase is a glutathione S-transferase (GST) superfamily protein capable of dehalogenating the toxic intermediate tetrachlorohydroquinone (TCHQ, 108). The product 108 is generated by the oxidative dehalogenation of the anthropogenic compound pentachlorophenol (109) followed by hydrogenation in the first two steps of the pathway catabolizing 109 in the aerobic bacterium Sphingomonas chlorophenolica (Figure 37A).390–392 TCHQ-dehalogenase, PcpC, catalyzes the dechlorination of 108 to trichlorohydroquinone (TriCHQ), and then to dichlorohydroquinone (DCHQ), which subsequently undergoes enzymatic oxidative ring opening, followed by two rounds of abiotic dehalogenation to feeding into the catabolic β-ketoadipate pathway (Figure 37A).393 The primary sequence of the active site of PcpC is identical to that of glutathione-dependent malelylacetoacetate (MAA) isomerase involved in the catabolism of aromatic amino acids,394 suggestive of the recent recruitment MAA isomerase into a bona fide pentachlorophenol degradation pathway.
Figure 37. Bacterial degradation of 109.

(A) Bacterial pathway for degradation of 109. (B) Mechanism for dehalogenation of 109 by GST-like PcpC. Keto-enol tautomerization is followed by conjugation to glutathione (GS−). Attack by an active site cysteine thiolate forms a protein-glutathione thioester and the dehalogenated product. The catalytic cycle is completed by the resolution of the protein-glutathione thioester by an exogenous glutathione thiolate.
A series of elegant studies have elucidated the thiol-mediated redox mechanism utilized by TCHQ-dehalogenase.394–398 Mirroring the initial steps proposed for dehalogenation of iodotyrosine by IYD, 108 is deprotonated to afford a keto intermediate that then loses a chloride as a leaving group to afford a diketo species, which is subsequently conjugated to a glutathione molecule by nucleophilic addition (Figure 37B). The substrate-glutathione thioester is degraded by attack of an active site cysteine thiolate. The active site cysteine-glutathione thioester is subsequently resolved by attack by a glutathione thiolate to return the enzyme to its catalytically active state. A second glutathione-dependent GST-like dehalogenase, 2,5-dichloroquinone dehalogenase LinD, has been characterized from Sphigobium spp., but to a lesser extent than PcpC.399 GST-like dehalogenases add to a growing number of reductive dehalogenases from aerobic organisms that challenge the status quo of reductive dehalogenases from anaerobic environments. The recruitment of primary metabolic enzymes to perform “non-essential” functions is a theme shared between the pathways of secondary metabolism and catabolic pathways involved in the degradation of xenobiotics, and hints at a vast diversity of strategies recruited by Nature to cope with the manifold organohalogens of both anthropogenic and natural origin.393
6.3 Oxidative dehalogenases
Oxidative dehalogenation involves the replacement of a halogen with a hydroxyl group derived from molecular oxygen or hydrogen peroxide. A diverse array of enzymes catalyze oxidative haloaromatic dehalogenations, including flavin-dependent oxygenases, cytochrome P450 oxidases, and dehaloperoxidases.400–402 The best studied oxidative dehalogenase is the pentachlorophenol-4-monooxgygenase, PcpB, an O2/NADPH-dependent flavoprotein monooxygenase that catalyzes the conversion of 109 to tetrachloroquinone in the bacterial catabolism of 109 (Figure 37A).403 In contrast to the other dehalogenating enzymes described in this review, many enzymes that catalyze oxidative dehalogenases are promiscuous against a broad range of substrates.404,405 Hence, in the absence of a physiological context, it is unclear whether the dehalogenation activity of many oxidative dehalogenases is an incidental laboratory observation or evolved specifically for the dehalogenation of man-made organopollutants. For example, the oxidative dehalogenation of 109 by PcpB is highly inefficient and the enzyme shows substrate promiscuity for the downstream product 108, suggesting its function in the degradation pathway of 109 could be happenstance.406 To maintain the focus of the review to address dehalogenation mechanisms in the context of substrate-specific enzymes, the reader is referred to a series of recent reviews on the topic of oxidative enzymatic dehalogenation.400–402
6.4 Hydrolytic dehalogenases
In contrast to previously described dehalogenation strategies, hydrolytic dehalogenases (HDHs) catalyze the redox neutral substitution of a halogen by a hydroxyl derived either from water or a vicinal alcohol.334 At present, HDHs that dehalogenate a variety of anthropogenic pollutants comprise the most diverse sub-class of dehalogenase enzymes. Compared to their RDH counterparts, the characterization of HDHs has proceeded at a rapid pace due to their facile heterologous expression. Indeed, the first reported HDH crystal structure, that of an α/β-hydrolase-superfamily haloalkane dehalogenase, preceded that of B12-dependent RDHs by nearly a quarter of a century.407 In subsequent years, structures of haloacid, 4-chorobenzoyl-CoA, haloalcohol, and 3-chloroacrylic acid dehalogenases have joined the gamut of structurally described HDHs.334
The mechanisms of HDHs can be divided on the basis of substrate specificity of the enzymes. Haloalkane, haloacid, and 4-chlorobenzoyl-CoA HDHs are united by the formation of a covalent bond between the substrate and an active site aspartate residue that results in elimination of a halide in the first half reaction (Figure 38A,B).334 The aspartyl-substrate oxoester is then hydrolyzed in the second half reaction by an activated water molecule that is deprotonated by an active site base. Recognition of the substrate halogen by an active site halide binding site is critical to substrate binding and stabilization of the transition state in the first half-reaction.408 Interestingly, the only salient difference between the abovementioned HDHs and a canonical hydrolase is the exchange of the hydrolytic serine/cysteine residue (as in a Ser-His-Asp/Glu catalytic triad) with an aspartate. A remarkable bacterial HDH hydrolyzes the carbon-fluorine bond in the highly toxic natural product fluoroacetate.409,410 Despite sequence similarity to other members of the haloacid dehalogenases, fluoroacetate dehalogenase (FAcD) shows highest affinity for the fluorinated substrate in comparison to other differentially halogenated acetates.17 While the mechanism of FAcD proceeds via the anticipated aspartyl oxoester, its unique ability among haloacid dehalogenases to catalyze hydrolysis of a C-F bond has been attributed to an active site halogen binding site tailored to the small ionic radius of fluorine.410
Figure 38. Structure-mechanism for HDHs with a catalytic triad.

(A) Proposed reaction mechanism for haloalkane/haloacid HDHs. (B) Overall structure (left) and active site (right) for a recently reported structure of a haloalkane HDH from a marine Rhodobacteraceae sp (PDB: 4C6H). The catalytic Asp residue is shown with the carbon atoms colored cyan. The His and Glu residue comprising of the catalytic triad are shown with carbon atoms colored green. The in situ generated product from the substrate 1-bromohexane used in the crystallization trials, 1-hexanol, is shown bound to the active site in two alternate conformations in yellow. A chloride ion bound in a hydrophobic pocket in the vicinity of the product is shown as a grey sphere and likely denotes the binding site for the substrate halogen atom. (C) Proposed reaction mechanism for 4-chlorobenzoyl-CoA HDH.
In contrast to the HDHs that catalyze a two-step intermolecular substitution reaction, haloalcohol dehalogenases catalyze single-step dehalogenative intramolecular substitution reactions for halohydrin substrates. In this mechanism the alcohol vicinal to departing halogen substituent is deprotonated by a Ser-Tyr-Lys/Arg catalytic triad followed by intramolecular substitution of the vicinal halogen leading to formation of an epoxide (Figure 39A–C).334,412 Halogenation of alkyl centers renders them inherently activated to inter- or intramolecular substitution reactions, a strategy routinely exploited in synthetic organic chemistry as well as natural product biosynthetic routes such as that for merochlorin biosynthesis. The dehalogenative substitution reactions catalyzed by HDHs are in stark contrast with the intricate radical chemistry employed by NHFe halogenases to halogenate unactivated alkyl centers. Interestingly, haloalcohol dehalogenases belong to the short chain dehydrogenase/reductase (SDR) superfamily of dinucleotide-binding redox enzymes. However, haloalcohol dehalogenases lack the dinucleotide binding motif, and structural analysis reveals that this domain has been substituted by bulky amino acid residue side chains that compose a halogen binding site.334,412 In addition to dehalogenation, the epoxide cyanolysis and azidolysis side activity of the HDHs have been utilized to generate β-hydroxynitriles and azides in conjunction with site-directed enzyme engineering efforts.413–416
Figure 39. Structure-mechanism for SDR-like HDHs.

(A) Proposed reaction mechanism for halohydrin dehalogenases and recruitment of a cyanide anion to resolve the dehalogenated epoxide product to generate β-hydroxynitriles. (B) Structural similarity of between halohydrin HDHs (in green, PDB: 1PWZ) and lanthipeptide oxidoreductase SDR ElxO (in brown, PDB: 4QEC). NAD+ bound to ElxO is shown in yellow sticks. (C) Active site of the halohydrin HDH HheC with an epoxide product bound to the active site (in yellow), and the side chains of the catalytic Tyr (in cyan), and the Ser and Arg (in green) residues. A chloride ion bound in the vicinity of the product is shown as a grey sphere.
Yet another dehalogenation mechanism is employed by the 3-chloroacrylic acid dehalogenase, CaaD, that adds catalytic base-activated water to the conjugated substrate double bond leading to the formation of a tetrahedral transition state that then leads to the departure of the halide.419 In essence, CaaD combines the two half reactions catalyzed by the haloalkane/haloacid HDHs into a single concerted reaction. While the role of the CaaD active site residues and the operative reaction mechanism has been a matter of extensive computational and experimental studies,420–424 CaaD is the first member of the tautomerase superfamily with hydratase activity.425 Moreover, CaaD dehalogenases acting upon both cis- and trans-3-chloroacryclic acid substrates have been characterized. A more recent addition to the group of hydrolytic dehalogenases is the 2-haloacrylate hydratase that unexpectedly requires reduced flavin (FADH2) for catalytic proficiency. While the role of the flavin in the reaction mechanism remains unknown, it has been suggested to either act as an acid-base catalyst as in the flavin-dependent isopentenyl diphosphate isomerase, or a transient electron donor as in the mechanism of flavin-dependent chorismate synthase.426 Further investigations of this intriguing enzyme promise to shed light on its surprising catalytic requirement.
A remarkable diversity of enzymes catalyzes variations on the theme of hydrolytic dehalogenation from the haloalkane dehalogenases that differ only modestly from their hydrolase counterparts, to haloacid dehalogenases that define a superfamily of their own. Hence, a careful study of these varied enzymes promises to provide insights into the evolution of enzymatic dehalogenation, in particular addressing the question of modern versus ancient origins of evolved dehalogenase function.404 For detailed account of hydrolytic dehalogenation, the reader is referred to several dedicated reviews on the topic.17,334
7. Future perspectives
Natural product biosynthetic schemes continue to provide examples of novel chemical transformations catalyzed by enzymes.427,428 Halogenation biochemistry is no different. Historically, as has been the underlying motivation of this review article, natural product chemistry has led to the discovery and characterization of novel halogenation biocatalysts, which, in turn, have inspired the development of novel halogenating chemical catalysts as well.429 This process has not been exhausted. A recent report describing the structure elucidation and biosynthetic gene cluster discovery of cyanobacterial bartolosides has uncovered a potentially novel diiron-dependent halogenase,430,431 with homologs present in other cyanobacteria, including in a cyanobacterial gene cluster encoding the production of structurally analogous halogenated cyclophane natural products.432 Furthermore, a recent report challenges our understanding of the catalytic scope of FDHs which were previously thought to only halogenate aromatic rings or electron rich activated aliphatic carbon centers. Discovery and characterization of a fungal bifunctional methyltransferase-halogenase enzyme established the gem-dichlorination on a non-activated carbon atom,433 a transformation previously within the realm of NHFe halogenases proceeding with the halide radical route. Another recently described FDH, Amm3,179 is a close homolog of the RiPP FDH MibH, perhaps postulating towards the involvement of peptidic substrates in the elaboration of marine halogenated pyrroloquinoline alkaloids,434 an assertion that is supported by the presence of numerous biosynthetic enzyme encoding genes with resemblance to RiPP dehydratases in the recently described amm biosynthetic gene cluster.179
Further novel halogenases await discovery. A recent report describing the regio-, stereo-, and enantioselective dihalogenation of allylic alcohols during the course of the synthesis of halogenated terpenes such as the anticancer candidate molecule halomon posits an as yet undiscovered enzymatic halogenation strategy for the biosyntheses of these ubiquitous marine macroalgal natural products.435 Enantioselective dichlorination has also allowed access to chlorosulfolipid marine natural products,436 among other recent efforts,437,438 bringing into focus the exquisite control exercised by these as yet undiscovered marine halogenases to furnish structures that are otherwise unrealized by the enzymatic halogenation routes characterized thus far. Without a doubt, polyhalogenated natural products continue to inspire synthetic chemistry efforts.439 Directed evolution of halogenases too is expanding the substrate scope of halogenating enzymes, pushing them as viable complementary catalysts in complex synthetic schemes.440–442
The inventory of experimentally characterized halogenases is dwarfed by both known halogenated natural products, as well as putative halogenases that are detected in sequence databases. While sequence databases are expanding at an exponential pace, the sequence space explored by halogenases is restricted, for the most part, to culturable bacteria. Two sets of contemporary technological advances promise to affect a paradigm shift in where the halogenases of the future will come from. First are techniques such as metagenomics and single-cell genomics that allow the interrogation of complex microbial communities without a priori constraints of culturing or enrichment.443–445 Coupling these to advances in synthesis of large DNA sequences, direct capture of natural product gene clusters from a complex pool of heterogeneous DNA, and iteratively teaching computational algorithms to find novel classes of natural product gene clusters will revolutionize our access to sequence space that has not yet been explored. Particularly with respect to natural product halogenation biochemistry, for instance, filter-feeding benthic marine invertebrates such as sponges are one of the most prolific producers where halogenated natural products can constitute up to 10% of the sponge dry weight.34,347,446,447 Marine sponges are the ‘petri-dishes of the oceans’ harboring an extraordinary diversity of endosymbiotic microbiome that has been postulated, and in many cases experimentally verified, to be the source of the natural product diversity.448–451 It is thus noteworthy that while the inventory of FDHs from sponge-microbiome consortia has attracted recent interest,452,453 to the best of our knowledge, no sponge-derived FDH, V-CPO, V-BPO, or SAM-dependent halogenase has been confirmed within genomic or biochemical context for the production of a halogenated natural product. A singular instance of homologs of NHFe halogenases detected in sponge microbiomes have been paired with natural product biosynthetic chemistry.81
The second advance is the interrogation of halogenation biochemistry, at the genetic and biochemical levels, for the production of halogenated natural products by eukaryotic sources. Notable examples from halogenases encoded within fungal natural product biosynthetic gene clusters have been mentioned above. Again, looking towards to the marine environment, an area that has been genetically unexplored, is the production of halogenated natural products by macroalgae and seaweeds. In particular, marine red and brown algae have been celebrated producers of bioactive halogenated natural products, some with curious accumulation of iodide from the seawater with postulated roles in self-defense against biofouling.454 In addition to pharmaceutical candidates, several thousand tons of ozone-damaging polyhalogenated methanes, such as bromoform and dibromomethane, are produced by marine eukaryotes every year that saturate the marine layer and contribute significantly to ozone-depletion.455–457 Nearly forty years ago, the pioneering work of Hager and coworkers hinted towards the contribution of hydrogen peroxide-dependent marine macroalgal haloperoxidases in the production of bromoform using biomimetic substrates.458,459 Despite the environmental and ecological significance, scant progress460–462 has been made to identify the genetic and biochemical bases for the production of polyhalogenated methanes in the oceans, and the physiological substrates for the abovementioned macroalgal haloperoxidases remain unidentified. In fact, while numerous other marine algal haloperoxidases have also been identified and their biochemical activity characterized with respect to biomimetic substrates and model bioconversions,189,194,212 to date, the dedicated genetic and biochemical contribution of even a single macroalgal halogenase in a natural product biosynthetic scheme has not been established. An untapped reservoir of eukaryotic halogenases presents itself that should become accessible as the scientific community realigns its focus to include increasingly more eukaryotic sources to interrogate for secondary metabolic schemes. A recent report describing the contribution of bromide as an essential micronutrient for the posttranslational sulfilimine formation bears testimony to the presence of halogenation biochemistry in all kingdoms of life.463
In light of halogenation biochemistry discussed thus far, it is interesting to note that the molecular basis for halide specificity for halogenases has not been elucidated. Numerous examples of marine brominated natural products have been presented, including 24, 29, 31, and 103, for which the biosynthetic gene clusters are known and experimentally validated.50,107 However, the molecular sieve that allows marine brominases to selectively incorporate bromine in natural product structures against a greater than 500-fold excess of chloride in seawater remains elusive. Characterizing this selectivity promises to open avenues for generating differentially halogenated natural products, further pushing forward the already impressive contribution of halogen atoms in conferring favorable bioactivities to natural product scaffolds.
Contrasting the cornucopia of halogenases discovered from natural products biosynthesis described in this review, to date, only a solitary dehalogenating enzyme- Bmp8, has been characterized from the context of a microbial natural product biosynthetic pathway. This is in addition to the two physiological deiodinases described from thyroid hormone maturation and iodide salvage. A common feature of these physiological dehalogenase enzymes is that they partially undo the handiwork of halogenation biocatalysts. Furthermore, apart from fluoroacetate hydrolytic dehalogenase, the enzymatic degradation of the >5,000 known organohalogen natural products remains almost entirely unexplored as a source of dehalogenating enzymes. What is perhaps most striking of all is that, for the most part, the cofactors utilized by known halogenation enzymes is a mutually exclusive set from the cofactors involved in enzymatic dehalogenation. Even in the rare case of the flavoenzyme reductive iodotyrosine dehalogenase, the FMN co-factor serve a reductive role in contrast to the oxidative role of the flavin co-factor found in FDHs. This co-factor gap might be inherent to chemical biology, but could just as well hint at the infancy of the field of biological dehalogenation. Despite the fact that enzymatic dehalogenation once superseded biological halogenation with respect to the diversity of enzyme strategies, the tables have quickly turned in the post-genomic era.464 In part, this knowledge gap maybe due to fact that the search for dehalogenases has, and continues to be biased toward a narrow spectrum of anthropogenic substrates. While clearly no dearth of dehalogenases has been discovered to date, how many more might be found if natural product biosynthesis and biodegradation are taken into account? Hinting at an answer to this question, studies have implicated a compelling biological contribution to global halogen cycling, which includes both the incorporation and removal of halogens from organic scaffolds.465,466 Moreover, recent metagenomics surveys of pristine unpolluted terrestrial environments for sequences related to known classes of halogenating and dehalogenating enzymes have revealed an astounding diversity of biocatalysts with unknown substrates.467,468
Acknowledgments
The authors’ laboratories are supported by the US National Science Foundation (OCE-1313747 to B.S.M.); the US National Institutes of Health (K99ES026620 to V.A., and P01-ES021921, R01-AI047818, and R01-CA127622 to B.S.M.); the Helen Hay Whitney Foundation postdoctoral fellowship to V.A; startup funds from the Department of Medicinal Chemistry and Pharmacognosy of the College of Pharmacy of UIC to A.S.E.; a Hans W. Vahlteich Research award to A.S.E.; and startup funds from the Department of Medicinal Chemistry in the College of Pharmacy at the University of Utah to J.M.W.
Biographies
Vinayak Agarwal received his Bachelors and Masters degrees in Biochemical Engineering from the Indian Institute of Technology, Delhi, before moving to the University of Illinois Urbana-Champaign for graduate studies with Dr. Satish K. Nair at the Center for Biophysics and Computational Biology. Graduating in 2012, he joined the laboratory of Dr. Bradley S. Moore at UC San Diego to interrogate marine halogenation biosynthetic pathways. He has received postdoctoral funding award from the Helen Hay Whitney Foundation (2014–2016) and the NIH Pathway to Independence Award (2016).
Zachary D. Miles obtained his Bachelor of Science degree from the University of Wisconsin-Madison in 2009. Soon after, he started his graduate work at the University of Arizona in the department of Chemistry and Biochemistry under the guidance of Prof. Vahe Bandarian, where he received both a National Science Foundation IGERT Program in Genomics Fellowship and a National Institutes of Health Biological Chemistry Training Program Fellowship. After graduating with his Ph.D. in 2014, he joined the laboratory of Prof. Bradley Moore and is currently studying the biosynthesis of marine meroterpenoids.
Jaclyn Winter received her B.Sc. in Chemistry and Molecular Genetics from the State University of New York at Fredonia in 2004 and completed her Ph.D. at UC San Diego under the mentorship of Dr. Bradley S. Moore in 2010. After graduate school, she completed postdoctoral training with Dr. Christian Hertweck at the Hans Knöll Institute and Leibniz Institute for Natural Products Research and Infection Biology in Jena, Germany (2010–2011) and with Dr. Yi Tang at UC Los Angeles (2011–2014). During her graduate and postdoctoral studies, she was a recipient of a NIH Marine Biotechnology Predoctoral Ruth L. Kirschstein National Research Service Award and the L’Oréal/AAAS USA Postdoctoral Fellowship for Women in Science. She is currently an Assistant Professor in Medicinal Chemistry in the College of Pharmacy at the University of Utah, Salt Lake City, UT.
Alessandra S. Eustáquio graduated from the University of São Paulo, Brazil, with a degree in Pharmacy and Biochemistry, before obtaining a PhD from the University of Tübingen, Germany, mentored by Prof. Lutz Heide. After being a Life Sciences Research Foundation postdoctoral fellow at Prof. Bradley S. Moore’s laboratory at the University of California San Diego, Alessandra joined Pfizer’s Natural Products group as a Principal Scientist, where she worked with natural product drug development for four years. Alessandra has been an Assistant Professor in the Department of Medicinal Chemistry and Pharmacognosy of the University of Illinois at Chicago since August 2015.
Abrahim El Gamal graduated from Stanford University in 2011 with a B.S. in Chemistry and M.S. in Chemical Engineering. Shortly thereafter he moved to the University of California, San Diego where he completed his Ph.D. work in the laboratory of Dr. Bradley S. Moore in the summer of 2016. During his graduate career, Abrahim was a recipient of a U.S. National Institutes of Health Marine Biotechnology Training Grant pre-doctoral fellowship. He is currently a postdoc with Dr. Chaitan Khosla at Stanford University.
Bradley Moore is Professor of Marine Chemical Biology at the Scripps Institution of Oceanography and Professor and Chair of Pharmaceutical Chemistry the Skaggs School of Pharmacy and Pharmaceutical Sciences at UC San Diego. He holds degrees in chemistry from the University of Hawaii (B.S. 1988) and Washington (Ph.D. 1994), was a postdoc at the University of Zurich (1994–1995), and held prior faculty appointments at the University of Washington (1996–1999) and Arizona (1999–2005). His research interests involve exploring and exploiting marine microbial genomes to discover new biosynthetic enzymes, secondary metabolic pathways, and natural products for their biomedical and biotechnological utility.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Gribble GW. Naturally occurring organohalogen compounds–a comprehensive survey. Fortschr Chem Org Naturst. 1996;68:1–423. [PubMed] [Google Scholar]
- 2.Gribble GW. Naturally Occurring Organohalogen Compounds – A Comprehensive Update. Springer; Vienna: 2010. [Google Scholar]
- 3.Gribble Gw. The natural production of organobromine compounds. Environ Sci Pollut Res Int. 2000;7:37–47. doi: 10.1065/espr199910.002. [DOI] [PubMed] [Google Scholar]
- 4.Emerson S, Hedges J. Chemical Oceanography and the Marine Carbon Cycle. 2008 [Google Scholar]
- 5.Lohman DC, Edwards DR, Wolfenden R. Catalysis by desolvation: the catalytic prowess of SAM-dependent halide-alkylating enzymes. J Am Chem Soc. 2013;135:14473–14475. doi: 10.1021/ja406381b. [DOI] [PubMed] [Google Scholar]
- 6.Wang LS, Zhou XF, Fredimoses M, Liao SR, Liu YH. Naturally occurring organoiodines. RSC Adv. 2014;4:57350–57376. [Google Scholar]
- 7.O’Hagan D, Harper DB. Fluorine-containing natural products. J Fluor Chem. 1999;100:127–133. [Google Scholar]
- 8.Dembitsky VM. Biogenic iodine and iodine-containing metabolites. Nat Prod Commun. 2006;1:139–175. [Google Scholar]
- 9.Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev. 2002;23:38–89. doi: 10.1210/edrv.23.1.0455. [DOI] [PubMed] [Google Scholar]
- 10.Harris CM, Kannan R, Kopecka H, Harris TM. The role of the chlorine substituents in the antibiotic vancomycin: preparation and characterization of mono- and didechlorovancomycin. J Am Chem Soc. 1985;107:6652–6658. [Google Scholar]
- 11.Groll M, Huber R, Potts BC. Crystal structures of Salinosporamide A (NPI-0052) and B (NPI-0047) in complex with the 20S proteasome reveal important consequences of beta-lactone ring opening and a mechanism for irreversible binding. J Am Chem Soc. 2006;128:5136–5141. doi: 10.1021/ja058320b. [DOI] [PubMed] [Google Scholar]
- 12.Kale AJ, McGlinchey RP, Lechner A, Moore BS. Bacterial self-resistance to the natural proteasome inhibitor salinosporamide A. ACS Chem Biol. 2011;6:1257–1264. doi: 10.1021/cb2002544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bister B, Bischoff D, Nicholson GJ, Stockert S, Wink J, Brunati C, Donadio S, Pelzer S, Wohlleben W, Sussmuth RD. Bromobalhimycin and chlorobromobalhimycins–illuminating the potential of halogenases in glycopeptide antibiotic biosyntheses. Chembiochem. 2003;4:658–662. doi: 10.1002/cbic.200300619. [DOI] [PubMed] [Google Scholar]
- 14.Dinos GP, Athanassopoulos CM, Missiri DA, Giannopoulou PC, Vlachogiannis IA, Papadopoulos GE, Papaioannou D, Kalpaxis DL. Chloramphenicol derivatives as antibacterial and anticancer agents: historic problems and current solutions. Antibiotics (Basel) 2016;5:20. doi: 10.3390/antibiotics5020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Sanchez-Rosello M, Acena JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. Fluorine in pharmaceutical industry: fluorine-containing drugs introduced to the market in the last decade (2001–2011) Chem Rev. 2014;114:2432–2506. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- 16.Jugder BE, Ertan H, Lee M, Manefield M, Marquis CP. Reductive dehalogenases come of age in biological destruction of organohalides. Trends Biotechnol. 2015;33:595–610. doi: 10.1016/j.tibtech.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 17.Kurihara T, Esaki N. Bacterial hydrolytic dehalogenases and related enzymes: Occurrences, reaction mechanisms, and applications. Chem Rec. 2008;8:67–74. doi: 10.1002/tcr.20141. [DOI] [PubMed] [Google Scholar]
- 18.Hager LP, Morris DR, Brown FS, Eberwein HChloroperoxidase., II Utilization of halogen anions. J Biol Chem. 1966;241:1769–1777. [PubMed] [Google Scholar]
- 19.Clutterbuck PW, Mukhopadhyay SL, Oxford AE, Raistrick H. Studies in the biochemistry of micro-organisms. Biochem J. 1940;34:664–677. doi: 10.1042/bj0340664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson SM, Paul IC, Rinehart KL, Srinivas R. Absolute configuration of caldariomycin. J Am Chem Soc. 1968;90:136–140. [Google Scholar]
- 21.Hofrichter M, Ullrich R. Heme-thiolate haloperoxidases: versatile biocatalysts with biotechnological and environmental significance. Appl Microbiol Biotechnol. 2006;71:276–288. doi: 10.1007/s00253-006-0417-3. [DOI] [PubMed] [Google Scholar]
- 22.Sundaramoorthy M, Terner J, Poulos TL. The crystal structure of chloroperoxidase: a heme peroxidase–cytochrome P450 functional hybrid. Structure. 1995;3:1367–1377. doi: 10.1016/s0969-2126(01)00274-x. [DOI] [PubMed] [Google Scholar]
- 23.Neumann CS, Fujimori DG, Walsh CT. Halogenation strategies in natural product biosynthesis. Chem Biol. 2008;15:99–109. doi: 10.1016/j.chembiol.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 24.Blasiak LC, Drennan CL. Structural perspective on enzymatic halogenation. Acc Chem Res. 2009;42:147–155. doi: 10.1021/ar800088r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vaillancourt FH, Yeh E, Vosburg DA, Garneau-Tsodikova S, Walsh CT. Nature’s inventory of halogenation catalysts: oxidative strategies predominate. Chem Rev. 2006;106:3364–3378. doi: 10.1021/cr050313i. [DOI] [PubMed] [Google Scholar]
- 26.Weichold V, Milbredt D, van Pee KH. Specific enzymatic halogenation-from the discovery of halogenated enzymes to their applications in vitro and in vivo. Angew Chem Int Ed Engl. 2016;55:6374–6389. doi: 10.1002/anie.201509573. [DOI] [PubMed] [Google Scholar]
- 27.van Pee KH, Dong CJ, Flecks S, Naismith J, Patallo EP, Wage T. Biological halogenation has moved far beyond haloperoxidases. Adv Microbiol. 2006;59:127–157. doi: 10.1016/S0065-2164(06)59005-7. [DOI] [PubMed] [Google Scholar]
- 28.Wever R, Plat H, Deboer E. Isolation procedure and some properties of the bromoperoxidase from the seaweed Ascophyllum nodosum. Biochim Biophys Acta. 1985;830:181–186. [Google Scholar]
- 29.Butler A, Walker JV. Marine haloperoxidases. Chem Rev. 1993;93:1937–1944. [Google Scholar]
- 30.Petty MA. An introduction to the origin and biochemistry of microbial halometabolites. Bacteriol Rev. 1961;25:111–130. doi: 10.1128/br.25.2.111-130.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rutledge PJ, Challis GL. Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat Rev Microbiol. 2015;13:509–523. doi: 10.1038/nrmicro3496. [DOI] [PubMed] [Google Scholar]
- 32.Lane AL, Moore BS. A sea of biosynthesis: marine natural products meet the molecular age. Nat Prod Rep. 2011;28:411–428. doi: 10.1039/c0np90032j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerwick WH, Moore BS. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem Biol. 2012;19:85–98. doi: 10.1016/j.chembiol.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blunt JW, Copp BR, Keyzers RA, Munro MH, Prinsep MR. Marine natural products. Nat Prod Rep. 2016;33:382–431. doi: 10.1039/c5np00156k. [DOI] [PubMed] [Google Scholar]
- 35.Denmark SE, Kuester WE, Burk MT. Catalytic, asymmetric halofunctionalization of alkenes–a critical perspective. Angew Chem Int Ed Engl. 2012;51:10938–10953. doi: 10.1002/anie.201204347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dong C, Flecks S, Unversucht S, Haupt C, van Pee KH, Naismith JH. Tryptophan 7-halogenase (PrnA) structure suggests a mechanism for regioselective chlorination. Science. 2005;309:2216–2219. doi: 10.1126/science.1116510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mole DJ, Webster SP, Uings I, Zheng X, Binnie M, Wilson K, Hutchinson JP, Mirguet O, Walker A, Beaufils B, et al. Kynurenine-3-monooxygenase inhibition prevents multiple organ failure in rodent models of acute pancreatitis. Nat Med. 2016;22:202–209. doi: 10.1038/nm.4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El Gamal A, Agarwal V, Diethelm S, Rahman I, Schorn MA, Sneed JM, Louie GV, Whalen KE, Mincer TJ, Noel JP, et al. Biosynthesis of coral settlement cue tetrabromopyrrole in marine bacteria by a uniquely adapted brominase-thioesterase enzyme pair. Proc Natl Acad Sci U S A. 2016;113:3797–3802. doi: 10.1073/pnas.1519695113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cole LJ, Gatti DL, Entsch B, Ballou DP. Removal of a methyl group causes global changes in p-hydroxybenzoate hydroxylase. Biochemistry. 2005;44:8047–8058. doi: 10.1021/bi050108x. [DOI] [PubMed] [Google Scholar]
- 40.Teufel R, Agarwal V, Moore BS. Unusual flavoenzyme catalysis in marine bacteria. Curr Opin Chem Biol. 2016;31:31–39. doi: 10.1016/j.cbpa.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walsh CT, Wencewicz TA. Flavoenzymes: versatile catalysts in biosynthetic pathways. Nat Prod Rep. 2013;30:175–200. doi: 10.1039/c2np20069d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eichhorn E, van der Ploeg JR, Leisinger T. Characterization of a two-component alkanesulfonate monooxygenase from Escherichia coli. J Biol Chem. 1999;274:26639–26646. doi: 10.1074/jbc.274.38.26639. [DOI] [PubMed] [Google Scholar]
- 43.Campbell ZT, Baldwin TO. Fre is the major flavin reductase supporting bioluminescence from Vibrio harveyi luciferase in Escherichia coli. J Biol Chem. 2009;284:8322–8328. doi: 10.1074/jbc.M808977200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yeh E, Cole LJ, Barr EW, Bollinger JM, Jr, Ballou DP, Walsh CT. Flavin redox chemistry precedes substrate chlorination during the reaction of the flavin-dependent halogenase RebH. Biochemistry. 2006;45:7904–7912. doi: 10.1021/bi060607d. [DOI] [PubMed] [Google Scholar]
- 45.Yeh E, Blasiak LC, Koglin A, Drennan CL, Walsh CT. Chlorination by a long-lived intermediate in the mechanism of flavin-dependent halogenases. Biochemistry. 2007;46:1284–1292. doi: 10.1021/bi0621213. [DOI] [PubMed] [Google Scholar]
- 46.Bitto E, Huang Y, Bingman CA, Singh S, Thorson JS, Phillips GN. The structure of flavin-dependent tryptophan 7-halogenase RebH. Proteins: Struct Funct Bioinf. 2008;70:289–293. doi: 10.1002/prot.21627. [DOI] [PubMed] [Google Scholar]
- 47.Zhu X, De Laurentis W, Leang K, Herrmann J, Ihlefeld K, van Pee KH, Naismith JH. Structural insights into regioselectivity in the enzymatic chlorination of tryptophan. J Mol Biol. 2009;391:74–85. doi: 10.1016/j.jmb.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flecks S, Patallo EP, Zhu X, Ernyei AJ, Seifert G, Schneider A, Dong C, Naismith JH, van Pee KH. New insights into the mechanism of enzymatic chlorination of tryptophan. Angew Chem Int Ed Engl. 2008;47:9533–9536. doi: 10.1002/anie.200802466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agarwal V, El Gamal AA, Yamanaka K, Poth D, Kersten RD, Schorn M, Allen EE, Moore BS. Biosynthesis of polybrominated aromatic organic compounds by marine bacteria. Nat Chem Biol. 2014;10:640–647. doi: 10.1038/nchembio.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Agarwal V, Moore BS. Enzymatic synthesis of polybrominated dioxins from the marine environment. ACS Chem Biol. 2014;9:1980–1984. doi: 10.1021/cb5004338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eppink MH, Cammaart E, Van Wassenaar D, Middelhoven WJ, van Berkel WJ. Purification and properties of hydroquinone hydroxylase, a FAD-dependent monooxygenase involved in the catabolism of 4-hydroxybenzoate in Candida parapsilosis CBS604. Eur J Biochem. 2000;267:6832–6840. doi: 10.1046/j.1432-1033.2000.01783.x. [DOI] [PubMed] [Google Scholar]
- 52.Eppink MH, Boeren SA, Vervoort J, van Berkel WJ. Purification and properties of 4-hydroxybenzoate 1-hydroxylase (decarboxylating), a novel flavin adenine dinucleotide-dependent monooxygenase from Candida parapsilosis CBS604. J Bacteriol. 1997;179:6680–6687. doi: 10.1128/jb.179.21.6680-6687.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meganathan R. Ubiquinone biosynthesis in microorganisms. FEMS Microbiol Lett. 2001;203:131–139. doi: 10.1111/j.1574-6968.2001.tb10831.x. [DOI] [PubMed] [Google Scholar]
- 54.Hammer PE, Hill DS, Lam ST, VanPee KH, Ligon JM. Four genes from Pseudomonas fluorescens that encode the biosynthesis of pyrrolnitrin. Appl Environ Microbiol. 1997;63:2147–2154. doi: 10.1128/aem.63.6.2147-2154.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kirner S, Hammer PE, Hill DS, Altmann A, Fischer I, Weislo LJ, Lanahan M, van Pee KH, Ligon JM. Functions encoded by pyrrolnitrin biosynthetic genes from Pseudomonas fluorescens. J Bacteriol. 1998;180:1939–1943. doi: 10.1128/jb.180.7.1939-1943.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hohaus K, Altmann A, Burd W, Fischer I, Hammer PE, Hill DS, Ligon JM, vanPee KH. NADH-dependent halogenases are more likely to be involved in halometabolite biosynthesis than haloperoxidases. Angew Chem Int Ed Engl. 1997;36:2012–2013. [Google Scholar]
- 57.Keller S, Wage T, Hohaus K, Holzer M, Eichhorn E, van Pee KH. Purification and partial characterization of tryptophan 7-halogenase (PrnA) from Pseudomonas fluorescens. Angew Chem Int Ed Engl. 2000;39:2300–2302. doi: 10.1002/1521-3773(20000703)39:13<2300::aid-anie2300>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 58.Yeh E, Garneau S, Walsh CT. Robust in vitro activity of RebF and RebH, a two-component reductase/halogenase, generating 7-chlorotryptophan during rebeccamycin biosynthesis. Proc Natl Acad Sci U S A. 2005;102:3960–3965. doi: 10.1073/pnas.0500755102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Pee KH, Patallo EP. Flavin-dependent halogenases involved in secondary metabolism in bacteria. Appl Microbiol Biotechnol. 2006;70:631–641. doi: 10.1007/s00253-005-0232-2. [DOI] [PubMed] [Google Scholar]
- 60.van Pee KH. Enzymatic chlorination and bromination. Methods Enzymol. 2012;516:237–257. doi: 10.1016/B978-0-12-394291-3.00004-6. [DOI] [PubMed] [Google Scholar]
- 61.van Pee KH. Biosynthesis of halogenated alkaloids. Alkaloids Chem Biol. 2012;71:167–210. doi: 10.1016/b978-0-12-398282-7.00002-3. [DOI] [PubMed] [Google Scholar]
- 62.Du YL, Williams DE, Patrick BO, Andersen RJ, Ryan KS. Reconstruction of cladoniamide biosynthesis reveals nonenzymatic routes to bisindole diversity. ACS Chem Biol. 2014;9:2748–2754. doi: 10.1021/cb500728h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chang FY, Brady SF. Cloning and characterization of an environmental DNA-derived gene cluster that encodes the biosynthesis of the antitumor substance BE-54017. J Am Chem Soc. 2011;133:9996–9999. doi: 10.1021/ja2022653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chang FY, Brady SF. Discovery of indolotryptoline antiproliferative agents by homology-guided metagenomic screening. Proc Natl Acad Sci U S A. 2013;110:2478–2483. doi: 10.1073/pnas.1218073110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ryan KS. Biosynthetic gene cluster for the cladoniamides, bis-indoles with a rearranged scaffold. PLoS One. 2011;6:e23694. doi: 10.1371/journal.pone.0023694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zeng J, Zhan JX. Characterization of a tryptophan 6-halogenase from Streptomyces toxytricini. Biotech Lett. 2011;33:1607–1613. doi: 10.1007/s10529-011-0595-7. [DOI] [PubMed] [Google Scholar]
- 67.Shepherd SA, Menon BR, Fisk H, Struck AW, Levy C, Leys D, Micklefield J. A structure-guided switch in the regioselectivity of a tryptophan halogenase. Chembiochem. 2016;17:821–824. doi: 10.1002/cbic.201600051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Milbredt D, Patallo EP, van Pee KH. A tryptophan 6-halogenase and an amidotransferase are involved in thienodolin biosynthesis. Chembiochem. 2014;15:1011–1020. doi: 10.1002/cbic.201400016. [DOI] [PubMed] [Google Scholar]
- 69.Xu L, Han T, Ge M, Zhu L, Qian X. Discovery of the new plant growth-regulating compound LYXLF2 based on manipulating the halogenase in Amycolatopsis orientalis. Curr Microbiol. 2016 doi: 10.1007/s00284-016-1052-6. [DOI] [PubMed] [Google Scholar]
- 70.Payne JT, Andorfer MC, Lewis JC. Methods Enzymol. Academic Press; [DOI] [PubMed] [Google Scholar]
- 71.Smith DR, Gruschow S, Goss RJ. Scope and potential of halogenases in biosynthetic applications. Curr Opin Chem Biol. 2013;17:276–283. doi: 10.1016/j.cbpa.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 72.van Pée KH, Milbredt D, Patallo EP, Weichold V, Gajewi M. In: Methods Enzymol. Sarah EOC, editor. Vol. 575. Academic Press; 2016. [DOI] [PubMed] [Google Scholar]
- 73.Poor CB, Andorfer MC, Lewis JC. Improving the stability and catalyst lifetime of the halogenase RebH by directed evolution. Chembiochem. 2014;15:1286–1289. doi: 10.1002/cbic.201300780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Menon BR, Latham J, Dunstan MS, Brandenburger E, Klemstein U, Leys D, Karthikeyan C, Greaney MF, Shepherd SA, Micklefield J. Structure and biocatalytic scope of thermophilic flavin-dependent halogenase and flavin reductase enzymes. Org Biomol Chem. 2016;14:9354–9361. doi: 10.1039/c6ob01861k. [DOI] [PubMed] [Google Scholar]
- 75.Frese M, Sewald N. Enzymatic halogenation of tryptophan on a gram scale. Angew Chem Int Ed Engl. 2015;54:298–301. doi: 10.1002/anie.201408561. [DOI] [PubMed] [Google Scholar]
- 76.Schnepel C, Minges H, Frese M, Sewald N. A high-throughput fluorescence assay to determine the activity of tryptophan halogenases. Angew Chem Int Ed Engl. 2016 doi: 10.1002/anie.201605635. [DOI] [PubMed] [Google Scholar]
- 77.Deb Roy A, Gruschow S, Cairns N, Goss RJ. Gene expression enabling synthetic diversification of natural products: chemogenetic generation of pacidamycin analogs. J Am Chem Soc. 2010;132:12243–12245. doi: 10.1021/ja1060406. [DOI] [PubMed] [Google Scholar]
- 78.Runguphan W, Qu X, O’Connor SE. Integrating carbon-halogen bond formation into medicinal plant metabolism. Nature. 2010;468:461–464. doi: 10.1038/nature09524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Runguphan W, O’Connor SE. Diversification of monoterpene indole alkaloid analogs through cross-coupling. Org Lett. 2013;15:2850–2853. doi: 10.1021/ol401179k. [DOI] [PubMed] [Google Scholar]
- 80.Frese M, Schnepel C, Minges H, Voß H, Feiner R, Sewald N. Modular combination of enzymatic halogenation of tryptophan with Suzuki–Miyaura cross-coupling reactions. ChemCatChem. 2016;8:1799–1803. [Google Scholar]
- 81.Flatt P, Gautschi J, Thacker R, Musafija-Girt M, Crews P, Gerwick W. Identification of the cellular site of polychlorinated peptide biosynthesis in the marine sponge Dysidea (Lamellodysidea) herbacea and symbiotic cyanobacterium Oscillatoria spongeliae by CARD-FISH analysis. Marine Biology. 2005;147:761–774. [Google Scholar]
- 82.Wang S, Xu Y, Maine EA, Wijeratne EM, Espinosa-Artiles P, Gunatilaka AA, Molnar I. Functional characterization of the biosynthesis of radicicol, an Hsp90 inhibitor resorcylic acid lactone from Chaetomium chiversii. Chem Biol. 2008;15:1328–1338. doi: 10.1016/j.chembiol.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 83.Reeves CD, Hu Z, Reid R, Kealey JT. Genes for the biosynthesis of the fungal polyketides hypothemycin from Hypomyces subiculosus and radicicol from Pochonia chlamydosporia. Appl Environ Microbiol. 2008;74:5121–5129. doi: 10.1128/AEM.00478-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zeng J, Zhan J. A novel fungal flavin-dependent halogenase for natural product biosynthesis. Chembiochem. 2010;11:2119–2123. doi: 10.1002/cbic.201000439. [DOI] [PubMed] [Google Scholar]
- 85.Zeng J, Lytle AK, Gage D, Johnson SJ, Zhan J. Specific chlorination of isoquinolines by a fungal flavin-dependent halogenase. Bioorg Med Chem Lett. 2013;23:1001–1003. doi: 10.1016/j.bmcl.2012.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wick J, Heine D, Lackner G, Misiek M, Tauber J, Jagusch H, Hertweck C, Hoffmeister D. A fivefold parallelized biosynthetic process secures chlorination of Armillaria mellea (Honey Mushroom) toxins. Appl Environ Microbiol. 2016;82:1196–1204. doi: 10.1128/AEM.03168-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Payne JT, Andorfer MC, Lewis JC. Regioselective arene halogenation using the FAD-dependent halogenase RebH. Angew Chem Int Ed Engl. 2013;52:5271–5274. doi: 10.1002/anie.201300762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shepherd SA, Karthikeyan C, Latham J, Struck AW, Thompson ML, Menon BRK, Styles MQ, Levy C, Leys D, Micklefield J. Extending the biocatalytic scope of regiocomplementary flavin-dependent halogenase enzymes. Chem Sci. 2015;6:3454–3460. doi: 10.1039/c5sc00913h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Holzer M, Burd W, Reissig HU, van Pee KH. Substrate specificity and regioselectivity of tryptophan 7-halogenase from Pseudomonas fluorescens BL915. Adv Syn Catal. 2001;343:591–595. [Google Scholar]
- 90.Winter JM, Sato M, Sugimoto S, Chiou G, Garg NK, Tang Y, Watanabe K. Identification and characterization of the chaetoviridin and chaetomugilin gene cluster in Chaetomium globosum reveal dual functions of an iterative highly-reducing polyketide synthase. J Am Chem Soc. 2012;134:17900–17903. doi: 10.1021/ja3090498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sato M, Winter JM, Kishimoto S, Noguchi H, Tang Y, Watanabe K. Combinatorial generation of chemical diversity by redox enzymes in chaetoviridin biosynthesis. Org Lett. 2016;18:1446–1449. doi: 10.1021/acs.orglett.6b00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Thanapipatsiri A, Gomez-Escribano JP, Song L, Bibb M, Al-Bassam M, Chandra G, Thamchaipenet A, Challis G. Discovery of unusual biaryl polyketides by activation of a silent Streptomyces venezuelae biosynthetic gene cluster. Chembiochem. 2016 doi: 10.1002/cbic.201600396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Neumann CS, Walsh CT, Kay RR. A flavin-dependent halogenase catalyzes the chlorination step in the biosynthesis of Dictyostelium differentiation-inducing factor 1. Proc Natl Acad Sci U S A. 2010;107:5798–5803. doi: 10.1073/pnas.1001681107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Austin MB, Saito T, Bowman ME, Haydock S, Kato A, Moore BS, Kay RR, Noel JP. Biosynthesis of Dictyostelium discoideum differentiation-inducing factor by a hybrid type I fatty acid-type III polyketide synthase. Nat Chem Biol. 2006;2:494–502. doi: 10.1038/nchembio811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Thomas MG, Burkart MD, Walsh CT. Conversion of L-proline to pyrrolyl-2-carboxyl-S-PCP during undecylprodigiosin and pyoluteorin biosynthesis. Chem Biol. 2002;9:171–184. doi: 10.1016/s1074-5521(02)00100-x. [DOI] [PubMed] [Google Scholar]
- 96.Lin S, Van Lanen SG, Shen B. Regiospecific chlorination of (S)-beta-tyrosyl-S-carrier protein catalyzed by SgcC3 in the biosynthesis of the enediyne antitumor antibiotic C-1027. J Am Chem Soc. 2007;129:12432–12438. doi: 10.1021/ja072311g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dorrestein PC, Yeh E, Garneau-Tsodikova S, Kelleher NL, Walsh CT. Dichlorination of a pyrrolyl-S-carrier protein by FADH2-dependent halogenase PltA during pyoluteorin biosynthesis. Proc Natl Acad Sci U S A. 2005;102:13843–13848. doi: 10.1073/pnas.0506964102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yamanaka K, Ryan KS, Gulder TA, Hughes CC, Moore BS. Flavoenzyme-catalyzed atropo-selective N,C-bipyrrole homocoupling in marinopyrrole biosynthesis. J Am Chem Soc. 2012;134:12434–12437. doi: 10.1021/ja305670f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mantovani SM, Moore BS. Flavin-linked oxidase catalyzes pyrrolizine formation of dichloropyrrole-containing polyketide extender unit in chlorizidine A. J Am Chem Soc. 2013;135:18032–18035. doi: 10.1021/ja409520v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang X, Parry RJ. Cloning and characterization of the pyrrolomycin biosynthetic gene clusters from Actinosporangium vitaminophilum ATCC 31673 and Streptomyces sp. strain UC 11065. Antimicrob Agents Chemother. 2007;51:946–957. doi: 10.1128/AAC.01214-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hofer I, Crusemann M, Radzom M, Geers B, Flachshaar D, Cai X, Zeeck A, Piel J. Insights into the biosynthesis of hormaomycin, an exceptionally complex bacterial signaling metabolite. Chem Biol. 2011;18:381–391. doi: 10.1016/j.chembiol.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 102.Walsh CT, Garneau-Tsodikova S, Howard-Jones AR. Biological formation of pyrroles: nature’s logic and enzymatic machinery. Nat Prod Rep. 2006;23:517–531. doi: 10.1039/b605245m. [DOI] [PubMed] [Google Scholar]
- 103.Al-Mourabit A, Zancanella MA, Tilvi S, Romo D. Biosynthesis, asymmetric synthesis, and pharmacology, including cellular targets, of the pyrrole-2-aminoimidazole marine alkaloids. Nat Prod Rep. 2011;28:1229–1260. doi: 10.1039/c0np00013b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Heide L, Westrich L, Anderle C, Gust B, Kammerer B, Piel J. Use of a halogenase of hormaomycin biosynthesis for formation of new clorobiocin analogues with 5-chloropyrrole moieties. Chembiochem. 2008;9:1992–1999. doi: 10.1002/cbic.200800186. [DOI] [PubMed] [Google Scholar]
- 105.Wynands I, van Pee KH. A novel halogenase gene from the pentachloropseudilin producer Actinoplanes sp. ATCC 33002 and detection of in vitro halogenase activity. FEMS Microbiol Lett. 2004;237:363–367. doi: 10.1016/j.femsle.2004.06.053. [DOI] [PubMed] [Google Scholar]
- 106.Nguyen DD, Wu CH, Moree WJ, Lamsa A, Medema MH, Zhao X, Gavilan RG, Aparicio M, Atencio L, Jackson C, et al. MS/MS networking guided analysis of molecule and gene cluster families. Proc Natl Acad Sci U S A. 2013;110:E2611–2620. doi: 10.1073/pnas.1303471110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ross AC, Gulland LE, Dorrestein PC, Moore BS. Targeted capture and heterologous expression of the Pseudoalteromonas alterochromide gene cluster in Escherichia coli represents a promising natural product exploratory platform. ACS Synth Biol. 2015;4:414–420. doi: 10.1021/sb500280q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Buedenbender S, Rachid S, Muller R, Schulz GE. Structure and action of the myxobacterial chondrochloren halogenase CndH: a new variant of FAD-dependent halogenases. J Mol Biol. 2009;385:520–530. doi: 10.1016/j.jmb.2008.10.057. [DOI] [PubMed] [Google Scholar]
- 109.Rachid S, Scharfe M, Blocker H, Weissman KJ, Muller R. Unusual chemistry in the biosynthesis of the antibiotic chondrochlorens. Chem Biol. 2009;16:70–81. doi: 10.1016/j.chembiol.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 110.Cadel-Six S, Dauga C, Castets AM, Rippka R, Bouchier C, Tandeau de Marsac N, Welker M. Halogenase genes in nonribosomal peptide synthetase gene clusters of Microcystis (cyanobacteria): sporadic distribution and evolution. Mol Biol Evol. 2008;25:2031–2041. doi: 10.1093/molbev/msn150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ishida K, Welker M, Christiansen G, Cadel-Six S, Bouchier C, Dittmann E, Hertweck C, Tandeau de Marsac N. Plasticity and evolution of aeruginosin biosynthesis in cyanobacteria. Appl Environ Microbiol. 2009;75:2017–2026. doi: 10.1128/AEM.02258-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pang AH, Garneau-Tsodikova S, Tsodikov OV. Crystal structure of halogenase PltA from the pyoluteorin biosynthetic pathway. J Struct Biol. 2015;192:349–357. doi: 10.1016/j.jsb.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 113.Agarwal V, Lin S, Lukk T, Nair SK, Cronan JE. Structure of the enzyme-acyl carrier protein (ACP) substrate gatekeeper complex required for biotin synthesis. Proc Natl Acad Sci U S A. 2012;109:17406–17411. doi: 10.1073/pnas.1207028109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Masoudi A, Raetz CR, Zhou P, Pemble CWt. Chasing acyl carrier protein through a catalytic cycle of lipid A production. Nature. 2014;505:422–426. doi: 10.1038/nature12679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sneed JM, Sharp KH, Ritchie KB, Paul VJ. The chemical cue tetrabromopyrrole from a biofilm bacterium induces settlement of multiple Caribbean corals. Proc Biol Sci. 2014;281 doi: 10.1098/rspb.2013.3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zehner S, Kotzsch A, Bister B, Sussmuth RD, Mendez C, Salas JA, van Pee KH. A regioselective tryptophan 5-halogenase is involved in pyrroindomycin biosynthesis in Streptomyces rugosporus LL-42D005. Chem Biol. 2005;12:445–452. doi: 10.1016/j.chembiol.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 117.Lang A, Polnick S, Nicke T, William P, Patallo EP, Naismith JH, van Pee KH. Changing the regioselectivity of the tryptophan 7-halogenase PrnA by site-directed mutagenesis. Angew Chem Int Ed Engl. 2011;50:2951–2953. doi: 10.1002/anie.201007896. [DOI] [PubMed] [Google Scholar]
- 118.Miller LM, Chatterjee C, van der Donk WA, Kelleher NL. The dehydratase activity of lacticin 481 synthetase is highly processive. J Am Chem Soc. 2006;128:1420–1421. doi: 10.1021/ja057203d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lee MV, Ihnken LA, You YO, McClerren AL, van der Donk WA, Kelleher NL. Distributive and directional behavior of lantibiotic synthetases revealed by high-resolution tandem mass spectrometry. J Am Chem Soc. 2009;131:12258–12264. doi: 10.1021/ja9033507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Thibodeaux CJ, Ha T, van der Donk WA. A price to pay for relaxed substrate specificity: a comparative kinetic analysis of the class II lanthipeptide synthetases ProcM and HalM2. J Am Chem Soc. 2014;136:17513–17529. doi: 10.1021/ja5089452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yu Y, Mukherjee S, van der Donk WA. Product formation by the promiscuous lanthipeptide synthetase ProcM is under kinetic control. J Am Chem Soc. 2015;137:5140–5148. doi: 10.1021/jacs.5b01409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Agarwal V, Diethelm S, Ray L, Garg N, Awakawa T, Dorrestein PC, Moore BS. Chemoenzymatic synthesis of acyl coenzyme A substrates enables in situ labeling of small molecules and proteins. Org Lett. 2015;17:4452–4455. doi: 10.1021/acs.orglett.5b02113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Peter DM, Vogeli B, Cortina NS, Erb TJ. A chemo-enzymatic road map to the synthesis of CoA esters. Molecules. 2016;21 doi: 10.3390/molecules21040517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dorrestein PC, Bumpus SB, Calderone CT, Garneau-Tsodikova S, Aron ZD, Straight PD, Kolter R, Walsh CT, Kelleher NL. Facile detection of acyl and peptidyl intermediates on thiotemplate carrier domains via phosphopantetheinyl elimination reactions during tandem mass spectrometry. Biochemistry. 2006;45:12756–12766. doi: 10.1021/bi061169d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mc Culloch I, Jaremko M, La Clair J, Burkart MD. Fluorescent mechanism-based probes for aerobic flavin-dependent enzyme activity. Chembiochem. 2016 doi: 10.1002/cbic.201600275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lin S, Van Lanen SG, Shen B. Characterization of the two-component, FAD-dependent monooxygenase SgcC that requires carrier protein-tethered substrates for the biosynthesis of the enediyne antitumor antibiotic C-1027. J Am Chem Soc. 2008;130:6616–6623. doi: 10.1021/ja710601d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lin S, Huang T, Horsman GP, Huang SX, Guo X, Shen B. Specificity of the ester bond forming condensation enzyme SgcC5 in C-1027 biosynthesis. Org Lett. 2012;14:2300–2303. doi: 10.1021/ol300720s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liu W, Christenson SD, Standage S, Shen B. Biosynthesis of the enediyne antitumor antibiotic C-1027. Science. 2002;297:1170–1173. doi: 10.1126/science.1072110. [DOI] [PubMed] [Google Scholar]
- 129.Van Lanen SG, Dorrestein PC, Christenson SD, Liu W, Ju J, Kelleher NL, Shen B. Biosynthesis of the beta-amino acid moiety of the enediyne antitumor antibiotic C-1027 featuring beta-amino acyl-S-carrier protein intermediates. J Am Chem Soc. 2005;127:11594–11595. doi: 10.1021/ja052871k. [DOI] [PubMed] [Google Scholar]
- 130.Yamanaka K, Reynolds KA, Kersten RD, Ryan KS, Gonzalez DJ, Nizet V, Dorrestein PC, Moore BS. Direct cloning and refactoring of a silent lipopeptide biosynthetic gene cluster yields the antibiotic taromycin A. Proc Natl Acad Sci U S A. 2014;111:1957–1962. doi: 10.1073/pnas.1319584111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pohanka A, Menkis A, Levenfors J, Broberg A. Low-abundance kutznerides from Kutzneria sp. 744. J Nat Prod. 2006;69:1776–1781. doi: 10.1021/np0604331. [DOI] [PubMed] [Google Scholar]
- 132.Broberg A, Menkis A, Vasiliauskas R. Kutznerides 1–4, depsipeptides from the actinomycete Kutzneria sp. 744 inhabiting mycorrhizal roots of Picea abies seedlings. J Nat Prod. 2006;69:97–102. doi: 10.1021/np050378g. [DOI] [PubMed] [Google Scholar]
- 133.Fujimori DG, Hrvatin S, Neumann CS, Strieker M, Marahiel MA, Walsh CT. Cloning and characterization of the biosynthetic gene cluster for kutznerides. Proc Natl Acad Sci U S A. 2007;104:16498–16503. doi: 10.1073/pnas.0708242104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Umezawa K, Ikeda Y, Uchihata Y, Naganawa H, Kondo S. Chloptosin, an apoptosis-inducing dimeric cyclohexapeptide produced by Streptomyces. J Org Chem. 2000;65:459–463. doi: 10.1021/jo991314b. [DOI] [PubMed] [Google Scholar]
- 135.Ji Z, Xu N, Gang Q, Wei S. Identification of pyrroloindoline-containing cyclic hexapeptides in the metabolites of Streptomyces alboflavus 313 by HPLC-DAD-ESI-MS/MS. J Antibiot (Tokyo) 2013;66:265–271. doi: 10.1038/ja.2013.7. [DOI] [PubMed] [Google Scholar]
- 136.Miller WR, Murray BE, Rice LB, Arias CA. Vancomycin-Resistant Enterococci: Therapeutic Challenges in the 21st Century. Infect Dis Clin North Am. 2016;30:415–439. doi: 10.1016/j.idc.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 137.Gerhard U, Mackay JP, Maplestone RA, Williams DH. The role of the sugar and chlorine substituents in the dimerization of vancomycin antibiotics. J Am Chem Soc. 1993;115:232–237. [Google Scholar]
- 138.Pinchman JR, Boger DL. Investigation into the functional impact of the vancomycin C-ring aryl chloride. Bioorg Med Chem Lett. 2013;23:4817–4819. doi: 10.1016/j.bmcl.2013.06.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pinchman JR, Boger DL. Probing the role of the vancomycin e-ring aryl chloride: selective divergent synthesis and evaluation of alternatively substituted E-ring analogues. J Med Chem. 2013;56:4116–4124. doi: 10.1021/jm4004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nadkarni SR, Patel MV, Chatterjee S, Vijayakumar EK, Desikan KR, Blumbach J, Ganguli BN, Limbert M. Balhimycin, a new glycopeptide antibiotic produced by Amycolatopsis sp. Y-86,21022. Taxonomy, production, isolation and biological activity. J Antibiot (Tokyo) 1994;47:334–341. doi: 10.7164/antibiotics.47.334. [DOI] [PubMed] [Google Scholar]
- 141.Pelzer S, Sussmuth R, Heckmann D, Recktenwald J, Huber P, Jung G, Wohlleben W. Identification and analysis of the balhimycin biosynthetic gene cluster and its use for manipulating glycopeptide biosynthesis in Amycolatopsis mediterranei DSM5908. Antimicrob Agents Chemother. 1999;43:1565–1573. doi: 10.1128/aac.43.7.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Puk O, Huber P, Bischoff D, Recktenwald J, Jung G, Sussmuth RD, van Pee KH, Wohlleben W, Pelzer S. Glycopeptide biosynthesis in Amycolatopsis mediterranei DSM5908: function of a halogenase and a haloperoxidase/perhydrolase. Chem Biol. 2002;9:225–235. doi: 10.1016/s1074-5521(02)00101-1. [DOI] [PubMed] [Google Scholar]
- 143.Puk O, Bischoff D, Kittel C, Pelzer S, Weist S, Stegmann E, Sussmuth RD, Wohlleben W. Biosynthesis of chloro-beta-hydroxytyrosine, a nonproteinogenic amino acid of the peptidic backbone of glycopeptide antibiotics. J Bacteriol. 2004;186:6093–6100. doi: 10.1128/JB.186.18.6093-6100.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.van Wageningen AM, Kirkpatrick PN, Williams DH, Harris BR, Kershaw JK, Lennard NJ, Jones M, Jones SJ, Solenberg PJ. Sequencing and analysis of genes involved in the biosynthesis of a vancomycin group antibiotic. Chem Biol. 1998;5:155–162. doi: 10.1016/s1074-5521(98)90060-6. [DOI] [PubMed] [Google Scholar]
- 145.Chiu HT, Hubbard BK, Shah AN, Eide J, Fredenburg RA, Walsh CT, Khosla C. Molecular cloning and sequence analysis of the complestatin biosynthetic gene cluster. Proc Natl Acad Sci U S A. 2001;98:8548–8553. doi: 10.1073/pnas.151246498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pootoolal J, Thomas MG, Marshall CG, Neu JM, Hubbard BK, Walsh CT, Wright GD. Assembling the glycopeptide antibiotic scaffold: The biosynthesis of A47934 from Streptomyces toyocaensis NRRL15009. Proc Natl Acad Sci U S A. 2002;99:8962–8967. doi: 10.1073/pnas.102285099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sosio M, Stinchi S, Beltrametti F, Lazzarini A, Donadio S. The gene cluster for the biosynthesis of the glycopeptide antibiotic A40926 by Nonomuraea species. Chem Biol. 2003;10:541–549. doi: 10.1016/s1074-5521(03)00120-0. [DOI] [PubMed] [Google Scholar]
- 148.Li TL, Huang F, Haydock SF, Mironenko T, Leadlay PF, Spencer JB. Biosynthetic gene cluster of the glycopeptide antibiotic teicoplanin: characterization of two glycosyltransferases and the key acyltransferase. Chem Biol. 2004;11:107–119. doi: 10.1016/j.chembiol.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 149.Banik JJ, Brady SF. Cloning and characterization of new glycopeptide gene clusters found in an environmental DNA megalibrary. Proc Natl Acad Sci U S A. 2008;105:17273–17277. doi: 10.1073/pnas.0807564105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Owen JG, Reddy BV, Ternei MA, Charlop-Powers Z, Calle PY, Kim JH, Brady SF. Mapping gene clusters within arrayed metagenomic libraries to expand the structural diversity of biomedically relevant natural products. Proc Natl Acad Sci U S A. 2013;110:11797–11802. doi: 10.1073/pnas.1222159110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Schmartz PC, Zerbe K, Abou-Hadeed K, Robinson JA. Bis-chlorination of a hexapeptide-PCP conjugate by the halogenase involved in vancomycin biosynthesis. Org Biomol Chem. 2014;12:5574–5577. doi: 10.1039/c4ob00474d. [DOI] [PubMed] [Google Scholar]
- 152.Welker M, von Dohren H. Cyanobacterial peptides - Nature’s own combinatorial biosynthesis. FEMS Microbiol Rev. 2006;30:530–563. doi: 10.1111/j.1574-6976.2006.00022.x. [DOI] [PubMed] [Google Scholar]
- 153.Rouhiainen L, Paulin L, Suomalainen S, Hyytiainen H, Buikema W, Haselkorn R, Sivonen K. Genes encoding synthetases of cyclic depsipeptides, anabaenopeptilides, in Anabaena strain 90. Mol Microbiol. 2000;37:156–167. doi: 10.1046/j.1365-2958.2000.01982.x. [DOI] [PubMed] [Google Scholar]
- 154.Trimurtulu G, Ohtani I, Patterson GML, Moore RE, Corbett TH, Valeriote FA, Demchik L. Total structures of cryptophycins, potent antitumor depsipeptides from the blue-green alga Nostoc sp. strain GSV 224. J Am Chem Soc. 1994;116:4729–4737. [Google Scholar]
- 155.Magarvey NA, Beck ZQ, Golakoti T, Ding YS, Huber U, Hemscheidt TK, Abelson D, Moore RE, Sherman DH. Biosynthetic characterization and chemoenzymatic assembly of the cryptophycins. Potent anticancer agents from Nostoc cyanobionts. ACS Chem Biol. 2006;1:766–779. doi: 10.1021/cb6004307. [DOI] [PubMed] [Google Scholar]
- 156.Rachid S, Krug D, Kunze B, Kochems I, Scharfe M, Zabriskie TM, Blocker H, Muller R. Molecular and biochemical studies of chondramide formation-highly cytotoxic natural products from Chondromyces crocatus Cm c5. Chem Biol. 2006;13:667–681. doi: 10.1016/j.chembiol.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 157.Crews P, Manes LV, Boehler M. Jasplakinolide, a cyclodepsipeptide from the marine sponge, Jaspis sp. Tetrahedron Lett. 1986;27:2797–2800. [Google Scholar]
- 158.Watts KR, Morinaka BI, Arnagata T, Robinson SJ, Tenney K, Bray WM, Gassner NC, Lokey RS, Media J, Valeriote FA, et al. Biostructural features of additional jasplakinolide (jaspamide) analogues. J Nat Prod. 2011;74:341–351. doi: 10.1021/np100721g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Fang X, Tiyanont K, Zhang Y, Wanner J, Boger D, Walker S. The mechanism of action of ramoplanin and enduracidin. Mol Biosyst. 2006;2:69–76. doi: 10.1039/b515328j. [DOI] [PubMed] [Google Scholar]
- 160.McCafferty DG, Cudic P, Frankel BA, Barkallah S, Kruger RG, Li W. Chemistry and biology of the ramoplanin family of peptide antibiotics. Biopolymers. 2002;66:261–284. doi: 10.1002/bip.10296. [DOI] [PubMed] [Google Scholar]
- 161.Floss HG, Yu TW. Rifamycin-mode of action, resistance, and biosynthesis. Chem Rev. 2005;105:621–632. doi: 10.1021/cr030112j. [DOI] [PubMed] [Google Scholar]
- 162.Yu TW, Bai L, Clade D, Hoffmann D, Toelzer S, Trinh KQ, Xu J, Moss SJ, Leistner E, Floss HG. The biosynthetic gene cluster of the maytansinoid antitumor agent ansamitocin from Actinosynnema pretiosum. Proc Natl Acad Sci U S A. 2002;99:7968–7973. doi: 10.1073/pnas.092697199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Spiteller P, Bai L, Shang G, Carroll BJ, Yu TW, Floss HG. The post-polyketide synthase modification steps in the biosynthesis of the antitumor agent ansamitocin by Actinosynnema pretiosum. J Am Chem Soc. 2003;125:14236–14237. doi: 10.1021/ja038166y. [DOI] [PubMed] [Google Scholar]
- 164.Chen JS, Su M, Shao L, Wang YX, Lin HM, Chen DJ. Investigation of halogenation during the biosynthesis of ramoplanin in Actinoplanes sp. ATCC33076. Appl Microbiol Biotechnol. 2016;100:289–298. doi: 10.1007/s00253-015-7014-2. [DOI] [PubMed] [Google Scholar]
- 165.Winn M, Fyans JK, Zhuo Y, Micklefield J. Recent advances in engineering nonribosomal peptide assembly lines. Nat Prod Rep. 2016;33:317–347. doi: 10.1039/c5np00099h. [DOI] [PubMed] [Google Scholar]
- 166.Yin X, Chen Y, Zhang L, Wang Y, Zabriskie TM. Enduracidin analogues with altered halogenation patterns produced by genetically engineered strains of Streptomyces fungicidicus. J Nat Prod. 2010;73:583–589. doi: 10.1021/np900710q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Han J, Chen J, Shao L, Zhang J, Dong X, Liu P, Chen D. Production of the ramoplanin activity analogue by double gene inactivation. PLoS One. 2016;11:e0154121. doi: 10.1371/journal.pone.0154121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Li XG, Tang XM, Xiao J, Ma GH, Xu L, Xie SJ, Xu MJ, Xiao X, Xu J. Harnessing the potential of halogenated natural product biosynthesis by mangrove-derived actinomycetes. Mar Drugs. 2013;11:3875–3890. doi: 10.3390/md11103875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Liao L, Chen R, Jiang M, Tian X, Liu H, Yu Y, Fan C, Chen B. Bioprospecting potential of halogenases from Arctic marine actinomycetes. BMC Microbiol. 2016;16:34. doi: 10.1186/s12866-016-0662-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Podzelinska K, Latimer R, Bhattacharya A, Vining LC, Zechel DL, Jia Z. Chloramphenicol biosynthesis: the structure of CmlS, a flavin-dependent halogenase showing a covalent flavin-aspartate bond. J Mol Biol. 2010;397:316–331. doi: 10.1016/j.jmb.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 171.He J, Magarvey N, Piraee M, Vining LC. The gene cluster for chloramphenicol biosynthesis in Streptomyces venezuelae ISP5230 includes novel shikimate pathway homologues and a monomodular non-ribosomal peptide synthetase gene. Microbiology. 2001;147:2817–2829. doi: 10.1099/00221287-147-10-2817. [DOI] [PubMed] [Google Scholar]
- 172.Fernandez-Martinez LT, Borsetto C, Gomez-Escribano JP, Bibb MJ, Al-Bassam MM, Chandra G. New insights into chloramphenicol biosynthesis in Streptomyces venezuelae ATCC 10712. Antimicrob Agents Chemother. 2014;58:7441–7450. doi: 10.1128/AAC.04272-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Castiglione F, Lazzarini A, Carrano L, Corti E, Ciciliato I, Gastaldo L, Candiani P, Losi D, Marinelli F, Selva E, et al. Determining the structure and mode of action of microbisporicin, a potent lantibiotic active against multiresistant pathogens. Chem Biol. 2008;15:22–31. doi: 10.1016/j.chembiol.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 174.Maffioli SI, Iorio M, Sosio M, Monciardini P, Gaspari E, Donadio S. Characterization of the congeners in the lantibiotic NAI-107 complex. J Nat Prod. 2014;77:79–84. doi: 10.1021/np400702t. [DOI] [PubMed] [Google Scholar]
- 175.Foulston LC, Bibb MJ. Microbisporicin gene cluster reveals unusual features of lantibiotic biosynthesis in actinomycetes. Proc Natl Acad Sci U S A. 2010;107:13461–13466. doi: 10.1073/pnas.1008285107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, et al. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Prod Rep. 2013;30:108–160. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Cruz JC, Iorio M, Monciardini P, Simone M, Brunati C, Gaspari E, Maffioli SI, Wellington E, Sosio M, Donadio S. Brominated variant of the lantibiotic NAI-107 with enhanced antibacterial potency. J Nat Prod. 2015;78:2642–2647. doi: 10.1021/acs.jnatprod.5b00576. [DOI] [PubMed] [Google Scholar]
- 178.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Jordan PA, Moore BS. Biosynthetic pathway connects cryptic ribosomally synthesized posttranslationally modified peptide genes with pyrroloquinoline alkaloids. Cell Chem Biol. 2016 doi: 10.1016/j.chembiol.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Vilter H. Peroxidases from Phaeophyceae - a vanadium(V)-dependent peroxidase from Ascophyllum nodosum .5. Phytochemistry. 1984;23:1387–1390. [Google Scholar]
- 181.Butler A, Sandy M. Mechanistic considerations of halogenating enzymes. Nature. 2009;460:848–854. doi: 10.1038/nature08303. [DOI] [PubMed] [Google Scholar]
- 182.Messerschmidt A, Wever R. X-ray structure of a vanadium-containing enzyme: chloroperoxidase from the fungus Curvularia inaequalis. Proc Natl Acad Sci U S A. 1996;93:392–396. doi: 10.1073/pnas.93.1.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Isupov MN, Dalby AR, Brindley AA, Izumi Y, Tanabe T, Murshudov GN, Littlechild JA. Crystal structure of dodecameric vanadium-dependent bromoperoxidase from the red algae Corallina officinalis. J Mol Biol. 2000;299:1035–1049. doi: 10.1006/jmbi.2000.3806. [DOI] [PubMed] [Google Scholar]
- 184.Weyand M, Hecht H, Kiess M, Liaud M, Vilter H, Schomburg D. X-ray structure determination of a vanadium-dependent haloperoxidase from Ascophyllum nodosum at 2.0 A resolution. J Mol Biol. 1999;293:595–611. doi: 10.1006/jmbi.1999.3179. [DOI] [PubMed] [Google Scholar]
- 185.Agarwal V, Borisova SA, Metcalf WW, van der Donk WA, Nair SK. Structural and mechanistic insights into C-P bond hydrolysis by phosphonoacetate hydrolase. Chem Biol. 2011;18:1230–1240. doi: 10.1016/j.chembiol.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zalatan JG, Fenn TD, Brunger AT, Herschlag D. Structural and functional comparisons of nucleotide pyrophosphatase/phosphodiesterase and alkaline phosphatase: implications for mechanism and evolution. Biochemistry. 2006;45:9788–9803. doi: 10.1021/bi060847t. [DOI] [PubMed] [Google Scholar]
- 187.Zalatan JG, Herschlag D. Alkaline phosphatase mono- and diesterase reactions: comparative transition state analysis. J Am Chem Soc. 2006;128:1293–1303. doi: 10.1021/ja056528r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Holtz KM, Stec B, Kantrowitz ER. A model of the transition state in the alkaline phosphatase reaction. J Biol Chem. 1999;274:8351–8354. doi: 10.1074/jbc.274.13.8351. [DOI] [PubMed] [Google Scholar]
- 189.Butler A, Carter-Franklin JN. The role of vanadium bromoperoxidase in the biosynthesis of halogenated marine natural products. Nat Prod Rep. 2004;21:180–188. doi: 10.1039/b302337k. [DOI] [PubMed] [Google Scholar]
- 190.Schneider CJ, Zampella G, DeGioa L, Pecoraro VL. Vanadium: the versatile metal. Vol. 974 American Chemical Society; 2007. [Google Scholar]
- 191.Winter JM, Moore BS. Exploring the chemistry and biology of vanadium-dependent haloperoxidases. J Biol Chem. 2009;284:18577–18581. doi: 10.1074/jbc.R109.001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Messerschmidt A, Prade L, Wever R. Implications for the catalytic mechanism of the vanadium-containing enzyme chloroperoxidase from the fungus Curvularia inaequalis by X-ray structures of the native and peroxide form. Biol Chem. 1997;378:309–315. doi: 10.1515/bchm.1997.378.3-4.309. [DOI] [PubMed] [Google Scholar]
- 193.Gribble GW. Amazing organohalogens. Am Sci. 2004;92:342–349. [Google Scholar]
- 194.Carter-Franklin JN, Butler A. Vanadium bromoperoxidase-catalyzed biosynthesis of halogenated marine natural products. J Am Chem Soc. 2004;126:15060–15066. doi: 10.1021/ja047925p. [DOI] [PubMed] [Google Scholar]
- 195.Fenical W. Halogenation in rhodophyta - Review. J Phycol. 1975;11:245–259. [Google Scholar]
- 196.Kladi M, Vagias C, Roussis V. Volatile halogenated metabolites from marine red algae. Phytochem Rev. 2004;3:337–366. [Google Scholar]
- 197.Cabrita MT, Vale C, Rauter AP. Halogenated compounds from marine algae. Mar Drugs. 2010;8:2301–2317. doi: 10.3390/md8082301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Paul C, Pohnert G. Production and role of volatile halogenated compounds from marine algae. Nat Prod Rep. 2011;28:186–195. doi: 10.1039/c0np00043d. [DOI] [PubMed] [Google Scholar]
- 199.Hewson WD, H LP. Bromoperoxidases and halogenated lipids in marine algae. J Phycol. 1980;16:340–345. [Google Scholar]
- 200.Yamada H, I N, Murakami S, Izumi Y. New bromoperoxidase from Coralline algae that brominates phenol compounds. Agric Biol Chem. 1985;49:2961–2967. [Google Scholar]
- 201.Hemrika W, Renirie R, Macedo-Ribeiro S, Messerschmidt A, Wever R. Heterologous expression of the vanadium-containing chloroperoxidase from Curvularia inaequalis in Saccharomyces cerevisiae and site-directed mutagenesis of the active site residues His(496), Lys(353), Arg(360), and Arg(490) J Biol Chem. 1999;274:23820–23827. doi: 10.1074/jbc.274.34.23820. [DOI] [PubMed] [Google Scholar]
- 202.Fenical W. Halogenation in the rhodophyta. J Phycol. 1975;11:245–259. [Google Scholar]
- 203.Faulkner DJ. Biomimetic synthesis of marine natural products. Pure Appl Chem. 1976;48:25–28. [Google Scholar]
- 204.Wanke TP, A C, Zatelli GA, Vieira LFO, Lhullier C, Falkenberg M. C15 acetogenins from the Laurencia complex: 50 years of research - and overview. Rev Bras Farmacogn. 2015;25:569–587. [Google Scholar]
- 205.Fukuzawa AAM, Takasugi Y, Nakamura M, Tamura M, Murai A. Enzymatic bromo-ether cyclization of laurediols with bromoperoxidase. Chem Lett. 1994:2307–2310. [Google Scholar]
- 206.Ishihara J, S Y, Kanoh N, Takasugi Y, Fukuzawa A, Murai A. Conversion of prelaureatin into laurallene, a bromo-allene compound, by enzymatic and chemical bromo-etherfication reactions. Tetrahedron. 1997;53:8371–8382. [Google Scholar]
- 207.Kaneko K, Washio K, Umezawa T, Matsuda F, Morikawa M, Okino T. cDNA cloning and characterization of vanadium-dependent bromoperoxidases from the red alga Laurencia nipponica. Biosci Biotechnol Biochem. 2014;78:1310–1319. doi: 10.1080/09168451.2014.918482. [DOI] [PubMed] [Google Scholar]
- 208.Camilli A, Bassler BL. Bacterial small-molecule signaling pathways. Science. 2006;311:1113–1116. doi: 10.1126/science.1121357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Wever RHW, Messerschmidt AHR, Wieghardt K, Poulos T, editors. John Wiley & Sons Ltd: Chichester, United Kingdom. 2001. [Google Scholar]
- 210.Cosse A, Potin P, Leblanc C. Patterns of gene expression induced by oligoguluronates reveal conserved and environment-specific molecular defense responses in the brown alga Laminaria digitata. New Phytol. 2009;182:239–250. doi: 10.1111/j.1469-8137.2008.02745.x. [DOI] [PubMed] [Google Scholar]
- 211.Michels JJ, Allain EJ, Borchardt SA, Hu P, McCoy WF. Degradation pathway of homoserine lactone bacterial signal molecules by halogen antimicrobials identified by liquid chromatography with photodiode array and mass spectrometric detection. J Chromatogr A. 2000;898:153–165. doi: 10.1016/s0021-9673(00)00849-9. [DOI] [PubMed] [Google Scholar]
- 212.Sandy M, Carter-Franklin JN, Martin JD, Butler A. Vanadium bromoperoxidase from Delisea pulchra: enzyme-catalyzed formation of bromofuranone and attendant disruption of quorum sensing. Chem Commun (Camb) 2011;47:12086–12088. doi: 10.1039/c1cc15605e. [DOI] [PubMed] [Google Scholar]
- 213.Dworjanyn SA, Wright JT, Paul NA, de Nys R, Steinberg PD. Cost of chemical defence in the red alga. Delisea pulchra Oikos. 2006;113:13–22. [Google Scholar]
- 214.Dworjanyn SA, de Nys R, Steinberg PD. Chemically mediated antifouling in the red alga Delisea pulchra. Mar Ecol Prog Ser. 2006;318:153–163. [Google Scholar]
- 215.Liu TN, M’Timkulu T, Geigert J, Wolf B, Neidleman SL, Silva D, Hunter-Cevera JC. Isolation and characterization of a novel nonheme chloroperoxidase. Biochem Biophys Res Commun. 1987;142:329–333. doi: 10.1016/0006-291x(87)90277-4. [DOI] [PubMed] [Google Scholar]
- 216.van Schijndel JW, Vollenbroek EG, Wever R. The chloroperoxidase from the fungus Curvularia inaequalis; a novel vanadium enzyme. Biochim Biophys Acta. 1993;1161:249–256. doi: 10.1016/0167-4838(93)90221-c. [DOI] [PubMed] [Google Scholar]
- 217.Barnett P, Hemrika W, Dekker HL, Muijsers AO, Renirie R, Wever R. Isolation, characterization, and primary structure of the vanadium chloroperoxidase from the fungus Embellisia didymospora. J Biol Chem. 1998;273:23381–23387. doi: 10.1074/jbc.273.36.23381. [DOI] [PubMed] [Google Scholar]
- 218.Ortiz-Bermudez P, Hirth KC, Srebotnik E, Hammel KE. Chlorination of lignin by ubiquitous fungi has a likely role in global organochlorine production. Proc Natl Acad Sci U S A. 2007;104:3895–3900. doi: 10.1073/pnas.0610074104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Winter JM, Moffitt MC, Zazopoulos E, McAlpine JB, Dorrestein PC, Moore BS. Molecular basis for chloronium-mediated meroterpene cyclization: cloning, sequencing, and heterologous expression of the napyradiomycin biosynthetic gene cluster. J Biol Chem. 2007;282:16362–16368. doi: 10.1074/jbc.M611046200. [DOI] [PubMed] [Google Scholar]
- 220.Simpson TJ. Applications of multinuclear NMR to structural and biosynthetic studies of polyketide microbial metabolites. Chem Soc Rev. 1987;16:123–160. [Google Scholar]
- 221.Geris R, Simpson TJ. Meroterpenoids produced by fungi. Nat Prod Rep. 2009;26:1063–1094. doi: 10.1039/b820413f. [DOI] [PubMed] [Google Scholar]
- 222.Kuzuyama T, Seto H. Diversity of the biosynthesis of the isoprene units. Nat Prod Rep. 2003;20:171–183. doi: 10.1039/b109860h. [DOI] [PubMed] [Google Scholar]
- 223.Littlechild J, Garcia-Rodriguez E, Dalby A, Isupov M. Structural and functional comparisons between vanadium haloperoxidase and acid phosphatase enzymes. J Mol Recognit. 2002;15:291–296. doi: 10.1002/jmr.590. [DOI] [PubMed] [Google Scholar]
- 224.Raugei S, Carloni P. Structure and function of vanadium haloperoxidases. J Phys Chem B. 2006;110:3747–3758. doi: 10.1021/jp054901b. [DOI] [PubMed] [Google Scholar]
- 225.Renirie R, Hemrika W, Wever R. Peroxidase and phosphatase activity of active-site mutants of vanadium chloroperoxidase from the fungus Curvularia inaequalis. Implications for the catalytic mechanisms. J Biol Chem. 2000;275:11650–11657. doi: 10.1074/jbc.275.16.11650. [DOI] [PubMed] [Google Scholar]
- 226.Bernhardt P, Okino T, Winter JM, Miyanaga A, Moore BS. A stereoselective vanadium-dependent chloroperoxidase in bacterial antibiotic biosynthesis. J Am Chem Soc. 2011;133:4268–4270. doi: 10.1021/ja201088k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Wagner C, Molitor IM, König GM. Critical view on the monochlorodimedone assay utilized to detect haloperoxidase activity. Phytochemistry. 2008;69:323–332. doi: 10.1016/j.phytochem.2007.07.020. [DOI] [PubMed] [Google Scholar]
- 228.Kaysser L, Bernhardt P, Nam SJ, Loesgen S, Ruby JG, Skewes-Cox P, Jensen PR, Fenical W, Moore BS. Merochlorins A-D, cyclic meroterpenoid antibiotics biosynthesized in divergent pathways with vanadium-dependent chloroperoxidases. J Am Chem Soc. 2012;134:11988–11991. doi: 10.1021/ja305665f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Sakoulas G, Nam SJ, Loesgen S, Fenical W, Jensen PR, Nizet V, Hensler M. Novel bacterial metabolite merochlorin A demonstrates in vitro activity against multi-drug resistant methicillin-resistant. Staphylococcus aureus PLoS One. 2012;7:e29439. doi: 10.1371/journal.pone.0029439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Teufel R, Kaysser L, Villaume MT, Diethelm S, Carbullido MK, Baran PS, Moore BS. One-pot enzymatic synthesis of merochlorin A and B. Angew Chem Int Ed Engl. 2014;53:11019–11022. doi: 10.1002/anie.201405694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Diethelm S, Teufel R, Kaysser L, Moore BS. A multitasking vanadium-dependent chloroperoxidase as an inspiration for the chemical synthesis of the merochlorins. Angew Chem Int Ed Engl. 2014;53:11023–11026. doi: 10.1002/anie.201405696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Pepper HP, George JH. Biomimetic total synthesis of (+/−)-merochlorin A. Angew Chem Int Ed Engl. 2013;52:12170–12173. doi: 10.1002/anie.201307200. [DOI] [PubMed] [Google Scholar]
- 233.Meier R, Strych S, Trauner D. Biomimetic synthesis of (+/−)-merochlorin B. Org Lett. 2014;16:2634–2637. doi: 10.1021/ol500800z. [DOI] [PubMed] [Google Scholar]
- 234.Palenik B, Ren Q, Dupont CL, Myers GS, Heidelberg JF, Badger JH, Madupu R, Nelson WC, Brinkac LM, Dodson RJ, et al. Genome sequence of Synechococcus CC9311: Insights into adaptation to a coastal environment. Proc Natl Acad Sci U S A. 2006;103:13555–13559. doi: 10.1073/pnas.0602963103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Johnson TL, Palenik B, Brahamsha B. Characterization of a functional vanadium-dependent bromoperoxidase in the marine cyanobacterium Synechococcus sp. CC9311. J Phycol. 2011;47:792–801. doi: 10.1111/j.1529-8817.2011.01007.x. [DOI] [PubMed] [Google Scholar]
- 236.Metcalf WW, van der Donk WA. Biosynthesis of phosphonic and phosphinic acid natural products. Annu Rev Biochem. 2009;78:65–94. doi: 10.1146/annurev.biochem.78.091707.100215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Horsman GP, Zechel DL. Phosphonate biochemistry. Chem Rev. 2016 doi: 10.1021/acs.chemrev.6b00536. [DOI] [PubMed] [Google Scholar]
- 238.Macedo-Ribeiro S, Hemrika W, Renirie R, Wever R, Messerschmidt A. X-ray crystal structures of active site mutants of the vanadium-containing chloroperoxidase from the fungus Curvularia inaequalis. J Biol Inorg Chem. 1999;4:209–219. doi: 10.1007/s007750050306. [DOI] [PubMed] [Google Scholar]
- 239.Fournier JB, Rebuffet E, Delage L, Grijol R, Meslet-Cladiere L, Rzonca J, Potin P, Michel G, Czjzek M, Leblanc C. The Vanadium iodoperoxidase from the marine flavobacteriaceae species Zobellia galactanivorans reveals novel molecular and evolutionary features of halide specificity in the vanadium haloperoxidase enzyme family. Appl Environ Microbiol. 2014;80:7561–7573. doi: 10.1128/AEM.02430-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Hasan Z, Renirie R, Kerkman R, Ruijssenaars HJ, Hartog AF, Wever R. Laboratory-evolved vanadium chloroperoxidase exhibits 100-fold higher halogenating activity at alkaline pH: catalytic effects from first and second coordination sphere mutations. J Biol Chem. 2006;281:9738–9744. doi: 10.1074/jbc.M512166200. [DOI] [PubMed] [Google Scholar]
- 241.Martinez VM, De Cremer G, Roeffaers MB, Sliwa M, Baruah M, De Vos DE, Hofkens J, Sels BF. Exploration of single molecule events in a haloperoxidase and its biomimic: localization of halogenation activity. J Am Chem Soc. 2008;130:13192–13193. doi: 10.1021/ja804606m. [DOI] [PubMed] [Google Scholar]
- 242.Ohshiro T, Littlechild J, Garcia-Rodriguez E, Isupov MN, Iida Y, Kobayashi T, Izumi Y. Modification of halogen specificity of a vanadium-dependent bromoperoxidase. Protein Sci. 2004;13:1566–1571. doi: 10.1110/ps.03496004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Soedjak HS, Butler A. Characterization of vanadium bromoperoxidase from Macrocystis and Fucus: reactivity of vanadium bromoperoxidase toward acyl and alkyl peroxides and bromination of amines. Biochemistry. 1990;29:7974–7981. doi: 10.1021/bi00486a028. [DOI] [PubMed] [Google Scholar]
- 244.Colpas GJ, H BJ, Kampf JW, Pecoraro VL. Functional models for vanadium haloperoxidases: reactivity and mechanism of halide oxidation. J Am Chem Soc. 1996;118:3469–3478. [Google Scholar]
- 245.Verhaeghe E, Buisson D, Zekri E, Leblanc C, Potin P, Ambroise Y. A colorimetric assay for steady-state analyses of iodo- and bromoperoxidase activities. Anal Biochem. 2008;379:60–65. doi: 10.1016/j.ab.2008.04.041. [DOI] [PubMed] [Google Scholar]
- 246.Orjala J, Gerwick WH. Barbamide, a chlorinated metabolite with molluscicidal activity from the caribbean cyanobacterium Lyngbya majuscula. J Nat Prod. 1996;59:427–430. doi: 10.1021/np960085a. [DOI] [PubMed] [Google Scholar]
- 247.Sitachitta N, Rossi J, Roberts MA, Gerwick WH, Fletcher MD, Willis CL. Biosynthesis of the marine cyanobacterial metabolite barbamide. 1. Origin of the trichloromethyl group. J Am Chem Soc. 1998;120:7131–7132. [Google Scholar]
- 248.Galonic DP, Vaillancourt FH, Walsh CT. Halogenation of unactivated carbon centers in natural product biosynthesis: trichlorination of leucine during barbamide biosynthesis. J Am Chem Soc. 2006;128:3900–3901. doi: 10.1021/ja060151n. [DOI] [PubMed] [Google Scholar]
- 249.Vaillancourt FH, Yin J, Walsh CT. SyrB2 in syringomycin E biosynthesis is a nonheme Fe(II) alpha-ketoglutarate- and O2-dependent halogenase. Proc Natl Acad Sci U S A. 2005;102:10111–10116. doi: 10.1073/pnas.0504412102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Ueki M, Galonic DP, Vaillancourt FH, Garneau-Tsodikova S, Yeh E, Vosburg DA, Schroeder FC, Osada H, Walsh CT. Enzymatic generation of the antimetabolite g,g-dichloroaminobutyrate by NRPS and mononuclear iron halogenase action in a streptomycete. Chem Biol. 2006;13:1183–1191. doi: 10.1016/j.chembiol.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 251.Pratter SM, Ivkovic J, Birner-Gruenberger R, Breinbauer R, Zangger K, Straganz GD. More than just a halogenase: modification of fatty acyl moieties by a trifunctional metal enzyme. Chembiochem. 2014;15:567–574. doi: 10.1002/cbic.201300345. [DOI] [PubMed] [Google Scholar]
- 252.Choi H, Mevers E, Byrum T, Valeriote FA, Gerwick WH. Lyngbyabellins K–N from two Palmyra Atoll collections of the marine cyanobacterium Moorea bouillonii. Eur J Org Chem. 2012;2012:5141–5150. doi: 10.1002/ejoc.201200691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Jiang W, Heemstra JR, Jr, Forseth RR, Neumann CS, Manaviazar S, Schroeder FC, Hale KJ, Walsh CT. Biosynthetic chlorination of the piperazate residue in kutzneride biosynthesis by KthP. Biochemistry. 2011;50:6063–6072. doi: 10.1021/bi200656k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Neumann CS, Walsh CT. Biosynthesis of (−)-(1S,2R)-allocoronamic acyl thioester by an Fe(II)-dependent halogenase and a cyclopropane-forming flavoprotein. J Am Chem Soc. 2008;130:14022–14023. doi: 10.1021/ja8064667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Vaillancourt FH, Yeh E, Vosburg DA, O’Connor SE, Walsh CT. Cryptic chlorination by a non-haem iron enzyme during cyclopropyl amino acid biosynthesis. Nature. 2005;436:1191–1194. doi: 10.1038/nature03797. [DOI] [PubMed] [Google Scholar]
- 256.Kelly WL, Boyne MT, 2nd, Yeh E, Vosburg DA, Galonic DP, Kelleher NL, Walsh CT. Characterization of the aminocarboxycyclopropane-forming enzyme CmaC. Biochemistry. 2007;46:359–368. doi: 10.1021/bi061930j. [DOI] [PubMed] [Google Scholar]
- 257.Takayama K, Wang C, Besra GS. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin Microbiol Rev. 2005;18:81–101. doi: 10.1128/CMR.18.1.81-101.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Huang W, Xu H, Li Y, Zhang F, Chen XY, He QL, Igarashi Y, Tang GL. Characterization of yatakemycin gene cluster revealing a radical S-adenosylmethionine dependent methyltransferase and highlighting spirocyclopropane biosynthesis. J Am Chem Soc. 2012;134:8831–8840. doi: 10.1021/ja211098r. [DOI] [PubMed] [Google Scholar]
- 259.Watanabe K, Hotta K, Nakaya M, Praseuth AP, Wang CCC, Inada D, Takahashi K, Fukushi E, Oguri H, Oikawa H. Escherichia coli allows efficient modular incorporation of newly isolated quinomycin biosynthetic enzyme into echinomycin biosynthetic pathway for rational design and synthesis of potent antibiotic unnatural natural product. J Am Chem Soc. 2009;131:9347–9353. doi: 10.1021/ja902261a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Thibodeaux CJ, Chang Wc, Liu Hw. Enzymatic chemistry of cyclopropane, epoxide, and aziridine biosynthesis. Chem Rev. 2012;112:1681–1709. doi: 10.1021/cr200073d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Gu L, Wang B, Kulkarni A, Geders TW, Grindberg RV, Gerwick L, Hakansson K, Wipf P, Smith JL, Gerwick WH, et al. Metamorphic enzyme assembly in polyketide diversification. Nature. 2009;459:731–735. doi: 10.1038/nature07870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Hillwig ML, Fuhrman HA, Ittiamornkul K, Sevco TJ, Kwak DH, Liu X. Identification and characterization of a welwitindolinone alkaloid biosynthetic gene cluster in the stigonematalean cyanobacterium. Hapalosiphon welwitschii Chembiochem. 2014;15:665–669. doi: 10.1002/cbic.201300794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Micallef ML, Sharma D, Bunn BM, Gerwick L, Viswanathan R, Moffitt MC. Comparative analysis of hapalindole, ambiguine and welwitindolinone gene clusters and reconstitution of indole-isonitrile biosynthesis from cyanobacteria. BMC Microbiol. 2014;14:213. doi: 10.1186/s12866-014-0213-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Stratmann K, Moore RE, Bonjouklian R, Deeter JB, Patterson GML, Shaffer S, Smith CD, Smitka TA. Welwitindolinones, unusual alkaloids from the blue-green algae Hapalosiphon welwitschii and Westiella intricata - relationship to fischerindoles and hapalindoles. J Am Chem Soc. 1994;116:9935–9942. [Google Scholar]
- 265.Hillwig ML, Liu X. A new family of iron-dependent halogenases acts on freestanding substrates. Nat Chem Biol. 2014;10:921–923. doi: 10.1038/nchembio.1625. [DOI] [PubMed] [Google Scholar]
- 266.Hillwig ML, Zhu Q, Liu X. Biosynthesis of ambiguine indole alkaloids in cyanobacterium Fischerella ambigua. ACS Chem Biol. 2014;9:372–377. doi: 10.1021/cb400681n. [DOI] [PubMed] [Google Scholar]
- 267.Hillwig ML, Zhu Q, Ittiamornkul K, Liu X. Discovery of a promiscuous non-heme iron halogenase in ambiguine alkaloid biogenesis: implication for an evolvable enzyme family for late-stage halogenation of aliphatic carbons in small molecules. Angew Chem Int Ed Engl. 2016;55:5780–5784. doi: 10.1002/anie.201601447. [DOI] [PubMed] [Google Scholar]
- 268.Blasiak LC, Vaillancourt FH, Walsh CT, Drennan CL. Crystal structure of the non-haem iron halogenase SyrB2 in syringomycin biosynthesis. Nature. 2006;440:368–371. doi: 10.1038/nature04544. [DOI] [PubMed] [Google Scholar]
- 269.Wong C, Fujimori DG, Walsh CT, Drennan CL. Structural analysis of an open active site conformation of nonheme iron halogenase CytC3. J Am Chem Soc. 2009;131:4872–4879. doi: 10.1021/ja8097355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Khare D, Wang B, Gu L, Razelun J, Sherman DH, Gerwick WH, Hakansson K, Smith JL. Conformational switch triggered by alpha-ketoglutarate in a halogenase of curacin A biosynthesis. Proc Natl Acad Sci U S A. 2010;107:14099–14104. doi: 10.1073/pnas.1006738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Mitchell AJ, Zhu Q, Maggiolo AO, Ananth NR, Hillwig ML, Liu X, Boal AK. Structural basis for halogenation by iron- and 2-oxo-glutarate-dependent enzyme WelO5. Nat Chem Biol. 2016;12:636–640. doi: 10.1038/nchembio.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Clifton IJ, McDonough MA, Ehrismann D, Kershaw NJ, Granatino N, Schofield CJ. Structural studies on 2-oxoglutarate oxygenases and related double-stranded beta-helix fold proteins. J Inorg Biochem. 2006;100:644–669. doi: 10.1016/j.jinorgbio.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 273.Aik W, McDonough MA, Thalhammer A, Chowdhury R, Schofield CJ. Role of the jelly-roll fold in substrate binding by 2-oxoglutarate oxygenases. Curr Opin Struct Biol. 2012;22:691–700. doi: 10.1016/j.sbi.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 274.Koehntop KD, Emerson JP, Que L., Jr The 2-His-1-carboxylate facial triad: a versatile platform for dioxygen activation by mononuclear non-heme iron(II) enzymes. J Biol Inorg Chem. 2005;10:87–93. doi: 10.1007/s00775-005-0624-x. [DOI] [PubMed] [Google Scholar]
- 275.Elkins JM, Ryle MJ, Clifton IJ, Dunning Hotopp JC, Lloyd JS, Burzlaff NI, Baldwin JE, Hausinger RP, Roach PL. X-ray crystal structure of Escherichia coli taurine/alpha-ketoglutarate dioxygenase complexed to ferrous iron and substrates. Biochemistry. 2002;41:5185–5192. doi: 10.1021/bi016014e. [DOI] [PubMed] [Google Scholar]
- 276.Busche A, Gottstein D, Hein C, Ripin N, Pader I, Tufar P, Eisman EB, Gu L, Walsh CT, Sherman DH. Characterization of molecular interactions between ACP and halogenase domains in the curacin A polyketide synthase. ACS Chem Biol. 2012;7:378–386. doi: 10.1021/cb200352q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Vaillancourt FH, Vosburg DA, Walsh CT. Dichlorination and bromination of a threonyl-S-carrier protein by the non-heme Fe(II) halogenase SyrB2. Chembiochem. 2006;7:748–752. doi: 10.1002/cbic.200500480. [DOI] [PubMed] [Google Scholar]
- 278.Grgurina I, Barca A, Cervigni S, Gallo M, Scaloni A, Pucci P. Relevance of chlorine-substituent for the antifungal activity of syringomycin and syringotoxin, metabolites of the phytopathogenic bacterium Pseudomonas syringae pv. syringae. Experientia. 1994;50:130–133. doi: 10.1007/BF01984950. [DOI] [PubMed] [Google Scholar]
- 279.Zhu Q, Hillwig ML, Doi Y, Liu X. Aliphatic halogenase enables late-stage C-H functionalization: selective synthesis of a brominated fischerindole slkaloid with enhanced antibacterial activity. Chembiochem. 2016;17:466–470. doi: 10.1002/cbic.201500674. [DOI] [PubMed] [Google Scholar]
- 280.Matthews ML, Neumann CS, Miles LA, Grove TL, Booker SJ, Krebs C, Walsh CT, Bollinger JM., Jr Substrate positioning controls the partition between halogenation and hydroxylation in the aliphatic halogenase, SyrB2. Proc Natl Acad Sci U S A. 2009;106:17723–17728. doi: 10.1073/pnas.0909649106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Matthews ML, Chang WC, Layne AP, Miles LA, Krebs C, Bollinger JM., Jr Direct nitration and azidation of aliphatic carbons by an iron-dependent halogenase. Nat Chem Biol. 2014;10:209–215. doi: 10.1038/nchembio.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Martinez S, Hausinger RP. Catalytic mechanisms of Fe(II)- and 2-oxoglutarate-dependent oxygenases. J Biol Chem. 2015;290:20702–20711. doi: 10.1074/jbc.R115.648691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Bollinger JM, Jr, Chang Wc, Matthews ML, Martinie RJ, Boal AK, Krebs C. 2-Oxoglutarate-Dependent Oxygenases. The Royal Society of Chemistry; 2015. [Google Scholar]
- 284.Fujimori DG, Barr EW, Matthews ML, Koch GM, Yonce JR, Walsh CT, Bollinger JM, Jr, Krebs C, Riggs-Gelasco PJ. Spectroscopic evidence for a high-spin Br-Fe(IV)-oxo intermediate in the alpha-ketoglutarate-dependent halogenase CytC3 from Streptomyces. J Am Chem Soc. 2007;129:13408–13409. doi: 10.1021/ja076454e. [DOI] [PubMed] [Google Scholar]
- 285.Galonic DP, Barr EW, Walsh CT, Bollinger JM, Jr, Krebs C. Two interconverting Fe(IV) intermediates in aliphatic chlorination by the halogenase CytC3. Nat Chem Biol. 2007;3:113–116. doi: 10.1038/nchembio856. [DOI] [PubMed] [Google Scholar]
- 286.Matthews ML, Krest CM, Barr EW, Vaillancourt FH, Walsh CT, Green MT, Krebs C, Bollinger JM. Substrate-triggered formation and remarkable stability of the C-H bond-cleaving chloroferryl intermediate in the aliphatic halogenase, SyrB2. Biochemistry. 2009;48:4331–4343. doi: 10.1021/bi900109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Kulik HJ, Drennan CL. Substrate placement influences reactivity in non-heme Fe(II) halogenases and hydroxylases. J Biol Chem. 2013;288:11233–11241. doi: 10.1074/jbc.M112.415570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Martinie RJ, Livada J, Chang WC, Green MT, Krebs C, Bollinger JM, Jr, Silakov A. Experimental correlation of substrate position with reaction outcome in the aliphatic halogenase, SyrB2. J Am Chem Soc. 2015;137:6912–6919. doi: 10.1021/jacs.5b03370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Borowski T, Noack H, Radon M, Zych K, Siegbahn PE. Mechanism of selective halogenation by SyrB2: a computational study. J Am Chem Soc. 2010;132:12887–12898. doi: 10.1021/ja101877a. [DOI] [PubMed] [Google Scholar]
- 290.de Visser SP, Latifi R. Carbon dioxide: a waste product in the catalytic cycle of alpha-ketoglutarate dependent halogenases prevents the formation of hydroxylated by-products. J Phys Chem B. 2009;113:12–14. doi: 10.1021/jp8097632. [DOI] [PubMed] [Google Scholar]
- 291.Wong SD, Srnec M, Matthews ML, Liu LV, Kwak Y, Park K, Bell CB, 3rd, Alp EE, Zhao J, Yoda Y, et al. Elucidation of the Fe(IV)=O intermediate in the catalytic cycle of the halogenase SyrB2. Nature. 2013;499:320–323. doi: 10.1038/nature12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Zhang Z, Ren J, Harlos K, McKinnon CH, Clifton IJ, Schofield CJ. Crystal structure of a clavaminate synthase-Fe(II)-2-oxoglutarate-substrate-NO complex: evidence for metal centered rearrangements. FEBS Lett. 2002;517:7–12. doi: 10.1016/s0014-5793(02)02520-6. [DOI] [PubMed] [Google Scholar]
- 293.Brown S, O’Connor SE. Halogenase engineering for the generation of new natural product analogues. Chembiochem. 2015;16:2129–2135. doi: 10.1002/cbic.201500338. [DOI] [PubMed] [Google Scholar]
- 294.Dong C, Huang F, Deng H, Schaffrath C, Spencer JB, O’Hagan D, Naismith JH. Crystal structure and mechanism of a bacterial fluorinating enzyme. Nature. 2004;427:561–565. doi: 10.1038/nature02280. [DOI] [PubMed] [Google Scholar]
- 295.Huang S, Ma L, Tong MH, Yu Y, O’Hagan D, Deng H. Fluoroacetate biosynthesis from the marine-derived bacterium Streptomyces xinghaiensis NRRL B-24674. Org Biomol Chem. 2014;12:4828–4831. doi: 10.1039/c4ob00970c. [DOI] [PubMed] [Google Scholar]
- 296.Deng H, Ma L, Bandaranayaka N, Qin Z, Mann G, Kyeremeh K, Yu Y, Shepherd T, Naismith JH, O’Hagan D. Identification of fluorinases from Streptomyces sp. MA37, Norcardia brasiliensis, and Actinoplanes sp. N902–109 by genome mining. Chembiochem. 2014;15:364–368. doi: 10.1002/cbic.201300732. [DOI] [PubMed] [Google Scholar]
- 297.Cadicamo CD, Courtieu J, Deng H, Meddour A, O’Hagan D. Enzymatic fluorination in Streptomyces cattleya takes place with an inversion of configuration consistent with an SN2 reaction mechanism. Chembiochem. 2004;5:685–690. doi: 10.1002/cbic.200300839. [DOI] [PubMed] [Google Scholar]
- 298.Deng H, Cobb SL, McEwan AR, McGlinchey RP, Naismith JH, O’Hagan D, Robinson DA, Spencer JB. The fluorinase from Streptomyces cattleya is also a chlorinase. Angew Chem Int Ed Engl. 2006;45:759–762. doi: 10.1002/anie.200503582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Eustaquio AS, Pojer F, Noel JP, Moore BS. Discovery and characterization of a marine bacterial SAM-dependent chlorinase. Nat Chem Biol. 2008;4:69–74. doi: 10.1038/nchembio.2007.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Gulder TA, Moore BS. Salinosporamide natural products: Potent 20 S proteasome inhibitors as promising cancer chemotherapeutics. Angew Chem Int Ed Engl. 2010;49:9346–9367. doi: 10.1002/anie.201000728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Eustaquio AS, McGlinchey RP, Liu Y, Hazzard C, Beer LL, Florova G, Alhamadsheh MM, Lechner A, Kale AJ, Kobayashi Y, et al. Biosynthesis of the salinosporamide A polyketide synthase substrate chloroethylmalonyl-coenzyme A from S-adenosyl-L-methionine. Proc Natl Acad Sci U S A. 2009;106:12295–12300. doi: 10.1073/pnas.0901237106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W. Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus salinospora. Angew Chem Int Ed Engl. 2003;42:355–357. doi: 10.1002/anie.200390115. [DOI] [PubMed] [Google Scholar]
- 303.Zhu X, Robinson DA, McEwan AR, O’Hagan D, Naismith JH. Mechanism of enzymatic fluorination in Streptomyces cattleya. J Am Chem Soc. 2007;129:14597–14604. doi: 10.1021/ja0731569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Senn HM, O’Hagan D, Thiel W. Insight into enzymatic C-F bond formation from QM and QM/MM calculations. J Amer Chem Soc. 2005;127:13643–13655. doi: 10.1021/ja053875s. [DOI] [PubMed] [Google Scholar]
- 305.Deng H, O’Hagan D. The fluorinase, the chlorinase and the duf-62 enzymes. Curr Opin Chem Biol. 2008;12:582–592. doi: 10.1016/j.cbpa.2008.06.036. [DOI] [PubMed] [Google Scholar]
- 306.Eustaquio AS, Harle J, Noel JP, Moore BS. S-Adenosyl-L-methionine hydrolase (adenosine-forming), a conserved bacterial and archaeal protein related to SAM-dependent halogenases. Chembiochem. 2008;9:2215–2219. doi: 10.1002/cbic.200800341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Deng H, Botting CH, Hamilton JT, Russell RJ, O’Hagan D. S-adenosyl-L-methionine:hydroxide adenosyltransferase: a SAM enzyme. Angew Chem Int Ed Engl. 2008;47:5357–5361. doi: 10.1002/anie.200800794. [DOI] [PubMed] [Google Scholar]
- 308.Deng H, McMahon SA, Eustaquio AS, Moore BS, Naismith JH, O’Hagan D. Mechanistic insights into water activation in SAM hydroxide adenosyltransferase (duf-62) Chembiochem. 2009;10:2455–2459. doi: 10.1002/cbic.200900369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Fontecave M, Atta M, Mulliez E. S-adenosylmethionine: nothing goes to waste. Trends Biochem Sci. 2004;29:243–249. doi: 10.1016/j.tibs.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 310.Frey PA, Magnusson OT. S-Adenosylmethionine: a wolf in sheep’s clothing, or a rich man’s adenosylcobalamin? Chem Rev. 2003;103:2129–2148. doi: 10.1021/cr020422m. [DOI] [PubMed] [Google Scholar]
- 311.Xu G, Wang BG. Independent evolution of six families of halogenating enzymes. PLoS One. 2016;11:e0154619. doi: 10.1371/journal.pone.0154619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Williams PG, Buchanan GO, Feling RH, Kauffman CA, Jensen PR, Fenical W. New cytotoxic salinosporamides from the marine actinomycete Salinispora tropica. J Org Chem. 2005;70:6196–6203. doi: 10.1021/jo050511+. [DOI] [PubMed] [Google Scholar]
- 313.Muller K, Faeh C, Diederich F. Fluorine in pharmaceuticals: looking beyond intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 314.O’Hagan D. Understanding organofluorine chemistry. An introduction to the C-F bond. Chem Soc Rev. 2008;37:308–319. doi: 10.1039/b711844a. [DOI] [PubMed] [Google Scholar]
- 315.Hong H, Spiteller D, Spencer JB. Incorporation of fluoroacetate into an aromatic polyketide and its influence on the mode of cyclization. Angew Chem Int Ed Engl. 2008;47:6028–6032. doi: 10.1002/anie.200801100. [DOI] [PubMed] [Google Scholar]
- 316.Eustaquio AS, Moore BS. Mutasynthesis of fluorosalinosporamide, a potent and reversible inhibitor of the proteasome. Angew Chem Int Ed Engl. 2008;47:3936–3938. doi: 10.1002/anie.200800177. [DOI] [PubMed] [Google Scholar]
- 317.Eustaquio AS, O’Hagan D, Moore BS. Engineering fluorometabolite production: fluorinase expression in Salinispora tropica yields fluorosalinosporamide. J Nat Prod. 2010;73:378–382. doi: 10.1021/np900719u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Walker MC, Thuronyi BW, Charkoudian LK, Lowry B, Khosla C, Chang MC. Expanding the fluorine chemistry of living systems using engineered polyketide synthase pathways. Science. 2013;341:1089–1094. doi: 10.1126/science.1242345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Walker MC, Chang MC. Natural and engineered biosynthesis of fluorinated natural products. Chem Soc Rev. 2014;43:6527–6536. doi: 10.1039/c4cs00027g. [DOI] [PubMed] [Google Scholar]
- 320.O’Hagan D, Deng H. Enzymatic fluorination and biotechnological developments of the fluorinase. Chem Rev. 2015;115:634–649. doi: 10.1021/cr500209t. [DOI] [PubMed] [Google Scholar]
- 321.Vranken C, Fin A, Tufar P, Hofkens J, Burkart MD, Tor Y. Chemoenzymatic synthesis and utilization of a SAM analog with an isomorphic nucleobase. Org Biomol Chem. 2016;14:6189–6192. doi: 10.1039/c6ob00844e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Thomsen M, Vogensen SB, Buchardt J, Burkart MD, Clausen RP. Chemoenzymatic synthesis and in situ application of S-adenosyl-L-methionine analogs. Org Biomol Chem. 2013;11:7606–7610. doi: 10.1039/c3ob41702f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Singh S, Zhang J, Huber TD, Sunkara M, Hurley K, Goff RD, Wang G, Zhang W, Liu C, Rohr J, et al. Facile chemoenzymatic strategies for the synthesis and utilization of S-adenosyl-(L)-methionine analogues. Angew Chem Int Ed Engl. 2014;53:3965–3969. doi: 10.1002/anie.201308272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Bothwell IR, Luo M. Large-scale, protection-free synthesis of Se-adenosyl-L-selenomethionine analogues and their application as cofactor surrogates of methyltransferases. Org Lett. 2014;16:3056–3059. doi: 10.1021/ol501169y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Harper DB. The global chloromethane cycle: biosynthesis, biodegradation and metabolic role. Nat Prod Rep. 2000;17:337–348. doi: 10.1039/a809400d. [DOI] [PubMed] [Google Scholar]
- 326.Bayer TS, Widmaier DM, Temme K, Mirsky EA, Santi DV, Voigt CA. Synthesis of methyl halides from biomass using engineered microbes. J Am Chem Soc. 2009;131:6508–6515. doi: 10.1021/ja809461u. [DOI] [PubMed] [Google Scholar]
- 327.Itoh N, Toda H, Matsuda M, Negishi T, Taniguchi T, Ohsawa N. Involvement of S-adenosylmethionine-dependent halide/thiol methyltransferase (HTMT) in methyl halide emissions from agricultural plants: isolation and characterization of an HTMT-coding gene from Raphanus sativus (daikon radish) BMC Plant Biol. 2009;9:116. doi: 10.1186/1471-2229-9-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Wuosmaa AM, Hager LP. Methyl chloride transferase: a carbocation route for biosynthesis of halometabolites. Science. 1990;249:160–162. doi: 10.1126/science.2371563. [DOI] [PubMed] [Google Scholar]
- 329.Attieh JM, Hanson AD, Saini HS. Purification and characterization of a novel methyltransferase responsible for biosynthesis of halomethanes and methanethiol in Brassica oleracea. J Biol Chem. 1995;270:9250–9257. doi: 10.1074/jbc.270.16.9250. [DOI] [PubMed] [Google Scholar]
- 330.Schmidberger JW, James AB, Edwards R, Naismith JH, O’Hagan D. Halomethane biosynthesis: structure of a SAM-dependent halide methyltransferase from Arabidopsis thaliana. Angew Chem Int Ed Engl. 2010;49:3646–3648. doi: 10.1002/anie.201000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Rhew RC, Ostergaard L, Saltzman ES, Yanofsky MF. Genetic control of methyl halide production in Arabidopsis. Curr Biol. 2003;13:1809–1813. doi: 10.1016/j.cub.2003.09.055. [DOI] [PubMed] [Google Scholar]
- 332.Vetter W. Marine halogenated natural products of environmental relevance. Rev Environ Contam Toxicol. 2006;188:1–57. doi: 10.1007/978-0-387-32964-2_1. [DOI] [PubMed] [Google Scholar]
- 333.Smidt H, de Vos WM. Anaerobic microbial dehalogenation. Annu Rev Microbiol. 2004;58:43–73. doi: 10.1146/annurev.micro.58.030603.123600. [DOI] [PubMed] [Google Scholar]
- 334.de Jong RM, Dijkstra BW. Structure and mechanism of bacterial dehalogenases: different ways to cleave a carbon-halogen bond. Curr Opin Struct Biol. 2003;13:722–730. doi: 10.1016/j.sbi.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 335.Fetzner S. Bacterial dehalogenation. Appl Microbiol Biotechnol. 1998;50:633–657. doi: 10.1007/s002530051346. [DOI] [PubMed] [Google Scholar]
- 336.Miot F, Dupuy C, Dumont J, Rousset B. Chapter 2 Thyroid hormone synthesis and secretion. 2000 [PubMed] [Google Scholar]
- 337.Schweizer U, Schlicker C, Braun D, Kohrle J, Steegborn C. Crystal structure of mammalian selenocysteine-dependent iodothyronine deiodinase suggests a peroxiredoxin-like catalytic mechanism. Proc Natl Acad Sci U S A. 2014;111:10526–10531. doi: 10.1073/pnas.1323873111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Thomas SR, McTamney PM, Adler JM, Laronde-Leblanc N, Rokita SE. Crystal structure of iodotyrosine deiodinase, a novel flavoprotein responsible for iodide salvage in thyroid glands. J Biol Chem. 2009;284:19659–19667. doi: 10.1074/jbc.M109.013458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Hesch RD, Brunner G, Soling HD. Conversion of thyroxine (T4) and triiodothyronine (T3) and the subcellular localisation of the converting enzyme. Clin Chim Acta. 1975;59:209–213. doi: 10.1016/0009-8981(75)90031-5. [DOI] [PubMed] [Google Scholar]
- 340.Visser TJ, Van Der Does-Tobé I, Docter R, Hennemann G. Conversion of thyroxine into tri-iodothyronine by rat liver homogenate. Biochem J. 1975;150:489–493. doi: 10.1042/bj1500489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Visser TJ, Leonard JL, Kaplan MM, Larsen PR. Different pathways of iodothyronine 5′-deiodination in rat cerebral cortex. Biochem Biophys Res Commun. 1981;101:1297–1304. doi: 10.1016/0006-291x(81)91588-6. [DOI] [PubMed] [Google Scholar]
- 342.Köhrle J. The deiodinase family: selenoenzymes regulating thyroid hormone availability and action. Cell Mol Life Sci. 2000;57:1853–1863. doi: 10.1007/PL00000667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Schweizer U, Steegborn C. New insights into the structure and mechanism of iodothyronine deiodinases. J Mol Endocrinol. 2015;55:R37–52. doi: 10.1530/JME-15-0156. [DOI] [PubMed] [Google Scholar]
- 344.Berry MJ, Maia AL, Kieffer JD, Harney JW, Larsen PR. Substitution of cysteine for selenocysteine in type I iodothyronine deiodinase reduces the catalytic efficiency of the protein but enhances its translation. Endocrinology. 1992;131:1848–1852. doi: 10.1210/endo.131.4.1396330. [DOI] [PubMed] [Google Scholar]
- 345.Koh CS, Didierjean C, Navrot N, Panjikar S, Mulliert G, Rouhier N, Jacquot JP, Aubry A, Shawkataly O, Corbier C. Crystal structures of a poplar thioredoxin peroxidase that exhibits the structure of glutathione peroxidases: insights into redox-driven conformational changes. J Mol Biol. 2007;370:512–529. doi: 10.1016/j.jmb.2007.04.031. [DOI] [PubMed] [Google Scholar]
- 346.Poole LB. The catalytic mechanism of peroxiredoxins. Subcell Biochem. 2007;44:61–81. doi: 10.1007/978-1-4020-6051-9_4. [DOI] [PubMed] [Google Scholar]
- 347.Agarwal V, Li J, Rahman I, Borgen M, Aluwihare LI, Biggs JS, Paul VJ, Moore BS. Complexity of naturally produced polybrominated diphenyl ethers revealed via mass spectrometry. Environ Sci Technol. 2015;49:1339–1346. doi: 10.1021/es505440j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Liu H, Lohith K, Rosario M, Pulliam TH, O’Connor RD, Bell LJ, Bewley CA. Polybrominated diphenyl ethers: structure determination and trends in antibacterial activity. J Nat Prod. 2016;79:1872–1876. doi: 10.1021/acs.jnatprod.6b00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Calcul L, Chow R, Oliver AG, Tenney K, White KN, Wood AW, Fiorilla C, Crews P. NMR strategy for unraveling structures of bioactive sponge-derived oxy-polyhalogenated diphenyl ethers. J Nat Prod. 2009;72:443–449. doi: 10.1021/np800737z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Butt CM, Wang D, Stapleton HM. Halogenated phenolic contaminants inhibit the in vitro activity of the thyroid-regulating deiodinases in human liver. Toxicol Sci. 2011;124:339–347. doi: 10.1093/toxsci/kfr117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Ucan-Marin F, Arukwe A, Mortensen AS, Gabrielsen GW, Letcher RJ. Recombinant albumin and transthyretin transport proteins from two gull species and human: chlorinated and brominated contaminant binding and thyroid hormones. Environ Sci Technol. 2010;44:497–504. doi: 10.1021/es902691u. [DOI] [PubMed] [Google Scholar]
- 352.Rokita SE, Adler JM, McTamney PM, Watson JA., Jr Efficient use and recycling of the micronutrient iodide in mammals. Biochimie. 2010;92:1227–1235. doi: 10.1016/j.biochi.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Moreno JC, Klootwijk W, van Toor H, Pinto G, D’Alessandro M, Leger A, Goudie D, Polak M, Gruters A, Visser TJ. Mutations in the iodotyrosine deiodinase gene and hypothyroidism. N Engl J Med. 2008;358:1811–1818. doi: 10.1056/NEJMoa0706819. [DOI] [PubMed] [Google Scholar]
- 354.Stanbury JB. The requirement of monoiodotyrosine deiodinase for triphosphopyridine nucleotide. J Biol Chem. 1957;228:801–811. [PubMed] [Google Scholar]
- 355.Stanbury JB, Morris ML. Deiodination of diiodotyrosine by cell-free systems. J Biol Chem. 1958;233:106–108. [PubMed] [Google Scholar]
- 356.Rosenberg IN, Goswami A. Purification and characterization of a flavoprotein from bovine thyroid with iodotyrosine deiodinase activity. J Biol Chem. 1979;254:12318–12325. [PubMed] [Google Scholar]
- 357.Goswami A, Rosenberg IN. Characterization of a flavoprotein iodotyrosine deiodinase from bovine thyroid. Flavin nucleotide binding and oxidation-reduction properties. J Biol Chem. 1979;254:12326–12330. [PubMed] [Google Scholar]
- 358.Moreno JC. Identification of novel genes involved in congenital hypothyroidism using serial analysis of gene expression. Horm Res. 2003;60(Suppl 3):96–102. doi: 10.1159/000074509. [DOI] [PubMed] [Google Scholar]
- 359.Moreno JC, de Vijlder JJ, Vulsma T, Ris-Stalpers C. Genetic basis of hypothyroidism: recent advances, gaps and strategies for future research. Trends Endocrinol Metab. 2003;14:318–326. doi: 10.1016/s1043-2760(03)00137-1. [DOI] [PubMed] [Google Scholar]
- 360.Phatarphekar A, Buss JM, Rokita SE. Iodotyrosine deiodinase: a unique flavoprotein present in organisms of diverse phyla. Mol Biosyst. 2014;10:86–92. doi: 10.1039/c3mb70398c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Kitamura S, Kuwasako M, Ohta S, Tatsumi K. Reductive debromination of (alpha-bromoiso-valeryl)urea by intestinal bacteria. J Pharm Pharmacol. 1999;51:79–84. doi: 10.1211/0022357991771999. [DOI] [PubMed] [Google Scholar]
- 362.Roldan MD, Perez-Reinado E, Castillo F, Moreno-Vivian C. Reduction of polynitroaromatic compounds: the bacterial nitroreductases. FEMS Microbiol Rev. 2008;32:474–500. doi: 10.1111/j.1574-6976.2008.00107.x. [DOI] [PubMed] [Google Scholar]
- 363.Hecht HJ, Erdmann H, Park HJ, Sprinzl M, Schmid RD. Crystal structure of NADH oxidase from Thermus thermophilus. Nat Struct Biol. 1995;2:1109–1114. doi: 10.1038/nsb1295-1109. [DOI] [PubMed] [Google Scholar]
- 364.Taga ME, Larsen NA, Howard-Jones AR, Walsh CT, Walker GC. BluB cannibalizes flavin to form the lower ligand of vitamin B12. Nature. 2007;446:449–453. doi: 10.1038/nature05611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Hu J, Chuenchor W, Rokita SE. A switch between one- and two-electron chemistry of the human flavoprotein iodotyrosine deiodinase is controlled by substrate. J Biol Chem. 2015;290:590–600. doi: 10.1074/jbc.M114.605964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Kunishima M, Friedman JE, Rokita SE. Transition-state stabilization by a mammalian reductive dehalogenase. J Am Chem Soc. 1999;121:4722–4723. [Google Scholar]
- 367.McTamney PM, Rokita SE. A mammalian reductive deiodinase has broad power to dehalogenate chlorinated and brominated substrates. J Am Chem Soc. 2009;131:14212–14213. doi: 10.1021/ja906642n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Ahn YB, Rhee SK, Fennell DE, Kerkhof LJ, Hentschel U, Haggblom MM. Reductive dehalogenation of brominated phenolic compounds by microorganisms associated with the marine sponge Aplysina aerophoba. Appl Environ Microbiol. 2003;69:4159–4166. doi: 10.1128/AEM.69.7.4159-4166.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Jeon JR, Murugesan K, Nam IH, Chang YS. Coupling microbial catabolic actions with abiotic redox processes: a new recipe for persistent organic pollutant (POP) removal. Biotechnol Adv. 2013;31:246–256. doi: 10.1016/j.biotechadv.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 370.Field JA, Sierra-Alvarez R. Microbial degradation of chlorinated dioxins. Chemosphere. 2008;71:1005–1018. doi: 10.1016/j.chemosphere.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 371.Field JA, Sierra-Alvarez R. Microbial degradation of chlorinated benzenes. Biodegradation. 2008;19:463–480. doi: 10.1007/s10532-007-9155-1. [DOI] [PubMed] [Google Scholar]
- 372.El Gamal A, Agarwal V, Rahman I, Moore BS. Enzymatic reductive dehalogenation controls the biosynthesis of marine bacterial pyrroles. J Am Chem Soc. 2016;138:13167–13170. doi: 10.1021/jacs.6b08512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Lovell FM. Structure of a bromine-rich marine antibiotic. J Am Chem Soc. 1966;88:4510–&. [Google Scholar]
- 374.Burkholder PR, Pfister RM, Leitz FH. Production of a pyrrole antibiotic by a marine bacterium. Appl Microbiol. 1966;14:649–653. doi: 10.1128/am.14.4.649-653.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Gribble GW. Occurrence of halogenated alkaloids. Alkaloids Chem Biol. 2012;71:1–165. doi: 10.1016/b978-0-12-398282-7.00001-1. [DOI] [PubMed] [Google Scholar]
- 376.Gribble GW. A recent survey of naturally occurring organohalogen compounds. Environ Chem. 2015;12:396–405. [Google Scholar]
- 377.Ni S, Fredrickson JK, Xun L. Purification and characterization of a novel 3-chlorobenzoate-reductive dehalogenase from the cytoplasmic membrane of Desulfomonile tiedjei DCB-1. J Bacteriol. 1995;177:5135–5139. doi: 10.1128/jb.177.17.5135-5139.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Banerjee R, Ragsdale SW. The many faces of vitamin B-12: Catalysis by cobalamin-dependent enzymes. Annu Rev Biochem. 2003;72:209–247. doi: 10.1146/annurev.biochem.72.121801.161828. [DOI] [PubMed] [Google Scholar]
- 379.Hug LA, Maphosa F, Leys D, Loffler FE, Smidt H, Edwards EA, Adrian L. Overview of organohalide-respiring bacteria and a proposal for a classification system for reductive dehalogenases. Philos Trans Royal Soc B. 2013;368 doi: 10.1098/rstb.2012.0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Chen K, Huang LL, Xu CF, Liu XM, He J, Zinder SH, Li SP, Jiang JD. Molecular characterization of the enzymes involved in the degradation of a brominated aromatic herbicide. Mol Microbiol. 2013;89:1121–1139. doi: 10.1111/mmi.12332. [DOI] [PubMed] [Google Scholar]
- 381.Mac Nelly A, Kai M, Svatos A, Diekert G, Schubert T. Functional heterologous production of reductive dehalogenases from Desulfitobacterium hafniense strains. Appl Environ Microbiol. 2014;80:4313–4322. doi: 10.1128/AEM.00881-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Suyama A, Iwakiri R, Kai K, Tokunaga T, Sera N, Furukawa K. Isolation and characterization of Desulfitobacterium sp. strain Y51 capable of efficient dehalogenation of tetrachloroethene and polychloroethanes. Biosci Biotechnol Biochem. 2001;65:1474–1481. doi: 10.1271/bbb.65.1474. [DOI] [PubMed] [Google Scholar]
- 383.Bommer M, Kunze C, Fesseler J, Schubert T, Diekert G, Dobbek H. Structural basis for organohalide respiration. Science. 2014;346:455–458. doi: 10.1126/science.1258118. [DOI] [PubMed] [Google Scholar]
- 384.Payne KA, Quezada CP, Fisher K, Dunstan MS, Collins FA, Sjuts H, Levy C, Hay S, Rigby SE, Leys D. Reductive dehalogenase structure suggests a mechanism for B12-dependent dehalogenation. Nature. 2015;517:513–516. doi: 10.1038/nature13901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.DeWeerd KA, Mandelco L, Tanner RS, Woese CR, Suflita JM. Desulfomonile tiedjei gen. nov. and sp. nov., a novel anaerobic, dehalogenating, sulfate-reducing bacterium. Arch Microbiol. 1990;154:23–30. [Google Scholar]
- 386.Schmitz RPH, Wolf J, Habel A, Neumann A, Ploss K, Svatos A, Boland W, Diekertt G. Evidence for a radical mechanism of the dechlorination of chlorinated propenes mediated by the tetrachloroethene reductive dehalogenase of Sulfurospirillum multivorans. Environ Sci Technol. 2007;41:7370–7375. doi: 10.1021/es071026u. [DOI] [PubMed] [Google Scholar]
- 387.Suflita JM, Horowitz A, Shelton DR, Tiedje JM. Dehalogenation: a novel pathway for the anaerobic biodegradation of haloaromatic compounds. Science. 1982;218:1115–1117. doi: 10.1126/science.218.4577.1115. [DOI] [PubMed] [Google Scholar]
- 388.Neumann A, Wohlfarth G, Diekert G. Tetrachloroethene dehalogenase from Dehalospirillum multivorans: cloning, sequencing of the encoding genes, and expression of the pceA gene in Escherichia coli. J Bacteriol. 1998;180:4140–4145. doi: 10.1128/jb.180.16.4140-4145.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Krzmarzick MJ, Crary BB, Harding JJ, Oyerinde OO, Leri AC, Myneni SC, Novak PJ. Natural niche for organohalide-respiring Chloroflexi. Appl Environ Microbiol. 2012;78:393–401. doi: 10.1128/AEM.06510-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Xun L, Orser CS. Purification and properties of pentachlorophenol hydroxylase, a flavoprotein from Flavobacterium sp. strain ATCC 39723. J Bacteriol. 1991;173:4447–4453. doi: 10.1128/jb.173.14.4447-4453.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Xun L, Topp E, Orser CS. Purification and characterization of a tetrachloro-p-hydroquinone reductive dehalogenase from a Flavobacterium sp. J Bacteriol. 1992;174:8003–8007. doi: 10.1128/jb.174.24.8003-8007.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Xu L, Resing K, Lawson SL, Babbitt PC, Copley SD. Evidence that pcpA encodes 2,6-dichlorohydroquinone dioxygenase, the ring cleavage enzyme required for pentachlorophenol degradation in Sphingomonas chlorophenolica strain ATCC 39723. Biochemistry. 1999;38:7659–7669. doi: 10.1021/bi990103y. [DOI] [PubMed] [Google Scholar]
- 393.Copley SD. Evolution of efficient pathways for degradation of anthropogenic chemicals. Nat Chem Biol. 2009;5:559–566. doi: 10.1038/nchembio.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Anandarajah K, Kiefer PM, Jr, Donohoe BS, Copley SD. Recruitment of a double bond isomerase to serve as a reductive dehalogenase during biodegradation of pentachlorophenol. Biochemistry. 2000;39:5303–5311. doi: 10.1021/bi9923813. [DOI] [PubMed] [Google Scholar]
- 395.McCarthy DL, Navarrete S, Willett WS, Babbitt PC, Copley SD. Exploration of the relationship between tetrachlorohydroquinone dehalogenase and the glutathione S-transferase superfamily. Biochemistry. 1996;35:14634–14642. doi: 10.1021/bi961730f. [DOI] [PubMed] [Google Scholar]
- 396.Kiefer PM, Jr, Copley SD. Characterization of the initial steps in the reductive dehalogenation catalyzed by tetrachlorohydroquinone dehalogenase. Biochemistry. 2002;41:1315–1322. doi: 10.1021/bi0117504. [DOI] [PubMed] [Google Scholar]
- 397.Kiefer PM, Jr, McCarthy DL, Copley SD. The reaction catalyzed by tetrachlorohydroquinone dehalogenase does not involve nucleophilic aromatic substitution. Biochemistry. 2002;41:1308–1314. doi: 10.1021/bi0117495. [DOI] [PubMed] [Google Scholar]
- 398.Warner JR, Lawson SL, Copley SD. A mechanistic investigation of the thiol-disulfide exchange step in the reductive dehalogenation catalyzed by tetrachlorohydroquinone dehalogenase. Biochemistry. 2005;44:10360–10368. doi: 10.1021/bi050666b. [DOI] [PubMed] [Google Scholar]
- 399.Miyauchi K, Suh SK, Nagata Y, Takagi M. Cloning and sequencing of a 2,5-dichlorohydroquinone reductive dehalogenase gene whose product is involved in degradation of gamma-hexachlorocyclohexane by Sphingomonas paucimobilis. J Bacteriol. 1998;180:1354–1359. doi: 10.1128/jb.180.6.1354-1359.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Isin EM, Guengerich FP. Complex reactions catalyzed by cytochrome P450 enzymes. Biochim Biophys Acta. 2007;1770:314–329. doi: 10.1016/j.bbagen.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 401.Borja J, Taleon DM, Auresenia J, Gallardo S. Polychlorinated biphenyls and their biodegradation. Proc Biochem. 2005;40:1999–2013. [Google Scholar]
- 402.Arora PK, Bae H. Role of dehalogenases in aerobic bacterial degradation of chlorinated aromatic compounds. J Chem. 2014;2014:10. [Google Scholar]
- 403.Crawford RL, Jung CM, Strap JL. The recent evolution of pentachlorophenol (PCP)-4-monooxygenase (PcpB) and associated pathways for bacterial degradation of PCP. Biodegradation. 2007;18:525–539. doi: 10.1007/s10532-006-9090-6. [DOI] [PubMed] [Google Scholar]
- 404.Valverde C, Orozco A, Becerra A, Jeziorski MC, Villalobos P, Solis JC. Halometabolites and cellular dehalogenase systems: an evolutionary perspective. Int Rev Cytol. 2004;234:143–199. doi: 10.1016/S0074-7696(04)34004-0. [DOI] [PubMed] [Google Scholar]
- 405.Franzen S, Thompson MK, Ghiladi RA. The dehaloperoxidase paradox. Biochim Biophys Acta. 2012;1824:578–588. doi: 10.1016/j.bbapap.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 406.Hlouchova K, Rudolph J, Pietari JM, Behlen LS, Copley SD. Pentachlorophenol hydroxylase, a poorly functioning enzyme required for degradation of pentachlorophenol by Sphingobium chlorophenolicum. Biochemistry. 2012;51:3848–3860. doi: 10.1021/bi300261p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Franken SM, Rozeboom HJ, Kalk KH, Dijkstra BW. Crystal structure of haloalkane dehalogenase: an enzyme to detoxify halogenated alkanes. EMBO J. 1991;10:1297–1302. doi: 10.1002/j.1460-2075.1991.tb07647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Bohac M, Nagata Y, Prokop Z, Prokop M, Monincova M, Tsuda M, Koca J, Damborsky J. Halide-stabilizing residues of haloalkane dehalogenases studied by quantum mechanic calculations and site-directed mutagenesis. Biochemistry. 2002;41:14272–14280. doi: 10.1021/bi026427v. [DOI] [PubMed] [Google Scholar]
- 409.Kates JR, Jones RF. Fluoroacetate inhibition of amino acids during photosynthesis of Chlamydomonas reinhardti. Science. 1964;143:145–146. doi: 10.1126/science.143.3602.145. [DOI] [PubMed] [Google Scholar]
- 410.Chan PW, Yakunin AF, Edwards EA, Pai EF. Mapping the reaction coordinates of enzymatic defluorination. J Am Chem Soc. 2011;133:7461–7468. doi: 10.1021/ja200277d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Novak HR, Sayer C, Isupov MN, Gotz D, Spragg AM, Littlechild JA. Biochemical and structural characterisation of a haloalkane dehalogenase from a marine Rhodobacteraceae. FEBS Lett. 2014;588:1616–1622. doi: 10.1016/j.febslet.2014.02.056. [DOI] [PubMed] [Google Scholar]
- 412.van Hylckama Vlieg JE, Tang L, Lutje Spelberg JH, Smilda T, Poelarends GJ, Bosma T, van Merode AE, Fraaije MW, Janssen DB. Halohydrin dehalogenases are structurally and mechanistically related to short-chain dehydrogenases/reductases. J Bacteriol. 2001;183:5058–5066. doi: 10.1128/JB.183.17.5058-5066.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Schallmey M, Floor RJ, Hauer B, Breuer M, Jekel PA, Wijma HJ, Dijkstra BW, Janssen DB. Biocatalytic and structural properties of a highly engineered halohydrin dehalogenase. Chembiochem. 2013;14:870–881. doi: 10.1002/cbic.201300005. [DOI] [PubMed] [Google Scholar]
- 414.de Jong RM, Tiesinga JJ, Villa A, Tang L, Janssen DB, Dijkstra BW. Structural basis for the enantioselectivity of an epoxide ring opening reaction catalyzed by halo alcohol dehalogenase HheC. J Am Chem Soc. 2005;127:13338–13343. doi: 10.1021/ja0531733. [DOI] [PubMed] [Google Scholar]
- 415.Janssen DB, Majeric-Elenkov M, Hasnaoui G, Hauer B, Lutje Spelberg JH. Enantioselective formation and ring-opening of epoxides catalysed by halohydrin dehalogenases. Biochem Soc Trans. 2006;34:291–295. doi: 10.1042/BST20060291. [DOI] [PubMed] [Google Scholar]
- 416.Hopmann KH, Himo F. Cyanolysis and azidolysis of epoxides by haloalcohol dehalogenase: theoretical study of the reaction mechanism and origins of regioselectivity. Biochemistry. 2008;47:4973–4982. doi: 10.1021/bi800001r. [DOI] [PubMed] [Google Scholar]
- 417.de Jong RM, Tiesinga JJ, Rozeboom HJ, Kalk KH, Tang L, Janssen DB, Dijkstra BW. Structure and mechanism of a bacterial haloalcohol dehalogenase: a new variation of the short-chain dehydrogenase/reductase fold without an NAD(P)H binding site. EMBO J. 2003;22:4933–4944. doi: 10.1093/emboj/cdg479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Ortega MA, Velasquez JE, Garg N, Zhang Q, Joyce RE, Nair SK, van der Donk WA. Substrate specificity of the lanthipeptide peptidase ElxP and the oxidoreductase ElxO. ACS Chem Biol. 2014;9:1718–1725. doi: 10.1021/cb5002526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.de Jong RM, Bazzacco P, Poelarends GJ, Johnson WH, Jr, Kim YJ, Burks EA, Serrano H, Thunnissen AM, Whitman CP, Dijkstra BW. Crystal structures of native and inactivated cis-3-chloroacrylic acid dehalogenase. Structural basis for substrate specificity and inactivation by (R)-oxirane-2-carboxylate. J Biol Chem. 2007;282:2440–2449. doi: 10.1074/jbc.M608134200. [DOI] [PubMed] [Google Scholar]
- 420.Azurmendi HF, Wang SC, Massiah MA, Poelarends GJ, Whitman CP, Mildvan AS. The roles of active-site residues in the catalytic mechanism of trans-3-chloroacrylic acid dehalogenase: a kinetic, NMR, and mutational analysis. Biochemistry. 2004;43:4082–4091. doi: 10.1021/bi030241u. [DOI] [PubMed] [Google Scholar]
- 421.Sevastik R, Whitman CP, Himo F. Reaction mechanism of cis-3-chloroacrylic acid dehalogenase: a theoretical study. Biochemistry. 2009;48:9641–9649. doi: 10.1021/bi900879a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Schroeder GK, Johnson WH, Jr, Huddleston JP, Serrano H, Johnson KA, Whitman CP. Reaction of cis-3-chloroacrylic acid dehalogenase with an allene substrate, 2,3-butadienoate: hydration via an enamine. J Am Chem Soc. 2012;134:293–304. doi: 10.1021/ja206873f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Poelarends GJ, Serrano H, Huddleston JP, Johnson WH, Jr, Whitman CP. A mutational analysis of active site residues in trans-3-chloroacrylic acid dehalogenase. FEBS Lett. 2013;587:2842–2850. doi: 10.1016/j.febslet.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Schroeder GK, Huddleston JP, Johnson WH, Jr, Whitman CP. A mutational analysis of the active site loop residues in cis-3-chloroacrylic acid dehalogenase. Biochemistry. 2013;52:4204–4216. doi: 10.1021/bi4004414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Huddleston JP, Burks EA, Whitman CP. Identification and characterization of new family members in the tautomerase superfamily: analysis and implications. Arch Biochem Biophys. 2014;564:189–196. doi: 10.1016/j.abb.2014.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Mowafy AM, Kurihara T, Kurata A, Uemura T, Esaki N. 2-haloacrylate hydratase, a new class of flavoenzyme that catalyzes the addition of water to the substrate for dehalogenation. Appl Environ Microbiol. 2010;76:6032–6037. doi: 10.1128/AEM.00334-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Walsh CT. A chemocentric view of the natural product inventory. Nat Chem Biol. 2015;11:620–624. doi: 10.1038/nchembio.1894. [DOI] [PubMed] [Google Scholar]
- 428.Khosla C. Quo vadis, enzymology? Nat Chem Biol. 2015;11:438–441. doi: 10.1038/nchembio.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Rodriguez RA, Pan CM, Yabe Y, Kawamata Y, Eastgate MD, Baran PS. Palau’chlor: a practical and reactive chlorinating reagent. J Am Chem Soc. 2014;136:6908–6911. doi: 10.1021/ja5031744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Leao PN, Nakamura H, Costa M, Pereira AR, Martins R, Vasconcelos V, Gerwick WH, Balskus EP. Biosynthesis-assisted structural elucidation of the bartolosides, chlorinated aromatic glycolipids from cyanobacteria. Angew Chem Int Ed Engl. 2015;54:11063–11067. doi: 10.1002/anie.201503186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Afonso TB, Costa MS, Rezende de Castro R, Freitas S, Silva A, Schneider MP, Martins R, Leao PN. Bartolosides E-K from a marine coccoid cyanobacterium. J Nat Prod. 2016;79:2504–2513. doi: 10.1021/acs.jnatprod.6b00351. [DOI] [PubMed] [Google Scholar]
- 432.Preisitsch M, Heiden SE, Beerbaum M, Niedermeyer TH, Schneefeld M, Herrmann J, Kumpfmuller J, Thurmer A, Neidhardt I, Wiesner C, et al. Effects of halide ions on the carbamidocyclophane biosynthesis in Nostoc sp. CAVN2. Mar Drugs. 2016;14:21. doi: 10.3390/md14010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Chankhamjon P, Tsunematsu Y, Ishida-Ito M, Sasa Y, Meyer F, Boettger-Schmidt D, Urbansky B, Menzel KD, Scherlach K, Watanabe K, et al. Regioselective dichlorination of a non-activated aliphatic carbon atom and phenolic bismethylation by a multifunctional fungal flavoenzyme. Angew Chem Int Ed Engl. 2016;55:11955–11959. doi: 10.1002/anie.201604516. [DOI] [PubMed] [Google Scholar]
- 434.Antunes EM, Copp BR, Davies-Coleman MT, Samaai T. Pyrroloiminoquinone and related metabolites from marine sponges. Nat Prod Rep. 2005;22:62–72. doi: 10.1039/b407299p. [DOI] [PubMed] [Google Scholar]
- 435.Bucher C, Deans RM, Burns NZ. Highly selective synthesis of halomon, plocamenone, and isoplocamenone. J Am Chem Soc. 2015;137:12784–12787. doi: 10.1021/jacs.5b08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Landry ML, Hu DX, McKenna GM, Burns NZ. Catalytic enantioselective dihalogenation and the selective synthesis of (−)-deschloromytilipin A and (−)-danicalipin A. J Am Chem Soc. 2016;138:5150–5158. doi: 10.1021/jacs.6b01643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Bailey AM, Wolfrum S, Carreira EM. Biological investigations of (+)-danicalipin A enabled through synthesis. Angew Chem Int Ed Engl. 2016;55:639–643. doi: 10.1002/anie.201509082. [DOI] [PubMed] [Google Scholar]
- 438.Fischer S, Huwyler N, Wolfrum S, Carreira EM. Synthesis and biological evaluation of bromo- and fluorodanicalipin A. Angew Chem Int Ed Engl. 2016;55:2555–2558. doi: 10.1002/anie.201510608. [DOI] [PubMed] [Google Scholar]
- 439.Chung WJ, Vanderwal CD. Stereoselective halogenation in natural product synthesis. Angew Chem Int Ed Engl. 2016;55:4396–4434. doi: 10.1002/anie.201506388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Sun H, Yeo WL, Lim YH, Chew X, Smith DJ, Xue B, Chan KP, Robinson RC, Robins EG, Zhao H, et al. Directed evolution of a fluorinase for improved fluorination efficiency with a non-native substrate. Angew Chem Int Ed Engl. 2016;55:14277–14280. doi: 10.1002/anie.201606722. [DOI] [PubMed] [Google Scholar]
- 441.Payne JT, Poor CB, Lewis JC. Directed evolution of RebH for site-selective halogenation of large biologically active molecules. Angew Chem Int Ed Engl. 2015;54:4226–4230. doi: 10.1002/anie.201411901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Andorfer MC, Park HJ, Vergara-Coll J, Lewis JC. Directed evolution of RebH for catalyst-controlled halogenation of indole C-H bonds. Chem Sci. 2016;7:3720–3729. doi: 10.1039/c5sc04680g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Lasken RS. Genomic sequencing of uncultured microorganisms from single cells. Nat Rev Microbiol. 2012;10:631–640. doi: 10.1038/nrmicro2857. [DOI] [PubMed] [Google Scholar]
- 444.Katz M, Hover BM, Brady SF. Culture-independent discovery of natural products from soil metagenomes. J Ind Microbiol Biotechnol. 2016;43:129–141. doi: 10.1007/s10295-015-1706-6. [DOI] [PubMed] [Google Scholar]
- 445.Piel J. Approaches to capturing and designing biologically active small molecules produced by uncultured microbes. Annu Rev Microbiol. 2011;65:431–453. doi: 10.1146/annurev-micro-090110-102805. [DOI] [PubMed] [Google Scholar]
- 446.Unson MD, Holland ND, Faulkner DJ. A brominated secondary metabolite synthesized by the cyanobacterial symbiont of a marine sponge and accumulation of the crystalline metabolite in the sponge tissue. Mar Biol. 1994;119:1–11. [Google Scholar]
- 447.Becerro MA, Paul VJ. Effects of depth and light on secondary metabolites and cyanobacterial symbionts of the sponge Dysidea granulosa. Mar Ecol Prog Ser. 2004;280:115–128. [Google Scholar]
- 448.Abdelmohsen UR, Bayer K, Hentschel U. Diversity, abundance and natural products of marine sponge-associated actinomycetes. Nat Prod Rep. 2014;31:381–399. doi: 10.1039/c3np70111e. [DOI] [PubMed] [Google Scholar]
- 449.Hentschel U, Piel J, Degnan SM, Taylor MW. Genomic insights into the marine sponge microbiome. Nat Rev Microbiol. 2012;10:641–654. doi: 10.1038/nrmicro2839. [DOI] [PubMed] [Google Scholar]
- 450.Freeman MF, Vagstad AL, Piel J. Polytheonamide biosynthesis showcasing the metabolic potential of sponge-associated uncultivated ‘Entotheonella’ bacteria. Curr Opin Chem Biol. 2015;31:8–14. doi: 10.1016/j.cbpa.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 451.Thomas T, Moitinho-Silva L, Lurgi M, Bjork JR, Easson C, Astudillo-Garcia C, Olson JB, Erwin PM, Lopez-Legentil S, Luter H, et al. Diversity, structure and convergent evolution of the global sponge microbiome. Nat Commun. 2016;7:11870. doi: 10.1038/ncomms11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Bayer K, Scheuermayer M, Fieseler L, Hentschel U. Genomic mining for novel FADH(2)-dependent halogenases in marine sponge-associated microbial consortia. Mar Biotechnol. 2013;15:63–72. doi: 10.1007/s10126-012-9455-2. [DOI] [PubMed] [Google Scholar]
- 453.Ozturk B, de Jaeger L, Smidt H, Sipkema D. Culture-dependent and independent approaches for identifying novel halogenases encoded by Crambe crambe (marine sponge) microbiota. Sci Rep. 2013;3:2780. doi: 10.1038/srep02780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Küpper CF, Schweigert N, Ar Gall E, Legendre J-M, Vilter H, Kloareg B. Iodine uptake in Laminariales involves extracellular, haloperoxidase-mediated oxidation of iodide. Planta. 1998;207:163–171. [Google Scholar]
- 455.Saiz-Lopez A, von Glasow R. Reactive halogen chemistry in the troposphere. Chem Soc Rev. 2012;41:6448–6472. doi: 10.1039/c2cs35208g. [DOI] [PubMed] [Google Scholar]
- 456.Fan SM, Jacob DJ. Surface ozone depletion in arctic spring sustained by bromine reactions on aerosols. Nature. 1992;359:522–524. [Google Scholar]
- 457.Salawitch RJ. Atmospheric chemistry: biogenic bromine. Nature. 2006;439:275–277. doi: 10.1038/439275a. [DOI] [PubMed] [Google Scholar]
- 458.Theiler R, Cook JC, Hager LP, Siuda JF. Halohydrocarbon synthesis by bromoperoxidase. Science. 1978;202:1094–1096. doi: 10.1126/science.202.4372.1094. [DOI] [PubMed] [Google Scholar]
- 459.Beissner RS, Guilford WJ, Coates RM, Hager LP. Synthesis of brominated heptanones and bromoform by a bromoperoxidase of marine origin. Biochemistry. 1981;20:3724–3731. doi: 10.1021/bi00516a009. [DOI] [PubMed] [Google Scholar]
- 460.Ohsawa N, Ogata Y, Okada N, Itoh N. Physiological function of bromoperoxidase in the red marine alga, Corallina pilulifera: production of bromoform as an allelochemical and the simultaneous elimination of hydrogen peroxide. Phytochemistry. 2001;58:683–692. doi: 10.1016/s0031-9422(01)00259-x. [DOI] [PubMed] [Google Scholar]
- 461.Quack B, Atlas E, Petrick G, Stroud V, Schauffler S, Wallace DWR. Oceanic bromoform sources for the tropical atmosphere. Geophys Res Lett. 2004;31 [Google Scholar]
- 462.Moore RM, Webb M, Tokarczyk R, Wever R. Bromoperoxidase and iodoperoxidase enzymes and production of halogenated methanes in marine diatom cultures. J Geophys Res-Oceans. 1996;101:20899–20908. [Google Scholar]
- 463.McCall AS, Cummings CF, Bhave G, Vanacore R, Page-McCaw A, Hudson BG. Bromine is an essential trace element for assembly of collagen IV scaffolds in tissue development and architecture. Cell. 2014;157:1380–1392. doi: 10.1016/j.cell.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.van Pee KH, Unversucht S. Biological dehalogenation and halogenation reactions. Chemosphere. 2003;52:299–312. doi: 10.1016/S0045-6535(03)00204-2. [DOI] [PubMed] [Google Scholar]
- 465.Oberg G. The natural chlorine cycle - fitting the scattered pieces. Appl Microbiol Biotechnol. 2002;58:565–581. doi: 10.1007/s00253-001-0895-2. [DOI] [PubMed] [Google Scholar]
- 466.Leri AC, Hakala JA, Marcus MA, Lanzirotti A, Reddy CM, Myneni SCB. Natural organobromine in marine sediments: New evidence of biogeochemical Br cycling. Global Biogeochem Cyc. 2010;24 [Google Scholar]
- 467.Weigold P, El-Hadidi M, Ruecker A, Huson DH, Scholten T, Jochmann M, Kappler A, Behrens S. A metagenomic-based survey of microbial (de)halogenation potential in a German forest soil. Sci Rep. 2016;6:28958. doi: 10.1038/srep28958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Hug LA, Edwards EA. Diversity of reductive dehalogenase genes from environmental samples and enrichment cultures identified with degenerate primer PCR screens. Front Microbiol. 2013;4:341. doi: 10.3389/fmicb.2013.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]