

Abstract
Zika virus (ZIKV)-infection is associated with neurological disorders of both the central and peripheral nervous systems (PNS), yet few studies have directly examined PNS-infection. Here we show that intraperitoneally or intraventricularly-injected ZIKV in the mouse could infect and impact peripheral neurons in vivo. Moreover, ZIKV productively infects stem cell-derived human neural crest cells and peripheral neurons in vitro, leading to increased cell death, transcriptional dysregulation and cell-type specific molecular pathology.
Zika virus (ZIKV), a flavivirus transmitted primarily by Aedes mosquitoes, has spread to a growing number of countries. While most ZIKV-infected patients exhibit few or relatively mild symptoms including mild fever, skin rash, conjunctivitis, muscle and joint pain, malaise or headache, prenatal ZIKV infection may cause microcephaly and other serious brain anomalies in fetuses or infants1,2. Recent studies of ZIKV pathogenesis in the central nervous system (CNS) have shown that ZIKV crosses the placenta and causes microcephaly by targeting cortical neural progenitor cells, inducing cell death and impairing neurodevelopment3–8. Other pronounced symptoms of ZIKV infection are retro-orbital pain, abdominal pain, and diarrhea9, which are associated with the peripheral nervous system (PNS), and more specifically sensory and enteric neurons. In particular, peripheral neuropathy without any CNS symptom in a ZIKV patient has been reported10,11, which is supported by persistent ZIKV detection in PNS of rhesus macaques12. In addition, ZIKV causes Guillain-Barré syndrome (GBS)1, another PNS disorder. In contrast to CNS model systems3–6, PNS model systems for investigating ZIKV pathology are limited. In this study, we investigated PNS infection of ZIKV using both in vivo and in vitro model systems.
We adopted the type-I interferon receptor deficient (A129) mice to generate a ZIKV infection model in vivo. ZIKV SZ01 was injected intraperitoneally into 5 weeks old A129 mice and inspected 3 days later. To examine the potential for ZIKV infection in the PNS, we examined the dorsal root ganglia (DRG) and small intestine from ZIKV-infected and mock-treated mice. We detected the presence of ZIKV in both DRG and small intestine with ZIKV antisera (Supplementary Fig. 1a,b). In addition, robust ZIKV signals co-localized with Tuj1, indicating that mouse peripheral neurons in the DRG and gut were infected by ZIKV (Supplementary Fig. 1a,b). To test whether PNS infection of ZIKV could also occur in a microcephaly mouse model5, we injected ZIKV SZ01 into the lateral ventricle of wild-type mice at embryonic day 13.5 (E13.5) brains and analyzed tissue at postnatal day 1 (P1). We also detected ZIKV in the DRG, small intestine, and spinal cord with co-localization with Tuj1, NeuN, or cleaved Caspase 3 (cl-Casp3; Fig. 1 and Supplementary Fig. 1c,d), demonstrating that brain-injected ZIKV could infect and impact peripheral neurons in the mouse DRG and gut in vivo.
Figure 1.
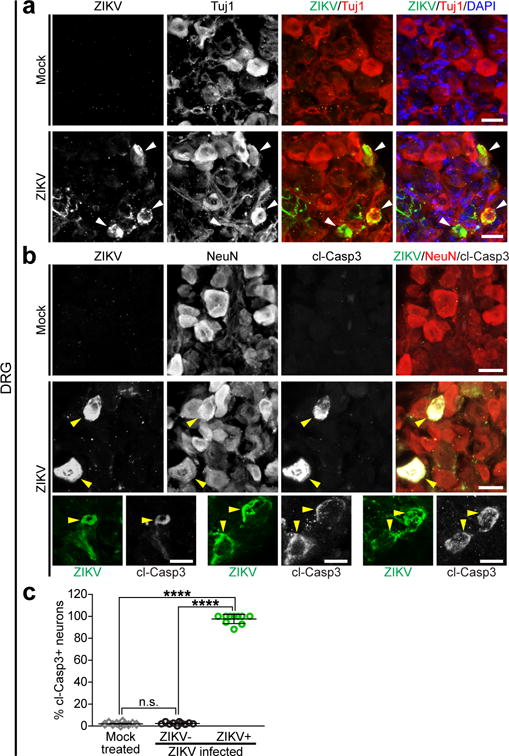
ZIKV infection results in apoptosis of somatosensory neurons in mouse peripheral nervous system. Mouse embryos were infected by intraventricular injection of 650 PFUs of ZIKVSZ01 or mock at E13.5 and analyzed at P1. (a) Representative images of mouse dorsal root ganglia (DRG)-sections stained with the indicated antibodies. White arrowheads, ZIKV-infected DRG neurons. (b) Sample images of DRG-sections stained with the indicated antibodies. Cleaved-Caspase 3, cl-Casp3. Yellow arrowheads, ZIKV-infected apoptotic DRG neurons. Scale bars, 20 μm. (c) The percentage of apoptotic neurons in the mock-treated and ZIKV-infected DRGs. All error bars represent mean ± s.e.m. (****P < 0.0001; n.s., not significant; unpaired Student’s t-test; n = 10 for Mock-treated, n = 10 for ZIKV-infected). The n represents the number of analyzed DRGs from 4 pups.
These in vivo observations prompted us to use a human stem cell-based model to directly examine ZIKV infection and its molecular pathology. Neural crest cells are migratory multipotent progenitors that give rise to various cell types, including neurons and glia of the PNS13. We previously developed a highly efficient protocol14 to differentiate human pluripotent stem cells (hPSC) into human neural crest cells (hNCCs), which can be further differentiated into human peripheral neurons (hPNs) (Supplementary Fig. 2). We used a clinically isolated ZIKV strain from the 2015 Puerto Rico Zika outbreak, PRVABC59 (hereafter ZIKVPR), which is closely related to epidemic strains circulating in the Americas that have been linked to in utero ZIKV infection15. We performed infections at a low or moderate multiplicity of infection (MOI = 0.04 or 0.4) for 2 h. Infection rates were quantified 65 h later with immunocytochemistry using an anti-ZIKV envelope protein (ZIKVE) antibody. The HNK1/AP2α-expressing hNCCs were readily infected by ZIKVPR (Fig. 2a,b). Similar to previous CNS model systems for ZIKV infection3,16, the staining signal for ZIKVE was concentrated in the perinuclear structures of hNCCs (Fig. 2a) and ZIKVPR infection reduced the cell viability of hNCCs compared to mock-infected cells (Fig. 2c and Supplementary Fig. 3a). These results are in accordance with a recent paper demonstrating that ZIKV infects cranial NCCs, resulting in reduced viability17.
Figure 2.
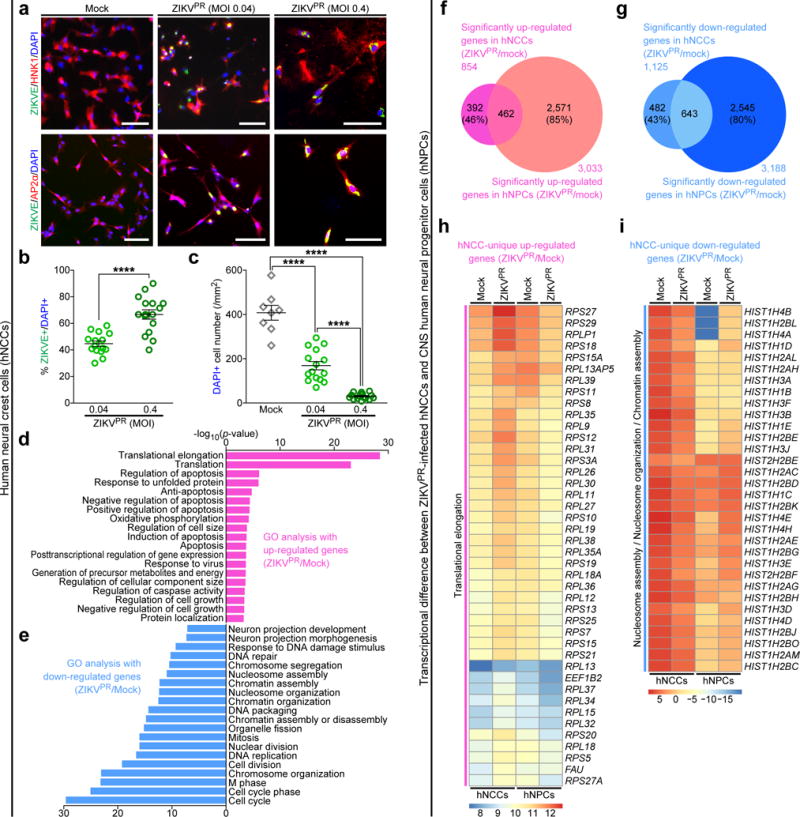
ZIKV efficiently infects human pluripotent stem cell-derived neural crest cells. hNCCs were treated with ZIKVPR (MOI of 0.04 or 0.4) or mock for 65 h. (a) Representative images of hNCCs immunostained with the indicated antibodies. Scale bars, 100 μm. (b) Quantification of the percentage of ZIKVE+ hNCCs, relative to the number of DAPI+ cells (n = 15 for MOI of 0.04, n = 16 for MOI of 0.4; ****P < 0.0001; unpaired Student’s t-test). (c) The number of DAPI+ hNCCs per mm2 (n = 8 for Mock, n = 15 for MOI of 0.04, n = 16 for MOI of 0.4; ****P < 0.0001; unpaired Student’s t-test). All error bars represent mean ± s.e.m. The number of cell cultures is indicated for each group (n). (d,e) Genes with significant differential expression between infected and uninfected-hNCCs were subjected to GO analyses. (d) All significant terms are shown for up-regulated genes. (e) Top 20 most significant terms are shown for down-regulated genes. (f,g) Weighted Venn diagrams between ZIKVPR-infected hNCCs and CNS hNPCs, showing overlap of (f) significantly up-regulated genes or (g) significantly down-regulated genes. (h,i) Heatmaps showing the expression levels (log2 FPKM) of specific genes in mock and ZIKVPR-infected hNCCs and hNPCs. FPKM, fragments per kilobase of transcript per million mapped reads.
To investigate the impact of ZIKVPR infection on hNCCs at the molecular level, we employed global transcriptome analyses (RNA-seq). Our genome-wide analyses identified a large number of differentially expressed genes upon viral infection (Supplementary Fig. 4a,b and Supplementary Table 1). Gene ontology (GO) analyses of ZIKVPR-infected hNCCs revealed a particular enrichment of up-regulated genes in apoptotic, cell-death-related terms, which was more pronounced than in infected CNS human neural progenitor cells (hNPCs)3,16, and enrichment in translation-terms (Fig. 2d). The downregulated genes were enriched in cell-cycle-related terms (Fig. 2e), which is consistent with our previous findings in CNS hNPCs3,16. Notably, transcriptome-wide comparison of gene expression to ZIKVPR-infected CNS hNPCs revealed some similarities and differences (Fig. 2f–i, Supplementary Fig. 5 and Supplementary Table 2). Between ZIKVPR-infected hNCCs and CNS hNPCs, significant numbers of genes are commonly up- and down-regulated (462 and 643 genes, respectively), but there are still large numbers of genes that were differentially expressed (874 genes in hNCCs and 5,116 genes in CNS hNPCs; Fig. 2f,g and Supplementary Fig. 5). Further GO analyses showed that genes in the “translational elongation” term are uniquely up-regulated in ZIKVPR-infected hNCCs (false discovery rate (FDR) = 8.4E-39; Fig. 2h and Supplementary Fig. 5b) and genes in “nucleosome organization” term are uniquely down-regulated in ZIKVPR-infected hNCCs compared to CNS hNPCs (FDR = 3.9E-8; Fig. 2i and Fig. 2i and Supplementary Fig. 5b). This suggests that molecular pathology in ZIKVPR-infected hNCCs is distinct from that of CNS hNPCs.
A number of recent studies have shown that in the CNS ZIKV targets NPCs with little direct infection of neurons both in culture and in vivo3–5,18–20. In contrast, ZIKVPR readily infected PRPH+TUJ1+ hPNs (Fig. 3a,b). ZIKVE was concentrated in perinuclear structures of the hPSC-derived hPNs (Fig. 3a), similar to a recent report that infectious ZIKV particles are located in the disrupted endoplasmic reticulum of CNS neurons in the human fetal brain1. ZIKVPR infection in hPNs led to reduced cell numbers compared to the mock-infected cells (Fig. 3c and Supplementary Fig. 3b). Mechanistically, ZIKVPR infection increased CASP3 activation in hNCCs and hPNs 65 h after infection, as compared to the mock infection, suggesting increased cell death (Supplementary Fig. 3c–f). Genome-wide transcription analyses of ZIKVPR-infected hPNs identified a large number of differentially expressed genes upon viral infection (Supplementary Fig. 4c,d and Supplementary Table 3). GO analyses of ZIKVPR-infected hPNs revealed that differential gene expression patterns were similar to that of ZIKVPR-infected hNCCs (Fig. 2d,e and Fig. 3d,e), mainly associated with apoptotic cell death and cell cycle. In ZIKVPR-infected hPNs and hNCCs, large numbers of transcripts were differentially expressed (1,983 genes in hPNs and 945 genes in hNCCs), while a subset of these genes are commonly up- and down-regulated (419 and 615 genes, respectively; Supplementary Fig. 6 and 7). Further GO analyses with the list of differentially expressed genes unique to ZIKVPR-infected hPNs showed several enriched terms for down-regulated genes, including “regulation of cell development” and “regulation of neurogenesis” (Supplementary Fig. 6g and 7a), suggesting that ZIKVPR causes significant developmental and cellular changes in hPNs. In particular, ZIKVPR disrupted “WNT signaling pathway” in infected hPNs (Supplementary Fig. 7b), highlighting drastic molecular perturbations upon ZIKVPR-infection. To verify our RNA-seq results, we focused on genes associated with apoptosis and epigenetic regulation, using qRT-PCR, western blotting, and immunocytochemical analyses (Supplementary Fig. 8). We confirmed that the expression and phosphorylation levels of c-Jun are uniquely increased in ZIKVPR-infected hPNs (Supplementary Fig. 8c,e,f,i,j), suggesting that c-Jun phosphorylation-associated apoptosis is one of the possible cell death pathways in ZIKVPR-infected hPNs (Supplementary Fig. 9). We also confirmed that the expression and acetylation levels of histone H3 are uniquely decreased in ZIKVPR-infected hNCCs (Supplementary Fig. 8d,g,h), indicating that the dysregulation of histone protein expression and acetylation may be associated with cell death of ZIKVPR-infected hNCCs (Supplementary Fig. 9). Overall, our unbiased global transcriptome datasets not only support our findings of massive cell death in infected hNCCs and hPNs, but also provide a valuable resource of cell type-specific molecular pathology of ZIKAPR for the field.
Figure 3.
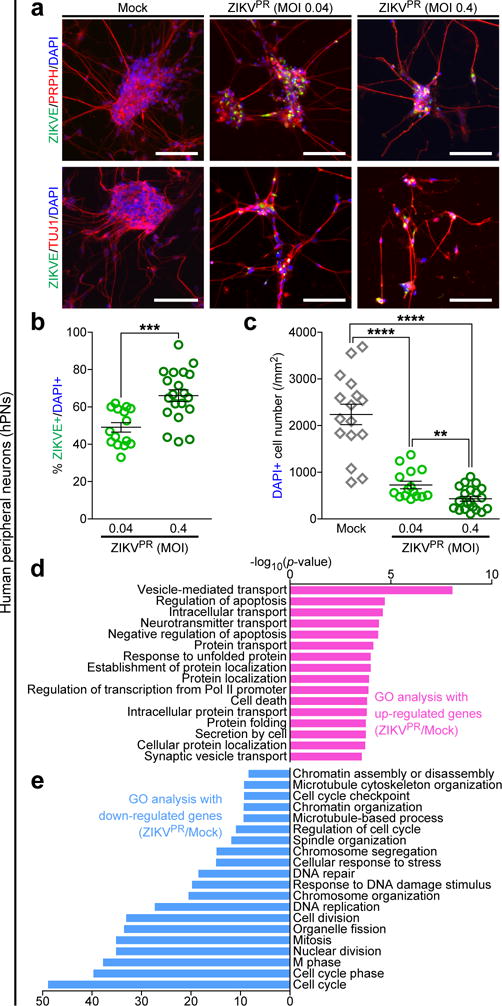
Effective ZIKV infection in human peripheral neurons causes significant cell death and pathogenic transcriptional dysregulations. hPNs were treated with ZIKVPR (MOI of 0.04 or 0.4) or mock for 65 h. (a) Representative images of hPNs immunostained with the indicated antibodies. Scale bars, 100 μm. (b) Quantification of the percentage of ZIKVE+ hPNs, relative to the number of DAPI+ cells (n = 15 for MOI of 0.04, n = 20 for MOI of 0.4; ***P < 0.001; unpaired Student’s t-test). (c) The number of DAPI+ hPNs per mm2 (n = 16 for Mock, n = 15 for MOI of 0.04, n = 20 for MOI of 0.4; **P < 0.01; ****P < 0.0001; unpaired Student’s t-test). The number of cell cultures is indicated for each group (n). All error bars represent mean ± s.e.m. (d,e) Genes with significant differential expression between infected and uninfected-hPNs were subjected to GO analyses. (d) All significant terms are shown for up-regulated genes. (e) Top 20 most significant terms are shown for down-regulated genes.
While it has been established that ZIKV can cause neural developmental defects during brain development6, whether ZIKV can efficiently infect hPNs was not previously known. Here, we demonstrated that either intraperitoneal injection or fetal mouse brain injection of ZIKV leads to infections in the PNS. Both human stem cell-derived hNCCs and hPNs are permissive to ZIKV infection, presenting massive cell death compared to CNS cells. In addition, we found ZIKV infection in human stem cell-derived Schwann cells, but not in skeletal muscle cells (Supplementary Fig. 10), which suggests a tissue specific vulnerability to ZIKV infection. Our results are also supported by recent human case studies and a primate model study, which showed PNS ZIKV-pathology10–12. We could also detect high levels of ZIKV in mouse spinal cord after intraventricular injection and secretion of infectious ZIKV particles from infected hNCCs and hPNs in culture (Supplementary Fig. 11). It remains to be determined how ZIKV can be transmitted from the abdominal cavity or the brain to PNS regions. Potential routes include blood stream, cerebrospinal fluid and inter-neuronal transmission. It is also important to elucidate how PNS ZIKV infection is relevant to peripheral neuropathies.
In summary, our study provides an efficient humanized model to study ZIKV infection in the PNS, showing similarities and differences in the molecular pathophysiology of ZIKV compared to the developing CNS. Our study opens a new avenue to further investigate cellular and molecular mechanisms, and our humanized peripheral neuron model can facilitate the discovery and validation of therapeutic agents for ZIKV-related PNS symptoms.
Online METHODS
ZIKV preparation and animal infection
The Asian lineage ZIKV SZ01 strain (GenBank accession no: KU866423) was isolated from a patient who had returned to China from Samoa and was amplified in C6/36 cells21. The ZIKV replicated efficiently in C6/36 cells and the viral growth curve was determined by qRT-PCR assay. The type-I interferon receptor deficient (A129) mice were used to generate a ZIKV intraperitoneal infection model. Around 5× 105 plaque-forming unit (PFU) of ZIKV SZ01 or culture medium was injected intraperitoneally into 5-week-old A129 mice and inspected 3 days later. To generate the microcephaly mouse model, we anesthetized pregnant ICR mice at embryonic day 13.5 (E13.5), exposed the uterine horns and injected 1 μL of ZIKV SZ01 (6.5×105 PFU/mL) or culture medium (RPMI medium 1640 basic + 2% FBS) into the lateral ventricles of embryos using a calibrated micropipette, as described previously5. For each pregnant dam, two thirds of the embryos received ZIKV infection while the rest were injected with culture medium to provide littermate controls. After virus injection, the embryos were placed back into the abdominal cavity of dams and wound was closed. Tissue was harvested and analyzed at P1. All experimental procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee at Beijing Institute of Microbiology and Epidemiology and conducted in a biological safety protection laboratory. The experimenters were blinded to treatment. None of the viable animals were excluded from our analyses.
Immunohistochemistry
The trunk tissues with dorsal root ganglion (DRG) and spinal cord from the infected pups and their littermate controls were collected at P1. The tissues were fixed in 4% paraformaldehyde overnight, dehydrated with 20% sucrose in PBS and embedded in OCT compound for cryostat sectioning. The sections with DRG and spinal cord were immunostained with human convalescent serum of ZIKV-infected patients5 and the commercial primary antibodies (Supplementary Table 4). For measuring the percentage of apoptotic DRG neurons labeled by cl-Casp3, we counted 10 tissue slices (30 μm/slice) from 4 pairs of animals under fluorescence microscopy. We didn’t dissect the DRG out of trunk tissues from P1 mice but directly sectioned the trunk tissue containing spinal cord and DRG. We found that there were around 10 slices containing DRG tissues after observing more than 30 slices for each group and counted the infected/apoptotic neurons for quantification. Some slices contain more than 150 DRG neurons but others had less than 50 DRG neurons. We counted all of the slices with DRG neurons. Preparations of enteric ganglia were generated by peeling off the small intestinal villus layer of a freshly-dissected segment of the Jejunum. The stretched surface along with the attached myenteric were fixed overnight in 4% PFA in PBS. The whole mounts were used for immunofluorescence staining. The experimenters were blinded to treatment. None of the viable animals were excluded from our analyses. We also did not exclude any data points for quantification.
hPSC culture and PNS differentiation
Undifferentiated H9 hESCs (passages 40–50) were cultured on mitotically inactivated mouse embryonic fibroblasts (MEFs, Applied Stem Cell) in the hESC medium, containing DMEM/F12, 20% knockout serum replacement (KSR), 0.1 mM MEM-NEAA, 2 mM L-glutamine, 55 μM β-mercaptoethanol (Life Technologies) and 10 ng/mL FGF2 (R&D Systems). All cells were maintained at 37°C and 5% CO2 in a humidified incubator. For neural differentiation, hES cells were plated at 5–20 × 103 cells on a confluent layer of irradiated (50 Gy) stromal cells (MS-5) in 60-mm cell culture plates in a medium containing DMEM/F12, 20% KSR, 0.1 mM MEM-NEAA, 2 mM L-glutamine, 55 μM β-mercaptoethanol (KSR medium) as described previously14,22,23. After 16 days in KSR medium, cultures were switched to N2 medium14,22–24. Medium was changed every 2 days, and growth factors were added as described previously14,22–24. Briefly, differentiation was initiated by adding 100 nM LDN193189 (abcam) and 10 μM SB431542 (Cayman Chemical) in KSR medium. Other small-molecule compounds used in neural induction and differentiation were as follows: 0.2 mM ascorbic acid, 0.2 mM dibutyryl-cyclic AMP (Sigma-Aldrich), and 1 μM purmorphamine (PMP, Cayman Chemical). Recombinant growth factors were as follows: 50 ng/ml sonic hedgehog (Shh), 100 ng/ml FGF8 (R&D Systems), and 10 ng/ml BDNF (PeproTech). Rosette structures were harvested mechanically at day 22–28 of differentiation (termed passage 0; P0) and gently re-plated on 15 μg/ml polyornithine / 1 μg/ml laminin / 10 ng/ml fibronectin (PO/Lam/FN)-coated culture dishes in N2 medium (termed passage 1; P1). P1 cultures were supplemented with AA, PMP, Shh, FGF8, and BDNF. After 6–7 d of P1 culture, cells were mechanically triturated after exposure to Ca2+/Mg2+-free Hanks’ balanced salt solution (CMF-HBSS, 20 min at 25 °C) and labeled with antibodies for flow cytometry (Supplementary Table 4). FACS sorting (p75+/HNK1+) was performed on a MoFlo (Dako). Sorted cells were plated on culture dishes pre-coated with PO/Lam/FN (10–30 × 103 cells/cm2). hPSC-derived hNCCs were maintained in the ‘hNCC media’, which contains neurobasal media supplemented with 2 mM L-glutamine, 1X B-27, 1X N-2 supplements (Life Technologies), 20 ng/ml FGF2, and 20 ng/ml EGF (R&D Systems). For directed differentiation of hPSC–derived NCCs toward hPNs, FGF2/EGF-expanded hNCCs were differentiated with the ‘hPN media’, which contains neurobasal media supplemented with 2 mM L-glutamine, 1X B-27, 1X N-2 supplements, 0.2 mM ascorbic acid, 0.2 mM dibutyryl-cyclic AMP, 10 ng/mL BDNF, 10 ng/mL NGF (PeproTech), and 10 ng/mL GDNF (R&D Systems) at least for 2 weeks23. For directed differentiation of hPSC–derived NCCs toward Schwann cells, we followed a previously described protocol14,22. Myogenic differentiation and myotube formation followed a previously described protocol25.
hPSC culture and CNS hNPC differentiation
Human iPSC lines had been fully characterized and passaged on MEF feeder layers26. All studies followed institutional IRB and ISCRO guidelines and protocols approved by Johns Hopkins University School of Medicine. Human iPSCs were differentiated into forebrain-specific hNPCs and immature neurons following a previously established protocol26. Briefly, hiPSCs colonies were detached from the feeder layer with 1 mg/mL collagenase treatment for 1 hour and suspended in embryonic body (EB) medium, consisting of FGF-2-free iPSC medium supplemented with 2 μM Dorsomorphin and 2 μM A-83, in non-treated polystyrene plates for 4 days with a daily medium change. After 4 days, EB medium was replaced by neural induction medium (hNPC medium) consisting of DMEM/F12, N2 supplement, NEAA, 2 μg/mL heparin and 2 μM cyclopamine. The floating EBs were then transferred to matrigel-coated 6-well plates at day 7 to form neural tube-like rosettes. The attached rosettes were kept for 15 days with hNPC medium change every other day. On day 22, the rosettes were picked mechanically and transferred to low attachment plates (Corning) to form neurospheres in hNPC medium containing B27. The neurospheres were then dissociated with Accutase at 37 °C for 10 minutes and placed onto matrigel-coated 6-well plates at day 24 to form monolayer hNPCs in hNPC medium containing B27. These hNPCs expressed forebrain-specific progenitor markers, including NESTIN, PAX6, EMX-1, FOXG1 and OTX226.
Preparation of ZIKVPR and cell infection
The ZIKV PRVABC59 strain (ZIKVPR) was obtained from ATCC (Manassas, VA) and subsequently amplified in Aedes albopictus clone C6/36 cells (ATCC). Briefly, C6/36 cells were inoculated with viral inoculum for 1 h at 28 °C in a low volume of medium (3 mL per T-75 flask), with rocking every 15 min, before the addition of an additional 17 mL medium. Virus-inoculated cells were then incubated at 28 °C for 6–7 d before harvesting the supernatant. C6/36-amplified ZIKVPR titer was determined by infecting Vero cells for 48 h with a methylcellulose overlay and analyzed for focus-forming units per mL (FFU/mL). In mock infections, an equal volume of spent uninfected C6/36 culture medium was used. hPSC-derived hNCCs were seeded at a density of 50 cells/mm2 and maintained for 2–4 days prior to ZIKV infection. hNCC-derived hPNs were seeded at a density of 1700 cells/mm2 and matured for 2–3 weeks prior to ZIKV infection. Then these cells were infected with ZIKVPR at MOI of 0.04 or 0.4 and analyzed at 65 h post infection after three times washing the cells with culture media or PBS. The experimenter was not blinded to treatment. None of cell cultures were excluded from our analyses.
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde and stained with the primary antibodies (Supplementary Table 4) after permeabilization with 0.1% Triton X-100/0.5% BSA/PBS solution. Appropriate Alexa Fluor® 488 or 568 labeled secondary antibody (Life Technologies) and DAPI (Roche Applied Science) nuclear counter-staining were used for visualization. The stained samples were analyzed using fluorescence microscopy (Eclipse TE2000-E, Nikon). The numbers of ZIKVE+, Ki-67+, or DAPI+ cells per mm2 were counted under fluorescence microscopy. Cell viability 65 h after ZIKVPR infection was represented as percentage fold change (relative to mock) of DAPI+ cell number (/mm2) in ZIKVPR-infected cells (mean ± s.e.m.). The experimenter was not blinded to treatment. None of cell cultures were excluded from our analyses.
RNA isolation, RNA-seq library preparation, and sequencing
RNA-seq libraries were prepared from isolated total RNA, and were sequenced as previously described16. Briefly, total cellular RNA was purified from cell pellets using the TRIzol Reagent (Life Technologies) according to the manufacturer’s instructions. RNA-seq libraries were generated from 1 μg of total RNA from duplicated or triplicated samples (2 or 3 cell cultures) per condition using the TruSeq LT RNA Library Preparation Kit v2 (Illumina) following the manufacturer’s protocol. An Agilent 2100 BioAnalyzer and DNA1000 kit (Agilent) were used to quantify amplified cDNA and to control the quality of the libraries. Illumina HiSeq2500 was used to perform 100-cycle single-read sequencing. Image processing and sequence extraction were performed using the standard cloud-based Illumina pipeline in BaseSpace. The experimenter was blinded to treatment.
Bioinformatic analyses
Paired-end RNA-seq reads were first aligned to human transcriptome annotations and genome assembly (hg19) using TopHat v2.0.1327. The numbers of mapped reads for each condition can be found in Supplementary Table 1 and 3. FPKM (fragments per kilobase of transcript per million mapped reads) values were calculated by Cufflinks v2.2.128. Pairwise comparisons between infected and mock conditions were performed to detect differentially expressed (DE) genes using Cuffdiff v2.2.128. In hPNs and hNCCs, DE genes are defined as ones with a q-value less than 0.05; and in CNS hNPCs, DE genes are defined as the ones with a q-value less than 0.05, and with absolute log2 fold change of expression levels no less than 0.57 (the same significance cut-off used by Cuffdiff for the RNA-seq data of hNCCs). Gene ontology (GO) analyses on biological processes were performed by the Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.729. To identify significantly enriched GO terms, a Benjamini–Hochberg procedure was used to control the false discovery rate (FDR) at 0.05. Pathway enrichment analyses and disease-related gene enrichment analyses were performed by the WEB-based Gene SeT AnaLysis Toolkit (WebGestalt) update 201530 with adjusted p-value no greater than 0.05. The experimenter was blinded to treatment.
qRT-PCR
Total RNA was extracted by using the TRIzol Reagent, and reverse transcribed by using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). qRT-PCR mixtures were prepared with KAPA SYBR® FAST qPCR Kit Master Mix (Kapa Biosystems) and reactions were done with the Mastercycler ep Realplex2 (Eppendorf). Each transcript level was assessed by qRT-PCR normalized to GAPDH expression. Primers for qRT-PCR were listed in Supplementary Table 5. The experimenter was not blinded to treatment. None of cell cultures were excluded from our analyses.
Western blot analyses
Cells were lysed in 1X RIPA buffer (Cell Signaling Technology) and supplemented with 1% SDS, 10% glycerol, and 1 mM PMSF. After sonicating to reduce the viscosity, cell lysates were mixed with Benzonase (Sigma-Aldrich) and incubated for 15 min at 37 °C. The samples were clarified by centrifugation at 15,000 × g for 30 min at 14 °C, boiled at 98 °C for 2 min in 1X Laemmli sample buffer (Sigma-Aldrich) supplemented with 20 mM DTT, resolved by SDS-PAGE, and transferred to nitrocellulose membranes (Bio-Rad). WB analyses were performed with the indicated antibodies (Supplementary Table 4). The bands were quantified by densitometry using ImageJ (NIH). The experimenter was not blinded to treatment.
Viral titer by focus forming assay
Cells were infected with ZIKVPR at MOI of 0.04 for 65 h before harvesting the supernatant. Human cell amplified ZIKVPR titer was determined by infecting Vero cells for 48 h with a methylcellulose overlay and analyzed for FFUs per mL (FFU/mL) as previously described31. The experimenter was not blinded to treatment.
Statistical analysis
All data were tested for normal distribution. Values are from at least three independent experiments with multiple biological replicates each, and reported as mean ± s.e.m. Differences between two samples were analyzed for significance using the unpaired two-tailed Student’s t-test in Prism (GraphPad). No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications3,5,16,18,22,23,26,31. Experimenters using in vivo samples were always blinded to treatment. No special randomization procedures were used for assigning groups.
Supplementary Material
Acknowledgments
We thank Dr. Kimberly M. Christian for comments. We acknowledge the services of Hao Zhang at Flow Cytometry Core Facility (JHSPH), Weining Tang at Omega Bioservices (Omega Bio-tek, Inc.), and Mike Zwick and Ben Isett at the Emory Integrated Genomics Core (EIGC). This work was supported by grants from the Robertson Investigator Award from New York Stem Cell Foundation (G.L.), Maryland Stem Cell Research Funding (MSCRF; G.L., H.S., G.M.), NIH R01NS093213 (G.L.), R01MH105128 and R35NS097370 (G.M.), U19AI131130 (G.M., Z.W., H.T., and P.J.), U19MH106434, P01NS097206, R37NS047344 (H.S.), R21AI119530 (H.T.), NSF (China) (31430037), MOST (China) “973” program (2014CB942801/2012YQ03026006; Y.W., Z.X.), FSU Zika seed funding (H.T.), and by Adelson Medical Research Foundation (G.M.). The authors (G.L., Y.O., G.M., H.S.) acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation.
Footnotes
ACCESSION CODES
The accession number for the RNA-seq data reported in this paper is GEO: GSE87750.
AUTHOR CONTRIBUTIONS
Y.O.: conception and study design, performing experiments, data analysis, data assembly, and interpretation of data, and writing manuscript; F.Z., Y.W., E.M.L., Q.-F.W., I.Y.C., H.L., F.M., R.L., L.H., T.X., C.L., Z.W. and C.F.Q.: performing experiments and/or data analysis; H.W., H.T., Z.X., P.J., H.S., G.M., and G.L.: study design and data analysis; H.T., Z.X., P.J., H.S., G.M., and G.L.: conception and study design, data analysis and interpretation, and writing manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
A Supplementary Methods Checklist is available.
Code availability. TopHat v2.0.1327 and Cuffdiff v2.2.128 used for data analysis are freely available at https://ccb.jhu.edu/software/tophat/index.shtml and http://cole-trapnell-lab.github.io/cufflinks/cuffdiff/. R packages ggplot2 and pheatmap used for data visualization are freely available at http://ggplot2.org/ and the Comprehensive R Archive Network (CRAN).
Data availability. The RNA-seq data reported in this paper has been deposited in GEO, GSE87750. The remaining data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
- 1.Mlakar J, et al. N Engl J Med. 2016;374:951–958. doi: 10.1056/NEJMoa1600651. [DOI] [PubMed] [Google Scholar]
- 2.Rasmussen SA, Jamieson DJ, Honein MA, Petersen LR. N Engl J Med. 2016;374:1981–1987. doi: 10.1056/NEJMsr1604338. [DOI] [PubMed] [Google Scholar]
- 3.Tang H, et al. Cell Stem Cell. 2016;18:587–590. doi: 10.1016/j.stem.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cugola FR, et al. Nature. 2016;534:267–271. doi: 10.1038/nature18296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li C, et al. Cell Stem Cell. 2016;19:120–126. doi: 10.1016/j.stem.2016.04.017. [DOI] [PubMed] [Google Scholar]
- 6.Ming GL, Tang H, Song H. Cell Stem Cell. 2016;19:690–702. doi: 10.1016/j.stem.2016.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li H, Saucedo-Cuevas L, Shresta S, Gleeson JG. Neuron. 2016;92:949–958. doi: 10.1016/j.neuron.2016.11.031. [DOI] [PubMed] [Google Scholar]
- 8.Wen Z, Song H, Ming GL. Genes Dev. 2017;31:849–861. doi: 10.1101/gad.298216.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brasil P, et al. PLoS Negl Trop Dis. 2016;10:e0004636. doi: 10.1371/journal.pntd.0004636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Medina MT, et al. J Neurol Sci. 2016;369:271–272. doi: 10.1016/j.jns.2016.08.044. [DOI] [PubMed] [Google Scholar]
- 11.Cleto TL, de Araujo LF, Capuano KG, Rego Ramos A, Prata-Barbosa A. Pediatr Neurol. 2016;65:e1–e2. doi: 10.1016/j.pediatrneurol.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 12.Hirsch AJ, et al. PLoS Pathog. 2017;13:e1006219. doi: 10.1371/journal.ppat.1006219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuchs S, Sommer L. Neurodegener Dis. 2007;4:6–12. doi: 10.1159/000100354. [DOI] [PubMed] [Google Scholar]
- 14.Lee G, Chambers SM, Tomishima MJ, Studer L. Nat Protoc. 2010;5:688–701. doi: 10.1038/nprot.2010.35. [DOI] [PubMed] [Google Scholar]
- 15.Faria NR, et al. Science. 2016;352:345–349. doi: 10.1126/science.aaf5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang F, et al. Nucleic Acids Res. 2016;44:8610–8620. doi: 10.1093/nar/gkw765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bayless NL, Greenberg RS, Swigut T, Wysocka J, Blish CA. Cell Host Microbe. 2016;20:423–428. doi: 10.1016/j.chom.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qian X, et al. Cell. 2016;165:1238–1254. doi: 10.1016/j.cell.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, et al. Cell Stem Cell. 2016;19:593–598. doi: 10.1016/j.stem.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Onorati M, et al. Cell Rep. 2016;16:2576–2592. doi: 10.1016/j.celrep.2016.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng YQ, et al. Sci China Life Sci. 2016;59:428–430. doi: 10.1007/s11427-016-5043-4. [DOI] [PubMed] [Google Scholar]
- 22.Lee G, et al. Nat Biotechnol. 2007;25:1468–1475. doi: 10.1038/nbt1365. [DOI] [PubMed] [Google Scholar]
- 23.Oh Y, et al. Cell Stem Cell. 2016;19:95–106. doi: 10.1016/j.stem.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perrier AL, et al. Proc Natl Acad Sci U S A. 2004;101:12543–12548. doi: 10.1073/pnas.0404700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi IY, et al. Cell Rep. 2016;15:2301–2312. doi: 10.1016/j.celrep.2016.05.016. [DOI] [PubMed] [Google Scholar]
- 26.Wen Z, et al. Nature. 2014;515:414–418. doi: 10.1038/nature13716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim D, et al. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trapnell C, et al. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang da W, Sherman BT, Lempicki RA. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, Duncan D, Shi Z, Zhang B. Nucleic Acids Res. 2013;41:W77–83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu M, et al. Nat Med. 2016;22:1101–1107. doi: 10.1038/nm.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.