

Introduction and Clinical Overview
IgG4-related disease (IgG4-RD) is a systemic, multi-organ disorder with heterogeneous clinical features but distinctive presentations.1, 2 The classic onset of this disease is subacute, taking the form of tumor-like lesions that develop gradually. These lesions are generally identified either on physical examination (e.g., salivary gland enlargement) or imaging (e.g., a renal, lung, or pancreatic mass), frequently at a time when the patient feels well. In the first years following recognition of IgG4-RD, the diagnosis was often made by pathologists in the complete absence of clinical suspicion for the disease. More recently, because of growing familiarity with typical IgG4-RD presentations and the use of blood IgG4 testing, presumptive diagnoses are often rendered by clinicians before the performance of diagnostic tests. Biopsy confirmation of the diagnosis remains important in most cases.
IgG4-RD is not a new disease. Cases of the disease are well documented in medical history going back at least to the late 1800s3 , but the individual organ manifestations were considered to be distinct disease entities for more than a century, each confined to single organs and given eponymic designations: e.g., Riedel’s thyroiditis (circa 1883), Küttner’s tumor (dacryoadenitis involving the submandibular gland, circa 1896), and Ormond’s disease (circa 1960).4, 5, 6 In 2001, Hamano, Kawa, and colleagues demonstrated that an elevated serum IgG4 level was useful in distinguishing “sclerosing pancreatitis,” now termed type 1 (IgG4-related) autoimmune pancreatitis, from other diseases of the pancreatic and biliary tract.7
In 2003, Kamisawa and Nakajima recognized similar pathology findings in extra-pancreatic organs of patients with sclerosing pancreatitis.8 They described a disease typified by elevated serum IgG4 levels, responsiveness to glucocorticoids, multi-organ involvement, and a fibroinflammatory infiltrate. The name “IgG4-related autoimmune disease” was given to this disorder initially because of the abundance of IgG4-expressing plasma cells within affected tissues, thereby unifying seemingly disparate conditions. The involvement of bile ducts, major salivary glands, lymph nodes, and retroperitoneal tissue was also described in this original case series. Since that time, IgG4-RD has been reported to affect virtually every organ in the body. The lacrimal gland, lung, and kidney are other common sites of disease involvement [Figure 1, A–B].2 The aorta, pachymeninges, and bile ducts comprise exceptions to the tumefactive clinical presentation, presumably because of their tubular structures and/or thin walls.9, 10, 11
Figure 1.
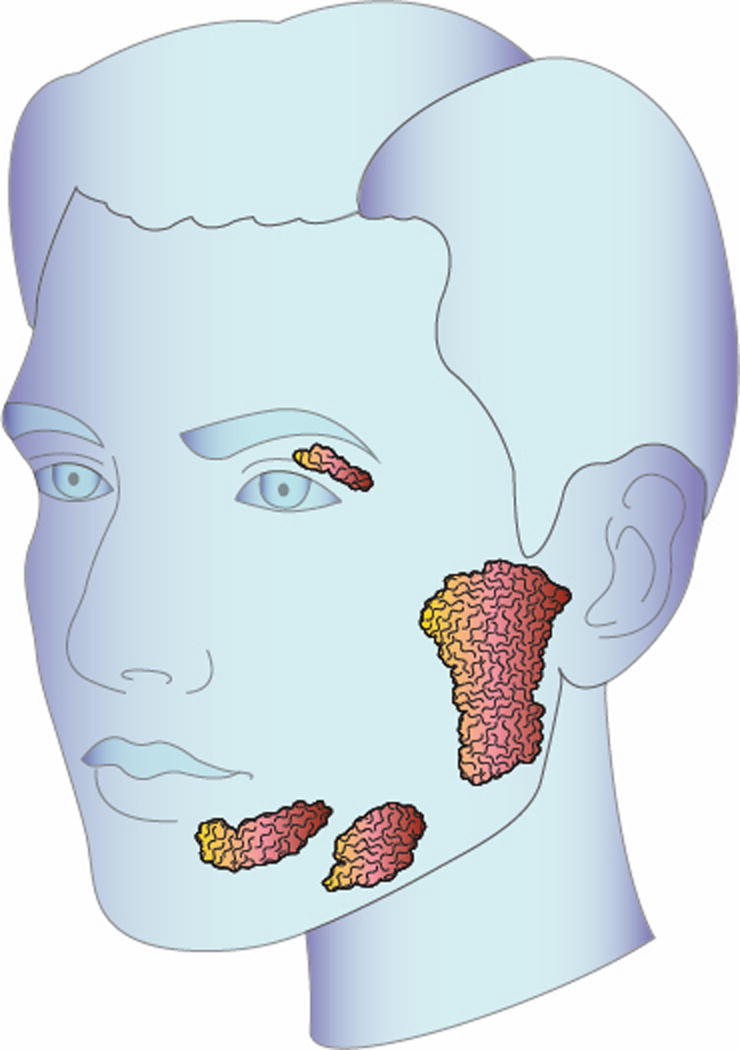
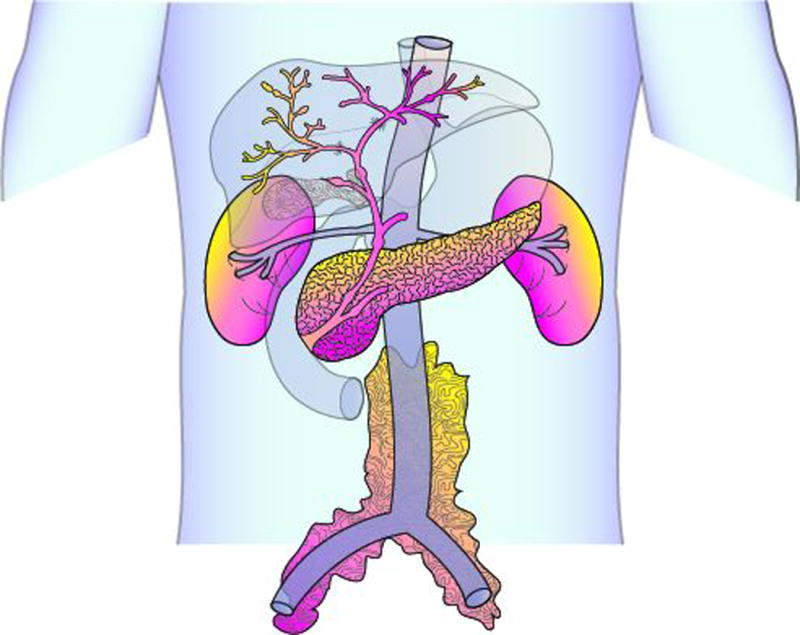
(A) Head and neck illustration highlighting involvement of the lacrimal and major salivary glands. Lacrimal and salivary gland enlargement is most often bilateral in distribution. (B) Abdomen illustration highlighting typical organ involvement including the pancreas, bile ducts, kidneys, and retroperitoneal tissue. Radiographically, the retroperitoneal fibrosis often extends inferiorly to encase the iliac vessels. Not depicted here, the aorta and lung are other common sites of disease involvement.
These diverse clinical features are linked by shared pathological findings across involved organs: a lymphoplasmacytic infiltrate; storiform fibrosis (“storiform” from storea, the Latin word for woven mat); obliterative phlebitis; and enrichment with IgG4-expressing plasma cells.13 Such features, though strongly suggestive of IgG4-RD, should never be regarded in isolation as diagnostic of this condition. Rather, careful correlation of pathology features with clinical, serologic, and imaging findings is essential to diagnosis. The American College of Rheumatology/European League against Rheumatism (ACR/EULAR) Classification Criteria for IgG4-Related Disease, scheduled for completion in 2017, underscore this point firmly. International consensus statements on the nomenclature of IgG4-RD, its pathologic findings, and clinical management have been published.12, 13, 14 The IgG4-RD Responder Index, designed to serve as a clinical trials outcome measure, is now undergoing a worldwide validation study.15, 16
Much has been learned about the histopathologic, clinical, cellular and molecular aspects of IgG-RD since the first description in 2003. The number of publications on IgG4-RD has surged from fewer than 300 prior to 2011 to more than 1,700 at the start of 2017. Because of the rapid evolution in understanding, keeping abreast of the most current thought about clinical features and mechanistic details of IgG4-RD has been challenging. This review serves as a concise summary of the current state of knowledge on IgG4-RD, emphasizing how dialogue between the clinic and laboratory feeds both new discovery and patient care.
What Targeted Treatment Has Taught Us
Most IgG4-RD patients respond to glucocorticoids, but glucocorticoids often fail to induce treatment-free remission.14 Few data support the use of conventional disease modifying anti-rheumatic drugs such as azathioprine, methotrexate, and mycophenolate mofetil.14 Because the serum IgG4 elevations suggested a role of the humoral immune system, we employed B cell depletion (rituximab) to treat a small series of patients. This intervention led to swift clinical responses and sharply reduced circulating IgG4 levels within weeks.17 Many patients could be treated without glucocorticoids, excluding the 100 mg of methylprednisolone administered with each infusion.17
One patient from this early experience who had cutaneous IgG4-RD underwent both pre- and post-treatment skin biopsies, permitting the opportunity to assess the effects of B-cell depletion at the tissue level. As in the circulation, IgG4-expressing plasma cells in tissue diminished substantially after rituximab [Figure 2A–B].18 Moreover, a dramatic reduction in myofibroblast activation and density was seen, indicating a relationship between activated B cells and the generation of fibrosis in IgG4-RD.18 We therefore investigated the B cell compartment more deeply, exploiting the tool of next-generation sequencing. In contrast to previously available sequencing technology, next generation sequencing platforms have the capacity to sequence billions of nucleotides within a matter of hours. This has revolutionized the study of disease mechanisms by providing a practical means of interrogating entire genomes, transcriptomes, microbiomes as well as immune repertoires. For example, by sequencing every B-cell receptor mRNA molecule present in a given infiltrating or circulating B cell population, we can precisely understand the extent of somatic hypermutation it has undergone, the connectivity to another cell population and degree of clonal restriction it contains. The application of this technology to IgG4-RD is described here.
Figure 2.

Immunohistochemical staining for IgG4 (A and B) and CD3 (C and D) from the same cutaneous site pre- (A and C) and post- (B and D) rituximab treatment demonstrating a marked decline in tissue infiltrating B and T cells.
The Prominence of Plasmablasts
Plasmablasts – circulating, antibody-secreting cells – are the progenitors of long-lived plasma cells (Figure 3). Plasmablasts are characterized as CD19dim expressing cells of the B lymphocyte lineage that demonstrate high expression of CD27 and CD38. Plasmablasts lack CD20, distinguishing them from memory B cells. Although described in certain rheumatologic disease19, 20, plasmablasts appear to play particularly important roles in IgG4-RD. Levels of these circulating cells were substantially expanded in 84 IgG4-RD patients compared to healthy controls.21 Moreover, most of the plasmablasts in these patients had undergone class switching to IgG4. The immunoglobulin heavy chain repertoire in IgG4-RD plasmablasts was demonstrated by next-generation sequencing to be oligoclonally-restricted, a finding strongly suggestive of an antigen-driven process.21 In addition, extensive somatic hypermutation within the rearranged variable regions supported the notion of T cell/B cell collaboration.21 Further, plasmablast levels declined within 2–4 weeks of B cell depletion therapy, correlating with clinical improvement.21
Figure 3.
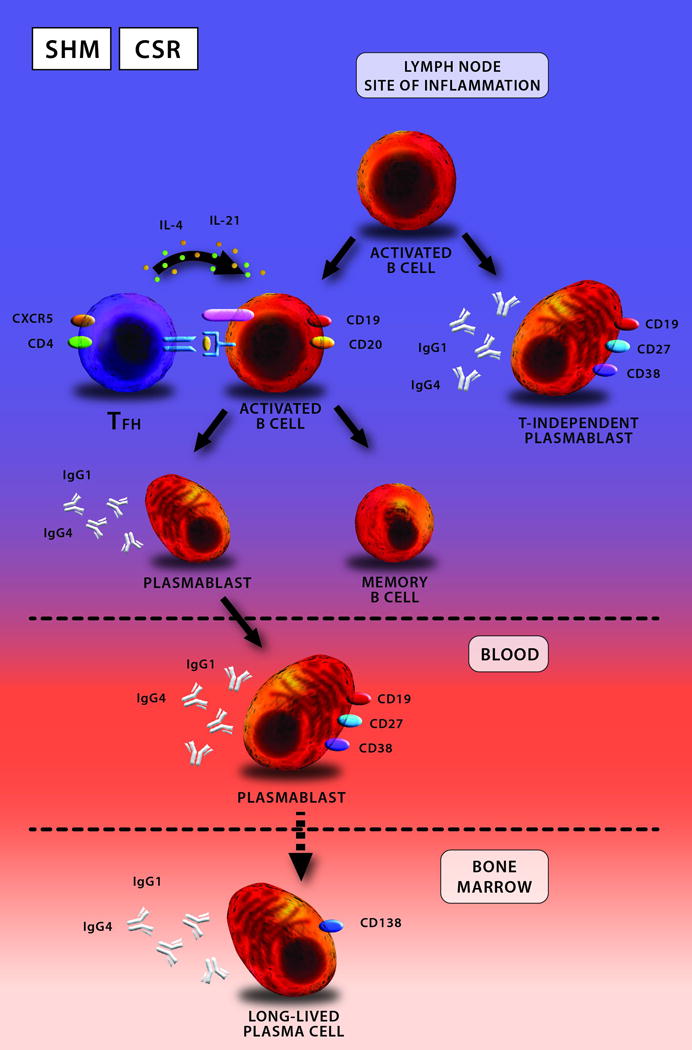
T-B collaboration, mediated by IL-4 and IL-21, is at the center of driving the differentiation of activated B cells into antibody-secreting cells. During this interaction, multiple processes occur including class-switch recombination (CSR) and somatic hypermutation (SHM), resulting in affinity maturation. T-dependent plasmablasts circulate for a short period of time before a small fraction of them home to the bone marrow where they differentiate into long-lived plasma cells. The initial decline in serum IgG4 levels following treatment is attributed to the loss of T-independent plasmablasts in the tissue and T-dependent plasmablasts in the blood. In the case of CD20-targeted therapy, the loss of plasmablasts occurs secondary to destroying their parent activated B cells. The slow and gradual decline in serum IgG4 levels is hypothetically due to a reduction in the bone marrow resident, long-lived plasma cells.
Key: TFH = follicular helper T cell; IgG1 = immunoglobulin G1, IgG4 = immunoglobulin G4, , CD = cluster of differentiation.
There are two major implications of these findings. First, plasmablasts are a useful biomarker of disease activity in IgG4-RD. Expanded plasmablast populations are evident even in patients with clinical and histopathological features consistent with IgG4-RD who have normal serum IgG4 concentrations.22 In an examination of 37 patients with IgG4-RD, circulating plasmablasts were elevated compared to both healthy and disease controls, yielding a sensitivity and specificity of 87% and 91%, respectively.22 Moreover, the extent of plasmablast expansion correlated with the number of organs involved.22 Blood plasmablast levels also decline following glucocorticoid treatment. However, elevated blood plasmablast levels are not specific for IgG4-RD. Infections, other primary B cell-mediated diseases, and infusion reactions – among other causes – can lead to plasmablast expansion. Thus, clinical correlation is required for proper interpretation. Moreover, reliable assays for plasmablasts are not yet widely available.
The second implication pertains to how plasmablasts may be involved in the disease mechanism of IgG4-RD. Following rituximab treatment, the rapid decline in circulating plasmablasts – cells that lack CD20 expression – is attributable both to their short lifespans and to elimination of their cellular precursors, namely activated naïve and memory B-cells. After the pool of their precursors has been eliminated by CD20-targeted therapy, short-lived plasmablasts cannot be repopulated at the conclusion of their natural lifespan.
A Brief Overview of the Cellular Components of the Humoral Immune Response
B cell differentiation into an antibody-secreting cell begins with activation of a naïve B cell followed by the germinal center reaction orchestrated by a follicular helper T (TFH) cell (Figure 3). This activation may occur in a lymph node or in a tertiary lymphoid organ within an affected tissue. The resulting antigen-specific B cell follows one of three fates. First, it can become a memory B cell, poised to respond to future cognate antigen exposure. Second, it can become a short-lived, extra-follicular plasmablast that arises in a manner independent of TFH cells and is found abundantly within affected tissues. Or third, it can become a T cell-dependent plasmablast, with epigenetic imprinting enabling it to persist for extended periods of time, homing to bone marrow and differentiating further into long-lived plasma cells. Memory B cells have the capability of repopulating both the short-lived extra-follicular plasmablasts and the T cell-dependent plasmablasts.
Circulating plasmablasts are comprised of both short-lived and T-dependent plasmablasts, but these types of cells are difficult to distinguish from each other with traditional markers.23 Of note, both types of plasmablasts are CD20-negative. Either can differentiate into plasma cells but only T-cell-dependent plasmablasts have the ability to undergo terminal differentiation into long-lived plasma cells that reside in the bone marrow.24 We generally refer to cells that express at least some CD19 and some surface immunoglobulin as plasmablasts, reserving the term “plasma cells” for those that are CD19-negative but express CD138 on their surface. Circulating plasmablasts express high levels of HLA-DR21, highlighting their potential role in antigen presentation.
Following infectious antigen exposures, plasmablasts circulate for less than 2 weeks.25 In immune-mediated conditions such as systemic lupus erythematosus, however, persistence of circulating plasmablasts is documented long afterwards, suggesting continuous generation as a result of chronic antigen exposure.19 A small fraction of circulating plasmablasts expresses certain chemokine receptors that facilitate their homing back to the bone marrow. There, a minority of these cells manage to secure appropriate bone marrow niches necessary for development and long-term maintenance. Such cells differentiate into long-lived plasma cells and help maintain durable humoral immunity.
The persistence of significant serum IgG4 elevations during clinical remission suggests that the IgG4 molecule itself may not participate directly in pathogenesis. The beneficial effect of B cell depletion in IgG4-RD (and possibly in other conditions) may stem not from reduction in immunoglobulin levels, therefore, but rather from the removal of activated B cells from the reservoir of antigen-presenting cells at disease sites. The efficacy of B cell-depletion therapies therefore likely stems from mechanisms other than simply reducing serum IgG4 concentrations. Specifically, reductions in the secretion of cytokines from activated B cells and interruption of the presentation of antigens contribute further to the impact of B cell depletion (Figure 4).
Figure 4.
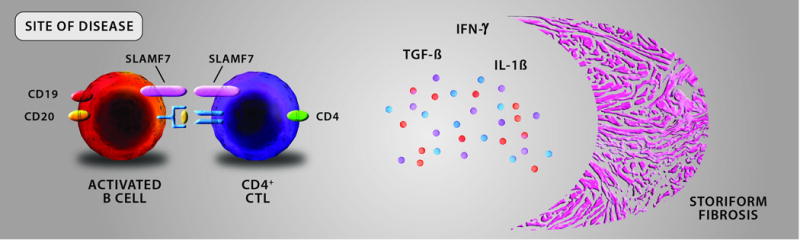
At the site of disease, activated B cells are thought to possess a potent antigen-presenting capacity driving the expansion of effector T cells. The T cells implicated in the disease pathogenesis are cytotoxic CD4+ T cells (CD4+ CTL) rather than any traditional helper T cell subset seen in other rheumatologic diseases (i.e. Th1, Th17). CD4+ CTLs carry both cytotoxic and pro-fibrotic functions, the latter thought to occur by the secretion of transforming growth factor-beta (TGF-β), interferon-gamma (IFN-γ) and interleukin-1 beta (IL-1β). The resultant fibrosis takes a “storiform” pattern for unclear reasons and this conglomeration of activated B cells, CD4+ CTLs, and fibrosis forms the fibro-inflammatory lesions observed microscopically and tumefactive lesions observed clinically.
Key: CD = cluster of differentiation; CD4+ CTL = CD4+ cytotoxic T lymphocyte; SLAMF7 = signaling lymphocyte activating marker family 7; TGF-β = transforming growth factor-beta; IFN-γ = interferon-gamma, IL-1β = interleukin-1 beta.
What is the Role of IgG4 Itself?
The precise role of IgG4 in disease pathophysiology – if any – remains unclear. Although elevated serum IgG4 levels were the abnormality that originally called this disease to medical attention, in fact the serum level of IgG4 is elevated in only about two thirds of IgG4-RD patents.26 Moreover, serum IgG4 levels are often so high as to trigger concern for a plasma cell dyscrasia, but the performance of IgG4 as a serum biomarker of disease activity is imperfect.22
On the tissue side, IgG4 is the dominant immunoglobulin subclass expressed by plasma cells within affected disease sites13. However, the presence of large numbers of IgG4+ plasma cells at an involved site is not specific for the diagnosis of IgG4-RD. Significant IgG4+ plasma cell infiltrates have been observed in multiple inflammatory, infectious, and malignant conditions27.
Two earlier case series, supported by a prospective, open-label rituximab trial, suggested that CD20-targeted B cell depletion is effective in IgG4-RD.17, 28, 29 Rituximab led to sustained clinical remission without concomitant glucocorticoid use at 12 months in nearly half of the subjects in the open-label trial. Despite dramatic clinical responses and steady declines in serum IgG4 levels, however, only 42% completely normalized their levels by 12 months.29 This observation underscores two additional points. First, IgG4 itself may not play a critical role in disease pathogenesis. Second, serum IgG4 elevations likely derive not only from short-lived plasmablasts but also from long-lived plasma cells.
IgG4 has traditionally been considered a non-inflammatory, perhaps anti-inflammatory, antibody, especially in conditions mediated by IgE.30 As an example, serum IgG4 concentrations are known to increase over the professional lives of beekeepers, presumably as a guard against anaphylaxis.31 The same observations have been made in patients treated with desensitizing immunotherapy, following which the induction of IgG4 responses corresponds to reduced allergic symptoms.32
Certain structural and functional attributes of the IgG4 molecule make it an unlikely driver of inflammation. Its Fc portion provides low binding affinity for activating Fc receptors but a relatively preserved affinity for binding to FcγRIIb, an inhibitory Fc receptor.33 In addition, the Fc portion binds poorly to C1q, the first component of the complement cascade. Finally, the hinge region of the IgG4 molecule has unique amino acid moieties that facilitate dissociation of its originally-paired heavy chains, permitting re-association with similarly split IgG4 molecules of different antigen specificity.34 This process, known as “Fab-arm exchange,” renders newly-formed IgG4 molecules functionally bispecific and incapable of cross-linking antigen or forming immune complexes. Such properties allow the IgG4 molecule to serve as an “antigen sink,” competing with other IgG subclasses to bind antigen at sites of inflammation without inciting further inflammation, thereby blunting the inflammatory response of chronic antigenic exposure.
It remains possible that a “disease-causing” IgG fraction produced by a subset of antibody-secreting cells contributes to tissue injury, perhaps through immune complexes or Fc-receptor engagement. Many IgG4-RD patients studied have concurrent elevations in other IgG subclasses (e.g., IgG1) that are, unlike IgG4, more capable of these effector functions.
Clues from an Animal Model
An animal model has shed light on the potential interactions of IgG subclasses in contributing to IgG4-RD and offered a potential mechanism whereby IgG4 might contribute to disease.35 Purified IgG subclass molecules from the serum of patients with IgG4-RD and controls were transferred to neonatal mice.35 This transfer induced pathologic changes in both the pancreas and submandibular glands, two organs commonly affected by IgG4-RD in humans. Although transfer of either purified IgG1 or IgG4 alone produced the disease phenotype, the transfer of both molecules together led to reduced IgG1 deposition and a decrease in the histopathologic severity of disease within affected organs.35
The induction of disease by IgG4 transfer in this model appears contrary to the concept of IgG4 as an anti-inflammatory molecule, yet a subset of IgG4 antibodies not subject to Fab arm exchange may have been responsible for this observation. Indeed, the extent to which Fab-arm exchange occurs in vivo is unknown. It is also unclear how human IgG4 interacts with mouse Fc receptors. Further, it remains possible that under some conditions, IgG4 participates in the formation of immune complexes, which contribute to tissue injury. For example, there is clear evidence of IgG4 immune complex deposition in IgG4-related tubulointerstitial nephritis and some suggestion that a similar process contributes to IgG4-related autoimmune pancreatitis.36, 37 Some experimental data support the concept of differing capacity to bind complement between the IgG4 molecules in IgG4-RD compared to those from healthy donors.38 The relationship of this finding to post-translational modifications of the Fc portion of the IgG4 molecule remains to be elucidated.
Th2 Cells, Atopy and IgG4-RD
The dominant cell type at sites of IgG4-RD is the T-cell, but neither the precise phenotype of these T-cells nor their potential role in IgG4-RD were known for some years. 39 Early studies suggested – based on characteristic cytokine gene signatures (i.e., IL-4, IL-5, IL-13) – that type 2 helper T (Th2) cells were the predominant T-cell subset both in the circulation and at disease sites.40, 41 The frequent presence of atopic disease (i.e., eczema, allergic rhinitis, or asthma), peripheral blood eosinophilia, and elevated serum IgE levels lent further credence to the Th2 hypothesis.
A crucial limitation in those early studies, however, was the failure to define the cellular source of the cytokine gene signature. No study had definitively identified Th2 cells in disease tissue, primarily because no study had stained for both cell-surface CD4 and intracellular GATA-3, the lineage-defining transcription factor for this subset of T cells.
Recent examination of Th2 cells in IgG4-RD demonstrated that their expansion in the circulation occurred only in the portion of patients with concurrent atopic disease, suggesting atopy as a confounding factor in previous investigations.42 Subsequent studies analyzing the T cell repertoire from subjects with active IgG4-RD who also had a history of atopy documented no clonal expansions of Th2 cells but rather indicated diverse polyclonality, consistent with a lifetime of exposure to environmental allergens.43 In contrast, the same study detected a highly-restricted repertoire in a separate T-cell population: cytotoxic CD4+ T-cells (CD4+ CTLs).43
Looking Beyond Th2 Cells: CD4+ CTLs
Other lines of evidence converged on a type of T cell other than Th2 and TFH cells. Careful analysis of skin biopsies shown in Figure 2 yielded other key information.18 Not only were CD20+ B cells depleted from the tissue five weeks after rituximab treatment, but CD3+ cells were also dramatically reduced following anti-CD20 therapy [Figure 2C, D]. We therefore focused further on the pathogenic role of the T cell compartment.
Circulating effector T cells (CD4+CD27loCD62Llo) were sorted and underwent gene expression analysis.43 The effector gene signature consisted of both cytolytic (e.g., granzyme, granulysin, and perforin) and myeloid (e.g., interleukin-1β) features.43 In addition, these cells were found to secrete two other pro-fibrotic cytokines, transforming growth factor-beta (TGF-β) and interferon-gamma (IFN-γ). Such cells, termed CD4+ cytotoxic T lymphocytes (CTLs), have been described in the setting of chronic viral infections such as in HIV.44 The gene expression analysis additionally revealed marked upregulation of the signaling lymphocytic activation marker member 7 (SLAMF7), a surface protein of the SLAM/CD2 family. SLAM receptors, broadly expressed on hematopoietic cells and possessing the capability of acting as “self-ligands,” (i.e., the SLAM protein on one cell binds to the SLAM protein on the other), thereby permitting cell-cell interaction.45 Whether these receptors stimulate or inhibit the immune cell function depends on the presence or absence of an intra-cellular adaptor of the SLAM-associated protein (SAP) family.45 The specific function of SLAMF7 in the setting of IgG4-RD may pertain to CD4+ CTL-B cell interactions, because both the CD4+CTL and cells of the B-lymphocyte lineage express this molecule.21, 43
Four strong pieces of evidence support CD4+ CTLs as principal drivers of IgG4-RD. These cells: 1) undergo large clonal expansions; 2) infiltrate tissues affected by the disease in large numbers; 3) actively secrete cytokines in these tissues, indicating recent antigen activation; and, 4) decline with rituximab-induced disease remission.43, 46 A current pathophysiological model of IgG4-RD is shown in Figures 3 and 4.
T Follicular Helper Cells
The germinal center reaction, with T-B collaboration at its core, results in B cell proliferation, differentiation, somatic hypermutation, and class-switch recombination (Figure 3).47 TFH cells secreting interleukin 21 (IL-21) are found within IgG4-RD-affected tissues.48 The observation of clonally-expanded, class-switched plasmablasts in IgG4-RD strongly suggests that TFH cells contribute to this disease in some manner, implicitly assuming a role for class-switched antibodies. TFH cells were first examined in IgG4-RD in the blood.49
Circulating CD4+ TFH cells have been classified into types 1, 2 and 17, corresponding to the nomenclature adopted for helper T cells. This classification, based upon differential expression of surface chemokine receptors, has not been confirmed in TFH cells residing in human lymphoid organs.50 Circulating TFH2 cells are expanded in the circulation of patients with IgG4-RD when compared to controls. Their levels also correlate well with circulating plasmablasts, serum IgG4, and interleukin-4 (IL-4) levels.49
The functional significance of TFH2 cells in IgG4-RD was examined through co-culture experiments with sorted TFH subsets and naïve B cells.51 TFH2 cells from IgG4-RD patients induced naïve B cells to differentiate into IgG4-producing plasmablasts more efficiently in vitro than did the same cells from healthy donor subjects.51 In addition, expansions of both circulating TFH1 and TFH2 populations were observed in IgG4-RD subjects. This degree of expansion correlated with disease activity, and TFH2 cell expansions declined after glucocorticoid treatment.51 These two studies support the role of TFH2 cells in orchestrating the B cell immune response in IgG4-RD. However, given the challenge of interpreting TFH data from the blood, where the TFH cells represent memory cells rather than effector cell populations, these findings must be validated at the tissue level.
From Bench to Bedside
Next generation sequencing studies have implicated both plasmablasts and CD4+ CTLs in the disease pathogenesis by the identification of restricted repertoires within both cell populations. Identification of clonal restriction is a major step beyond the associations of cell populations with disease states permitted by flow cytometry. Combining data from next generation sequencing, single cell RNA sequencing, and protein expression has illuminated a variety of potential targeted therapeutic strategies.
B cell lineage targeted treatments
B cell depletion
Substantial experience indicates that B-cell depletion strategies, particularly with CD20-targeted therapy, are effective treatments in IgG4-RD.17, 18, 28, 29 Rituximab is an appealing alternative to glucocorticoids both for the induction and maintenance of remission in IgG4-RD52. In addition to the prospective, open-label study29, rituximab has been analyzed retrospectively in case series of patients with IgG4-related autoimmune pancreatitis, cholangitis, orbital pseudotumor, and large vessel vasculitis.53, 54, 55, 14 Despite the evidence to date for excellent clinical efficacy, obtaining this medication is often a logistical challenge in the absence of regulatory approval.
B cell inhibition
XmAb5871 is a novel molecule with an antigen-binding site specific for CD19, thereby targeting cells of the B-cell lineage. However, it differs from the antibody-dependent cellular cytotoxicity (ADCC) mechanism of anti-CD20-directed therapy.56 XmAb5871 capitalizes upon the natural inhibitory mechanism of FcγRIIb, the only Fc receptor expressed by B cells, which acts as a negative regulator in conditions of antigen excess and immune complex formation.57 When FcγRIIb is co-ligated with CD19, B cell function is inhibited and plasmablasts are rendered dormant.
The Fc portion of XmAb5871 was engineered to have 400-fold increased affinity for FcγRIIb, permitting it to compete effectively for this binding site.58 This molecule has demonstrated efficacy in suppressing B cell activation and proliferation as well as disease activity in rheumatoid arthritis.59 We recently completed enrollment on an open-label, single-arm study on its efficacy in IgG4-RD.60 Preliminary analyses of this approach are promising and have been reported.61
Targeting T cells
CD4+ T cells offer a variety of potential therapeutic targets.
Co-Stimulatory Blockade
Abatacept, a fusion-protein composed of the extra-cellular domain of cytotoxic T lymphocyte associated protein 4 (CTLA-4) and the IgG1 constant domain, ameliorates autoimmune disease by binding to CD80/86 on antigen-presenting cells, thereby interfering with ligation to the co-stimulatory CD28 molecule on T-cells.62 A case report described an IgG4-RD patient with dacryoadenitis, sialoadenitis and autoimmune pancreatitis treated successfully with abatacept.63 Whether this effect was mediated by interfering with CD28 co-stimulation of CD4+ CTLs or from a more direct effect on activated B cells, macrophages or TFH cells, which also express CD80/86, is not clear.
CD4+ Cytotoxic T Lymphocytes
CD4+ CTLs have strong appeal as a potential therapeutic target. Elotuzumab, an immuno-stimulatory mAb directed against SLAMF7, is approved for the treatment of refractory multiple myeloma64. The proposed mode of action of elotuzumab is via the activation of NK cells and resultant ADCC of the SLAMF7-expressing cellular target.65 The SLAMF7 receptor relies on an intra-cellular SAP adaptor known as EAT-2 to exert its stimulatory effect. The lack of EAT-2 expression in myeloma cells suggests that the binding of SLAMF7 is not activating in these cells, permitting NK-mediated cytotoxicity without direct activation of the targeted myeloma cells.62 The potential of anti-SLAMF7 antibodies to target CD4+ CTLs in IgG4-RD remains untested and the expression of SAP members (including EAT-2) in CD4+ CTLs requires further study.
T Follicular Helper Cells
Therapies targeting TFH cells have been contemplated and pursued in a variety of immune-mediated conditions. Targeting certain surface markers that are specific for TFH cells and only expressed after antigen activation are currently being explored in SLE and Sjögren’s syndrome with AMG-557, a neutralizing mAb directed against the inducible T cell co-stimulatory ligand (ICOS-L).66, 67 Further studies on TFH cells in IgG4-RD are needed before the pursuit of such therapeutic strategies could be justified.
Disruption of Antigen Presentation
Another potential mean of intervening on the disease process may include the direct disruption of antigen presentation. With the suspected role of activated B cells as antigen presenters in IgG4-RD (Figure 4), a molecule to extinguish this process specifically within B cells could stultify that mechanism of disease without fully disabling the immune system’s ability to process and present infectious antigens by dendritic cells and macrophages.
A concise summary of the potential therapeutic agents mentioned here is displayed in Table 1.
Table 1.
| Agent | Mechanism of Action | Type of Clinical Trial (number of subjects) |
Trial Status |
|---|---|---|---|
| Rituximab | CD20-directed ADCC | Open-label, prospective (n = 30) | Completed |
| XmAb5871 | CD19-directed B cell inhibition | Open-label, prospective (n = 21) | Enrolling |
| Abatacept | Co-stimulation blockade | Case report (n = 1) | - |
| Elotuzumab | SLAMF7-directed ADCC | Unstudied | - |
| AMG-557 | ICOS-directed Tfh inhibition | Unstudied | - |
Persistent Knowledge Gaps
Antigens driving IgG4-RD
Observations of restricted repertoires of both plasmablasts and CD4+ CTLs indicate that IgG4-RD is an antigen-driven disease. The numbers of antigens that can trigger this disease, their precise identities, and whether they are self or foreign (e.g., viral) all remain unknown. Plasmablast studies have yielded interesting insights.21 Single-cell sorted plasmablasts were used to clone and express monoclonal antibodies from dominantly-expanded plasmablasts and found by immunofluorescence to be self-reactive to eukaryotic cells.21 The passive transfer model of purified human IgG to neonatal mice also suggests autoreactivity.35 These observations remain the most compelling evidence that IgG4-RD might be driven by self-antigen. However, given the established association of CD4+ CTLs with chronic viral infections and the highly-restricted repertoire of these cells, the possibility that the disease arises from chronic viral antigen stimulation has not been excluded. The identification of causative antigens will help clarify the role of T-B collaboration in IgG4-RD and whether the observed clonal expansions of B and T cells are related by a common antigen.
Conclusions
Through clinical observation and cutting-edge scientific techniques, considerable strides have been made towards addressing fundamental questions about IgG4-RD. In a short period of time, major roles in disease pathogenesis have been identified for activated B cells, plasmablasts, CD4+ CTLs, and TFH cells. We now understand, at least in part, the potential mechanisms underlying the observed efficacy of anti-CD20 therapy and how to explain changes in serum IgG4 levels following treatment. Building upon our current mechanistic understanding is likely to identify new potential targets for treatment, yielding in turn opportunities for additional mechanistic insights.
Acknowledgments
Grant Support: Drs. Perugino and Wallace is funded under NIH T32AR007258. Dr. Wallace is supported by a Scientist Development Award by the Rheumatology Research Foundation and an NIH Loan Repayment Award. The work was also supported by an Autoimmunity Center of Excellence (ACE) Award for IgG4-Related Disease (Dr. Pillai, NIH-NIAID 5U19AI110495) and by the philanthropic support of Drs. Shankaran Nair and Edward A. Fox.
Footnotes
The authors report no potential conflicts of interest.
References
- 1.Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366:539–51. doi: 10.1056/NEJMra1104650. [DOI] [PubMed] [Google Scholar]
- 2.Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet. 2015;385:1460–71. doi: 10.1016/S0140-6736(14)60720-0. [DOI] [PubMed] [Google Scholar]
- 3.Mikulicz J. Über eine eigenartige symmetrische erkrankung der tränen und mundspeicheldrüsen. Stuttgart: Beitr.z.Chir.Fesrschr.f. Theodor Billroth. 1892:610–30. [Google Scholar]
- 4.Riedel B. Die chronische, zur Bildung eisenharter Tumoren führende Entzündung der Schilddrüse. Verhandlungen der deutschen Gesellschaft für Chirurgie. 1896;25:101–105. [Google Scholar]
- 5.Kuttner H. Uberentzundiche Tumoren der submaaxilspeichdeldruse. Bruns Beitr Chir. 1896;15:815–834. [Google Scholar]
- 6.Ormond JK. Idiopathic retroperitoneal fibrosis: an established clinical entity. JAMA. 1960;174:1561–1568. doi: 10.1001/jama.1960.03030120001001. [DOI] [PubMed] [Google Scholar]
- 7.Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344(10):732–738. doi: 10.1056/NEJM200103083441005. [DOI] [PubMed] [Google Scholar]
- 8.Kamisawa T, Funata N, Hayashi Y, Eishi Y, Koike M, Tsuruta K, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol. 2003;38(10):982–984. doi: 10.1007/s00535-003-1175-y. [DOI] [PubMed] [Google Scholar]
- 9.Perugino CA, Wallace ZS, Meyersohn N, Oliveira G, Stone JR, Stone JH. Large vessel involvement by IgG4-related disease. Medicine (Baltimore) 2016;95(28):e3344. doi: 10.1097/MD.0000000000003344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wallace ZS, Carruthers MN, Khosroshahi A, Carruthers R, Shinagore S, Stemmer-Rachamimov A, et al. IgG4-related disease and hypertrophic pachymeningitis. Medicine (Baltimore) 2013;92(4):206–216. doi: 10.1097/MD.0b013e31829cce35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zen Y, Kawakami H, Kim JH. IgG4-related sclerosing cholangitis: all we need to know. J Gastroenterol. 2016;51(4):295–312. doi: 10.1007/s00535-016-1163-7. [DOI] [PubMed] [Google Scholar]
- 12.Stone JH, Khosroshahi A, Deshpande V, Chan JK, Heathcote JG, Aalberse R, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum. 2012;64:3061–3067. doi: 10.1002/art.34593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Stone JH, et al. Consensus statement on the pathology of IgG4-related disease. Modern Pathology. 2012;25:1181–1192. doi: 10.1038/modpathol.2012.72. [DOI] [PubMed] [Google Scholar]
- 14.Khosroshahi A, Wallace ZA, Crow JL, Akamizu T, Azumi A, Carruthers MN, et al. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheum. 2015;67:1688–99. doi: 10.1002/art.39132. [DOI] [PubMed] [Google Scholar]
- 15.Carruthers MN, Stone JH, Deshpande V, Khosroshahi A. Development of an IgG4-RD responder index. Int J Rheumatol. 2012;2012:259408. doi: 10.1155/2012/259408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wallace ZS, Khosroshahi Z, Carruthers M, Campochiaro C, Choi HK, Culver E, et al. An international, multi-specialty validation study of the IgG4-related disease responder index. (under review) doi: 10.1002/acr.23543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khosroshahi A, Bloch D, Deshpande V, Stone JH. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG-related systemic disease. Arthritis Rheum. 2010;62:1755–1762. doi: 10.1002/art.27435. [DOI] [PubMed] [Google Scholar]
- 18.Della-Torre E, Feeney E, Deshpande V, Mattoo H, Mahajan V, Kulikova M, et al. B-cell depletion attenuates serological biomarkers of fibrosis and myofibroblast activation in IgG4-related disease. Ann Rheum Dis. 2015;74:2236–2243. doi: 10.1136/annrheumdis-2014-205799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tripton CM, Fucile CF, Darce J, Chida A, Ichikawa T, Gregoretti I, et al. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat Immunol. 2015;16:755–765. doi: 10.1038/ni.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kerkman PF, Rombouts Y, van der Voort EL, Trouw LA, Huizinga TW, Toes RE, et al. Circulating plasmablasts/plasma cells as a source of anti-citrullinated protein antibodies in patients with rheumatoid arthritis. Ann Rheum Dis. 2013;72:1259–1263. doi: 10.1136/annrheumdis-2012-202893. [DOI] [PubMed] [Google Scholar]
- 21.Mattoo H, Mahajan V, Della-Torre E, Sekigami Y, Carruthers M, Wallace ZS, et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol. 2014;134:679–687. doi: 10.1016/j.jaci.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wallace ZS, Mattoo H, Carruthers M, Mahajan VS, Della Torre E, Lee H, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis. 2015;74:190–195. doi: 10.1136/annrheumdis-2014-205233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amanna IJ, Slifka MK. Mechanisms that determine plasma cell lifespan and the duration of humoral immunity. Immunol Rev. 2010;236:125–138. doi: 10.1111/j.1600-065X.2010.00912.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taillardet M, Haffar G, Mondière P, Asensio MJ, Gheit H, Burdin N, et al. The thymus-independent immunity conferred by a pneumococcal polysaccharide is mediated by long-lived plasma cells. Blood. 2009;114(20):4432–4440. doi: 10.1182/blood-2009-01-200014. [DOI] [PubMed] [Google Scholar]
- 25.Odendahl M, Mei H, Hoyer BF, Jacobi AM, Hansen A, Muehlinghaus G, et al. Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood. 2005;105:1614–1621. doi: 10.1182/blood-2004-07-2507. [DOI] [PubMed] [Google Scholar]
- 26.Wallace ZS, Deshpande V, Mattoo H, Mahajan V, Kulikova M, Pillai S, et al. IgG4-related disease clinical and laboratory features in one hundred twenty-five patients. Arthritis Rheum. 2015;67:2466–2475. doi: 10.1002/art.39205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strehl JD, Hartmann A, Agaimy A. Numerous IgG4-positive plasma cells are ubiquitous in diverse localised non-specific chronic inflammatory conditions and need to be distinguished from IgG4-related systemic disorders. J Clin Pathol. 2011;64:237–243. doi: 10.1136/jcp.2010.085613. [DOI] [PubMed] [Google Scholar]
- 28.Khosroshahi A, Bloch D, Deshpande V, Stone JH. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG-related systemic disease. Arthritis Rheum. 2010;62:1755–1762. doi: 10.1002/art.27435. [DOI] [PubMed] [Google Scholar]
- 29.Carruthers MN, Topazian MD, Khosroshahi A, Witzig TE, Wallace ZS, Hart PH, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis. 2015;74:1171–1177. doi: 10.1136/annrheumdis-2014-206605. [DOI] [PubMed] [Google Scholar]
- 30.Aalberse RC, Stapel SO, Schuurman J, Rispens T. Immunoglobulin G4: an odd antibody. Clin Exp Allergy. 2009;39:469–477. doi: 10.1111/j.1365-2222.2009.03207.x. [DOI] [PubMed] [Google Scholar]
- 31.Aalberse RC, Van der Gaag R, Van Leeuwen J. Serologic aspects of IgG4 antibodies I Prolonged immunization results in an IgG4-restricted response. J Immunol. 1983;130:722–726. [PubMed] [Google Scholar]
- 32.Nouri-Aria KT, Wachholz PA, Francis JN, Jacobson MR, Walker SM, Wilcock LK, et al. Grass pollen immunotherapy induces mucosal and peripheral IL-10 responses and blocking IgG activity. J Immunol. 2004;172:3252–3259. doi: 10.4049/jimmunol.172.5.3252. [DOI] [PubMed] [Google Scholar]
- 33.Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113:3716–3725. doi: 10.1182/blood-2008-09-179754. [DOI] [PubMed] [Google Scholar]
- 34.Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:520. doi: 10.3389/fimmu.2014.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shiokawa M, Kodama Y, Kuriyama K, Yoshimura K, Tomono T, Morita T, et al. Pathogenicity of IgG in patients with IgG4-related disease. Gut. 2016 Aug;65:1322–1332. doi: 10.1136/gutjnl-2015-310336. [DOI] [PubMed] [Google Scholar]
- 36.Cornell LD, Chicano SL, Deshpande V, et al. Pseudotumors due to IgG4 immune-complex tubulointerstitial nephritis associated with autoimmune pancreatocentric disease. Am J Surg Pathol. 2007;31:1586–1597. doi: 10.1097/PAS.0b013e318059b87c. [DOI] [PubMed] [Google Scholar]
- 37.Detlefsen S, Brasen JH, Zamboni G, Capelli P, Kloppel G. Deposition of complement C3c, immunoglobulin (Ig)G4 and IgG at the basement membrane of pancreatic ducts and acini in autoimmune pancreatitis. Histopathology. 2010;57:825–835. doi: 10.1111/j.1365-2559.2010.03717.x. [DOI] [PubMed] [Google Scholar]
- 38.Sugimoto M, Watanabe H, Asano T, Sato S, Takagi T, Kobayashi H, et al. Possible participation of IgG4 in the activation of complement in IgG4-related disease with hypocomplementemia. Mod Rheumatol. 2016;26:251–258. doi: 10.3109/14397595.2015.1076924. [DOI] [PubMed] [Google Scholar]
- 39.Mahajan VS, Mattoo H, Deshpande V, Pillai SS, Stone JH. IgG4-related disease. Annu Rev Pathol. 2014;9:315–47. doi: 10.1146/annurev-pathol-012513-104708. [DOI] [PubMed] [Google Scholar]
- 40.Saito Y, Kagami S, Kawashima S, Takahashi K, Ikeda K, Hirose K, et al. Roles of CRTH2+CD4+ T-cells in immunoglobulin G4-related lacrimal gland enlargement. International Archives of Allergy and Immunology. 2012;158(Suppl):42–46. doi: 10.1159/000337761. [DOI] [PubMed] [Google Scholar]
- 41.Zen Y, Fujii T, Harada K, Kawano M, Yamada K, Takahira M, et al. Th2 and regulatory immune reactions are increased in immunoglobulin G4-related sclerosing pancreatitis and cholangitis. Hepatology. 2007;45:1538–1546. doi: 10.1002/hep.21697. [DOI] [PubMed] [Google Scholar]
- 42.Mattoo H, Della-Torre E, Mahajan V, Stone J, Pillai S. Circulating Th2 memory cells in IgG4 Related Disease are restricted to a defined subset of subjects with atopy. Allergy. 2014;69:399–402. doi: 10.1111/all.12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mattoo H, Mahajan VS, Maehara T, Deshpande V, Della-Torre E, Wallace ZS, et al. Clonal expansion of CD4+ cytotoxic T lymphocytes in patients with IgG4-related disease. J Allergy Clin Immunol. 2016;138:825–838. doi: 10.1016/j.jaci.2015.12.1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson S, Eller M, Teigler JE, Maloveste SM, Schultz BT, Soghoian DZ, et al. Cooperativity of HIV-specific cytolytic CD4 T-cells and CD8 T-cells in control of HIV viremia. J Virol. 2015;89:7494–505. doi: 10.1128/JVI.00438-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu N, Veillette A. SLAM family receptors in normal immunity and immune pathologies. Curr Opin Immunol. 2016;38:45–51. doi: 10.1016/j.coi.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 46.Maehara T, Mattoo H, Ohta M, Mahajan V, Moriyama M, Yamauchi M, et al. Lesional CD4+ IFN-γ+ cytotoxic T lymphocytes in IgG4-related dacryoadenitis and sialoadenitis. Ann Rheum Dis. 2017;76:377–385. doi: 10.1136/annrheumdis-2016-209139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crotty S. A brief history of T-cell help to B-cells. Nat Rev Immunol. 2015 Mar;15:185–189. doi: 10.1038/nri3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maehara T, Moriyama M, Nakashima H, Miyake K, Hayashida JN, Tanaka A, et al. Interleukin-21 contributes to germinal center formation and immunoglobulin G4 production in IgG4-related dacryoadenitis and sialoadenitis, so-called Mikulicz’s disease. Ann Rheum Dis. 2012;71:2011–2019. doi: 10.1136/annrheumdis-2012-201477. [DOI] [PubMed] [Google Scholar]
- 49.Akiyama M, Suzuki K, Yamaoka K, Yasuoka H, Takeshita M, Kaneko Y, et al. Number of circulating follicular helper 2 T-cells correlates with IgG4 and interleukin-4 and plasmablast numbers in IgG4-related disease. Arthritis Rheum. 2015;67:2476–2481. doi: 10.1002/art.39209. [DOI] [PubMed] [Google Scholar]
- 50.Morita R, Schmitt N, Bentebibel SE, Ranganathan R, Bourdery L, Zurawski G, et al. Human blood CXCR5+ CD4+ T-cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. 2011;34:108–121. doi: 10.1016/j.immuni.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Akiyama M, Yasuoka H, Yamaoka K, Suzuki K, Kaneko Y, Kondo H, et al. Enhanced IgG4 production by follicular helper 2 T-cells and the involvement of follicular helper 1 T-cells in the pathogenesis of IgG4-related disease. Arthritis Res Ther. 2016;18:167. doi: 10.1186/s13075-016-1064-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perugino CA, Stone JH. Treatment of IgG4-related disease: current and future approaches. Z Rheumatol. 2016;75:681–686. doi: 10.1007/s00393-016-0142-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hart PA, Topazian MD, Witzig TE, Clain JE, Gleeson FC, Klebig RR, et al. Treatment of relapsing autoimmune pancreatitis with immunomodulators and rituximab: the Mayo Clinic experience. Gut. 2013;62:1607–1615. doi: 10.1136/gutjnl-2012-302886. [DOI] [PubMed] [Google Scholar]
- 54.Topazian M, Witzig TE, Smyrk TC, Pulido JS, Levy MJ, Kamath PS, et al. Rituximab therapy for refractory biliary strictures in immunoglobulin G4-associated cholangitis. Clin Gastroenterology Hepatol. 2008;6:364–366. doi: 10.1016/j.cgh.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 55.Witzig TE, Inwards DJ, Habermann TM, Dogan A, Kurtin PJ, Gross JB, et al. Treatment of benign orbital pseudolymphomas with the monoclonal anti-CD20 antibody rituximab. Mayo Clin Proc. 2007;82:692–699. [PubMed] [Google Scholar]
- 56.Glennie MJ, French RR, Cragg MS, Taylor RP. Mechanisms of killing by anti-CD20 monoclonal antibodies. Mol Immunol. 2007;44:3823–3837. doi: 10.1016/j.molimm.2007.06.151. [DOI] [PubMed] [Google Scholar]
- 57.Espeli M, Smith KG, Clatworthy MR. FcγRIIb and autoimmunity. Immunol Rev. 2016;269:194–211. doi: 10.1111/imr.12368. [DOI] [PubMed] [Google Scholar]
- 58.Horton HM, Chu SY, Ortiz EC, Pong E, Cemerski S, Leung IW, et al. Antibody-mediated coengagement of FcγRIIb and B-cell receptor complex suppresses humoral immunity in systemic lupus erythematosus. J Immunol. 2011;186:4223–4233. doi: 10.4049/jimmunol.1003412. [DOI] [PubMed] [Google Scholar]
- 59.Chu SY, Yeter K, Kotha R, Pong E, Miranda Y, Phung s, et al. Suppression of rheumatoid arthritis B-cells by XmAb5871, an anti-CD19 antibody that coengages B-cell antigen receptor complex and Fcγ receptor IIb inhibitory receptor. Arthritis Rheumatol. 2014;66:1153–1164. doi: 10.1002/art.38334. [DOI] [PubMed] [Google Scholar]
- 60.https://clinicaltrials.gov/ct2/show/NCT02725476
- 61.Stone JH, Wallace ZS, Perugino CA, Fernandes AD, Foster PA, Zack DJ. A trial of XmAb5871, a reversible inhibitor of CD19+ cells, in IgG4-related disease. Oral presentation at the meeting of the American College of Rheumatology; Washington, DC: Nov, 2016. [Google Scholar]
- 62.Cutolo M, Sulli A, Paolino S, Pizzorni C. CTLA-4 blockade in the treatment of rheumatoid arthritis: an update. Expert Rev Clin Immunol. 2016;12:417–425. doi: 10.1586/1744666X.2016.1133295. [DOI] [PubMed] [Google Scholar]
- 63.Yamamoto M, Takahashi H, Takano K, Shimizu Y, Sakurai N, Suzuki C, et al. Efficacy of abatacept for IgG4-related disease over 8 months. Ann Rheum Dis. 2016;75:1576–1578. doi: 10.1136/annrheumdis-2016-209368. [DOI] [PubMed] [Google Scholar]
- 64.Lonial S, Dimopoulos M, Palumbo A, White D, Grosicki S, Spicka I, et al. ELOQUENT-2 investigators Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373:621–631. doi: 10.1056/NEJMoa1505654. [DOI] [PubMed] [Google Scholar]
- 65.Veillette A, Guo H. CS1, a SLAM family receptor involved in immune regulation, is a therapeutic target in multiple myeloma. Crit Rev Oncol Hematol. 2013;88(1):168–177. doi: 10.1016/j.critrevonc.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 66.Sullivan BA, Tsuji W, Kivitz A, Peng J, Arnold GE, Boedigheimer MJ, et al. Inducible T-cell co-stimulator ligand (ICOSL) blockade leads to selective inhibition of anti-KLH IgG responses in subjects with systemic lupus erythematosus. Lupus Sci Med. 2016;3:e000146. doi: 10.1136/lupus-2016-000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.https://www.clinicaltrials.gov/ct2/show/NCT02334306