

Summary
Mycobacterium tuberculosis (Mtb) has a proteasome system that is essential for its ability to cause lethal infections in mice. A key component of the system is the proteasomal adenosine triphosphatase (ATPase) Mpa, which captures, unfolds, and translocates protein substrates into the Mtb proteasome core particle for degradation. Here, we report the crystal structures of near full-length hexameric Mtb Mpa in apo and ADP-bound forms. Surprisingly, the structures revealed a ubiquitin-like β-grasp domain that precedes the proteasome-activating carboxyl terminus. This domain, which was only found in bacterial proteasomal ATPases, buries the carboxyl terminus of each protomer in the central channel of the hexamer and hinders the interaction of Mpa with the proteasome core protease. Thus, our work reveals the structure of a bacterial proteasomal ATPase in the hexameric form, and the structure finally explains why Mpa is unable to stimulate robust protein degradation in vitro in the absence of other, yet-to-be-identified co-factors.
Graphical abstract
A portrait of the bacterial proteasomal ATPase hexamer Mpa. The N-terminal coiled coil domain is responsible for recognizing and recruiting the pupylated protein substrate for degradation by the 20S core protease.
Introduction
Proteasomes are regulated proteases that are ubiquitous in eukaryotes and archaea and are found in some bacterial genera (Hanna & Finley, 2007). In eukaryotes, a doomed protein substrate is usually ubiquitylated and recognized by a multi-subunit complex called the 19S regulatory particle. The regulatory particle includes a hexamer of six distinct ATPases (Rpt1-6) that unfolds and translocates proteins through its axial channel into a barrel-shaped protease called the 20S proteasome, or core particle (CP) (Finley et al., 2016, Groll et al., 1997, Lowe et al., 1995, Hu et al., 2006). While prokaryotes do not make ubiquitin, some archaea are proposed to use a ubiquitin-like protein conjugation system termed SAMPylation (Maupin-Furlow, 2013). Like ubiquitin, SAMPs (small archaeal modifier proteins) have a β-grasp fold, and their covalent linkage to proteins requires a carboxyl (C)-terminal glycine. The archaeal homohexameric proteasomal ATPase PAN (proteasome activating nucleotidase) may be responsible for unfolding a SAMPylated protein and delivering it into a proteasome (Fu et al., 2016).
Actinobacteria have evolved a proteasome system that is analogous to yet distinct from the archaeal and eukaryotic systems [reviewed in (Jastrab & Darwin, 2015)]. In mycobacteria and other related genera such as Streptomyces and Rhodococcus, a protein substrate is modified by the covalent attachment of a small protein called Pup (prokaryotic ubiquitin-like protein) (Pearce et al., 2008, Compton et al., 2015, Yun et al., 2012). Unlike ubiquitin or SAMP, which are highly structured, Pup is intrinsically disordered and uses a C-terminal glutamate instead of a glycine to form an isopeptide bond with a lysine of a substrate (Pearce et al., 2008, Burns et al., 2009, Sutter et al., 2010, Chen et al., 2009, Liao et al., 2009). A hexameric mycobacterial proteasome ATPase (Mpa) directly recognizes pupylated substrates and unfolds and translocates them into the mycobacterial 20S CP for degradation (Darwin et al., 2005, Burns et al., 2009, Sutter et al., 2010, Wang et al., 2009). Proteolysis by the Pup–proteasome system is essential for lethal Mtb infections in mice for numerous reasons (Samanovic & Darwin, 2016). Most importantly, mutations in the Pup-proteasome genes result in Mtb virulence attenuation by increasing bacterial susceptibility to nitric oxide, an antimicrobial free radical released by macrophages (Darwin et al., 2003). The Pup-proteasome system also directly or indirectly regulates copper resistance, which is required for the robust growth of Mtb in vivo (Festa et al., 2011, Wolschendorf et al., 2011, Ward et al., 2010). Thus, inhibiting the activity of this regulated degradation system could provide a new therapeutic target for tuberculosis, the leading cause of death worldwide by a single infectious agent (http://www.who.int/mediacentre/factsheets/fs104/en/).
An Mpa protomer consists of several distinct domains (Finley et al., 2016). First, each amino (N)-terminal α-helix of a protomer within a hexameric ring dimerizes with the helix of an adjacent protomer to form a coiled-coil structure. This pairing of adjacent helices results in the formation of three extended dimers, breaking the six-fold symmetry at the N-termini (Zhang et al., 2009, Djuranovic et al., 2009, Wang et al., 2010). The coiled-coil domain in Mpa acts as a specific receptor for Pup: upon binding to Mpa, Pup, which is mostly disordered in solution, becomes helical (Wang et al., 2010). Deletion of the coiled-coil region abolishes the ability of Mpa to unfold a model substrate (Striebel et al., 2010).
Following the coiled-coil domain are two tandem oligonucleotide/oligosaccharide-binding (OB) domains forming a double ring; this contrasts with the archaeal PAN and the eukaryotic Rpt1-6 proteins, each of which has a single OB domain (Zhang et al., 2009, Djuranovic et al., 2009, Wang et al., 2010). In the absence of the N-terminal coiled-coil, the OB domains alone are sufficient to maintain the hexameric state of Mpa (Wang et al., 2009).
The C-terminal half of all proteasomal ATPases is largely an ATPase associated with various cellular activities (AAA) domain, which hydrolyzes ATP to drive the unfolding and translocation of proteins into the 20S CP. Finally, at its C-terminus, Mpa has a glycine-glutamine-tyrosine-leucine (GQYL) motif that is essential for activating proteolysis by the mycobacterial proteasome (Pearce et al., 2006, Jastrab et al., 2015). Replacing G, Y or L, but not Q, in the GQYL motif with an alanine in Mpa or another bacterial proteasome activator (Bpa/PafE) abolishes the activator-assisted proteolysis by Mtb 20S CPs (Jastrab et al., 2015, Delley et al., 2014). This sequence is the functional equivalent of a motif found at the C-termini of PAN, Rpt2, Rpt3, and Rpt5, which have C-terminal hydrophobic-tyrosine-X (HbYX) motifs that are required for proteasomal activation (Smith et al., 2007, Unverdorben et al., 2014, Luan et al., 2016). While the eukaryotic Rpt1-6 hexamer co-purifies robustly with 20 SCPs, prokaryotic ATPases like Mpa and PAN bind weakly to their respective 20S CPs (Smith et al., 2011, Wang et al., 2009).
We noticed that Mpa is longer than the PAN and Rpt proteins, and the majority of the extra length is at the C-terminus. To determine the function of this uncharacterized region, we determined the atomic structure of the near-complete Mtb Mpa hexamer by X-ray crystallography. In addition to revealing a three-ring architecture composed of a double OB ring on top of an ATPase ring, we found a ubiquitin-like β-grasp domain preceding the C-terminus of Mpa that prevents the GQYL motif from docking with the 20S CP. Thus, our study revealed a crucial feature that distinguishes bacterial proteasome activators from those found in the other domains of life.
RESULTS
Overall features in the crystal structure of an engineered Mpa hexamer
Wild-type Mpa is composed of 609 amino acids. To facilitate crystallization, we removed the N-terminal 94 or 97 residues that contains the coiled-coil domain (Wang et al., 2010). The gel filtration profile indicated that the engineered Mpa formed hexamers in solution (Figure S1). We crystallized and determined Mpa structures in apo and ADP-bound forms at 3.49 Å and 2.9 Å resolution, respectively (Table 1). The apo Mpa crystal structure was determined in P212121 space group with two distinct hexamers in the asymmetric unit (Figure S2A). The ADP-bound structure was obtained by co-crystallizing Mpa in the presence of 5 mM ATPγS in the P321 space group. In the P321 crystal, the central chamber of the Mpa hexamer coincided with the crystallographic three-fold axis (Figure S2B). We found that ATPγS was hydrolyzed to ADP in the crystals such that each of the two protomers in the asymmetric unit was bound to one ADP (Figure S3). ATPγS is slowly hydrolysable in solution, so it was unsurprising that we and others observed ADP in the crystals (Satyshur et al., 2007, Zebisch & Strater, 2008, Watanabe et al., 2015). The apo Mpa structure was largely superimposable with the ADP-bound structure, with an RMSD value ranging from 0.75 to 1.95 Å (Figure S4).
Table 1.
Data collection and Refinement statistics
| Mpa-ADP | Mpa-apo | |
|---|---|---|
| Data collection | ||
| Wave length (Å) | 0.9791 | 1.100 |
| Space group | P321 | P212121 |
| Cell dimensions (Å) | a =b=111.93, c=196.07, α=β= 90°, γ=120° | a =115.27, b=202.59, c=303.01, α=β=γ= 90° |
| Resolution range (Å) | 43.45–2.9 (3.06–2.9)a | 70.2–3.49 (3.58–3.49) |
| Rmerge(%) a | 11.2 (70.3) | 6.9(72.1) |
| I/σI | 19.73 (1.93) | 9.9(1.7) |
| Completeness (%) | 100 (100) | 98.7 (99.3) |
| Total reflections | 698900(66742) | 277696(13888) |
| Multiplicity | 21.7(21.2) | 3.1(3.1) |
|
| ||
| Refinement | ||
| Resolution (Å) | 43.45–2.90 | 70.2–3.49 |
| Unique reflection | 32237(3155) | 89013(4499) |
| Rwork/Rfreeb | 0.230/0.266 | 0.237/0.282 |
| Non-H atoms | 7179 | 44299 |
| Protein | 7102 | 44299 |
| Ligand | 56 | 0 |
| Average B-factors (Å2) | 62.47 | 130.08 |
| Macromolecules | 62.73 | |
| Ligands | 38.46 | |
| Root-mean-square deviation | ||
| Bond length (Å) | 0.008 | 0.006 |
| Bond angle (º) | 1.31 | 1.134 |
| Ramachandran statistics (%) | ||
| Favored | 97 | 91.5 |
| Allowed | 2.9 | 7.8 |
| Outliers (%) | 0 | 0.7 |
| Molprobity score | 1.55 | 2.93 |
| Clashscore | 3.14 | 15.12 |
Numbers in parentheses are for the highest resolution shell.
5% of the reflections were randomly selected for Rfree and omitted from the refinement process.
We found that the engineered Mpa hexamer is 100 Å high and about 130 Å wide (Figure 1A). The hexamer can be divided into two main sections: the double OB ring section on the distal face and the ATPase ring section on the proximal face, with “distal” and “proximal” referring to the position relative to the 20S CP in an assembled proteasome. These two rings are co-axial, i.e., the central six-fold axis of the OB ring coincides with that of the ATPase ring. As we reported before (Wang et al., 2009), each of the two OB domains in an Mpa protomer is composed of a five-stranded β-barrel, and the OB double ring in a hexamer has an outer diameter of 70 Å and an axial channel of 25 Å at the distal face. The three glycines that replaced the 17-residue loop (Ala-194 to Arg-210) outside the second OB domain are disordered. Another 10-residue loop outside the large AAA subdomain (Glu-316 to Ala-325) is also disordered. The interface between the OB ring and the ATPase ring is formed by extensive electrostatic interactions (Figure 1B). A negatively charged patch (Glu-166, Asp-181, Glu-182, and Glu-183) on the proximal face of the OB2 ring (left image) interacts with a positively charged patch (Arg-347, Arg-350, Arg-355, and Arg-357) of the ATPase ring (right image).
Figure 1. Crystal structure of theengineered Mpa hexamer.

(A) The ADP-bound hexamericMpain side view (left panel) and top view (right panel). The hexamer can be divided into an OB ring section and an ATPase ring section. The six Mpaprotomers are colored differently and labeled A through F, with ADP molecules shown in stick form. “Distal” and “proximal” refer to relative orientation with respect to the 20S CP. The disordered loops are shown as dashed black curves. (B) Surface potential of the Mpa hexamer. The middle panel shows a side view of the hexamer; the dashed rectangle outlines the interface between the rings. Left, the proximal (bottom) OB ring surface;right, the distal (top) surface of ATPase ring. The negatively charged Asp-181 to Glu-183 residues on the bottom of the OB ring interact with the positively charged Arg-347 to Arg-355 residues on the top of the ATPase ring.
The AAA ATPase domain of Mpa is composed of two distinct subdomains: a large α/β region and a small helical region (Figure 2A). The α/β-domain has a five-stranded β-sheet core flanked by five α-helices on one side and three α-helices on the other. Two loops in the α/β-domain facing the hexamer channel, labeled as pore loops 1 and 2, are disordered. Pore loop 1 connects α-helix 5 and α-helix 6 and contains the conserved aromatic and hydrophobic (Ar-ϕ) motif. The small helical subdomain contains five α-helices. These two AAA subdomains closely resemble those of PAN and Rpt1 (Zhang et al., 2009, Unverdorben et al., 2014), with a RMSD of 1.77 Å for 153 Cα atoms and 1.60 Å for 152 Cα atoms, respectively (Figure 2B). Assembly of the hexamer requires extensive interactions between neighboring protomers, which buries a total of about 5,000 Å2 of surface area; interactions between neighboring OB domains contribute about 1,500 Å2, while the ATPase domains contribute about 3,500 Å2.
Figure 2. Mpa has a unique β-grasp-fold domain near the C-terminus.
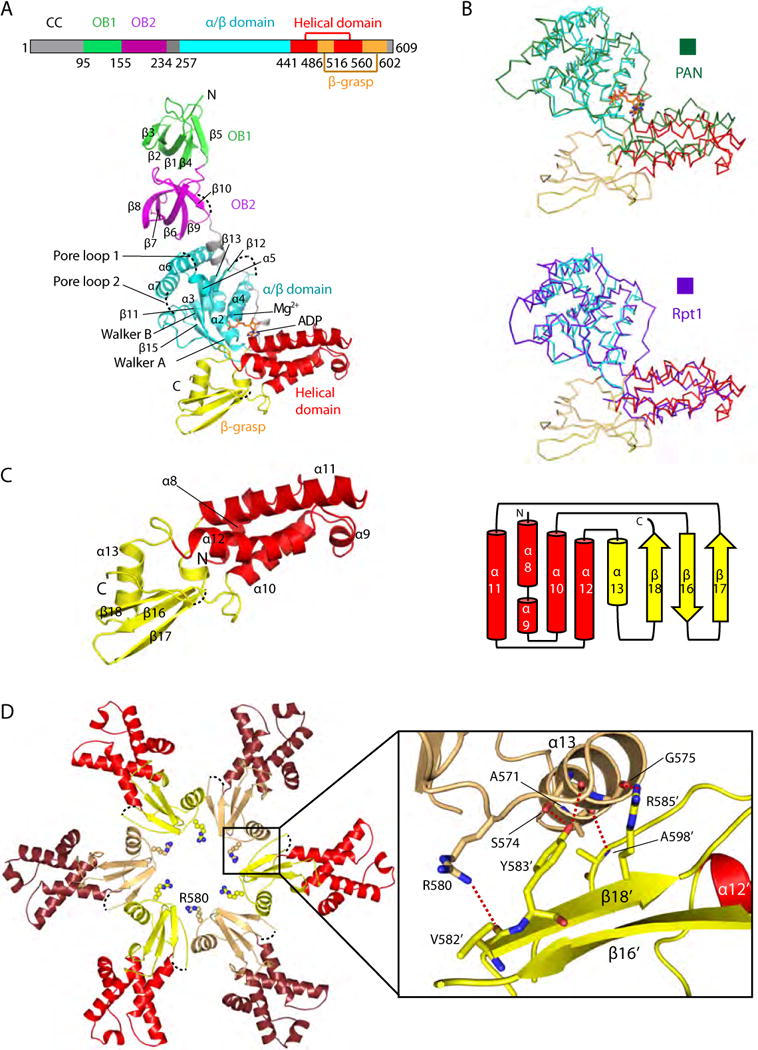
(A) Top: Domain mapof Mpa. Bottom: Structure of a singleMpaprotomer, with the OB1 domain in green, OB2 in purple, the large AAA subdomain in cyan, the small AAA subdomain in red, and the β-grasp-like domain in yellow. The secondary structure elements are labeled in the OB and ATPase regions. ADP bound to the hinge region between the two AAA subdomains is shown in stick form. Four disordered loops, including pore loop 1 (the Ar-Φloop) and pore loop 2, are shown as dashed black curves. (B) Superimposition of the ATPase region of Mpa with that of PAN (top panel, dark green ribbon) or of the Rpt1 (bottom panel, purple ribbon). The ATPase domains of Mpa, PAN, and Rpt1 are similar, but the C-terminal β-grasp fold is found only in Mpa. (C) Left, structure of the C-terminal β-grasp domain of Mpa; right, topology diagram. (D) The insertion domains interact with each other in anMpa hexamer to form the exit port lined by Arg-580. Five hydrogen bonds at the interface between neighboring β-grasp domains are shown as dashed red lines in the inset window.
A β-grasp-like domain near the C-terminus in bacterial proteasomal ATPases
We noticed that bacterial proteasomal ATPases are longer than PAN and Rpt1-6, and most of the additional sequence is found near the C-termini of these proteins (Figure S5). Upon the structural determination of the C-terminal domain of Mpa, we found an unexpected β-grasp-like fold, comprising one α-helix (α13) and a three-strand β-sheet (β16–β18) (Figure 2C). The first two β-strands (β16, β17) are between α10 and α11 of the small AAA subdomain, and the third β-strand (β18) and an α-helix (α13) are appended to the end of that subdomain (Figure 2C). The β-grasp domains of neighboring protomers interact extensively via a network of five H-bonds between α13 of one protomer and β18 of the neighbor: two H-bonds between Ser-574 and Tyr-583′, one H-bond between the carbonyl oxygen of Gly-575 and Arg-585′, one H-bond between Arg-580 and Val-582′, and finally, one H-bond between Ala-571 and Ala-598′. The interactions cause the β-grasp domains of the hexamer to form the last constriction of diameter of 22 Å along the axial channel. Six Arg-580 side chains in the loops connecting α13 and β18 line the constriction at the exit port (Figure 2D).
Charged residues lining the axial channel are important for substrate degradation
The structure of the Mpa hexamer revealed an axial channel that resembled the shape of a vase, with a diameter that varies widely from the top substrate entrance to the bottom substrate exit port (Figure 3A). In the upper OB ring region, Glu-145 and Arg-120 in the OB1 domain form a funnel shape with an opening of 25 Å. Lys-225 and His-179 in the OB2 domains form the narrowest portion of the axial channel, a 15 Å wide neck. The entry point to the upper chamber of the ATPase ring is 26 Å wide and is lined with pore loop 1 preceded by a conserved Lys-340. The central portion of the ATPase channel is the widest point, 37 Å, and is lined with Arg-417 and Asp-419 in β-hairpin loops (β14–β15). These arginines are highly conserved among proteasomal ATPases(Figure S5). Between the neck and widest point of the channel is the disordered pore loop 2 that contains the conserved Arg-380, Val-384, and Ser-385 (Figure S5). This unresolved loop likely makes the channel narrower than it appears in our structure. In the lower β-grasp domain, Arg-597, R580, and Asp-600 line the channel.
Figure 3. The Mpa hexamer has a highly charged axial channel.
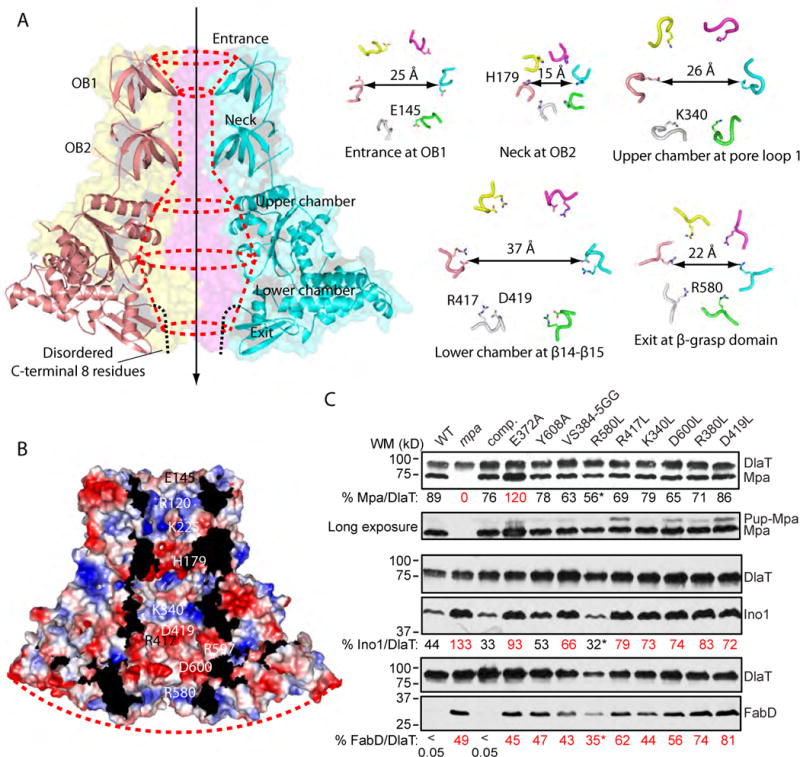
(A) The vase-like shape of the of Mpa hexamercentral chamber. The entry port and the neckare formed by the OB domain. The upper and lower chambers are defined by pore loop 1 and loop β14-β15 in the ATPase domain. The exit port is defined by loop α13-β18 in the β-grasp domain. The dashed black curves indicate the disordered C-terminal eight amino acids. (B) Surface potential of the translocation chamber of Mpa hexamer. The red dash indicates the convex shape of the C-terminal surface. (C) Immunoblot analysis of proteasome substratesin Mtb strains expressing mutant alleles of mpa. Charged residues lining the axial channel of the Mpa ring were mutatedtoleucine (L) or glycine (G). E372A is a previously characterized Walker B motif mutant, and Y608A is has a mutation in the essential GQYL motif at the C-terminus of Mpa.FabD and Ino1 are Mtb proteasome substrates. DlaTwas the loading control. Just below the first panel is a longer exposure of the immunoblot to Mpa that shows a potential pupylated species of Mpa in several strains. The amount of Mpa, Ino1, and FabD is shown as a percentage of the DlaT loading control and was quantified from the same immunoblot using Image J software. Lanes labeled in red show test protein that had at least a 50% difference from either the wild type (WT) or mpa+ complemented (“comp.”) strains. (*) indicates a sample that had overall lower protein amounts. For all panels, strains contain the plasmid pMV306 with either no cloned insert (“WT” and “mpa” lanes) or the indicated mutant mpa alleles (each expressed in the mpa null mutant). See Table S1 for strain numbers and genotypes. See Experimental Procedures for additional details.
A prominent feature of the channel is that it is lined with many charged residues (Figure 3B). Although the channel should be filled with water, the conserved charged residues likely contact the substrate peptide and consequently influence the efficiency of substrate degradation. We assessed the importance of these residues on the degradation of two proteasomal substrates in Mtb, malonyl coA-acyl carrier protein transacylase (FabD) and inositol 1-phosphate synthetase (Ino1) in vivo (Figure 3C). Eight single or double point mutations were introduced into a complementation plasmid with mpa expressed from its native promoter; the resulting plasmids were then integrated onto the Mtb chromosome of an mpa-null mutant to determine whether they could restore the degradation of FabD and Ino1. Because we chose to mutate residues located in the solvent-exposed internal channel and not in between Mpa protomers, the mutations were not expected to significantly alter the overall structure or hexameric state of the Mpa ring. We verified that the point mutations did not alter the ability of mutant Mpa to form hexamers relative to WT Mpa in total Mtb cell lysates using native gels (Figure S6). We found that most of the mutant proteins retained substantial ATPase activity, but there was a 20–55% reduction in activity in several of the proteins (Figure S7). While this likely contributed to the reduced degradation observed in vivo, we cannot rule out that there were also defects in substrate translocation for ATPase-independent reasons since we observed fairly dramatic decreases in degradation.
As previously shown, a mutation in the Walker B box of Mpa (E372A) led to increased amounts of Mpa as well as FabD and Ino1 (Figure 3C) (Pearce et al., 2006, Festa et al., 2010). This observation had been also shown for mutations in the GQYL motif (Y608A) (Pearce et al., 2006, Darwin et al., 2005, Wang et al., 2009). Under the conditions tested here, MpaY608A, did not accumulate above WT levels, but it was unable to facilitate FabD or Ino1 degradation (Figure 3C). Most of the other mutant proteins also appeared at levels comparable to the strain complemented with WT mpa. Importantly, all but one of the mutations (MpaR580L) resulted in less degradation of FabD and Ino1 (Figure 3C). Because proteolysis of substrates in the strain producing MpaR580L looked similar to proteolysis in the wild type strain, this residue is not likely to be directly involved in the translocation of unfolded substrates. Almost all of the other residues selected for mutagenesis significantly interfered with protein degradation.
The nucleotide-binding site in the Mpa crystal structure
In the ADP-bound Mpa crystal structure, we observed that every Mpa protomer was bound to ADP, yielding a stoichiometry of six nucleotides per Mpa hexamer (Figure 4A, B; Figure S3). As expected, nucleotide was found between the large α/β subdomain and the small helical subdomain of each AAA domain (Figure 4A, C). A highly basic cavity formed by the backbone amides of Gly-196 and Gly-298, and Lys-299 of the Walker A motif in the α/β subdomain, accommodates the phosphate groups. This structural feature explains our previous finding that mutating Lys-299 to Gln reduces the affinity of nucleotide for the binding site (Darwin et al., 2005). Ile-448 and Tyr-452 in the small helical subdomain coordinate the adenine group, while Leu-301 in the α/β subdomain and Gln-520 in the helical subdomain stabilizes the ribose group (Figure 4D). The α/β subdomain of a neighboring protomer contributes two closely spaced arginines to the nucleotide-binding site, Arg-427 and -430. Arg-427 may be responsible for sensing the presence of the ATP γ-phosphate; Arg-430 forms a salt bridge with Glu-372 in the Walker B motif.
Figure 4. The nucleotide-binding site of an Mpa hexamer.

(A) Surface view of an Mpa hexamer, viewed from the N-terminal surface. Each of the six nucleotide-binding pockets was occupied by an ADP in the co-crystal structure of an Mpa hexamer. (B) 2Fo-Fc density map of one ADP molecule displayed at a high threshold of 3σ. ADPis shown in stick form andMg2+as a blue sphere. (C) A selected region of the Mpa hexamer, showing that the nucleotide pocket is formed between the large and the small AAA subdomains of one protomor (in wheat) and the large AAA subdomain of a neighboring protomer (in yellow). (D) Interaction between ADP and Mpa. Adenosine forms a H-bond with Y452. The diphosphate is cradled by the Walker A motif and stabilized by the Mg2+ above. Both the large and the small AAA sub-domains of the same protomer contribute to nucleotide binding.
The C-terminal, proteasome-activating motif is concealed in Mpa
In order to deliver unfolded proteins into a 20S CP, the C-terminus of proteasomal ATPases needs to physically interact with the 20S CPs. In eukaryotes, the surface of the Rpt1-6 hexamer proximal to the 20S CP is concave, with the C-terminal HbYX motifs fully exposed and radially located at the same positions as the binding pockets in the α-ring of the 20S CP (Figure 5A, left panel) (Beck et al., 2012, Smith et al., 2007). These features are conducive to Rpt1-6 docking onto the convex surface of the 20S CP. In Mpa, C-terminal GQYL motifs activate degradation by the Mtb 20S CP (Darwin et al., 2005), presumably by inserting into similar α-ring pockets. Because the last resolved residue in our Mpa structure is Thr-601, the structure of the C-terminal eight residues that include the GQYL motif is unknown. However, we found that Thr-601 is located inside the axial channel. A modeled eight-residue peptide extending from Thr-601 would barely emerge from the exit port of the Mpa hexamer and would not be long enough to reach the binding pockets of the Mtb 20S CPα-ring (Figure 5A, right panel). The Mpa C-termini appeared to be recessed because of the presence of the β-grasp-like domains identified in this work (Figure 2A, 3A–B). This observation is consistent with a previous finding that Mpa interacts with Mtb 20S CPs very weakly. An interaction between Mpa and “open-gate” proteasomes (20SOG CP), in which the eight N-terminal residues of the α-subunits that block the peptide entry port were removed, could only be observed qualitatively using EM (Lin et al., 2008, Wang et al., 2009). In the current study, we attempted to quantify an interaction between Mpa engineered in this study and 20SOG CPs in the absence or presence of nucleotide such as ADP and AMPPNP, but we detected no binding signals (Figure 5B).
Figure 5. The Mpa C-terminal GQYL motifsare partially hidden inside the axial channel.

(A) Left panel: The C-terminal HbYX motifs in Rpt2, Rpt3, and Rpt5 (shown in cyan spheres) are fully exposed, facilitating interaction with the 20S CP (PDB ID 4CR2). Right panel: The last eight disordered residues of Mpa (Glu-602 to Leu-609, red lines) weremodeled in Coot by extending downward fromThr-601. They likely are partially buried in the axial channel due to the presence of theβ-grasp domain (yellow). The modeled GQYLis shown as green spheres. The positioning of last eight residues in Mpa is different from that in Rpts, making it difficult to bind the Mtb 20S CP. Dashed green circles in the Mtb 20S CPα-ring denote the binding pockets for the MpaGQYL motifs. (B) In vitro binding of WTMpa to Mtb 20SOGCPas measured by ITC at 25 °C in the absence of nucleotides or in the presence of ADP or AMPPNP. In vitro binding of MpaΔN97/C-ext to Mtb 20SOGCPwas measured in the absence of nucleotide. For MpaΔN97/C-ext, a linker peptide “GGGGS” was inserted between Thr-601 and Glu-602 of MpaΔN97.
We hypothesized that an Mpa hexamer with fully exposed GQYL motifs would bind to 20S CPs more strongly. This concept is supported by our recent report of the crystal structure of an ATP-independent Mtb proteasome activator called PafE (Bai et al., 2016), which interacts in vitro with WT Mtb 20S CPs (Jastrab et al., 2015, Delley et al., 2014). Unlike Mpa, PafE forms dodecamers with fully exposed C-terminal GQYL motifs (Jastrab et al., 2015, Bai et al., 2016). We reasoned that extension of the Mpa C-terminus would expose the GQYL motifs and facilitate binding to 20S CPs. We inserted five residues (GGGGS) preceding the GQYL motif to produce an Mpa variant with 13 unstructured C-terminal residues (MpaΔN97/C-ext). Isothermal titration calorimetry (ITC) demonstrated that MpaΔN97/C-ext was bound to Mtb 20SOG CPs with an estimated Kd of 2.2μM (Figure 5B). These results support our hypothesis that the β-grasp fold domains near the C-termini of the Mtb proteasomal ATPase hexamer prevent Mpa binding to Mtb 20S CPs by burying the C-terminal GQYL motif.
Next, we determined if extension of the Mpa C-terminus could enhance the degradation of a pupylated substrate by the Mtb proteasome. We used pupylated FabD (Pup~FabD) as the substrate in an in vitro degradation assay. Consistent with the previous study using pupylated PanB as the substrate (Striebel et al., 2010), we detected only very low in vitro activity for the WT proteasome in the presence of Mpa, irrespective of a C-terminal extension in Mpa (Figure 6A). When we used 20SOG CPs, we observed degradation of FabD-Pup with WT Mpa (Figure 6B). Importantly, using MpaC-ext, but not a Walker B mutant version of MpaC-ext, significantly accelerated degradation by 20SOG CPs(Figure 6B).
Figure 6. Mpa with an extended C-terminus enhances the degradation of a pupylated substrate by open-gate proteasomes.

(A) Left, in vitro degradation of the substrate Pup~FabD by 20SWT or 20SOG CPs in the presence of MpaWTor MpaC-extwas analyzed by SDS-PAGE, followed by immunoblotting with polyclonal antibodies to Mtb FabD-His6. Substrate stability was monitored by removing aliquots at indicated time points and adding sample buffer to stop reaction. Right, a quantitative view of the degradation assays in the left panels by calculating the percentage of Pup~FabD compared to PrcB-His6 (Control) using Image J software. All data points were normalized to protein present at t = 0 hours. (B) Degradation as described in (A) using 20SOG CPs in the presence of MpaC-ext or MpaC-ext/E372A.
Discussion
Proteasomes are important to many fundamental biological pathways, and the degradation of many proteins by a proteasome usually relies on hexameric ATPases. Despite the great importance of these ATP-driven machines, no crystal structure of a full-length hexameric proteasomal ATPase at atomic resolution has been reported (Finley et al., 2016). In this paper, we describe the crystal structure of a hexamer composed of nearly full-length Mpa protomers. A key finding of this study was the identification of a β-grasp domain at the C-terminal region of Mpa, which appears to be unique among bacterial proteasomal ATPases and influences the structure and function of Mpa.
Site-directed mutagenesis of the hydrophilic channel showed that several residues were needed for the efficient turnover of at least two model proteasome substrates. We presume these residues participate in the direct movement of substrates through the axial channel to the 20S CP. Most if not all of the mutations had some impact on ATPase activity, but we cannot rule out that these mutations might also disrupt substrate interactions needed to move a protein through the channel and into the 20S CP.
The most significant finding of our work was the identification of a β-grasp domain that appears responsible for the weak interaction between Mpa and 20S CPs in vitro (Figure 5). We speculate that the tucked C-termini of an Mpa ring may prevent unnecessary activation of 20S CPs when pupylated substrates are not present, allowing 20S CPs to participate in other biological pathways such as PafE-mediated proteasomal degradation (Jastrab et al., 2015). We currently do not know how the hidden GQYL motif may be exposed for 20S core binding. However, the extensive structural and functional knowledge of the bacterial HslUV protease system may provide some clues (Sauer & Baker, 2011). The HslU ATPase undergoes a nucleotide-hydrolysis-coupled rotation and a movement of its C-terminal small AAA subdomain relative to the large AAA subdomain, causing the retraction or exposure of C-terminal tails for binding to the HslV peptidase (Bochtler et al., 2000, Wang et al., 2001a, Sousa et al., 2000, Wang et al., 2001b, Wang, 2004). In Mpa, the β-grasp domain is within a small AAA subdomain (Fig 2C), so an ATP-hydrolysis-coupled conformational change of this subdomain may be capable of exposing the GQYL motifs. While this is a reasonable hypothesis, no one has reported the efficient Mpa-assisted degradation of a pupylated substrate in vitro. This suggests that our current biochemical knowledge of the Mtb Pup-Mpa-proteasome system is incomplete and that unknown cofactors or conditions may still be required for this process.
The archaeal PAN hexamer was reported to bind four nucleotides, with two neighboring protomers each binding one ATP, the next two protomers each binding one ADP, and the remaining two protomers being unoccupied (Kim et al., 2015). In the cryo-EM structure of the human 26S proteasome at an average resolution of 3.5 Å, each of the six RPT proteins contains a bound nucleotide. Although the resolution is insufficient to distinguish ATP and ADP, it was suggested that both ATP and ADP are present in the structure, because three RPTs resemble an ATP-bound conformation, and the other three RPTs are more similar to an ADP-bound state (Huang et al., 2016). In another cryo-EM structure at 3.9-Å resolution, all six subunits were also occupied by nucleotides, with RPT6 bound by ADP and the other five subunits all bound by ATP (Schweitzer et al., 2016). In a third cryo-EM structure, the human Rpt1-6 is fully occupied by six nucleotides (Chen et al., 2016). Therefore, a proteasomal ATPase can either be partially occupied or fully occupied by nucleotides. However, the structures of archaeal and human proteasomal ATPases are asymmetric, particularly the structures of the substrate translocating loops. In the apo- and ADP-bound forms, the structures of the Mpa hexamer are similar and nearly symmetric, but the substrate binding loops are disordered. It is possible that ATP binding and/or hydrolysis may introduce more asymmetry in the structure. But efforts to capture Mpa in these states have not succeeded so far, and these states await future studies, perhaps by high-resolution cryo-EM that does not rely on crystallization.
Our previous crystal structure of an Mpa1-234 hexamer included the NT coiled-coil and the double OB ring (Wang et al., 2010). The Mpa structure presented here contains all of the domains except for the coiled-coil. By overlapping the double OB ring (98 – 234) that is shared between the Mpa1-234 structure and the MpaΔN94 structure, we were able to build a plausible atomic model of a hexamer made of full-length Mpa monomers (Figure 7A). This structure reveals for the first time the true extent of this bacterial ATPase: despite the disordered N-terminal 50 residues, Mpa with three pairs of long coiled-coils has a dramatic height of over 160 Å, approaching the length of the 20S CP, which is 170 Å tall (Figure 7B). The extraordinary height of Mpa is achieved by the unusually long coiled-coils, an extra OB domain, and a C-terminal β-grasp domain. The coiled-coils in Mpa are essential for recruiting pupylated proteins to the proteasome (Figure 7B), whereas the shorter coiled-coils of PAN and Rpt1-6 do not have such a role. Further study is needed to understand the function of the extra OB domain, as well as the C-terminal β-grasp domain, of Mpa. A high-priority goal is to determine how the C-terminal GQYL motifs are exposed from an Mpa hexamer to engage the Mtb 20S CP for the degradation of pupylated substrates.
Figure 7. Proposed model of the full Mpa-proteasome complex.
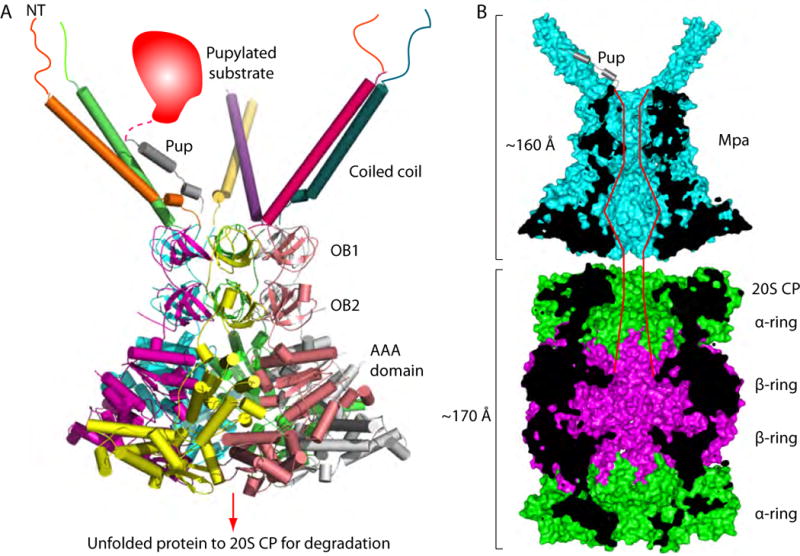
(A) An atomic model of the full-length Mpa hexamer generated by overlapping the shared OB domains in the crystal structures of Mpa1-234hexamer (PDB ID 3M9D) and of Mpa95-601hexamer. The unstructured N-terminal peptides are shown as lines. Only one of the three pairs of coiled coils can bind to a pupylated substrate at any given time. (B) Mpa and the 20S CP are vertically cut through the middle section to show the path of a protein substrate going through the Mpa axial channel to reach the proteolytic sites inside the β rings.
EXPERIMENTAL PROCEDURES
Molecular cloning, protein expression, and purification
For crystallization of Mpa in the ADP-bound state, we removed the N-terminal 94 residues and replaced the flexible loop outside the OB2 domain (194–210) by a triple glycine linker. We found that shortening the N-terminal 6×His-tag to a 3×His-tag was helpful for crystallization without removing the tag. We truncated the C-terminus by 7 residues, yielding a construct of HHH-Mpa95-193-GGG-211-602. The construct was confirmed by DNA sequencing and used to transform competent BL21(DE3) E. coli for protein expression. Transformed BL21(DE3) strains were cultured in Luria-Bertani broth medium with 100 μg/ml ampicillin at 37 °C for 4 h, which was when OD600 reached 0.8 to 1.0, and then induced at 30 °C for 6 h. Protein expression was induced by adding isopropyl-β-D-thiogalactopyranoside (IPTG) at a final concentration of 0.2 mM. Bacterial cells were collected by centrifugation at 4000 × g for 10 min at 4 °C, and the fresh cells were immediately resuspended in buffer A (25 mM Tris-HCl, pH 8.2, 500 mM NaCl, 5 mM MgCl2, 5%glycerol, 1 mM ATP, 10 mM imidazole). Cells were lysed by passing through a french press twice at 1800 bars, followed by centrifugation at 30000 × g for 60 min at 4 °C. Supernatant was collected and loaded onto a 5-ml Ni-NTA cartridge (Qiagen) pre-equilibrated with buffer A and eluted with an imidazole step gradient. The elution fractions containing Mpa were immediately loaded onto a Superdex 200 gel filtration column with buffer B (10 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.5 mM ATP). Fractions containing Mpa were collected and used directly for crystallization. Mpa hexamers were highly prone to nonspecific degradation and aggregation. Purification and crystallization were done within 24 h, and no sample concentration was performed beyond Ni-NTA affinity chromatography.
For Mpa crystallization in apo state, we removed the N-terminal 97 residues, yielding a construct of MpaΔN97. MpaΔN97 was expressed in E. coli strain ER2566 with a C-terminal 6×His-tag preceded by a thrombin cleavage site. To produce C-terminal intact protein samples for binding study to the 20S CP, MpaΔN97 was expressed with an internal 6×His-tag inserted at residue 199. MpaΔN97/C-ext used for the 20S CP binding study and MpaC-ext used for in vitro degradation assays were prepared by inserting GGGGS between Thr-601 and Glu-602 in MpaΔN97 and WT full-length Mpa, respectively. Protein expression was induced with 0.4 mM IPTG when the bacterial culture reached an OD600 value of 0.6–0.8. Induced cells were grown for another 16–20 h at 20°C, then were harvested and lysed with microfluidics in buffer 25 mM sodium phosphate pH 7.4, 500 mM NaCl, and 10% glycerol. The supernatant of the lysate was loaded onto a 5-ml Hitrap HP Ni-NTA column (GE Healthcare) and washed with lysis buffer containing 60 mM imidazole, followed by elution with a linear imidazole gradient. Fractions containing the target protein were collected. Further purification was performed using a Superdex 200 16/60 column (GE Healthcare) in a buffer of 10 mM HEPES, pH 7.4, plus 200 mM NaCl.
Crystallization, data reduction, and structure determination
We used the vapor diffusion sitting-drop method for crystallization of HHH-Mpa95-193-GGG-211-602 at 15 °C. The protein in buffer B at a concentration of 8–10 mg/ml was crystallized in solution containing 0.1 M Tris-HCl, pH 8.2, 20% PEG 400, 0.2M MgCl2, and 5 mM ATPγS. Streak seeding and macro seeding methods were used to improve the crystal quality. Due to the failure of initial molecular replacement trials, we soaked the crystals with Hg(AC)2. Most Mpa crystals were anisotropic and sensitive, so we only soaked Mpa crystals with 1 mM Hg(AC)2 in mother liquor for 10 min and then immediately harvested the crystals. Crystals were flash-frozen in mother liquor containing 25% glycerol as a cryo-protectant. The hanging drop vapor diffusion method was used for crystallization of MpaΔN97. In brief, a 0.5 μl droplet of protein sample was mixed with 0.5 μl of reservoir buffer, and the mixed droplet was incubated at 21 °C for diffusive equilibrium with 100μl reservoir solution. Protein concentration was 10 mg/ml in buffer containing 10 mM HEPES pH 7.4, 200 mM NaCl. Reservoir solution contains 100 mM Bis-Tris Propane, pH 7.0, 0.475 M NaCl, 18.2% PEG 4000, 20% glycerol. Crystals appeared in 7 days. Crystals were dehydrated for three weeks by replacing the mother liquor with 100 mM Bis-Tris propane pH 7.0, 0.6 M NaCl, 27–30% PEG 4000, 20% glycerol. Dehydrated crystals were directly flash frozen in liquid nitrogen. Crystal screening and data collection were carried out at the X25 and X29 beamlines of the National Synchrotron Light Source, Brookhaven National Laboratory, and at the BL17U beamline, Shanghai Synchrotron Radiation Facility. Diffraction images were indexed by IMOSFLM and scaled with SCALA. Most of the crystals were plate-like and highly anisotropic.
For the structure determination of HHH-Mpa95-193-GGG-211-602 co-crystallized with ATPγS, thousands of crystals were screened, leading to one complete data set to 2.9 Å resolution. Although the crystal was soaked with Hg2+, the diffraction data was inadvertently collected at the selenium peak (0.9791 Å). Phase determination using Hg-derived data failed, probably due to weak anomalous signal. We treated the data set as a native one and solved the structure by an advanced molecular replacement approach as implemented in the Phenix_Phaser-Rosetta program using a powerful computing cluster (DiMaio et al., 2013). The starting search models were the Mpa1- 234 hexamer (PDB ID: 3M9B) and PAN-Rpt5C (3WHK). The structure was largely built by ROSETTA and completed manually in COOT (Emsley & Cowtan, 2004).
We found the ATPγS used in co-crystallization was hydrolyzed to ADP in the crystal (Fig. S3A–B). AMg2+ ion was found to bind to the ADP. The soaked Hg2+ ion was identified some distance away from the nucleotide-binding pocket (Fig. S3C). The structure was refined in PHENIX with Cartesian simulated annealing and TLS refinement (Adams et al., 2010). The structure was refined in P321 at a resolution 2.9 Å, with Rfactor = 0.230 and Rfree=0.266. The apo Mpa structure was solved by molecular replacement using the ADP-bound Mpa model in PHASER (Mccoy et al., 2007) and PHENIX suite. The initial model was manually adjusted in COOT and refined by PHENIX and Refmac5 in CCP4 (Winn et al., 2011). Refinement statistics are summarized in Table 1.
Isothermal titration calorimetry
ITC experiments were performed in 20 mM Tris, pH 8.0, plus 150 mM NaCl, 2 mM MgCl2 at 25 °C using a VP-ITC microcalorimeter (MicroCal). In the absence and presence of 2 mM ADP or 2 mM AMPPNP, 30 μM MpaΔN97 was titrated into 2 μM 20SOG. In the absence of nucleotide, 40 μM MpaΔN97/C-ext was titrated into 2 μM 20SOG. The Origin 7.0 software was used to analyze the results.
In vivo and in vitro degradation assays
Substitutions were incorporated into the Mtb mpa gene using PCR, and mpa variants were cloned into the vector pMV306, which integrates into the chromosomal L5 attB site. An mpa mutant was transformed by electroporation with each plasmid, and bacteria were grown on selective Middlebrook 7H11 agar (Difco) supplemented with oleic acid, albumin, dextrose, and catalase (Middlebrook Enrichment, OADC, BBL). Mtb strains were grown to OD580 ~2, and 4 OD equivalents of bacteria from each culture were collected by centrifugation (2880 × g, 8 min) and washed with PBS plus 0.05% Tween-80. To prepare total cell lysates, bacteria were resuspended in 250 μl lysis buffer (10 mM Tris pH 8, 50 mM NaCl, 1 mM EDTA) and bead-beaten 3 × 30 sec. After letting the beads settle, supernatants were mixed with reducing SDS sample buffer and boiled. Lysates were separated by 12% SDS-PAGE and analyzed by immunoblotting using antibodies to Mpa, FabD, or Ino1 (Festa et al., 2010). Each blot was then stripped and reincubated with a primary antibody to dihydrolipoamide acyltransferase (DlaT) as a loading control (Tian et al., 2005).
For in vitro degradation assays, each reaction contained 6.9 μg 20S CP, 7.76 μg Mpa (WT or mutant variants), 2 μg Myc-Pup~FabD-his, 2.5 mM ATP, 20 mM MgCl2, 1 mM DTT and 50 mM NaCl in 50 mM Tris, pH 8 in a final volume of 100 μl. At indicated times, samples were withdrawn and added to SDS-denaturing sample buffer to stop the reactions. Samples were analyzed by SDS-PAGE, followed by immunoblotting with polyclonal antibodies to Mtb FabD-His6.
For both the in vivo and in vitro data, we quantified relative protein amounts in the immunoblots using Image J software (Schneider et al., 2012).
Native gel electrophoresis of mutant Mpa proteins
For each mpa mutant strain, we grew bacteria in 25 ml of 7H9 Middlebrook broth with 50μg/ml Hyg. Cultures were grown to an optical density at 580 nm (OD580) of about 2. We collected equivalent bacterial cell numbers by centrifugation and washed the bacteria once with TBST (25 mM Tris-HCl, pH 7.4,125 mM NaCl, 0.05% Tween 20). Bacteria were lysed by bead-beating in 500 μl of cold lysis buffer (50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 1 mM EDTA). We kept samples on ice at all times. The samples were transferred to a screw cap tunes with ~ 200 μl of zirconia beads, bead-beaten three times 30 s each with cooling on ice between beatings (beads and bead beater from BioSpec Products, Inc.). The lysates were centrifuged at 6,000 rpm for 4 min at 4°C in a microcentrifuge. Before removing from BSL3, we sterilized the samples through 0.45 μl syringe filters. For electrophoresis, 100 μl of each sample was mixed with native sample buffer (62.5 mM Tris-HCl, pH 6.8, 40% glycerol, 0.01% bromophenol blue). Proteins were separated on a 7.5% native gel (Bio-Rad, Inc), transferred to nitrocellulose, and analyzed by immunoblotting with antibodies to Mtb Mpa.
ATPase Assays
ATPase activity of Mpa and its mutants was determined by measuring the production of inorganic phosphate using the Malachite Green Phosphate Assay Kit (Sigma). The assays were started by adding 2.5 mM ATP to solution containing 2 μg WT Mpa or its mutants, 20 mM Tris-Cl pH 7.4, 10 mM MgCl2, 50 mM NaCl. The reactions were performed for 30 min at 37°C. Reaction products were collected every 10 min and added to 20 μl Malachite Green reagent in a 96-well plate. The amount of inorganic phosphate that was released was measured by the absorbance change at a wavelength of 620 nm.
Supplementary Material
Acknowledgments
We thank David Nadziejka for helpful comments on a draft version of this manuscript. This work was supported by NIH grants AI070285 (to H.L.), AI088075 (to K.H.D), and T32 AI007180 and F30 AI110067 (to J.B.J), and by National Key Basic Research Program of China 2013CB911500, NSFC31300600, Shenzhen Science and Technology innovation program grants ZDSY20120614-144410389, KQCX20130627-103353535, and JCYJ20160331-115853521 (to T.W.). K.H.D. holds an Investigator in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. X-ray diffraction data were collected at X25 and X29 of the National Synchrotron Light Source of Brookhaven National Laboratory and at BL17U of Shanghai Synchrotron Radiation Facility.
Footnotes
Accession codes
The coordinate and the structure factor files of apo and ADP-bound Mpa were deposited in PDB with accession number 5KZF and 5KWA, respectively.
We have no conflict of interest to declare.
AUTHOR CONTRIBUTIONS
K.H.D, J.B.J, T.W., and H.L. designed research. Y.W., D.L, Y.G, Y.H., S.Y., and T.W. solved the Mpa structure. K.H. and L.B. performed in vitro biochemical studies. J.B.J. and S.Z. did the protein accumulation assays and data analysis. S.Z. did the native gel analysis of Mpa mutants and the in vitro degradation assays. K.H., L.B, S.Z., K.H.D., T.W., and H.L. analyzed the data and wrote the paper with input from all authors.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai L, Hu K, Wang T, Jastrab JM, Darwin KH, Li H. Structural analysis of the dodecameric proteasome activator PafE in Mycobacterium tuberculosis. P Natl Acad Sci USA. 2016 doi: 10.1073/pnas.1512094113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck F, Unverdorben P, Bohn S, Schweitzer A, Pfeifer G, Sakata E, Nickell S, Plitzko JM, Villa E, Baumeister W, Forster F. Near-atomic resolution structural model of the yeast 26S proteasome. P Natl Acad Sci USA. 2012;109:14870–14875. doi: 10.1073/pnas.1213333109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochtler M, Hartmann C, Song HK, Bourenkov GP, Bartunik HD, Huber R. The structures of HsIU and the ATP-dependent protease HsIU-HsIV. Nature. 2000;403:800–805. doi: 10.1038/35001629. [DOI] [PubMed] [Google Scholar]
- Burns KE, Liu WT, Boshoff HIM, Dorrestein PC, Barry CE. Proteasomal Protein Degradation in Mycobacteria Is Dependent upon a Prokaryotic Ubiquitin-like Protein. J Biol Chem. 2009;284:3069–3075. doi: 10.1074/jbc.M808032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wu J, Lu Y, Ma YB, Lee BH, Yu Z, Ouyang Q, Finley DJ, Kirschner MW, Mao Y. Structural basis for dynamic regulation of the human 26S proteasome. P Natl Acad Sci USA. 2016;113:12991–12996. doi: 10.1073/pnas.1614614113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Solomon WC, Kang Y, Cerda-Maira F, Darwin KH, Walters KJ. Prokaryotic ubiquitin-like protein pup is intrinsically disordered. Journal of molecular biology. 2009;392:208–217. doi: 10.1016/j.jmb.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton CL, Fernandopulle MS, Nagari RT, Sello JK. Genetic and Proteomic Analyses of Pupylation in Streptomyces coelicolor. J Bacteriol. 2015;197:2747–2753. doi: 10.1128/JB.00302-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science. 2003;302:1963–1966. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- Darwin KH, Lin G, Chen Z, Li H, Nathan CF. Characterization of a Mycobacterium tuberculosis proteasomal ATPase homologue. Molecular microbiology. 2005;55:561–571. doi: 10.1111/j.1365-2958.2004.04403.x. [DOI] [PubMed] [Google Scholar]
- Delley CL, Laederach J, Ziemski M, Bolten M, Boehringer D, Weber-Ban E. Bacterial proteasome activator bpa (rv3780) is a novel ring-shaped interactor of the mycobacterial proteasome. PLoS One. 2014;9:e114348. doi: 10.1371/journal.pone.0114348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMaio F, Echols N, Headd JJ, Terwilliger TC, Adams PD, Baker D. Improved low-resolution crystallographic refinement with Phenix and Rosetta. Nat Methods. 2013;10:1102–1104. doi: 10.1038/nmeth.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djuranovic S, Hartmann MD, Habeck M, Ursinus A, Zwickl P, Martin J, Lupas AN, Zeth K. Structure and activity of the N-terminal substrate recognition domains in proteasomal ATPases. Molecular cell. 2009;34:580–590. doi: 10.1016/j.molcel.2009.04.030. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Festa RA, Jones MB, Butler-Wu S, Sinsimer D, Gerads R, Bishai WR, Peterson SN, Darwin KH. A novel copper-responsive regulon in Mycobacterium tuberculosis. Molecular microbiology. 2011;79:133–148. doi: 10.1111/j.1365-2958.2010.07431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festa RA, McAllister F, Pearce MJ, Mintseris J, Burns KE, Gygi SP, Darwin KH. Prokaryotic ubiquitin-like protein (Pup) proteome of Mycobacterium tuberculosis [corrected] PLoS One. 2010;5:e8589. doi: 10.1371/journal.pone.0008589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley D, Chen X, Walters KJ. Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends Biochem Sci. 2016;41:77–93. doi: 10.1016/j.tibs.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Liu R, Sanchez I, Silva-Sanchez C, Hepowit NL, Cao S, Chen S, Maupin-Furlow J. Ubiquitin-Like Proteasome System Represents a Eukaryotic-Like Pathway for Targeted Proteolysis in Archaea. MBio. 2016;7 doi: 10.1128/mBio.00379-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4 angstrom resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- Hanna J, Finley D. A proteasome for all occasions. FEBS Lett. 2007;581:2854–2861. doi: 10.1016/j.febslet.2007.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Lin G, Wang M, Dick L, Xu RM, Nathan C, Li H. Structure of the Mycobacterium tuberculosis proteasome and mechanism of inhibition by a peptidyl boronate. Molecular microbiology. 2006;59:1417–1428. doi: 10.1111/j.1365-2958.2005.05036.x. [DOI] [PubMed] [Google Scholar]
- Huang X, Luan B, Wu J, Shi Y. An atomic structure of the human 26S proteasome. Nature structural & molecular biology. 2016 doi: 10.1038/nsmb.3273. [DOI] [PubMed] [Google Scholar]
- Jastrab JB, Darwin KH. Bacterial Proteasomes. Annu Rev Microbiol. 2015;69:109–127. doi: 10.1146/annurev-micro-091014-104201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jastrab JB, Wang T, Murphy JP, Bai L, Hu K, Merkx R, Huang J, Chatterjee C, Ovaa H, Gygi SP, Li H, Darwin KH. An adenosine triphosphate-independent proteasome activator contributes to the virulence of Mycobacterium tuberculosis. P Natl Acad Sci USA. 2015;112:E1763–1772. doi: 10.1073/pnas.1423319112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YC, Snoberger A, Schupp J, Smith DM. ATP binding to neighbouring subunits and intersubunit allosteric coupling underlie proteasomal ATPase function. Nat Commun. 2015;6:8520. doi: 10.1038/ncomms9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao S, Shang Q, Zhang X, Zhang J, Xu C, Tu X. Pup, a prokaryotic ubiquitin-like protein, is an intrinsically disordered protein. Biochem J. 2009;422:207–215. doi: 10.1042/BJ20090738. [DOI] [PubMed] [Google Scholar]
- Lin G, Tsu C, Dick L, Zhou XK, Nathan C. Distinct specificities of Mycobacterium tuberculosis and mammalian proteasomes for N-acetyl tripeptide substrates. J Biol Chem. 2008;283:34423–34431. doi: 10.1074/jbc.M805324200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science. 1995;268:533–539. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- Luan B, Huang X, Wu J, Mei Z, Wang Y, Xue X, Yan C, Wang J, Finley DJ, Shi Y, Wang F. Structure of an endogenous yeast 26S proteasome reveals two major conformational states. P Natl Acad Sci USA. 2016;113:2642–2647. doi: 10.1073/pnas.1601561113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maupin-Furlow JA. Ubiquitin-like proteins and their roles in archaea. Trends Microbiol. 2013;21:31–38. doi: 10.1016/j.tim.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce MJ, Arora P, Festa RA, Butler-Wu SM, Gokhale RS, Darwin KH. Identification of substrates of the Mycobacterium tuberculosis proteasome. Embo J. 2006;25:5423–5432. doi: 10.1038/sj.emboj.7601405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce MJ, Mintseris J, Ferreyra J, Gygi SP, Darwin KH. Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science. 2008;322:1104–1107. doi: 10.1126/science.1163885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanovic MI, Darwin KH. Game of ‘Somes: Protein Destruction for Mycobacterium tuberculosis Pathogenesis. Trends Microbiol. 2016;24:26–34. doi: 10.1016/j.tim.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyshur KA, Worzalla GA, Meyer LS, Heiniger EK, Aukema KG, Misic AM, Forest KT. Crystal structures of the pilus retraction motor PilT suggest large domain movements and subunit cooperation drive motility. Structure. 2007;15:363–376. doi: 10.1016/j.str.2007.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer A, Aufderheide A, Rudack T, Beck F, Pfeifer G, Plitzko JM, Sakata E, Schulten K, Forster F, Baumeister W. Structure of the human 26S proteasome at a resolution of 3.9 A. P Natl Acad Sci USA. 2016;113:7816–7821. doi: 10.1073/pnas.1608050113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DM, Chang SC, Park S, Finley D, Cheng Y, Goldberg AL. Docking of the proteasomal ATPases’ carboxyl termini in the 20S proteasome’s alpha ring opens the gate for substrate entry. Molecular cell. 2007;27:731–744. doi: 10.1016/j.molcel.2007.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DM, Fraga H, Reis C, Kafri G, Goldberg AL. ATP Binds to Proteasomal ATPases in Pairs with Distinct Functional Effects, Implying an Ordered Reaction Cycle. Cell. 2011;144:526–538. doi: 10.1016/j.cell.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa MC, Trame CB, Tsuruta H, Wilbanks SM, Reddy VS, McKay DB. Crystal and solution structures of an HslUV protease-chaperone complex. Cell. 2000;103:633–643. doi: 10.1016/s0092-8674(00)00166-5. [DOI] [PubMed] [Google Scholar]
- Striebel F, Hunkeler M, Summer H, Weber-Ban E. The mycobacterial Mpa-proteasome unfolds and degrades pupylated substrates by engaging Pup’s N-terminus. Embo J. 2010;29:1262–1271. doi: 10.1038/emboj.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter M, Damberger FF, Imkamp F, Allain FHT, Weber-Ban E. Prokaryotic Ubiquitin-like Protein (Pup) Is Coupled to Substrates via the Side Chain of Its C-Terminal Glutamate. J Am Chem Soc. 2010;132:5610–+. doi: 10.1021/ja910546x. [DOI] [PubMed] [Google Scholar]
- Tian J, Bryk R, Shi S, Erdjument-Bromage H, Tempst P, Nathan C. Mycobacterium tuberculosis appears to lack alpha-ketoglutarate dehydrogenase and encodes pyruvate dehydrogenase in widely separated genes. Molecular microbiology. 2005;57:859–868. doi: 10.1111/j.1365-2958.2005.04741.x. [DOI] [PubMed] [Google Scholar]
- Unverdorben P, Beck F, Sledz P, Schweitzer A, Pfeifer G, Plitzko JM, Baumeister W, Forster F. Deep classification of a large cryo-EM dataset defines the conformational landscape of the 26S proteasome. P Natl Acad Sci USA. 2014;111:5544–5549. doi: 10.1073/pnas.1403409111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. Nucleotide-dependent domain motions within rings of the RecA/AAA(+) superfamily. J Struct Biol. 2004;148:259–267. doi: 10.1016/j.jsb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Wang J, Song JJ, Franklin MC, Kamtekar S, Im YJ, Rho SH, Seong IS, Lee CS, Chung CH, Eom SH. Crystal structures of the HslVU peptidase-ATPase complex reveal an ATP-dependent proteolysis mechanism. Structure. 2001a;9:177–184. doi: 10.1016/s0969-2126(01)00570-6. [DOI] [PubMed] [Google Scholar]
- Wang J, Song JJ, Seong IS, Franklin MC, Kamtekar S, Eom SH, Chung CH. Nucleotide-dependent conformational changes in a protease-associated ATPase HsIU. Structure. 2001b;9:1107–1116. doi: 10.1016/s0969-2126(01)00670-0. [DOI] [PubMed] [Google Scholar]
- Wang T, Darwin KH, Li H. Binding-induced folding of prokaryotic ubiquitin-like protein on the Mycobacterium proteasomal ATPase targets substrates for degradation. Nature structural & molecular biology. 2010;17:1352–1357. doi: 10.1038/nsmb.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Li H, Lin G, Tang C, Li D, Nathan C, Darwin KH, Li H. Structural insights on the Mycobacterium tuberculosis proteasomal ATPase Mpa. Structure. 2009;17:1377–1385. doi: 10.1016/j.str.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SK, Abomoelak B, Hoye EA, Steinberg H, Talaat AM. CtpV: a putative copper exporter required for full virulence of Mycobacterium tuberculosis. Molecular microbiology. 2010;77:1096–1110. doi: 10.1111/j.1365-2958.2010.07273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Kawashima T, Nishitani Y, Kanai T, Wada T, Inaba K, Atomi H, Imanaka T, Miki K. Structural basis of a Ni acquisition cycle for [NiFe] hydrogenase by Ni-metallochaperone HypA and its enhancer. P Natl Acad Sci USA. 2015;112:7701–7706. doi: 10.1073/pnas.1503102112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr D. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolschendorf F, Ackart D, Shrestha TB, Hascall-Dove L, Nolan S, Lamichhane G, Wang Y, Bossmann SH, Basaraba RJ, Niederweis M. Copper resistance is essential for virulence of Mycobacterium tuberculosis. P Natl Acad Sci USA. 2011;108:1621–1626. doi: 10.1073/pnas.1009261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun HY, Tamura N, Tamura T. Rhodococcus prokaryotic ubiquitin-like protein (Pup) is degraded by deaminase of pup (Dop) Biosci Biotechnol Biochem. 2012;76:1959–1966. doi: 10.1271/bbb.120458. [DOI] [PubMed] [Google Scholar]
- Zebisch M, Strater N. Structural insight into signal conversion and inactivation by NTPDase2 in purinergic signaling. P Natl Acad Sci USA. 2008;105:6882–6887. doi: 10.1073/pnas.0802535105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Hu M, Tian G, Zhang P, Finley D, Jeffrey PD, Shi Y. Structural insights into the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Molecular cell. 2009;34:473–484. doi: 10.1016/j.molcel.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.