

Abstract
The lack of reliable measures of alcohol intake is a major obstacle to the diagnosis and treatment of alcohol-related diseases. Epigenetic modifications such as DNA methylation may provide novel biomarkers of alcohol use. To examine this possibility, we performed an epigenome-wide association study of methylation of cytosine-phosphate-guanine dinucleotide (CpG) sites in relation to alcohol intake in 13 population-based cohorts (ntotal=13 317; 54% women; mean age across cohorts 42–76 years) using whole blood (9643 European and 2423 African ancestries) or monocyte-derived DNA (588 European, 263 African and 400 Hispanic ancestry) samples. We performed meta-analysis and variable selection in whole-blood samples of people of European ancestry (n=6926) and identified 144 CpGs that provided substantial discrimination (area under the curve=0.90–0.99) for current heavy alcohol intake (⩾42 g per day in men and ⩾28 g per day in women) in four replication cohorts. The ancestry-stratified meta-analysis in whole blood identified 328 (9643 European ancestry samples) and 165 (2423 African ancestry samples) alcohol-related CpGs at Bonferroni-adjusted P<1 × 10−7. Analysis of the monocyte-derived DNA (n=1251) identified 62 alcohol-related CpGs at P<1 × 10-7. In whole-blood samples of people of European ancestry, we detected differential methylation in two neurotransmitter receptor genes, the γ-Aminobutyric acid-A receptor delta and γ-aminobutyric acid B receptor subunit 1; their differential methylation was associated with expression levels of a number of genes involved in immune function. In conclusion, we have identified a robust alcohol-related DNA methylation signature and shown the potential utility of DNA methylation as a clinically useful diagnostic test to detect current heavy alcohol consumption.
Introduction
Each year, nearly 2.5 million deaths worldwide are attributable to alcohol use.1 Most alcohol-attributable diseases and injuries occur in people without a diagnosed alcohol use disorder.2, 3, 4, 5 Researchers have attempted to develop laboratory tests to detect heavy drinkers who are more reliable than self-reported alcohol intake (e.g. alcohol screening questionnaires). In addition, a biomarker would be useful in epidemiologic studies of health effects of alcohol as an objective measure to supplement and validate self-reported data. It could also prove useful in studies of other exposures where careful adjustment for alcohol intake is needed.6 Several biochemical measurements, such as serum alanine transaminase (ALT) and aspartate transaminase (AST) levels have been used to assess heavy alcohol use. However, the discriminatory ability of these biomarkers is far from ideal, with the area under the curve (AUC) to predict heavy alcohol consumption ranging from 0.21 to 0.67.7 The addition of four protein markers, AT-rich interactive domain-containing protein 4B (ARID4B), phosphatidylcholine-sterol acyltransferase (LCAT), hepatocyte growth factor-like protein (MST1) and ADP-ribosylation factor 6 (ARL6), improved AUC values for the detection of heavy drinkers to 0.73–0.86, leaving room for further improvement.7
Emerging evidence suggests that alcohol consumption influences epigenetic modifications,8, 9, 10 which in turn can affect gene expression levels.8, 11, 12 Methylation of the cytosine position in CpGs is among the best-characterized epigenetic modifications.13 To date, more than 20 studies have been conducted to identify alcohol-related DNA methylation signatures. Most of these studies, however, have focused on alcohol dependence in relation to ‘global’ methylation levels or preselected candidate genes14 and only a few studies have used epigenome-wide approaches.15, 16, 17, 18 The largest genome-wide study so far included about 700 individuals.16 To date, limited sample sizes have hindered the search for a robust alcohol-related DNA methylation signature. Hence, there is a need for a large-scale collaborative effort to determine the association of alcohol consumption with DNA methylation across the genome. Here we demonstrate that DNA methylation can be used as a highly predictive blood biomarker to detect heavy alcohol drinking. We also report meta-analysis results from epigenome-wide association studies (EWAS) in up to 13 317 individuals from 13 cohorts in which DNA methylation was measured in blood samples using the Infinium HumanMethylation450 BeadChip (Illumina, San Diego, CA, USA). Third, we determined the genetic contributions to alcohol-related methylation differences. Finally, we explored the functional implications of alcohol-related differential methylation by testing its association with gene expression in blood.
Materials and methods
Study population
This analysis included 13 317 participants from 13 population-based prospective cohorts of the Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium plus (CHARGE+) Consortium. These cohorts were sampled from free-living members of the community, but they were all not required to be healthy nor were they selected based on disease. During follow-up, some participants developed health conditions such as cardiovascular diseases (CVDs) and cancer. About 54% of participants were women and the average age was from 42 to 76 years old across the cohorts (Table 1). The patterns of alcohol consumption varied widely across the cohorts. For nine cohorts, fewer than one-third of participants reported no current alcohol intake and for four cohorts more than 50% of participants reported no current alcohol intake. The high proportion of non-drinkers in these four cohorts is in line with other studies of people of comparable age, birth cohort and gender mix. Heavy drinkers, defined below, represented 2–17% of participants across studies (Table 1). Informed consent for genetic studies was obtained from all subjects. The protocol for each study was approved by the institutional review board of each cohort.
Table 1. Characteristics of the study participants.
| Study | N | Men (%) | Age (years), mean (s.d.) | BMI mean (s.d.) | Current smoking (%) | g per day, Median (min, max) | Non-drinkers (%) | Light drinkers (%) | At-risk drinkers (%) | Heavy drinkers (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| European ancestry whole blood (n=9643) | ||||||||||
| CHS | 185 | 84 (45) | 76 (5) | 27 (5) | 16 (9) | 0 (0, 99) | 104 (56) | 59 (32) | 7 (4) | 15 (8) |
| EPIC-Norfolk | 1275 | 650 (51) | 60 (9) | 27 (4) | 191 (15) | 3 (0, 98) | 271 (21) | 865 (68) | 79 (6) | 60 (5) |
| FHS | 2427 | 1095 (45) | 66 (9) | 28 (5) | 304 (13) | 4 (0, 181) | 693 (29) | 1260 (52) | 280 (11) | 194 (8) |
| InCHIANTI | 499 | 225 (45) | 63 (16) | 27 (4) | 94 (19) | 8 (0, 161) | 106 (21) | 265 (53) | 70 (14) | 58 (12) |
| KORA F4 | 1797 | 877 (49) | 60 (9) | 28 (5) | 262 (15) | 7 (0, 150) | 534 (29) | 751 (42) | 282 (16) | 230 (13) |
| LBC1936 | 920 | 465 (51) | 70 (1) | 28 (4) | 103 (11) | 7 (0,158) | 181 (20) | 574 (62) | 104 (11) | 61 (7) |
| NAS | 623 | 623 (100) | 72 (7) | 28 (4) | 27 (4) | 6 (0, 93) | 148 (24) | 385 (62) | 52 (8) | 38 (6) |
| PIVUS | 818 | 412 (50) | 70 (0.2) | 27 (4) | 75 (9.2) | 6.7 (0, 61) | 142 (17) | 639 (78) | 32 (4) | 5 (1) |
| RS | 502 | 241 (48) | 58 (7) | 27 (5) | 137 (27) | 14 (0, 88) | 10 (2) | 366 (73) | 84 (17) | 42 (8) |
| TwinsUK | 597 | 0 (0) | 56 (9) | 27 (5) | 57 (10) | 2 (0,59) | 189 (31) | 375 (63) | 22 (4) | 11 (2) |
| African ancestry whole blood (n=2423) | ||||||||||
| ARIC | 2003 | 721 (36) | 56 (6) | 30 (6) | 490 (24) | 0 (0, 301) | 1519 (76) | 67 (3) | 69 (3) | 348 (17) |
| CHS | 190 | 66 (35) | 73 (5) | 29 (5) | 29 (15.3) | 0 (0, 74) | 123 (65) | 61(32) | 2 (1) | 4 (2) |
| GTP | 230 | 76 (33) | 42 (12) | 32 (8) | 74 (39) | 14 (0,143) | 45 (20) | 113 (49) | NA | 72 (31) |
| CD14+ monocytes (n=1251) | ||||||||||
| MESA | 1251 | 606 (48) | 60 (9) | 30 (6) | 114 (9%) | 8 (0, 191) | 691 (55) | 444 (36) | 65 (5) | 51 (4) |
Abbreviations: ARIC, The Atherosclerosis Risk in Communities study; BMI, body mass index; CHS, The Cardiovascular Health Study; EPIC-Norfolk, The European Prospective Investigation into Cancer-Norfolk study; FHS, The Framingham Heart Study; GTP, The Grady Trauma Project; KORA F4, The Cooperative Health Research in the Region of Augsburg study; InCHIANTI, Invecchiare in Chianti; LBC1936, The Lothian Birth Cohort 1936; MESA, The Multi-Ethnic Study of Atherosclerosis; NAS, The Normative Aging Study; PIVUS, The Prospective Investigation of the Vasculature in Uppsala Seniors Study; RS, The Rotterdam Study; TwinsUK, The TwinsUK Study. The drinking categories were defined based on grams of alcohol consumed per day (g per day): non-drinkers, g per day=0; light drinkers, 0<–≤28 g per day in men and 0<–≤14 g per day in women; at-risk drinkers, 28<–<42 g per day in men and 14<–<28 g per day in women; and heavy drinkers, g per day ≥42 in men and ≥28 in women. The Monocyte samples included mixed samples of European (47%), African (21%) and Hispanic (32%) ancestries.
Alcohol traits
Alcohol consumption was measured by self-administered questionnaires or structured interview with a trained psychologist at the same period when blood samples were obtained for DNA methylation quantification. Alcohol consumption measured the total consumption of beer, wine and spirits. For American cohorts, a drink was defined as 12 ounces of beer, 4–5 ounces of wine or 1.5 ounces of liquor, where one drink is equivalent to ~14 g of ethanol. For European cohorts, a slightly different definition of ‘a drink’ and its conversion to grams of ethanol was used (Supplementary Information: pp 14–16, 19–22 in Description of study samples). The continuous exposure variable was defined as the average grams of ethanol consumed per day (g per day) over the course of a year during the period when the blood sample was collected for DNA methylation quantification. The continuous variable was further categorized into four drinking categories. ‘Non-drinkers’ were subjects with no alcohol consumption (i.e., g per day=0); ‘light drinkers’ were subjects who consumed 0<g per day⩽28 in men and 0<g per day⩽14 in women; ‘at risk-drinkers’ were subjects who consumed 28<g per day<42 in men and 14<g per day<28 in women; ‘heavy drinkers’ were subjects who consumed ⩾42 g per day in men and ⩾28 g per day in women.
To explore the effects of ‘former’ alcohol drinking on DNA methylation, we examined alcohol consumption at prior examinations for all current non-drinkers (n=693, see Table 1) in the Framingham Heart Study (FHS) because information on prior drinking was not available in the majority of other cohorts. We classified non-drinkers in FHS into ‘never’ drinkers and ‘former’ drinkers. ‘Never’ drinkers were individuals who self-reported no alcohol consumption at any prior examination; ‘former’ drinkers were individuals who reported alcohol consumption at any prior examination. For ‘former’ drinkers, we calculated their alcohol consumption (‘g per day’) at each prior examination.
DNA methylation quantification and quality control
DNA was extracted from whole-blood (n=9643 European (EA) and 2423 African ancestry (AA)) and CD14+ monocyte (n=1251 of mixed EA (n=588), AA (n=263) and Hispanic ancestry (n=400) samples (Table 1 and Supplementary Information). Detailed information about DNA extraction, bisulfite conversion, methylation profiling, normalization and quality control (QC) procedures can be found in Supplementary Information. Study samples were excluded from analysis if they had a missing rate of >1–5% across methylation probles; poor single nucleotide polymorphism (SNP) matching compared with previous genotyping of the 65 SNPs included on the methylation array; or sample outliers identified by multidimensional scaling techniques. The methylation probes were excluded if they were the 65 SNP probes, or probes that were previously identified to map to multiple locations (n=29 233);19 had average detection P>0.01 (the detection P-value indicates the probe performance); had an underlying SNP within 10 bp of that probe or if the minor allele frequency (MAF) of the underlying SNP was >5% in the 1000 Genomes Project data (n=15 178).19 After these filtering procedures, ~440 000 DNA methylation probes remained for subsequent analyses.
Clinical and laboratory phenotypes
Age, height and weight were measured using standard protocols implemented at the time DNA samples were collected. Body mass index (BMI) was calculated as weight (kg) per height (m) squared. Smoking was determined by self-report. Current smokers were defined as smokers of at least 1 cigarette per day over the course of a year at the time of blood sample collection for methylation quantification.
In the FHS cohort, the serum AST and ALT levels were measured on fasting morning samples using the kinetic method20 at the same examination cycle when whole blood was obtained for DNA methylation measurement. ALT and AST were set to 5 if their measured levels were <5 U l−1. An individual was defined as having CVD if he or she had coronary heart disease, myocardial infarction, atherothrombotic infarction of brain or congestive heart failure. A cancer phenotype was defined if the person had any type of cancer. Both CVD and cancer phenotypes were defined at the time of blood collection for measuring DNA methylation.
Statistical analysis
Epigenome-wide association studies
In each cohort, the primary EWAS model used a DNA methylation β-value (the ratio of methylated probe intensity divided by the sum of the methylation and unmethylated probe intensity) as the outcome variable and the continuous alcohol trait (g per day) as the predictor variable of interest. Because it has been shown that many CpG sites are significantly associated with age,21 sex22 and BMI,23 these three variables were adjusted for in EWAS analysis. Furthermore, it has been known that DNA methylation profiling is susceptible to batch effects24 and by white cell composition in whole blood.25 Thus, ‘batch effects’ and ‘white cell blood counts’ were adjusted for to minimize confounding effects that may result in spurious association. To account for batch effects, ‘lab’, ‘experiment date’, ‘plate’, ‘row’ and ‘column’ were adjusted for in the analyses. The measured or imputed25 white blood cell counts for CD4 cells, CD8 cells, natural killer cells, B cells and monocytes were adjusted for in the analyses. Surrogate variables (to account for unknown confounders)26 or principal components (estimated from genotypes to account for population stratification)27were included in the EWAS model when applicable (pp 10, 11 and 15 of Description of study samples in Supplementary Information). In the secondary analysis, we used the categorical alcohol intake as the predictor adjusting for the same covariates described above. The non-drinker category was used as the reference group. We used a linear model in unrelated individuals or a linear mixed model in family samples to account for familial correlation in the association of DNA methylation and alcohol consumption.
It is unclear if smoking confounds the relationship between alcohol intake and DNA methylation or if smoking and alcohol intake are associated with common CpGs. Therefore, we performed an additional sensitivity analysis with and without current smoking status as a covariate in drinker-only samples. We compared the change in the regression coefficient for the continuous alcohol intake trait when including current smoking in the model compared with the regression coefficient when smoking was not included in the model using the following equation:
We performed sensitivity tests in the FHS data to investigate if CVD and cancer confounded the relationship between DNA methylation and alcohol consumption. The sensitivity test compared the regression coefficients and P-values between methylation and alcohol intake in a EWAS model that did not adjust for CVD or cancer status to a second model that adjusted for CVD or cancer status. Other covariates included age, sex, BMI, batch effects and white blood cell counts.
DNA methylation as a biomarker in predicting alcohol consumption
We performed the following four-step analyses to investigate if DNA methylation can be used as a biomarker in discriminating alcohol consumption categories (Figure 1).
Figure 1.

Overview of the study design. ARIC, The Atherosclerosis Risk in Communities study; BMI, body mass index; DNAm, DNA methylation value; FHS, the Framingham Heart Study; I (light/at-risk/heavy drinkers versus non-drinkers), the indicator variable for light drinkers versus non-drinkers, at-risk drinkers versus non-drinkers and heavy drinkers versus non-drinkers; KORA F4, The Cooperative Health Research in the Region of Augsburg study; LASSO, least absolute shrinkage and selection operator; LBC, The Lothian Birth Cohort; MESA, The Multi-Ethnic Study of Atherosclerosis; WBCs, white blood cell counts.
Step 1: To establish independent replication cohorts, we split the whole-blood DNA samples from 10 cohorts of (EA (n=9643; Table 1) into separate discovery and replication sets. The discovery set consisted of eight EA cohorts (n=6926), excluding the Lothian Birth Cohort 1936 (LBC1936) and Cooperative Health Research in the Region of Augsburg (KORA F4) study (Table 1). We performed a meta-analysis in the eight EA cohorts using an inverse-variance weighted random-effects model and selected CpGs at a relaxed threshold P<5 × 10−6.
Step 2: To minimize overfitting and to explore which CpGs are more important for including in a biomarker of alcohol consumption, we performed least absolute shrinkage and selection operator (LASSO) regression in the FHS cohort as a training set:
In the above formula, all CpGs at P<5 × 10−6 were included simultaneously in the LASSO analysis. Because alcohol consumption was right skewed and contained non-drinkers, the log-transformed alcohol consumption (log (g per day+1)) was used as the outcome. To minimize potential confounding effects in selecting a set of CpGs as a biomarker, we obtained the residuals for each CpG in a linear regression model (CpG=age+sex+BMI+batch effects+white blood cell counts). Here variables for ‘batch effects’ and ‘white blood cell counts’ were the same as the variables used in EWAS analysis. In the LASSO analysis, we selected four sets of CpGs using s=‘lambda.min’, ‘lambda.1se’, 0.08 and 0.12. The criterion s=‘lambda.min’ selected the largest number of CpGs and s=0.12 yielded the most parsimonious set of CpGs. We removed CpGs if they are not on the Infinium MethylationEPIC BeadChip (Illumina), which will replace the Illumina Infinium HumanMethylation450 BeadChip.
Step 3: In the FHS training set, we first estimated the proportion of variance in continuous alcohol consumption explained by the selected CpGs. The adjusted R2 was estimated for the ‘Null’ model log(g per day+1)=age+sex+BMI and the ‘Full’ model log (g per day+1)=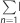 ResidCpG+age+sex+BMI. The proportion of variance explained by a set of CpGs was the difference of adjusted R2 between the ‘Full’ and ‘Null’ models:
ResidCpG+age+sex+BMI. The proportion of variance explained by a set of CpGs was the difference of adjusted R2 between the ‘Full’ and ‘Null’ models:  . Discrimination of heavy alcohol consumption from non-drinkers or light drinkers was our main focus. Therefore, we generated receiver-operating characteristic curves (ROC) in the FHS training cohort to evaluate the performance of these four sets of CpGs in classifying current heavy drinkers versus (1) non-drinkers, (2) light drinkers and (3) pooled individuals of light or non-drinkers. In addition, we evaluated if these CpGs can be used in classifying individuals in the following comparisons: (4) heavy drinkers versus at-risk-drinkers; (5) at-risk drinkers versus non-drinkers; (6) at-risk drinkers versus light drinkers; and (7) light drinkers versus non-drinkers. In all comparison pairs, the former category was the ‘disease’ group and the latter was the ‘control’ group. In ROC analysis, the expected probability of being ‘diseased’ was calculated using logistic regression in which the ‘disease’ (1/0) was used as the outcome variable, and age, sex and BMI without or with a set of CpGs (residuals) as independent variables. Sensitivity, specificity and the AUC for classifying ‘diseased’ individuals versus ‘controls’ were calculated. We also performed sensitivity tests to investigate the prediction performance from current smoking, ALT and AST.
. Discrimination of heavy alcohol consumption from non-drinkers or light drinkers was our main focus. Therefore, we generated receiver-operating characteristic curves (ROC) in the FHS training cohort to evaluate the performance of these four sets of CpGs in classifying current heavy drinkers versus (1) non-drinkers, (2) light drinkers and (3) pooled individuals of light or non-drinkers. In addition, we evaluated if these CpGs can be used in classifying individuals in the following comparisons: (4) heavy drinkers versus at-risk-drinkers; (5) at-risk drinkers versus non-drinkers; (6) at-risk drinkers versus light drinkers; and (7) light drinkers versus non-drinkers. In all comparison pairs, the former category was the ‘disease’ group and the latter was the ‘control’ group. In ROC analysis, the expected probability of being ‘diseased’ was calculated using logistic regression in which the ‘disease’ (1/0) was used as the outcome variable, and age, sex and BMI without or with a set of CpGs (residuals) as independent variables. Sensitivity, specificity and the AUC for classifying ‘diseased’ individuals versus ‘controls’ were calculated. We also performed sensitivity tests to investigate the prediction performance from current smoking, ALT and AST.
Step 4: We repeated the Step 3 analyses in two independent cohorts of whole-blood-derived DNA samples in people of EA (LBC1936 and KORA F4) for replication purposes. We also repeated the Step 3 analyses in whole-blood-derived DNA samples of people of AA (the Atherosclerosis Risk in Communities Study or ARIC) and in the monocyte-derived DNA samples (the Multi-Ethnic Study of Atherosclerosis or MESA) for both replication and generalization. The MESA samples included individuals of EA (n=588), AA (n=263) and Hispanic ancestry (n=400). We used all 1251 individuals in MESA to estimate the proportion of variance in alcohol consumption that was explained by the CpGs, but only used the 588 individuals of EA for the ROC analysis; there were too few heavy drinkers of AA or Hispanic ancestry for meaningful analysis.
The R statistical software (https://www.r-project.org/) was used for all analyses. Linear regression was performed using the function ‘lm’ for unrelated samples and ‘lme’ for family samples to account for family structure. LASSO was performed using the function ‘glenet’ in the ‘glenet’ R package with the parameter α=1 and 10-fold cross-validation to select CpGs. The ROC analysis used the ‘pROC’ package with the ‘lme’ function for logistic regression, and then the ‘predict’ function to predict the expected probability, and finally the ‘roc’ function to estimate sensitivity and specificity of a set of predictors for predicting ‘disease’ versus ‘control’ status.
Meta-analysis to identify DNA methylation signature
The inverse variance-weighted random-effects model28 was used in meta-analysis because of the heterogeneity in levels of alcohol consumption and population demographics (Table 1). The meta-analysis was performed in ancestry-stratified whole-blood-derived DNA samples (n=9643 EA and n=2423 AA) and, secondarily, in combined transethnic samples of whole-blood and monocyte-derived DNA (n=13 317). In the meta-analysis, a CpG was further removed if it was missing in five or more studies or its sample size was <20% of the total sample size. We used P<0.05/440 000 ~1 × 10−7 to establish significance.
We reported alcohol-related CpGs (P⩽1 × 10−7) in meta-analysis of ancestry-stratified whole-blood-derived DNA samples and in monocyte-derived DNA samples, and compared alcohol-related CpGs between ancestries and between whole-blood and monocyte samples. We also investigated the DNA methylation levels in several genes that were previously reported to be associated with alcohol metabolism29, 30 or alcohol-related neurotransmission.31, 32, 33, 34
DNA methylation in former and never drinkers
To investigate if DNA methylation signals differ between ‘former’ and ‘never’ drinkers, we performed three additional EWAS analyses with DNA methylation as the outcome variable and three binary traits as the independent variables (adjusting for age, sex, BMI, batch effects and white blood cell counts) in the FHS data. The first analysis using the binary trait ‘never’ versus ‘former’ as the independent variable, and the other two the binary traits ‘heavy’ versus ‘never’ or ‘heavy’ versus ‘former’ as the independent variable. A linear mixed-effects model was used to account for family structure.
Methylation quantitative trait loci analysis
Methylation quantitative trait locus analysis (meQTLs) was performed in three cohorts: FHS (n=2024), KORA F4 (n=1799) and the Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS) study (n=920). Genotyping, genotype imputation and QC details are described in the Supplementary Information. Using data imputed (allele dosage) to the 1000 Genomes (reference), we selected cis-SNPs (defined as ±100 kb) with imputation quality score >0.8 and minor allele frequency ⩾0.05. The meQTL mapping was performed between the significant alcohol-related CpGs (outcomes) and cis-SNPs (predictors), adjusting for age, sex, BMI, batch effects and white blood cell counts. The proportion of variance (r2) that can be explained by cis-SNPs or meQTLs for a CpG was also calculated in association analysis. We used a linear model in unrelated individuals or a linear mixed-effects model in family samples to account for familial correlation in association test between an SNP dosage and DNA methylation. Meta-analysis used the inverse-variance weighted random-effects model. We used P<0.05/n to establish significance, where n was the number of CpG–SNP pairs tested.
Association analysis between methylation and gene expression
Gene expression profiling and QC in FHS (n=1924) and KORA F4 (n=707) are detailed in the Supplementary Information. To perform the association analysis, the FHS samples were divided into discovery (n=966) and replication (n=958) sets by independent pedigrees. In both FHS and KORA F4 samples, residuals of gene expression levels (ResidGene) or CpG β-values (ResidCpG) were obtained by adjusting for age, sex, BMI, batch effects and white blood cell counts. Here batch effects and cell proportion differentials (if calculated) were expression-specific or methylation-specific values. The association analysis was then performed between ResidGene and ResidCpG. A linear model was used in unrelated samples and a linear mixed model was used in family data to account for family structure. The proportion of variance in a transcript that was explained by a CpG was also calculated. Because FHS and KORA F4 used different expression arrays, we only used CpG–gene name pairs that could be matched between the two studies. Therefore, we used the Z-score method35 for meta-analysis. We used P<0.05/n to establish statistical significance, where n was the number of CpG-gene transcript pairs tested.
Functional inference and pathway analysis
Genomic features of the alcohol-related CpGs
The genomic location of a CpG provides functional insight into regulatory features.36 According to the annotation ‘HumanMethylation450_15017482_v.1.2.csv’ provided by Illumina, we compared the enrichment or depletion of several genomic features, including CpG islands, CpG shores and shelves, enhancers, DNA hypersensitivity sites and promoters in the set of alcohol-related CpGs (P<1 × 10−7) compared with the background universe of all CpG probes assessed on the microarray that passed QC. The difference in proportions of a genomic feature was compared by the Fisher’s two-sided test.
Gene ontology enrichment analysis and functional inference
We performed Gene Ontology (http://geneontology.org/page/go-enrichment-analysis) enrichment analysis for the genes that were annotated to the significant alcohol-related CpGs. We also examined the genes whose expression levels were significantly associated with the significant alcohol-related CpGs.
Results
A methylation biomarker of alcohol consumption
The meta-analysis of the discovery set that included the whole-blood-derived DNA of individuals of EA from eight cohorts (n=6926; Table 1) identified 361 CpGs at P<5−10−6. Of these 361 CpGs, 333 are on the new Infinium MethylationEPIC BeadChip. Using the FHS cohort as the training set, we selected 5 (s=0.12), 23 (s=0.08), 78 (s= ‘lambda.1se’) and 144 (s= ‘lambda.min’) CpGs out of the 333 CpGs with the LASSO regression (see Materials and Methods). All CpGs in the smaller lists are subsets of the largest set of 144 CpGs (s= ‘labmda.min’) (Supplementary Table 1). All selected CpGs were available in MESA and ARIC. Five CpGs in the 144 set and one in the 78 CpG set were unavailable in KORA F4 and LBC1936 (Supplementary Table 1).
The most parsimonious set of 5 CpGs explained a substantial proportion of interindividual variance in alcohol consumption in KORA F4 (6.4%), LBC1936 (10.4%), ARIC (5.2%), MESA (9.9%) and FHS (15.0%). The addition of more CpGs yielded larger proportions of explained variance in alcohol consumption. The largest set (144 CpGs) explained 13.1 (KORA F4), 12.0 (LBC1936), 13.8 (ARIC), 13.1 (MESA) and 27.3% (FHS) of variance in alcohol consumption (Table 2). Because the FHS was used as the training cohort to select CpGs, the estimated variance values obtained in the FHS were more optimistic compared with those obtained in the four replication cohorts.
Table 2. The proportion of variance in alcohol consumption explained by DNA methylation.
| Study |
Variance explained (%) |
||||
|---|---|---|---|---|---|
| Null | 5 CpGs | 23 CpGs | 78 CpGs | 144 CpGs | |
| KORA F4 | 12.5 | 6.4 | 7.4 | 11.4 | 13.1 |
| LBC1936 | 9.9 | 10.4 | 11.1 | 12.2 | 12.0 |
| ARIC | 20.0 | 5.2 | 6.0 | 12.4 | 13.8 |
| MESA | 11.6 | 9.9 | 10.5 | 11.7 | 13.1 |
| FHS | 7.8 | 15.0 | 18.9 | 24.6 | 27.3 |
Abbreviations: ARIC, Atherosclerosis Risk in Communities Study; BMI, body mass index; CpG, cytosine-phosphate-guanine dinucleotide; FHS, Framingham Heart Study; KORA F4, Cooperative Health Research in the Region of Augsburg; LASSO, least absolute shrinkage and selection operator; LBC1936, The Lothian Birth Cohort 1936; MESA, Multi-Ethnic Study of Atherosclerosis.
The meta-analysis using the whole-blood-derived DNA of individuals of European ancestry from eight discovery cohorts (n=6926, see Materials and methods) excluding KORA F4 and LBC1936 identified 361 CpGs with P<5 × 10−6. Of these 361 CpGs, 333 are on the new Infinium MethylationEPIC BeadChip. Using the FHS data as the training set, we selected 5 (s=0.12), 23 (s=0.08), 78 (s= ‘lambda.1se’) and 144 (s= ‘lambda.min’) CpGs with the LASSO regression (see Materials and Methods). The testing cohorts included two cohorts of European ancestry with the whole-blood-derived DNA samples (KORA F4 and LBC1936), the African ancestry cohort with the whole-blood-derived DNA samples (ARIC) and the cohort of monocyte-derived DNA samples (MESA) of mixed ancestry (see Table 1). We estimated the proportion of variance in alcohol consumption explained by a list of CpGs as the difference of adjusted R2- using a linear regression model that included age, sex and BMI (the ‘Null’ model) and a model that included a list of CpGs in addition to age, sex and BMI. All selected CpGs were available in MESA and ARIC. Five CpGs in the 144 set and one CpG in the 78 set were unavailable in KORA F4 and LBC1936 (see Supplementary Table 1). The estimated variance values using the FHS data are more optimistic compared with those obtained for the four replication cohorts.
In ROC analysis of ‘disease’ versus ‘control’ status (see Materials and Methods), including any CpGs in addition to clinical variables (age, sex and BMI) (the ‘Full’ model) resulted in a larger AUC value compared with the model with only clinical variables (the ‘Null’ model). The models with the two smaller sets of CpGs (5 CpGs and 23 CpGs) yielded good prediction (AUCFull >0.80) in all five cohorts for discriminating heavy drinkers versus non-drinkers; the models with the two larger sets of CpGs (78 CpGs and 144 CpGs) gave good prediction (AUC>0.80) in all five cohorts for discriminating heavy drinkers versus non-drinkers/light drinkers/at-risk drinkers, or in discriminating at-risk drinkers versus non-drinkers (Figure 2 and Supplementary Figure 1). For example, the addition of the 144 CpGs to the null model yielded a high AUC for discriminating heavy drinkers versus non-drinkers (AUCFull=0.90–0.99 compared with AUCNull=0.63–0.80) and heavy drinkers versus light drinkers (AUCFull=0.85–0.99 compared to AUCNull=0.53–0.61) across the five cohorts; the addition of 78 CpGs to the null model yielded slightly lower AUC values compared with addition of the 144 CpGs: AUCFull=0.88–0.99 in discriminating heavy drinkers versus non-drinkers and AUCFull=0.82–0.96 in discriminating heavy drinkers versus light drinkers. It is worth noting that in discriminating heavy drinkers versus non-drinkers/light drinkers, the performance of the 144 CpGs and 78 CpGs was better in MESA and LBC1936 compared with that in FHS (the training cohort); but the performance of these two sets of CpGs was lower in KORA F4 and ARIC (Figure 2). Unavailability of a few CpGs in LBC1936 did not seem to affect discrimination (Table 2 and Figure 2).
Figure 2.

A biomarker of heavy alcohol drinking. Four sets of cytosine-phosphate-guanine dinucleotides (CpGs) were selected at s=0.12 (5 CpGs), s=0.08 (23 CpGs), s=‘lambda.1se’ (78 CpGs) and s=‘lambda.min’ (144 CpGs) using least absolute shrinkage and selection operator (LASSO) in the Framingham Heart Study (FHS) cohort (the training cohort). ROC analysis was performed to classify heavy drinkers versus non-drinkers (left figure) and heavy drinkers versus light drinkers (right figure). ‘Non-drinkers’ were subjects with no alcohol consumption (i.e., g per day=0); ‘light drinkers’ were subjects who consumed 0<g per day⩽28 in men and 0<g per day⩽14 in women; ‘heavy drinkers’ were subjects who consumed ⩾42 g per day in men and ⩾28 g per day in women. ARIC, The Atherosclerosis Risk in Communities study; KORA F4, The Cooperative Health Research in the Region of Augsburg study; LBC1936, The Lothian Birth Cohort 1936; MESA, The Multi-Ethnic Study of Atherosclerosis.
Current smoking explained a very small proportion of variance in alcohol consumption. For example, the change in adjusted R2=0.003 in FHS and 0.01 in MESA when current smoking was included in the model in addition to age, sex and BMI. Similarly, ALT or AST explained a small proportion of variance in alcohol consumption in FHS: the change in adjusted R2=0.004 when either ALS or AST was added to the null model. Therefore, neither ALT nor AST was a good biomarker for alcohol consumption, which was confirmed in ROC analysis: AUCNull+ALT or Null+AST=0.67 when ALT or AST was added in the null model (AUCNull=0.66) in discriminating heavy drinkers versus non-drinkers in FHS.
Epigenome-wide methylation signature of alcohol intake
In the main text, we reported ancestry-stratified meta-analysis (P<1 × 10−7) for whole-blood-derived DNA in individuals of EA (n=9643) and AA (n=2423) (Table 1) using an inverse-variance weighted random-effects model (Figure 1). Meta-analysis of pooled samples (n=13 317) and several sensitivity tests including EWAS in only drinkers and the investigation of whether or not prevalent CVD or cancer confound the relationship between DNA methylation and alcohol consumption are included in the Supplementary Information.
Genome-wide pattern of DNA methylation associated with alcohol consumption
We identified hundreds of CpGs (P<1 × 10−7) whose differential methylation across the genome was associated with alcohol intake: 363 CpGs in whole-blood samples of individuals of EA (Table 3, Figure 3 and Supplementary Table 2), 165 CpGs in whole-blood samples of individuals of (AA (Supplementary Table 3 and Supplementary Figure 2) and 62 CpGs in monocyte-derived DNA samples (Supplementary Table 4 and Supplementary Figure 3). Additional CpGs at P<1 × 10−4 are reported in Supplementary Tables 5–7. Genomic inflation in meta-analysis was estimated at ~10% or less, indicating low additional risk of false-positive findings (Supplementary Table 8). The majority of the alcohol-related CpGs exhibited an inverse relationship between higher alcohol intake and lower methylation (Supplementary Tables 2–4).
Table 3. The 30 most significant CpGs in relation to continuous alcohol intake in meta-analysis of whole-blood samples of European ancestry.
| IlmnID | UCSC gene | Chr | Position | P-value | β | S.e. | UCSC CpG islands | Relation to UCSC CpG island | Enhancer |
|---|---|---|---|---|---|---|---|---|---|
| cg03523740 | TXLNA | 1 | 32 645 027 | 4.4E−15 | −0.00022 | 2.8E−05 | Chr 1:32 645 154–32 645 814 | N_Shore | |
| cg20970369 | DENND2D | 1 | 111 744 108 | 3.2E−12 | −0.00023 | 3.3E−05 | Chr 1:111 746 337–111 747 303 | N_Shelf | |
| cg16246545 | PHGDH | 1 | 120 255 941 | 1.5E−12 | −0.00061 | 8.6E−05 | Chr 1:120 254 844–120 255 499 | S_Shore | |
| cg19266329 | 1 | 145 456 128 | 1.7E−13 | −0.00028 | 3.8E−05 | TRUE | |||
| cg19238380 | LMNA | 1 | 156 093 948 | 2.2E−12 | −0.00029 | 4.2E−05 | TRUE | ||
| cg11194994 | PEA15 | 1 | 160 175 974 | 7.3E−15 | −0.00017 | 2.2E−05 | Chr 1:160 175 132–160 175 702 | S_Shore | |
| cg07502661 | 2 | 43 398 339 | 2.6E−12 | −0.00019 | 2.7E−05 | Chr 2:43 398 040–43398276 | S_Shore | ||
| cg00883689 | SPTBN1 | 2 | 54 802 904 | 3.2E−12 | −0.00028 | 4.1E−05 | TRUE | ||
| cg13729116 | LETM1 | 4 | 1 859 262 | 6.7E−18 | −0.00018 | 2.1E−05 | Chr 4:1 857 065–1 858 887 | S_Shore | |
| cg25518868 | DIAPH1 | 5 | 140 984 057 | 2.3E−12 | −0.00012 | 1.8E−05 | TRUE | ||
| cg05593667 | 6 | 35 490 744 | 4.4E−16 | −0.00025 | 3.1E−05 | ||||
| cg20732076 | TRERF1 | 6 | 42 335 231 | 1.5E−12 | −0.00015 | 2.1E−05 | TRUE | ||
| cg06189038 | GAL3ST4 | 7 | 99 767 134 | 4.6E−13 | −0.00016 | 2.2E−05 | Chr 7:99 768 884–99 769 559 | N_Shore | |
| cg12873476 | 8 | 142 402 728 | 2.8E−12 | −0.00023 | 3.3E−05 | Chr 8:142 401 533−142 402 494 | S_Shore | TRUE | |
| cg03599037 | C10orf58 | 10 | 82 172 508 | 4.5E−13 | −0.00014 | 1.9E−05 | Chr 10:82 168 064–82 168 917 | S_Shelf | |
| cg06603309 | KCNQ1 | 11 | 2 724 144 | 2.7E−14 | 0.00017 | 2.2E−05 | Chr 11:2 720 410–2 722 087 | S_Shelf | TRUE |
| cg11376147 | SLC43A1 | 11 | 57 261 198 | 9.8E−13 | −0.00026 | 3.6E−05 | TRUE | ||
| cg00271311 | CNTF | 11 | 58 389 290 | 1.6E−13 | −0.00022 | 2.9E−05 | |||
| cg09448652 | SNORD30 | 11 | 62 621 367 | 1.3E−12 | −0.00026 | 3.6E−05 | Chr 11:62 623 359–62 623 877 | N_Shore | |
| cg09737197 | CPT1A | 11 | 68 607 675 | 5.0E−13 | −0.00016 | 2.2E−05 | Chr 11:68 608 155–68609419 | N_Shore | |
| cg02583484 | HNRNPA1 | 12 | 54 677 008 | 1.6E−19 | −0.00039 | 4.4E−05 | Chr 12:54 673 322–54 673 550 | S_Shelf | |
| cg23654112 | TBC1D24 | 16 | 2525 928 | 3.0E−13 | −0.00014 | 1.9E−05 | Chr 16:2 521 086–2 525 929 | Island | |
| cg08916477 | SEPT1 | 16 | 30 391 350 | 4.0E−13 | −0.00016 | 2.2E−05 | Chr 16:30 389 035–30 390 631 | S_Shore | |
| cg06469895 | TERF2 | 16 | 69 418 206 | 1.5E−13 | −0.00020 | 2.8E−05 | Chr 16:69 419 316–69 420 086 | N_Shore | |
| cg00574412 | ABHD15 | 17 | 27 892 866 | 1.1E−12 | −0.00017 | 2.3E−05 | Chr 17:27 893 086–27 896 078 | N_Shore | |
| cg21626848 | SC65 | 17 | 39 969 267 | 3.1E−15 | −0.00023 | 2.9E−05 | Chr 17:39 967 407–39 968 604 | S_Shore | |
| cg08677210 | MSI2 | 17 | 55 550 613 | 3.3E−12 | −0.00013 | 1.9E−05 | TRUE | ||
| cg15253293 | 17 | 79 366 853 | 1.1E−15 | −0.00014 | 1.7E−05 | Chr 17:79 366 806–79374742 | Island | ||
| cg24217948 | SETBP1 | 18 | 42 261 980 | 2.1E−12 | −0.00028 | 3.9E−05 | Chr 18:42 258 983–42 260 795 | S_Shore | TRUE |
| cg13127741 | COMMD7 | 20 | 31 331 821 | 3.1E−12 | −0.00023 | 3.3E−05 | Chr 20:31 330 957–31 331 410 | S_Shore |
Abbreviation: CpG, cytosine-phosphate-guanine dinucleotide; S.e, standard error.
Epigenome-wide association and meta-analysis of the continuous alcohol intake was performed using all whole-blood-derived DNA samples of European ancestry. The DNA methylation proportion was the outcome variable, grams alcohol consumed per day (g per day) was the predictor variable, adjusting for age, sex, body mass index, technical covariates and white blood cell counts. The inverse-variance weighted random-effects model was performed in meta-analysis (See Supplementary Table 2 for a full set of significant CpGs). The annotation “HumanMethylation450_15017482_v.1.2.csv” provided by Illumina was used to annotate the CpG loci.
Figure 3.

Meta-analysis of epigenome-wide association of alcohol intake in European ancestry (EA) whole-blood samples: the Manhattan plot (top) and the volcano plot (bottom). The DNA methylation proportion was the outcome variable, grams alcohol consumed per day (g per day) was the predictor variable, adjusting for age, sex, body mass index, technical covariates and white blood cell counts. The inverse-variance weighted random-effects model was performed in meta-analysis using all whole blood DNA samples of EA.
Fewer alcohol-related CpGs (P<1 × 10−7) were identified in the analysis of the categorical alcohol trait that compared light drinkers, at-risk drinkers and heavy drinkers with non-drinkers (Supplementary Tables 9–11 and Supplementary Figures 4–6). Additional CpGs at P<1 × 10−4 are included in Supplementary Tables 12–14. The majority of the alcohol-related CpGs identified in the analysis of the categorical alcohol trait (Supplementary Tables 9–11) were also significant or nominally significant in the association with the continuous alcohol consumption trait (Supplementary Tables 2–4).
Transethnic replication of methylation signatures
Of the 363 alcohol-related CpGs in EA samples, 56 had P<0.00014 (0.05/363) in AA samples; of the 165 alcohol-related CpGs in AA samples, 59 had P<0.00030 (0.05/165) in EA samples. Effect estimates of the 518 (union of 363 and 165) unique CpGs were moderately correlated between EA and AA whole-blood samples: Pearson’s correlation r=0.64 (Figure 4a). For example, cg11376147 in solute carrier family 43 (SLC43A1) displayed P<1 × 10−7 in both EA and AA samples from whole blood (Figure 4b).
Figure 4.

Comparison of regression coefficients of the significant cytosine-phosphate-guanine dinucleotides (CpGs) in association analysis of the continuous alcohol trait (g per day): (a) between European and African whole-blood samples; (b) the Forest plot of effect estimates and standard errors of cg11376147 in all study cohorts; and (c) between European whole-blood and CD14+ monocyte samples. (a) Includes a list of CpGs with P<1 × 10−7 in EA or AA whole-blood samples and (c) includes a list of CpGs with P<1 × 10−7 in EA whole-blood samples or in monocyte samples of mixed ancestries. The Pearson’s correlation was r=0.64 between the effect estimates in (a) and r=0.72 in (c). MM, monocyte, mixed ancestries; WB AA, whole blood, African ancestry; WB EA, whole blood, European ancestry.
Methylation signature in whole-blood- and monocyte-derived DNA
Of the 363 alcohol-related CpGs in EA whole blood samples, 57 replicated (P<0.00014; 0.05/363) in monocyte samples. Of the 62 alcohol-related CpGs in monocytes, 13 replicated (P<0.0008; 0.05/62) in whole-blood EA samples. The Pearson’s correlation was 0.72 for the 417 unique (union of 363 and 62) CpGs between EA whole-blood samples and monocyte samples (Figure 4c). For example, cg11376147 in SLC43A1 also displayed P<1 × 10−7 for association with alcohol consumption in monocyte-derived DNA (Figure 4b).
Similar DNA methylation pattern in former and never drinkers
Based on alcohol consumption at prior examinations in FHS, we classified the 693 non-drinkers (Table 1) into ‘never’ (n=107) and ‘former’ drinkers (n=586). Furthermore, among the 586 ‘former’ drinkers, 91 were ‘former’ heavy drinkers, 66 were ‘former’ at-risk drinkers and 429 were ‘former’ light drinkers. The EWAS using the binary trait ‘never’ versus ‘former’ as the independent variable did not yield any significant results (Supplementary Figure 7). We compared the EWAS results between ‘heavy’ versus ‘never’ and ‘heavy’ versus ‘former’. For genome-wide methylation loci, the correlation was 0.32 for regression coefficients and 0.20 for –log10 (P-values); for loci with P–value <1 × 10−7 (n=92) in either ‘heavy’ versus ‘never’ or ‘heavy’ versus ‘former’ drinkers, the correlation was 0.91 for regression coefficients and 0.88 for –log10 (P-values) (Supplementary Figure 8). These results indicate that DNA methylation levels were not considerably different between ‘never’ drinkers and ‘former’ drinkers and that DNA methylation changes due to heavy alcohol consumption revert after individuals abstained from alcohol intake for several years (FHS examinations were ~4 years apart).
Evaluation of smoking in the association between alcohol intake and DNA methylation
It is unclear if current cigarette smoking confounds the association between DNA methylation and alcohol intake, or if smoking and alcohol intake are associated with common CpGs. Therefore, we performed an analysis using smoking as an additional covariate in the EWAS (see Materials and methods). We found that some alcohol-related CpGs displayed a large change (>10% change) in the size of their regression coefficients when smoking was included as an additional covariate in the analysis of whole-blood-derived DNA samples in individuals of EA (35 of the 363 CpGs at P<1 × 10−7) and AA (92 of the 165 CpGs at P<1 × 10−7) ancestries, but none of the CpGs in the monocyte-derived DNA samples changed appreciably after additionally adjusting for smoking (Supplementary Tables 2–4). Several of the identified CpGs that displayed a large change in effect estimates following adjustment for smoking have been previously reported to be associated with smoking, including the CpGs in the aryl-hydrocarbon receptor repressor37, 38 (Supplementary Tables 5 and 6). We excluded the 35 CpGs that showed large change in effect estimates after adjusting for smoking (Supplementary Table 2) in subsequent analyses that were performed using the whole-blood-derived DNA samples from individuals of EA ancestry.
Alcohol metabolism enzymes and alcohol-related DNA methylation
Several functional DNA sequence variants in the alcohol dehydrogenase (ADH) and aldehyde dehydrogenase (ALDH) family of genes are known for their effects on alcohol metabolism.29, 30 We checked CpGs in the introns, exons and regulatory regions in these gene families according to the annotation provided by Illumina. No CpGs in the ADH (30 CpGs in seven genes) or ALDH (340 CpGs in 19 genes) loci were differentially methylated (P<1 × 10−7) in relation to alcohol use (Supplementary Tables 5–7, 15, 16).
Neurotransmitter receptors and alcohol-related DNA methylation
γ-aminobutyric acid (GABA), a major neurotransmitter, and its receptors are known for their involvement in the acute and chronic behavioral effects of ethanol in humans and animal models.31, 32, 33, 34 A total of 607 CpGs were mapped to introns, exons and regulatory regions of 22 GABA receptor genes (Supplementary Table 17). In meta-analysis of whole-blood-derived DNA samples of EA individuals, cg04781796 (βalcohol=0.0002, P=1.9 × 10−9) and cg09577455 (βalcohol=−0.0015, P=3.0 × 10−8) were significantly associated with alcohol consumption (Supplementary Table 2). However, neither of these CpGs was significant in whole-blood-derived DNA samples of people of AA (cg04781796: βalcohol=0.00004, P=0.17; cg09577455: βalcohol=−0.00003, P=0.0016) or in monocyte-derived DNA samples (cg04781796: βalcohol=0.0001, P=0.22; cg09577455: βalcohol=−0.00002, P=0.82). The CpG cg04781796 is located in a CpG island (Chr 1: 1 959 414–1 959 867, hg19) that is intronic to the GABA-A receptor, delta (GABRD; Figure 5); and cg09577455 is located in the north shore of a CpG island (Chr 6: 29 595 298–29 595 795, hg19) that is intronic to GABA-B receptor subunit 1 (GABBR1, Chr 6; Supplementary Figure 9). CpG cg04781796 displayed higher methylation in relation to greater alcohol intake, whereas cg09577455 displayed lower methylation levels in association with increased alcohol intake.
Figure 5.

The γ-aminobutyric acid-A (GABA-A) receptor, delta (GABRD): the associations of the 36 cytosine-phosphate-guanine dinucleotides (CpGs) within GABRD, genomic and regulatory features and correlation of methylation measurements. The results were obtained in meta-analysis of the association analysis of 9643 whole-blood-derived DNA samples of European ancestry (EA) individuals. The correlation of these 36 CpGs was calculated using the methylation measurements at 36 CpGs, adjusting for age, sex, technical covariates and white cell blood counts in the Framingham Heart Study samples.
Genetic basis underlying the significant alcohol-related CpGs
We tested for association of the methylation levels of 328 CpGs (selected in meta-analysis of DNA of EA individuals in Supplementary Table 2) with nearby SNPs (±100KB, cis-SNPs or cis-meQTLs) in three cohorts (total number of individuals=4623 from FHS, KORA F4 and PIVUS) (see Materials and methods). A total of 105 759 SNP-CpG pairs were formed from these 328 CpGs with cis-SNPs. Meta-analysis of the FHS, KORA F4 and PIVUS samples identified 14 160 SNP–CpG pairs (170 unique CpGs and 12 857 unique cis-SNPs) at P<0.05/105 759~4.7 × 10−7 (Supplementary Table 18). We found that 16 CpGs had meQTLs that explained 20–61% of interindividual variance in methylation at the corresponding CpG (Supplementary Table 18). None of these 12 857 significant meQTLs was associated with alcohol-related traits (P<1 × 10−8) by querying these significant meQTLs in the Catalog of Published Genome-Wide Association Studies (http://www.genome.gov/gwastudies/, latest version released on May 12, 2015).
Association of alcohol-related DNA methylation with gene expression
We tested for associations between the 328 alcohol-related CpGs and blood gene expression levels in FHS (n=1924) and KORA F4 (n=707) for genes within 1 Mb of these 328 CpGs in both studies (see Materials and methods). Meta-analysis identified 110 CpG–gene pairs (83 unique CpGs and 100 unique genes) at P<0.05/7111=7 × 10−6, where 7111 is the number of tested CpG–gene pairs (Supplementary Table 19). Of the 110 significant pairs, 86 (78%) displayed negative correlations between methylation and mRNA levels.
We examined associations of the two significant alcohol-related CpGs in GABA receptor genes with expression of cis genes. At the GABRD locus, cg04781796 was not associated with expression of any genes in blood within 1 Mb, whereas cg09577455 in the GABBR1 locus was associated with the expression of the interferon-induced transmembrane protein 4 pseudogene (IFITM4P; P=2.4 × 10−6) (Supplementary Table 19). Owing to the important role of GABA receptors in alcohol-induced signal transduction and immune functions, we carried out additional association analyses between these two CpGs and gene transcripts beyond 1 Mb or on different chromosomes (i.e. trans associations) in both FHS and KORA F4. Of the 35 746 association pairs, 228 showed significant association (P<0.05/35 746~1.4 × 10−6) with methylation of cg04781796 (Supplementary Table 20) and 13 transcripts were associated with cg09577455 (Supplementary Table 21) in the meta-analysis.
Functional inference and pathway analysis
Genomic features of the alcohol-related CpGs
We found that the 328 alcohol-related CpG set was significantly enriched for CpG island shores (48% versus 24%, P=7.3 × 10−12) and enhancers (29% versus 22%, P=0.003) compared with all CpGs that passed QC in meta-analysis. In contrast, the 328 alcohol-associated CpG set was significantly depleted for CpG islands (16% versus 32%, P=1.1 × 10−6) and promoters (3% versus 7% P=0.009) (Supplementary Table 22). There was no significant difference in proportions of CpG island shelves and DNase I hypersensitive sites among the 328 alcohol-associated CpGs.
We found similar enrichment and depletion for the 144 CpGs that were selected in biomarker analysis. These 144 CpGs were significantly enriched for CpG island shores (47% versus 24%, P=4.5 × 10−11) and enhancers (30% versus 22%, P=0.002), but significantly depleted for CpG islands (13% versus 32%, P=1.0 × 10−6) and promoters (2% versus 7%, P=0.007).
Gene ontology enrichment analysis and functional inference
A total of 257 genes were annotated to the 328 alcohol-related CpGs. These 257 genes were enriched for 95 biological processes (Bonferroni-corrected P<0.05, Supplementary Table 23) including regulation of transcription, macromolecule metabolic process and cellular response to stress and chemicals. The most significant biological process constituted 32 genes enriched (>4-fold) for ‘negative regulation of transcription from RNA polymerase II promoter’ (Bonferroni-corrected P=2.3 × 10−7; Supplementary Table 24). For the 100 cis genes whose transcript levels were significantly associated with 83 CpGs (Supplementary Table 19), the analysis of biological processes showed that the most significantly enriched process was ‘negative regulation of transposition’ (Bonferroni-corrected P=8.0 × 10−4, Supplementary Table 25). Other enriched processes included defense response to virus (P=0.006) and DNA cytosine deamination (P=0.02). The trans-transcripts that were significantly associated with cg04781796 (GABRD) were enriched for pathways that are involved in immune functions such as lymphocyte activation (P=1.1 × 10−11) and immune system process (P=3.2 × 10−11; Supplementary Table 26). The trans-transcripts that were associated with cg09577455 in GABBR1 were also enriched for immune response (P=0.015; Supplementary Table 27).
Discussion
We conducted an EWAS of alcohol intake in 13 cohorts including 13 317 samples of whole-blood or monocyte-derived DNA from individuals of mostly EA and AA. We identified hundreds of differentially methylated CpGs (P<1 × 10−7) in relation to alcohol consumption. More than half of the alcohol-related methylation sites were associated with cis-genetic variants, supporting the hypothesis that DNA methylation sites are sensitive to both environmental and genetic influences.39 In addition, we developed a robust and replicable DNA methylation biomarker that provides substantial discrimination for current heavy alcohol intake.
A set of 144 CpGs was highly predictive for discriminating current heavy alcohol drinkers from non-drinkers (AUC>0.90) in all replication cohorts. As a biomarker, these selected CpGs performed better than commonly clinical variables and biomarkers in discriminating current heavy alcohol drinking.7 This is in line with the discriminatory power of DNA methylation for other complex traits, such as BMI.40 Therefore, a whole-blood DNA methylation biomarker has the potential to be developed into a commercially marketable diagnostic test to detect current heavy alcohol consumption. Such a test could be useful to supplement and validate self-reported alcohol consumption data, or in a forensic setting, or as a screening test.
The biomarker analysis and ancestry-stratified meta-analysis showed that a number of DNA methylation sites displayed consistent alcohol-related effects in whole-blood samples of people of EA and AA. However, the transancestry comparison also showed the lack of similarities of many CpG sites. We propose three explanations. First, some DNA methylation sites are truly ancestry-specific, which needs to be confirmed by future studies. Second, sample heterogeneity in alcohol consumption may explain a part of the non-concordance for some CpGs in AA and EA groups. For example, in ARIC, ~76% individuals were non-drinkers and ~17% were heavy drinkers, whereas in most EA cohorts, >60% of participants were light drinkers. Third, a large difference in sample sizes (EA n=9643 and AA n=2423) and the probability in sampling are additional reasons for the lack of replication when a Bonferroni-corrected threshold was used.
We provide evidence that alcohol-related DNA methylation is associated with gene expression in whole blood. Of note, we showed that whole-blood epigenetic changes in GABA receptor genes were significantly associated with the expression levels of a number of genes that are involved in immune function supporting the recent findings that GABA and its receptor have effects on immune cells through cross-talk between the nervous system and the immune system.41 However, as our data are cross-sectional and observational in nature, further research is needed to determine if these changes are causal or reactive. The gene set analysis is based on a crucial and unrealistic independence assumption pertaining to genes, which may not be valid for biological processes. Therefore, we should interpret the significant P-values with caution.42
In addition to the cross-sectional nature of this study, our findings were limited to DNA samples from mostly middle- and older-aged individuals of EA and AAs. Future studies are needed to investigate the generalizability of our findings to other age groups and ancestries. Nevertheless, as the largest study of its kind, this work identified a robust alcohol-related DNA methylation signature in blood and demonstrated that the alcohol-related methylation changes in blood are of sufficient magnitude to be interesting clinically, which addresses a gap within the field. Future studies are warranted to investigate whether alcohol-related methylation in blood affects GABA neurotransmitter function in the brain and to investigate how alcohol-related epigenetic modifications influence the beneficial and detrimental downstream consequences of alcohol-related health outcomes. Identifying how alcohol-induced DNA methylation changes modify gene expression and result in pathway activation or suppression may shed light on the molecular basis of alcohol addiction and alcohol-related diseases and reveal new therapeutic strategies.
Acknowledgments
Detailed acknowledgments were included in Supplementary Information.
Accession codes. The meta-analysis results for the continuous and categorical alcohol consumption traits can be downloaded from the database of Genotypes and Phenotypes (dbGaP) CHARGE Summary site under accession phs000930. DNA methylation measurements by several cohorts have been deposited in dbGap. Contact information for obtaining DNA methylation measurements from the European cohorts and some of the American cohorts is available upon request.
Footnotes
Supplementary Information accompanies the paper on the Molecular Psychiatry website (http://www.nature.com/mp)
Erik Ingelsson is an advisor and consultant for Precision Wellness Inc (Redwood City, CA). The remaining authors declare no conflict of interest.
Supplementary Material
References
- NIAAA. Alcohol Facts and Statistics. Available at: https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/alcohol-facts-and-statistics.
- Rehm J, Baliunas D, Borges GL, Graham K, Irving H, Kehoe T et al. The relation between different dimensions of alcohol consumption and burden of disease: an overview. Addiction 2010; 105: 817–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogeil RP, Room R, Matthews S, Lloyd B. Alcohol and burden of disease in Australia: the challenge in assessing consumption. Aust NZ J Public Health 2015; 39: 121–123. [DOI] [PubMed] [Google Scholar]
- Rehm J, Taylor B, Roerecke M, Patra J. Alcohol consumption and alcohol-attributable burden of disease in Switzerland, 2002. Int J Public Health 2007; 52: 383–392. [DOI] [PubMed] [Google Scholar]
- Ferreira-Borges C, Rehm J, Dias S, Babor T, Parry CD. The impact of alcohol consumption on African people in 2012: an analysis of burden of disease. Trop Med Int Health 2015; 21: 52–60. [DOI] [PubMed] [Google Scholar]
- Allen JP. Use of biomarkers of heavy drinking in health care practice. Mil Med 2003; 168: 364–367. [PubMed] [Google Scholar]
- Liangpunsakul S, Lai X, Ross RA, Yu Z, Modlik E, Westerhold C et al. Novel serum biomarkers for detection of excessive alcohol use. Alcohol Clin Exp Res 2015; 39: 556–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahs A, Curtis BJ, Waldschmidt TJ, Brown LA, Gauthier TW, Choudhry MA et al. Alcohol and epigenetic changes: summary of the 2011 Alcohol and Immunology Research Interest Group (AIRIG) meeting. Alcohol 2012; 46: 783–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng JT, Wu LS, Lee CS, Hsu PW, Cheng AT. Integrative epigenetic profiling analysis identifies DNA methylation changes associated with chronic alcohol consumption. Comput Biol Med 2015; 64: 299–306. [DOI] [PubMed] [Google Scholar]
- Leake I. Liver disease: alcohol causes epigenetic changes in hepatic stellate cells. Nat Rev Gastroenterol Hepatol 2014; 11: 704. [DOI] [PubMed] [Google Scholar]
- Nieratschker V, Batra A, Fallgatter AJ. Genetics and epigenetics of alcohol dependence. J Mol Psychiatry 2013; 1: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci 2011; 12: 623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson KD, Uzvolgyi E, Liang G, Talmadge C, Sumegi J, Gonzales FA et al. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res 1999; 27: 2291–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlaar N, Hutchison KE. Alcohol and the methylome: design and analysis considerations for research using human samples. Drug Alcohol Depend 2013; 133: 305–316. [DOI] [PubMed] [Google Scholar]
- Zhang R, Miao Q, Wang C, Zhao R, Li W, Haile CN et al. Genome-wide DNA methylation analysis in alcohol dependence. Addict Biol 2013; 18: 392–403. [DOI] [PubMed] [Google Scholar]
- Clark SL, Aberg KA, Nerella S, Kumar G, McClay JL, Chen W et al. Combined whole methylome and genomewide association study implicates CNTN4 in alcohol use. Alcohol Clin Exp Res 2015; 39: 1396–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Zhang R, Li W, Liao Y, Tang J, Miao Q et al. Genome-wide DNA methylation patterns in discordant sib pairs with alcohol dependence. Asia Pac Psychiatry 2013; 5: 39–50. [DOI] [PubMed] [Google Scholar]
- Philibert RA, Plume JM, Gibbons FX, Brody GH, Beach SR. The impact of recent alcohol use on genome wide DNA methylation signatures. Front Genet 2012; 3: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013; 8: 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomba R, Hwang SJ, O'Donnell CJ, Ellison RC, Vasan RS, D'Agostino RB Sr. et al. Parental obesity and offspring serum alanine and aspartate aminotransferase levels: the Framingham Heart Study. Gastroenterology 2008; 134: 953–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M, Pfeifer GP. Aging and DNA methylation. BMC Biol 2015; 13: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang FF, Cardarelli R, Carroll J, Fulda KG, Kaur M, Gonzalez K et al. Significant differences in global genomic DNA methylation by gender and race/ethnicity in peripheral blood. Epigenetics 2011; 6: 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick KJ, Nelson CP, Tsaprouni L, Sandling JK, Aissi D, Wahl S et al. DNA methylation and body-mass index: a genome-wide analysis. Lancet 2014; 383: 1990–1998. [DOI] [PubMed] [Google Scholar]
- Bock C. Analysing and interpreting DNA methylation data. Nat Rev Genet 2012; 13: 705–719. [DOI] [PubMed] [Google Scholar]
- Houseman EA, Kelsey KT, Wiencke JK, Marsit CJ. Cell-composition effects in the analysis of DNA methylation array data: a mathematical perspective. BMC Bioinform 2015; 16: 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, Zhuang J, Widschwendter M. Independent surrogate variable analysis to deconvolve confounding factors in large-scale microarray profiling studies. Bioinformatics 2011; 27: 1496–1505. [DOI] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006; 38: 904–909. [DOI] [PubMed] [Google Scholar]
- Borenstein M, Hedges LV, Higgins JP, Rothstein HR. A basic introduction to fixed-effect and random-effects models for meta-analysis. Res Synth Methods 2010; 1: 97–111. [DOI] [PubMed] [Google Scholar]
- Zakhari S. Overview: how is alcohol metabolized by the body? Alcohol Res Health 2006; 29: 245–254. [PMC free article] [PubMed] [Google Scholar]
- Takeshita T, Morimoto K, Mao XQ, Hashimoto T, Furuyama J. Phenotypic differences in low Km aldehyde dehydrogenase in Japanese workers. Lancet 1993; 341: 837–838. [PubMed] [Google Scholar]
- Olsen RW, Hanchar HJ, Meera P, Wallner M. GABAA receptor subtypes: the ‘one glass of wine’ receptors. Alcohol 2007; 41: 201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Porcu P, Werner DF, Matthews DB, Diaz-Granados JL, Helfand RS et al. The role of GABA(A) receptors in the acute and chronic effects of ethanol: a decade of progress. Psychopharmacology (Berl) 2009; 205: 529–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo G, Addolorato G, Agabio R, Carai MA, Pibiri F, Serra S et al. Role of GABA(B) receptor in alcohol dependence: reducing effect of baclofen on alcohol intake and alcohol motivational properties in rats and amelioration of alcohol withdrawal syndrome and alcohol craving in human alcoholics. Neurotox Res 2004; 6: 403–414. [DOI] [PubMed] [Google Scholar]
- Agabio R, Colombo G. GABAB receptor ligands for the treatment of alcohol use disorder: preclinical and clinical evidence. Front Neurosci 2014; 8: 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaykin DV. Optimally weighted Z-test is a powerful method for combining probabilities in meta-analysis. J Evol Biol 2011; 24: 1836–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JR, Busche S, Ge B, Kwan T, Pastinen T, Blanchette M. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol 2014; 15: R37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joubert BR, Haberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK et al. 450 K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect 2012; 120: 1425–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philibert RA, Beach SR, Brody GH. Demethylation of the aryl hydrocarbon receptor repressor as a biomarker for nascent smokers. Epigenetics 2012; 7: 1331–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Li X, Aryee MJ, Ekstrom TJ, Padyukov L, Klareskog L et al. GeMes, clusters of DNA methylation under genetic control, can inform genetic and epigenetic analysis of disease. Am J Hum Genet 2014; 94: 485–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Bonder MJ, Marioni RE, Zhu Z, McRae AF, Zhernakova A et al. Improving phenotypic prediction by combining genetic and epigenetic associations. Am J Hum Genet 2015; 97: 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, Mendu SK, Birnir B. GABA is an effective immunomodulatory molecule. Amino Acids 2013; 45: 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeman JJ, Buhlmann P. Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics 2007; 23: 980–987. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.