

Abstract
BACKGROUND
Chimeric CYP21A1P/CYP21A2 genes, caused by homologous recombination between CYP21A2 (cytochrome P450, family 21, subfamily A, polypeptide 2) and its highly homologous pseudogene CYP21A1P (cytochrome P450, family 21, subfamily A, polypeptide 1 pseudogene), are common in patients with congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency (21-OHD). A comprehensive junction site analysis of chimeric CYP21A1P/CYP21A2 genes is needed for optimizing genetic analysis strategy and determining clinical relevance.
METHODS
We conducted a comprehensive genetic analysis of chimeric CYP21A1P/CYP21A2 genes in a cohort of 202 unrelated 21-OHD patients. Targeted CYP21A2 mutation analysis was performed, and genotyping of chimeric CYP21A1P/CYP21A2 genes was cross-confirmed with Southern blot, RFLP, and multiplex ligation-dependent probe amplification analyses. Junction sites of chimera genes were determined by sequencing the long-PCR products amplified with primers CYP779f and Tena32F. An updated bioinformatics survey of Chi-like sequences was also performed.
RESULTS
Of 100 probands with a chimeric allele, 96 had a chimera associated with the severe classic salt-wasting form of CAH, and the remaining 4 carried an uncommon attenuated chimera with junction sites upstream of In2G (c.293−13A/C>G), which is associated with a milder phenotype. In addition to 6 of 7 reported chimeras, we identified a novel classic chimera (CH-8) and a novel attenuated chimera (CH-9). Attenuated chimeras explained prior genotype–phenotype discrepancies in 3 of the patients. Sequencing the CYP779f/Tena32F amplicons accurately differentiated between classic and attenuated chimeras. The bioinformatics survey revealed enrichment of Chi-like sequences within or in the vicinity of intron 2.
CONCLUSIONS
Junction site analysis can explain some genotype–phenotype discrepancies. Sequencing the well-established CYP779f/Tena32F amplicons is an unequivocal strategy for detecting attenuated chimeric CYP21A1P/CYP21A2 genes, which are clinically relevant.
Congenital adrenal hyperplasia (CAH)5 (OMIM 201910) due to 21-hydroxylase deficiency (21-OHD) is an autosomal recessive disorder of the adrenal cortex characterized by impairment of cortisol biosynthesis, with or without impairment of aldosterone biosynthesis (1). The cortisol synthesis block leads to corticotropin stimulation of the adrenal cortex with resulting androgen excess. A phenotypic spectrum exists, and the phenotype is classified into 3 subtypes according to clinical severity: classic salt-wasting (SW), classic simple virilizing (SV), and nonclassic (NC) (mild or late-onset) forms.
The CYP21A26 (cytochrome P450, family 21, subfamily A, polypeptide 2) gene encoding 21-hydroxylase is located on chromosome 6p23.1 and occurs in tandem with 3 other genes [RP1 (or RP2), C4A (or C4B), and TNXB (or TNXA)] that form a genetic module termed “RCCX” (i.e., RP-C4-CYP21-TNX) (2). The RP1 gene [synonym for the serine/threonine kinase 19 (STK19) gene] encodes a nuclear serine/threonine nuclear kinase; C4 encodes the immune effector protein complement component with isotypes encoded by C4A [complement component 4A (Rodgers blood group)] and C4B [complement component 4B (Chido blood group)]; and TNX encodes a member of the extracellular matrix protein family. The RCCX module is characterized by the high homology between the functional genes [RP1, CYP21A2, and TNXB (tenascin XB)] and the corresponding pseudogenes [RP2, synonym for serine/threonine kinase 19 pseudogene (STK19P); CYP21A1P, cytochrome P450, family 21, subfamily A, polypeptide 1 pseudogene; and TNXA, tenascin XA (pseudogene)].
Genetically caused 21-OHD is due to large gene deletions (approximately 30 kb), gene conversions, and point mutations (including small deletions and insertions) at the CYP21A2 gene (3, 4). The CYP21A2 and CYP21A1P genes are approximately 98% identical (5, 6). Deleterious defects harbored in the pseudogene can be transferred to the functional gene by homologous recombination, and such events produce common mutations that account for approximately 95% of all CYP21A2 mutations seen in CAH (7). Of these common mutations, 20%–30% are chimeric CYP21A1P/CYP21A2 genes generated by large gene-deletion or gene-conversion events (8, 9). In the last 3 decades, comprehensive studies have established a good correlation between genotype and phenotype in CAH patients across diverse ethnic groups and have provided valuable guidelines for genetic counseling (10–13). Nevertheless, some observed discrepancies are not explained by genetic screening via routine targeted mutation analysis and detection of the classic CYP21A1P/CYP21A2 chimera (8, 14, 15). Several possibilities might account for lack of genotype–phenotype concordance, including genetic variation in other genes that modify steroid action or salt balance, or the presence of uncommon chimeric genes (14, 16). To date, 7 types of chimeric CYP21A1P/CYP21A2 genes, which were termed chronologically after determination of the junction site, have been found. Six carry the pseudogene-specific mutation In2G (c.293−13A/C>G) in intron 2 (8, 9, 17–23) and thus are associated with a severe SW phenotype. This group of chimeras is common among CAH patients of Caucasian origin and has been referred to as the classic or common type of chimera (24). In addition, an uncommon chimeric gene, CH-4, has been identified. The CH-4 chimera has a junction site located between exon 1 and intron 2 upstream of In2G, and the chimeric enzyme retains partial 21-hydroxylase activity and produces a milder phenotype (8, 14, 15). The aim of our study was to carry out a comprehensive molecular genetic analysis of chimeric CYP21A1P/CYP21A2 genes, including precise determination of the junction sites, in a large cohort of CAH patients and to evaluate whether chimeric junction sites explain prior genotype–phenotype discrepancies. We also used junction site analysis to compare current strategies for detecting uncommon attenuated chimeric CYP21A1P/CYP21A2 genes.
Materials and Methods
PATIENTS
From 2006 to 2011, 252 patients with CAH due to 21-OHD (127 SW, 61 SV, and 64 NC patients) were enrolled in a Natural History Study at the NIH Clinical Center in Bethesda, MD (clinical trial no. NCT00250159). All patients and 262 parents from 202 unrelated families were genotyped. We report detailed molecular analyses of 100 probands who carried chimeric CYP21A1P/CYP21A2 genes. We recently reported the mutation profile of 182 unrelated patients with CAH, a subgroup of this cohort (25). The study was approved by the Eunice Kennedy Shriver National Institute of Child Health and Human Development Institutional Review Board. All adult participants and parents of participating children gave written informed consent. All minors gave their assent.
MOLECULAR ANALYSIS OF CYP21 GENES
DNA was extracted and CYP21A2 gene mutations were analyzed with standard methods (Esoterix). The 12 most common mutations [p.P30L (c.92C>T), In2G (c.293−13A/C>G), p.G110Efs (c.332_339del), p.I172N (c.518T>A), p.I236N (c.710T>A), p.V237E (c.713T>A), p.M239K (c.719T>A), p.V281L (c.844G>T), p.Leu307fs (c.923_924insT), p.Q318X (c.955C>T), p.R356W (c.1069C>T), and p.P453S (c.1360C>T)] were analyzed in a targeted mutation analysis strategy that used the multiplex minisequencing method (26). Twelve single-nucleotide polymorphisms across CYP21A2 were genotyped with the same method (F.K. Fujimura, unpublished data) used to infer possible chimeric CYP21A1P/CYP21A2 genes.
In our laboratory, Southern blotting was conducted according to an established protocol (27) to confirm chimeric CYP21A1P/CYP21A2 genes. Restriction enzymes TaqI and PshAI (New England Biolabs) were used to digest genomic DNA for Southern blotting. Alternatively, for samples without a DNA yield sufficient for Southern blotting, TaqI digestion of 8515-bp PCR fragments amplified with the primer pair CYP779f/Tena32F (28) was used to confirm chimeric CYP21A1P/CYP21A2 genes. Before TaqI digestion of the CYP779f/Tena32F amplicons, PCR products were purified (QIAquick PCR Purification Kit; Qiagen). In addition, multiplex ligation-dependent probe amplification (MLPA) with SALSA MLPA KIT P050-B2 CAH (MRC-Holland) was conducted to screen all of the probands and verify chimeric CYP21A1P/CYP21A2 genes. Probe hybridization and MLPA PCR were carried out according to the manufacturer’s guide. Amplification products were run on an ABI 3130×l Genetic Analyzer (Applied Biosystems/Life Technologies), and results were analyzed with Coffalyser software (version 9.4; MRC-Holland).
JUNCTION SITE ANALYSIS OF CHIMERIC
CYP21A1P/CYP21A2 GENES
Junction sites of confirmed chimeric CYP21A1P/CYP21A2 genes were analyzed with the PCR and by DNA sequencing. PCR products amplified with the primer pair CYP779f/Tena32F were sequenced to determine the junction site of each chimeric CYP21A1P/CYP21A2 gene with respect to the presence of CYP21A1P-specific sites from the 5′ end of the chimera. To identify a precise junction site that could not be located because of obstacles from a cluster of small insertion/deletions in intron 2, subcloning with the TA Cloning Kit (Life Technologies) followed by sequencing was carried out to differentiate between 2 alleles. The CYP779f/Tena32F PCR was carried out with Expand Long Range dNTPack (Roche Applied Science) according to the manufacturer’s instructions. Sequencing was conducted with ABI BigDye Terminator v3.1 chemistry on the ABI 3100 Genetic Analyzer (Applied Biosystems/Life Technologies), and alignment was performed with Sequencher 4.10.1 (Gene Codes Corporation) and Vector NTI Advance 11.0 (Life Technologies). The reference sequences of CYP21A2 and CYP21A1P are ENSG00000206338 and ENSG00000204338, respectively, from the Ensembl Genome Browser (http://uswest.ensembl.org/index.html), on which the nucleotide nomenclature at the cDNA level is based.
In addition, we used the most recent reference sequences (mentioned above) to perform an updated bioinformatics survey of the Chi sequence (5′-GCTGGTGG-3′) for the CYP21 genes.
CLASSIFICATION OF CHIMERIC CYP21A1P/CYP21A2 GENES
Chimeric genes were classified into 2 groups, classic and attenuated, depending on whether the junction site was upstream or downstream of the In2G mutation in intron 2. Chimeras harboring at least one In2G mutation are expected to be associated with the severe SW type of CAH. In contrast, chimeras carrying the weaker CYP21A1P promoter and the P30L (c.92C>T) mutation only are expected to be associated with a milder phenotype, which we have termed an “attenuated” chimera.
Results
MUTATION ANALYSIS
In our cohort of 202 unrelated patients with 21-OHD, we identified 6 of 7 known chimeric CYP21A1P/CYP21A2 genes and 2 novel chimeric genes (Table 1). Chimeric CYP21A1P/CYP21A2 genes were the most frequent type of mutation in our patients, with a total allele frequency of 31.4% (127 of 404 alleles), results that are consistent with those of other studies (9). As expected, harboring at least one In2G mutation (as in chimeras CH-1, CH-2, CH-3, CH-5, CH-6, and the newly identified CH-8) was associated with the SW type of CAH, thus constituting the classic group. In contrast, the known CH-4 chimera and a newly identified CH-9 chimera, which carry the weaker CYP21A1P promoter and the P30L (c.92C>T) mutation only, were associated with SV or NC CAH and were grouped into the attenuated group of chimeras. Of the 100 patients carrying chimera alleles, CH-4 and CH-9 were identified in 4 probands and explained the prior genotype-phenotype discrepancies in 3 probands (Table 2).
Table 1.
Chimeric CYP21A1P/CYP21A2 genes identified in 202 unrelated patients with 21-OHD.
| Categorya | Allele frequency, n (%)b | Junction sitec | Carriership of common mutationc |
|---|---|---|---|
| A. Classic | |||
| CH-1 | 46 (11.4) | G110Efs ˆ I172N | P30L, In2G, G110Efs |
| CH-2 | 1 (0.25) | D183E ˆ D234D | P30L, In2G, G110Efs, I172N |
| CH-3 | 8(2) | Q318X ˆ R356W | P30L, In2G, G110Efs, I172N, E6cluster, V281L, L307fx, Q318X |
| CH-5 | 44(10.9) | L307fx ˆ Q318X | P30L, In2G, G110Efs, I172N, E6cluster, L307fxd |
| CH-6 | 2 (0.5) | In2G ˆ G110Efs | P30L, In2G |
| CH-7 | 0 (0) | M239K ˆ L307fx | P30L, In2G, G110Efs, I172N, E6cluster |
| CH-8 | 22 (5.45) | R356W ˆ NDe | P30L, In2G, G110Efs, I172N, E6cluster, V281L, L307fx, Q318X, R356W |
| B. Attenuated | |||
| CH-4 | 3 (0.74) | c.138 ˆ c.292+45 | P30L |
| CH-9 | 1 (0.25) | c.293−74 ˆ c.293−67 | P30L |
Novel chimera alleles are shown in boldface.
N = 404 alleles.
Nomenclature at the protein level is based on conventional codon numbering. Nomenclature at the cDNA level, based on ENSG00000206338, is as follows: P30L (c.92C>T), In2G (c.293−13A/C>G), G110Efs (c.332_339del), I172N (c.518T>A), D183E (c.552C>G), D234D (c.705T>C), E6cluster [I236N (c.710T>A), V237E (c.713T>A), M239K (c.719T>A)], V281L (c.844G>T), L307fx (c.923_924insT), Q318X (c.955C>T), and R356W (c.1069C>T). E6cluster denotes 3 clustered mutations in exon 6.
c.884 is wild type in most CH-5 chimeras, except for 2 that contained mutant allele V281L.
ND denotes that the downstream site was not determined owing to a lack of distinguishable variants between CYP21 genes [Canturk et al. (29)].
Table 2.
Clinical data for patients with 21-OHD and attenuated chimeric CYP21A1P/CYP21A2 genes.
| Proband | Ethnicity | Sex | Mutations | Phenotype | Clinical information |
|---|---|---|---|---|---|
| 1 | Maternal: German/Irish | Male | 1. CH-5 | SV | Pubic hair at 4 years; bone age, 11 years, 6 months at chronologic age of 5 years; basal 17OHP,a 3174 ng/dL (95.3 nmol/L) |
| Paternal: Irish | 2. CH-4 | ||||
| 2 | Maternal: Italian | Male | 1. CH-4 | SV | Adult body odor and increased growth velocity at 5 years; basal 17OHP, 16 946 ng/dL (509 nmol/L) |
| Paternal: Irish/Scottish | 2. CH-1 | ||||
| 6 | Maternal: American Indian | Female | 1. In2Gb | SV | Diagnosed at 10 years with significant virilization including clitoromegaly, hirsutism, muscular habitus, and low voice; bone age, 15 years at chronologic age of 10 years; elevated urinary 17-ketosteroids |
| Paternal: French/Germar | 2. CH-4b | ||||
| 7 | German Czech and Austrian | Female | 1. V281L (c.844G>T)b | NC | Diagnosed at 28 years with hirsutism, male pattern baldness; basal 17OHP, 740 ng/dL (22 nmol/L); cosyntropin-stimulated 17OHP, 3460 ng/dL (104 nmol/L); precocious puberty with menarche at age 8 years |
| 2. CH-9b | |||||
| 7Sc | German Czech and Austrian | Female | 1. V281L(c.844G>Tb) | NC | Diagnosed at 62 years secondary to family genetic studies; basal 17OHP, 813 ng/dL (24 nmol/L); cosyntropin-stimulated 17OHP, 7190 ng/dL (216 nmol/L); menarche at age 9 years |
| 2. CH-9b |
17OHP, 17-hydroxyprogesterone.
Parental origin of allele is unknown because parents were not available.
S denotes sibling.
We identified 3 CH-4 alleles in 3 probands (probands 1, 2, and 6), for an allele frequency of 0.7% (3 of 404). No CYP21A2 gene was detected in proband 1 or 2, as is shown by the absence of a CYP21A2 band in a Southern blot (Fig. 1C) and a TaqI digestion assay of the 8515-bp PCR products (Fig. 1D). Each of these 2 patients carried 2 copies of a chimeric CYP21A1P/CYP21A2 gene. The reduced CYP21A2/CYP21A1P ratio in the Southern blot suggests that the mother of proband 1 (1M) and the parents of proband 2 (2M and 2F) are carriers of a chimeric allele (Fig. 1C). This supposition was confirmed by TaqI digestion assay (Fig. 1D). Sequencing data revealed that a CH-4 allele was transmitted from the father (1F) and the mother (2M) to probands 1 and 2, respectively (Fig. 2A). The junction site of CH-4 was located between c.138 (the middle of exon 1) and c.292+45 (the beginning of intron 2) (Fig. 2A), and a CYP21A1P-specific promoter (each of c.1−126T, c.1−113A, c.1−110C, c.1−103G, and c.1−4T was in a homozygous state; chromatograms not shown) was present in CH-4. Further sequencing analysis showed that proband 1 is a compound heterozygote for CH-4 and CH-5 and that proband 2 is a compound heterozygote for CH-4 and CH-1. The third CH-4 allele in our cohort was detected in proband 6 (Fig. 1D and Fig. 2A), who also carried the In2G mutation. Her parents were not available for the study.
Fig. 1. Schematic diagram of the RCCX module with Southern blotting and TaqI digestion analysis.
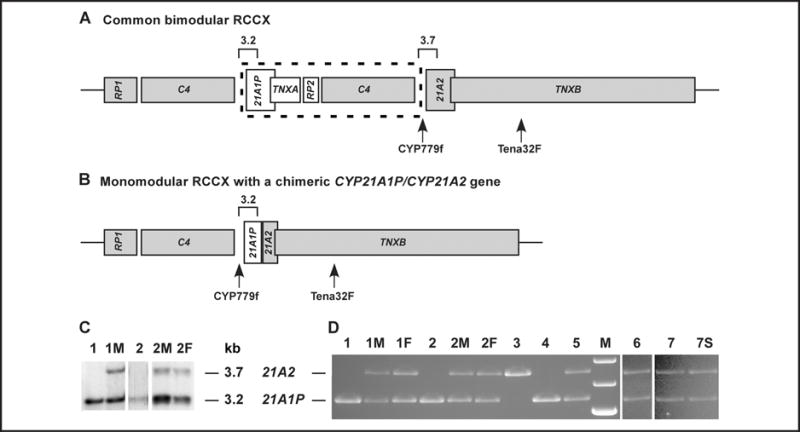
Shown are a common bimodular RCCX (A) and a monomodular RCCX with a chimeric CYP21A1P/CYP21A2 gene (B) in which junction sites can vary. Functional genes are in gray. Sizes and locations of the TaqI restriction fragments at CYP21 genes are annotated with open squares. C4 is a generalized symbol for the C4A, C4B, C4L, and C4S genes. A C4 gene can be C4A or C4B regarding protein isotype, and it can also be long (C4L) or short (C4S) regarding gene size. Only a C4L gene with unknown isotype is shown in the schematic. The size of the sequence in the dashed frame is approximately 30 kb. An 8515-bp PCR product amplified by primer pair CYP779f/Tena32F was digested with TaqI for genotyping of the chimeric gene. (C), Genotyping of chimeric gene by Southern blotting after TaqI digestion of genomic DNA. Probands 1 and 2 do not have a CYP21A2 band. Three parents (1M, 2M, and 2F) showed a reduced CYP21A2:CYP21A1P ratio, indicating a chimera allele. The relatively weak band of proband 2 was due to a low yield of extracted DNA. (D), Genotyping of a chimeric gene by TaqI digestion of the 8515-bp PCR product. Probands 1 and 2 do not have a CYP21A2 band. Four parents (1M, 1F, 2M, and 2F), proband 6, and 2 siblings from family 7 (proband 7 and patient 7S) are heterozygous for the chimera gene. Among 3 controls of known genotype, proband 3 is homozygous for the In2G (c.293−13A/C>G) mutation and presents structurally intact CYP21A2 genes. Probands 4 and 5 are homozygous and heterozygous, respectively, for a classic chimera gene. M, molecular-size markers (from top: 4.0 kb, 3.5 kb, and 3.0 kb); 21A2, functional gene CYP21A2; 21A1P, pseudogene CYP21A1P.
Fig. 2. Junction site analysis of attenuated chimeric CYP21A1P/CYP21A2 by DNA sequencing.
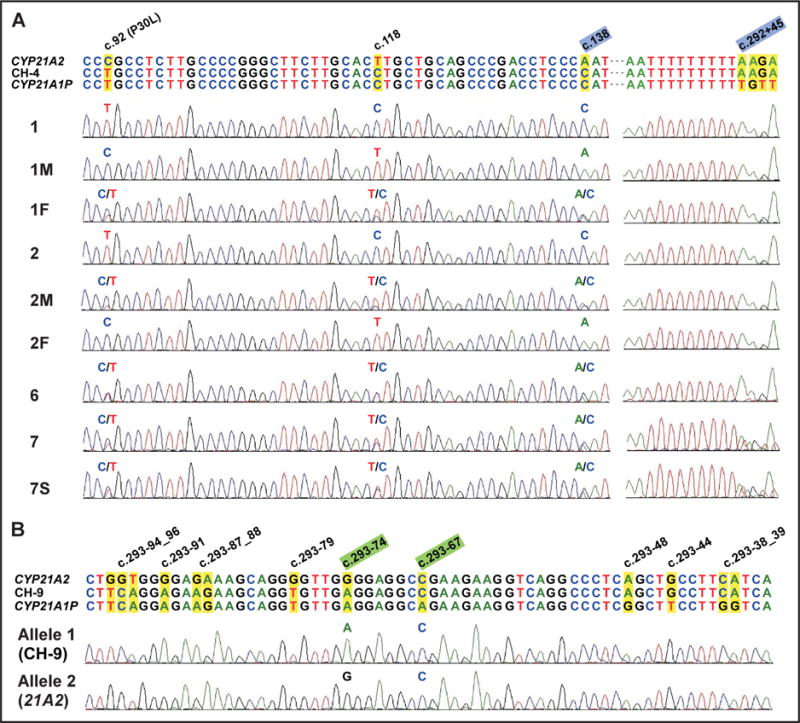
(A), Junction site of CH-4 is between c.138 and c.292+45 (highlighted in light blue). Probands 1 and 2 demonstrated a CYP21A1P-like sequence in exon 1 (homozygous for c.92, c.118, and c.138) and a CYP21A2-like sequence at the beginning of intron 2 (homozygous for c.292+45). Participants 1F, 2M, and proband 6 are heterozygous for 3 sites at exon 1, showing that they are carriers of the uncommon attenuated chimeric gene. Proband 7 and her sister (patient 7S) are heterozygous for both exon 1 sites and c.292+45, indicating that they carry a distinct chimeric allele with a junction site downstream of c.292+45. (B), Junction site of CH-9 is between c.293−74 and c.293−67 (highlighted in light green). CH-9 and CYP21A2 alleles, which share a CYP21A2-like sequence from c.293−67, were distinguished with TA cloning and sequencing for proband 7. 21A2, functional gene CYP21A2; 21A1P, pseudogene CYP21A1P.
In addition, we identified a novel attenuated chimera allele (CH-9) with a junction site between c.293−74 and c.293−67 in intron 2 of both proband 7 and her sister (patient 7S) (Fig. 1D and Fig. 2B). Upstream of the In2G mutation, as is seen in CH-4, the chimera with this novel junction site is also expected to produce a 21-hydroxylase with partial activity and thereby moderate the patient’s phenotype (Table 2). The parents were not available.
A novel classic chimeric CYP21A1P/CYP21A2 gene, CH-8, was identified in 8 patients, for an allele frequency of 5.45%. Its junction site was located downstream of the common mutation R356W (c.1069C>T). The location could not be narrowed down further because of the lack of unequivocally distinguishable sites between the CYP21A2 and CYP21A1P genes in the 3′ end region (29).
CH-5 and CH-1 are the most frequent chimeras in our cohort (Table 1). As has previously been shown (18), CH-5 carries a reversion to wild type in the V281L (c.844G>T) position. The majority of CH-5 alleles found in our cohort demonstrated the same haplotype; 2 contained a mutant allele at V281L (c.844G>T).
Neither the MLPA Kit P050-B2 CAH nor the PCR-based strategy with primer pair C/E (14) was able to distinguish attenuated chimeras CH-4 and CH-9 from classic chimera CH-6. Moreover, the C/E amplification produced false-negative results with respect to the presence of the 8-bp sequence (c.332_339), owing to a mismatch between mutation c.342C>T and primer E. The estimated frequency of the T allele is approximately 5% in our probands, who individually carry at least 1 copy of a chimeric CYP21A1P/CYP21A2 gene. Located at highly conserved areas, primers CYP779f and Tena32F, by contrast, were able to unequivocally amplify fragments that cover the entire length of the CYP21 genes and its 2-sided flanking sequence at the centromeric tail of the RCCX module, thus accurately identifying different types of chimeric CYP21A1P/CYP21A2 genes.
GENOTYPE-PHENOTYPE CORRELATIONS
Three probands carrying a chimeric CYP21A1P/CYP21A2 gene were previously thought to exhibit a genotype–phenotype discrepancy on the basis of the expected SW phenotype of a large CYP21A2 deletion. By carrying a weaker CYP21A1P promoter and a nonclassic mutation, P30L (c.92C>T), at exon 1 only, however, chimera CH-4 partially retains 21-hydroxylase activity (15), thus explaining the milder clinical phenotypes observed in these patients (Table 2). Similarly, with a junction site upstream of the In2G mutation (as seen in CH-4), the novel chimera gene CH-9 is also expected to retain partial 21-hydroxylase activity. This finding likely explains the mild NC phenotypes seen in proband 7 and her sister (patient 7S) (Table 2). Owing to the presence of the In2G mutation, the novel CH-8 chimera was associated with an SW phenotype.
BIOINFORMATICS SURVEY
We found no exact match for a Chi sequence throughout the CYP21 genes; however, we did find a Chi-like sequence (1 mismatch in 5′-GCTGGTGG-3′) at 6 sites within or in the vicinity of intron 2 (Fig. 3).
Fig. 3. Sequence alignment of CYP21 genes from promoter to exon 3.
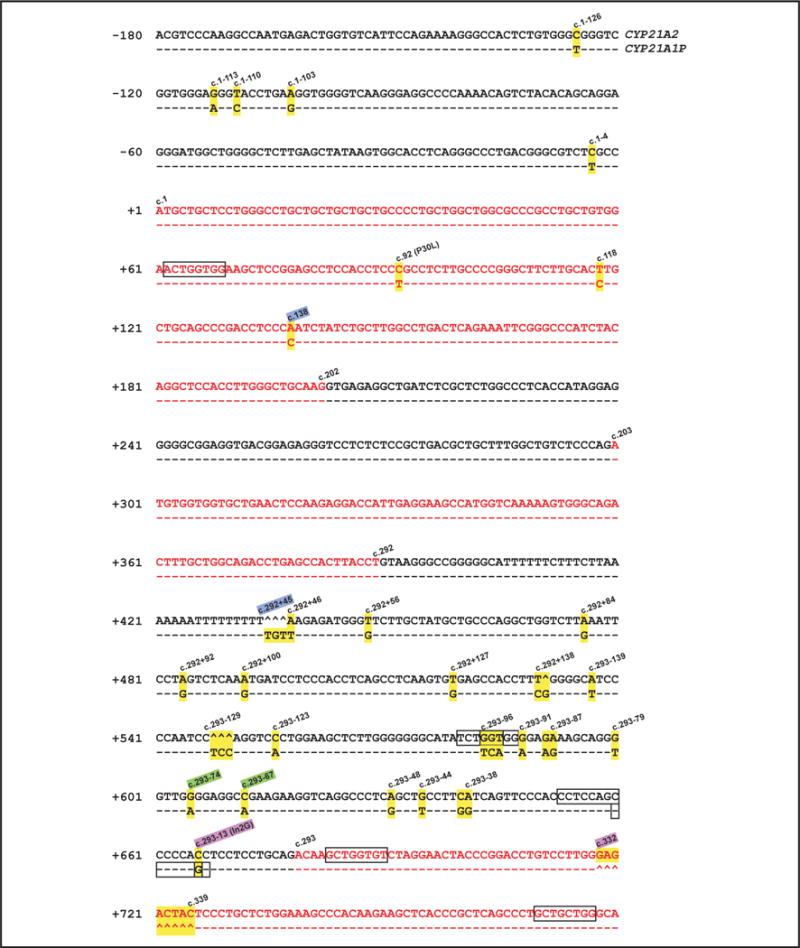
Located in this region, junction sites of 3 chimeric CYP21A1P/CYP21A2 genes (CH-4, −6, and −9) are demonstrated. CH-4, between c.138 and c.292 + 45 (highlighted in light blue); CH-6, between c.293−13 and c.332 (highlighted in pink); CH-9, between c.293−74 and c.293−67 (highlighted in light green). Conserved CYP21A1P sites, which are highlighted in yellow with the corresponding CYP21A2 sequence, are annotated. The coding sequence is presented in red. The hats (ˆ) denote nonexisting nucleotides, and dashed lines represent consensus sequence. Six sites of Chi-like sequence with only a 1-bp mismatch to 5′-GCTGGTGG-3′ or its complementary sequence (5′-CCACCAGC-3′) are framed in black.
Discussion
Our study is the first comprehensive and rigorous analysis of junction sites in chimeric CYP21A1P/CYP21A2 genes in a large cohort of North American patients with CAH due to 21-OHD. Genotype accurately predicts phenotype in approximately 90% of patients with CAH (10, 11, 25). Discrepancies between genotype and phenotype are continually being reported, however, including by our own group (25, 30). Chimeric genes with junction sites that impair 21-hydroxylase activity only mildly, which we have termed “attenuated” chimeric genes, provide one possible explanation for genotype–phenotype discrepancy; such attenuated chimeras explained a prior genotype–phenotype discrepancy for 3 of our patients. Our findings highlight the genomic complexity of the CYP21 locus and the fact that not all chimeric CYP21A1P/CYP21A2 genes severely impair 21-hydroxylase activity.
Chimeric CYP21A1P/CYP21A2 genes are a common mechanism leading to deleterious mutations in patients with 21-OHD and occur by homologous recombination between the 3′ end of the CYP21A1P gene and the 5′ end of CYP21A2. We have classified chimeric CYP21A1P/CYP21A2 genes into 2 categories, classic and attenuated, depending on the location of the junction sites relative to pseudogene mutation In2G within intron 2. The classic type of chimera contains the In2G mutation and produces a nonfunctional allele, which in the homozygous state is associated with the SW phenotype (24). Six different junction sites have been reported in the classic group of chimeras, which have been designated CH-1, CH-2, CH-3, CH-5, CH-6, and CH-7 (9, 20). We report a novel classic chimera, CH-8. In contrast, 21-hydroxylase enzyme activity is less severely impaired if the junction site occurs upstream of In2G. By carrying a weak CYP21A1P promoter and a nonclassic mutation, P30L (c.92C>T), at exon 1 only, the chimera partially retains activity 21-hydroxylase activity. These findings explain the milder clinical phenotypes in these patients. This type of uncommon CYP21A1P/CYP21A2 chimera (CH-4) has been described in a small number of patients with moderated phenotypes (9, 14, 15) and has been referred to as an “uncommon” chimera. We chose to classify this type of chimera as “attenuated” in order to incorporate the expected phenotype into the classification, thus improving this descriptive terminology and establishing a new classification scheme.
The present study determined the allele frequency of uncommon attenuated chimeric CYP21A1P/CYP21A2 genes associated with a milder phenotype for a large cohort of patients. CYP21A1P/CYP21A2 chimera CH-4 found in our study was first described in a patient with an SV form of CAH (15). L’Allemand et al. described another case of a Caucasian patient with a phenotype intermediate between the NC and SV forms of CAH (14). This patient was born a phenotypic female but had signs of clitoral hypertrophy at 6 months of age and had increased adrenal hormones characteristic of a classic patient. A recent report described a Brazilian patient who carried a similar chimera allele and also demonstrated a moderate SV phenotype in the presence of SW mutation In2G at the other allele. The junction site of the chimera in this patient was probably located at the beginning of intron 2, but no detailed sequencing data were provided (31). Similarly, our patients’ phenotypes were most consistent with the SV type of CAH. Vrzalova et al. recently described 1 patient homozygous for the CH-4 allele who was diagnosed with NC CAH (no clinical information was described), and 5 patients heterozygous for CH-4 and a classic chimera (CH-1 or CH-7) who had the SV type of CAH. Signs of precocious pseudopuberty developed in all patients (1 girl and 3 boys) with the genotype CH-4/CH-7; external genitalia virilization was also seen in the girl (9).
The CYP21A1P/CYP21A2 chimera found in proband 7 and her sibling (patient 7S) has a novel junction site between c.293−74 and c.293−67 that is also upstream of common mutation In2G (i.e., c.293−13A/C>G). Following the terminology in previous reports, we designated the novel chimera as CH-9. CH-9 also carries a weaker CYP21A1P promoter and a nonclassic mutation, P30L (c.92C>T), at exon 1. This chimera is predicted to have the same genetic consequence as that of CH-4. Thus, CH-9 falls into the group of attenuated chimeras along with CH-4. Proband 7 and sibling patient 7S have NC CAH, findings that are in accord with their carrying CH-9 on one allele and V281L, a common NC mutation, on the other. Interestingly, 2 patients with moderate SV phenotypes in the Brazilian study carried a second chimera allele (haplotype VIII) that seems to have a junction site similar to CH-9 (31). No sequencing analysis was presented to narrow down the precise junction sites, however. In addition, haplotype IV in the Brazilian study might represent the same kind of chimera as the CH-8 chimera identified and so designated in our study (31).
CH-7 was first identified in a Czech population (9). Strikingly, this chimera allele, which is the most frequent allele in the Czech population, was absent from our Caucasian patients of mixed ethnicity. In contrast, CH-5, one of the 2 most common chimeras in our cohort, has not been reported in the Czech patients. Additional studies in the Czech population may elucidate this discrepancy.
The high homology in the sequences of the active genes and the pseudogenes within the RCCX module predisposes the region to a high rate of nonallelic homologous recombination during meiosis (32). Specific genome-wide elements, such as a Chi sequence (5′-GCTGGTGG-3′), are important triggers for recombination events in eukaryotic cells. Enrichment of Chi-like sequences (1 mismatch in the 5′-GCTGGTGG-3′ sequence) within or in the vicinity of intron 2 provides one possible explanation for the junction site variations found in this region. Because intron 2 has been implicated as a hot spot for recombination and microconversion (32), a higher frequency of CH-4, CH-6, and CH-9 chimeras would be expected; however, our data suggest that they are rare chimera types. One alternative explanation may be due to intron 2 being the most variable region between the active genes and pseudogenes at the CYP21 locus, whereas recombination at regions other than intron 2 will generate chimeric products that are not distinguishable because of the high homology between the 2 genes.
Importantly, our study also provided us with the opportunity to compare existing strategies for detecting uncommon attenuated chimeric CYP21A1P/CYP21A2 genes. Both the MLPA methodology and PCR-based strategies, such as using primer pair C/E, have unavoidable limitations in their ability to detect chimeric CYP21A1P/CYP21A2 genes. MLPA is an efficient method for detecting large gene deletions and duplications and has been widely used in previous studies of CAH (9, 31, 33, 34). Current MLPA probes, however, are not able to distinguish attenuated chimeras CH-4 and CH-9 from classic chimera CH-6, owing to the lack of a probe for In2G, which is the crucial site for classifying chimeric CYP21A1P/CYP21A2 genes into the classic and attenuated types. The same limitation occurred with primer C/E amplification, in addition to the false-negative results for approximately 5% of the alleles due to the mismatch between mutation c.342C>T and primer E. The strategy of sequencing the CYP779f/Tena32F amplicons eliminated these potential errors in identifying chimera junction sites. Compared with other primer designs strictly targeting the CYP21 gene locus, this long-PCR strategy avoids nonspecific amplifications produced by high sequence homology between the active genes and pseudogenes at the CYP21 locus. In summary, sequencing the 8515-bp PCR fragments amplified with the well-established primer pair CYP779f/Tena32F combined with the MLPA method is an accurate strategy for detecting and determining junction sites of various chimeric CYP21A1P/CYP21A2 genes, especially attenuated alleles that are clinically relevant.
The junction site locations in the chimeric CYP21A1P/CYP21A2 genes are not all the same. Junction site location may influence gene functionality and the degree of 21-hydroxylase impairment. We have presented the most detailed chimera analysis completed to date, in a large cohort and with a rigorous methodology that included junction site sequencing. We propose a new classification scheme that distinguishes the chimeric configurations based on phenotypic consequences. Our data emphasize the importance of extensive molecular analysis in the diagnosis of CAH beyond routine mutation analysis. Evaluation of junction site locations of chimeric genes should be part of the genetic analysis for CYP21A2, especially when there is discordance between the observed phenotype and the phenotype predicted by routine CYP21A2 genotyping.
Acknowledgments
We are grateful to the patients for their participation in this study. We are also grateful to Dr. Frank Fujimura and Esoterix Laboratories for their excellent technical support and to Miki Nishitani for her assistance in manuscript preparation. We are indebted to Dr. Chack-Yung Yu (Department of Pediatrics, Ohio State University, Columbus, Ohio) for providing probes for Southern blotting. We thank NIA Core Laboratory staff for DNA extraction and sample processing, and we thank Tina Roberson for administrative assistance.
Research Funding: W. Chen, Intramural Research Program at the National Institute on Aging; Z. Xu, Intramural Research Program at the National Institute on Aging; A. Sullivan, Intramural Research Programs of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and National Institutes of Health Clinical Center; G.P. Finkielstain, Intramural Research Programs of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and National Institutes of Health Clinical Center; C. Van Ryzin, Intramural Research Programs of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and National Institutes of Health Clinical Center; D.P. Merke, Intramural Research Programs of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and National Institutes of Health Clinical Center, and Congenital Adrenal Hyperplasia Research, Education and Support (CARES) Foundation; N.B. McDonnell, Intramural Research Program at the National Institute on Aging.
Role of Sponsor: The funding organizations played no role in the design of study, choice of enrolled patients, review and interpretation of data, or preparation or approval of manuscript.
Footnotes
This research was previously presented at the Human Genetics and Genomics Gordon Research Conferences, Salve Regina University, Newport, RI, July 17–22, 2011.
Nonstandard abbreviations: CAH, congenital adrenal hyperplasia; 21-OHD, 21-hydroxylase deficiency; SW, salt-wasting; SV, simple virilizing; NC, nonclassic; TNX, tenascin X; In2G, c.293−13A/C>G; MLPA, multiplex ligation-dependent probe amplification; Chi sequence, 5′-GCTGGTGG-3′.
Human genes: CYP21A2, cytochrome P450, family 21, subfamily A, polypeptide 2; STK19, serine/threonine kinase 19 (synonym for RP1); C4A, complement component 4A (Rodgers blood group); TNXB, tenascin XB; STK19P, synonym for serine/threonine kinase 19 pseudogene (synonym for RP2); CYP21A1P, cytochrome P450, family 21, subfamily A, polypeptide 1 pseudogene; TNXA, tenascin XA (pseudogene).
Author Contributions: All authors confirmed they have contributed to the intellectual content of this paper and have met the following 3 requirements: (a) significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data; (b) drafting or revising the article for intellectual content; and (c) final approval of the published article.
Authors’ Disclosures or Potential Conflicts of Interest: Upon manuscript submission, all authors completed the Disclosures of Potential Conflict of Interest form. Potential conflicts of interest:
Employment or Leadership: D.P. Merke, United States Public Health Service.
Consultant or Advisory Role: None declared.
Stock Ownership: None declared.
Honoraria: None declared.
Expert Testimony: None declared.
References
- 1.Merke DP, Bornstein SR. Congenital adrenal hyperplasia. Lancet. 2005;365:2125–36. doi: 10.1016/S0140-6736(05)66736-0. [DOI] [PubMed] [Google Scholar]
- 2.Yang Z, Mendoza AR, Welch TR, Zipf WB, Yu CY. Modular variations of the human major histocompatibility complex class III genes for serine/threonine kinase RP, complement component C4, steroid 21-hydroxylase CYP21, and tenascin TNX (the RCCX module). A mechanism for gene deletions and disease associations. J Biol Chem. 1999;274:12147–56. doi: 10.1074/jbc.274.17.12147. [DOI] [PubMed] [Google Scholar]
- 3.Werkmeister JW, New MI, Dupont B, White PC. Frequent deletion and duplication of the steroid 21-hydroxylase genes. Am J Hum Genet. 1986;39:461–9. [PMC free article] [PubMed] [Google Scholar]
- 4.Lee HH. CYP21 mutations and congenital adrenal hyperplasia. Clin Genet. 2001;59:293–301. doi: 10.1034/j.1399-0004.2001.590501.x. [DOI] [PubMed] [Google Scholar]
- 5.White PC, New MI, Dupont B. Structure of human steroid 21-hydroxylase genes. Proc Natl Acad Sci U S A. 1986;83:5111–5. doi: 10.1073/pnas.83.14.5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Higashi Y, Yoshioka H, Yamane M, Gotoh O, Fujii-Kuriyama Y. Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome: a pseudogene and a genuine gene. Proc Natl Acad Sci U S A. 1986;83:2841–5. doi: 10.1073/pnas.83.9.2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med. 2003;349:776–88. doi: 10.1056/NEJMra021561. [DOI] [PubMed] [Google Scholar]
- 8.Concolino P, Mello E, Minucci A, Giardina E, Zuppi C, Toscano V, Capoluongo E. A new CYP21A1P/CYP21A2 chimeric gene identified in an Italian woman suffering from classical congenital adrenal hyperplasia form. BMC Med Genet. 2009;10:72. doi: 10.1186/1471-2350-10-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vrzalová Z, Hrubá Z, Hrabincová ES, Vrábelová S, Votava F, Koloušková S, Fajkusová L. Chimeric CYP21A1P/CYP21A2 genes identified in Czech patients with congenital adrenal hyperplasia. Eur J Med Genet. 2011;54:112–7. doi: 10.1016/j.ejmg.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 10.Krone N, Braun A, Roscher AA, Knorr D, Schwarz HP. Predicting phenotype in steroid 21-hydroxylase deficiency? J Clin Endocrinol Metab. 2000;85:1059–65. doi: 10.1210/jcem.85.3.6441. [DOI] [PubMed] [Google Scholar]
- 11.Stikkelbroeck NM, Hoefsloot LH, de Wijs IJ, Otten BJ, Hermus AR, Sistermans EA. CYP21 gene mutation analysis in 198 patients with 21-hydroxylase deficiency in The Netherlands: six novel mutations and a specific cluster of four mutations. J Clin Endocrinol Metab. 2003;88:3852–9. doi: 10.1210/jc.2002-021681. [DOI] [PubMed] [Google Scholar]
- 12.Jaaskelainen J, Levo A, Voutilainen R, Partanen J. Population-wide evaluation of disease manifestation in relation to molecular genotype in steroid 21-hydroxylase (CYP21) deficiency: good correlation in a well defined population. J Clin Endocrinol Metab. 1997;82:3293–7. doi: 10.1210/jcem.82.10.4271. [DOI] [PubMed] [Google Scholar]
- 13.Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. 1992;90:584–95. doi: 10.1172/JCI115897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.L’Allemand D, Tardy V, Gruters A, Schnabel D, Krude H, Morel Y. How a patient homozygous for a 30-kb deletion of the C4-CYP 21 genomic region can have a nonclassic form of 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2000;85:4562–7. doi: 10.1210/jcem.85.12.7018. [DOI] [PubMed] [Google Scholar]
- 15.Killeen AA, Sane KS, Orr HT. Molecular and endocrine characterization of a mutation involving a recombination between the steroid 21-hydroxylase functional gene and pseudogene. J Steroid Biochem Mol Biol. 1991;38:677–86. doi: 10.1016/0960-0760(91)90078-j. [DOI] [PubMed] [Google Scholar]
- 16.Gomes LG, Huang N, Agrawal V, Mendonca BB, Bachega TA, Miller WL. Extraadrenal 21-hydroxylation by CYP2C19 and CYP3A4: effect on 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2009;94:89–95. doi: 10.1210/jc.2008-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chu X, Braun-Heimer L, Rittner C, Schneider PM. Identification of the recombination site within the steroid 21-hydroxylase gene (CYP21) of the HLA-B47,DR7 haplotype. Exp Clin Immunogenet. 1992;9:80–5. [PubMed] [Google Scholar]
- 18.Helmberg A, Tabarelli M, Fuchs MA, Keller E, Dobler G, Schnegg I, et al. Identification of molecular defects causing congenital adrenal hyperplasia by cloning and differential hybridization of polymerase chain reaction-amplified 21-hydroxylase (CYP21) genes. DNA Cell Biol. 1992;11:359–68. doi: 10.1089/dna.1992.11.359. [DOI] [PubMed] [Google Scholar]
- 19.Lee HH, Lee YJ, Chan P, Lin CY. Use of PCR-based amplification analysis as a substitute for the Southern blot method for CYP21 deletion detection in congenital adrenal hyperplasia. Clin Chem. 2004;50:1074–6. doi: 10.1373/clinchem.2003.028597. [DOI] [PubMed] [Google Scholar]
- 20.Lee HH. The chimeric CYP21P/CYP21 gene and 21-hydroxylase deficiency. J Hum Genet. 2004;49:65–72. doi: 10.1007/s10038-003-0115-2. [DOI] [PubMed] [Google Scholar]
- 21.Lee HH. Chimeric CYP21P/CYP21 and TNXA/ TNXB genes in the RCCX module. Mol Genet Metab. 2005;84:4–8. doi: 10.1016/j.ymgme.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 22.Lee HH, Chang SF, Lee YJ, Raskin S, Lin SJ, Chao MC, et al. Deletion of the C4-CYP21 repeat module leading to the formation of a chimeric CYP21P/CYP21 gene in a 9.3-kb fragment as a cause of steroid 21-hydroxylase deficiency. Clin Chem. 2003;49:319–22. doi: 10.1373/49.2.319. [DOI] [PubMed] [Google Scholar]
- 23.White PC, New MI, Dupont B. HLA-linked congenital adrenal hyperplasia results from a defective gene encoding a cytochrome P-450 specific for steroid 21-hydroxylation. Proc Natl Acad Sci U S A. 1984;81:7505–9. doi: 10.1073/pnas.81.23.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.White PC, Vitek A, Dupont B, New MI. Characterization of frequent deletions causing steroid 21-hydroxylase deficiency. Proc Natl Acad Sci U S A. 1988;85:4436–40. doi: 10.1073/pnas.85.12.4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finkielstain GP, Chen W, Mehta SP, Fujimura FK, Hanna RM, Van Ryzin C, et al. Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2011;96:E161–72. doi: 10.1210/jc.2010-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krone N, Braun A, Weinert S, Peter M, Roscher AA, Partsch CJ, Sippell WG. Multiplex minisequencing of the 21-hydroxylase gene as a rapid strategy to confirm congenital adrenal hyperplasia. Clin Chem. 2002;48:818–25. [PubMed] [Google Scholar]
- 27.Chung EK, Wu YL, Yang Y, Zhou B, Yu CY. Human complement components C4A and C4B genetic diversities: complex genotypes and phenotypes. Curr Protoc Immunol. 2005:8. doi: 10.1002/0471142735.im1308s68. Chapter 13, Unit 13. [DOI] [PubMed] [Google Scholar]
- 28.Lee HH, Lee YJ, Lin CY. PCR-based detection of the CYP21 deletion and TNXA/TNXB hybrid in the RCCX module. Genomics. 2004;83:944–50. doi: 10.1016/j.ygeno.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 29.Canturk C, Baade U, Salazar R, Storm N, Portner R, Hoppner W. Sequence analysis of CYP21A1P in a German population to aid in the molecular biological diagnosis of congenital adrenal hyperplasia. Clin Chem. 2011;57:511–7. doi: 10.1373/clinchem.2010.156893. [DOI] [PubMed] [Google Scholar]
- 30.Wilson RC, Mercado AB, Cheng KC, New MI. Steroid 21-hydroxylase deficiency: Genotype may not predict phenotype. J Clin Endocrinol Metab. 1995;80:2322–9. doi: 10.1210/jcem.80.8.7629224. [DOI] [PubMed] [Google Scholar]
- 31.Coeli FB, Soardi FC, Bernardi RD, de Araujo M, Paulino LC, Lau IF, et al. Novel deletion alleles carrying CYP21A1P/A2 chimeric genes in Brazilian patients with 21-hydroxylase deficiency. BMC Med Genet. 2010;11:104. doi: 10.1186/1471-2350-11-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tusie-Luna MT, White PC. Gene conversions and unequal crossovers between CYP21 (steroid 21-hydroxylase gene) and CYP21P involve different mechanisms. Proc Natl Acad Sci U S A. 1995;92:10796–800. doi: 10.1073/pnas.92.23.10796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Concolino P, Mello E, Toscano V, Ameglio F, Zuppi C, Capoluongo E. Multiplex ligation-dependent probe amplification (MLPA) assay for the detection of CYP21A2 gene deletions/duplications in congenital adrenal hyperplasia: first technical report. Clin Chim Acta. 2009;402:164–70. doi: 10.1016/j.cca.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 34.Jang JH, Jin DK, Kim JH, Tan HK, Kim JW, Lee SY, et al. Multiplex ligation-dependent probe amplification assay for diagnosis of congenital adrenal hyperplasia. Ann Clin Lab Sci. 2011;41:44–7. [PubMed] [Google Scholar]