

Abstract
This study aims to investigate the prevalence of the highly leukotoxic JP2 sequence versus the minimally leukotoxic non-JP2 sequence of Aggregatibacter actinomycetemcomitans within a cohort of 180 young African Americans, with and without localized aggressive periodontitis (LAP), in north Florida. The study included patients aged 5 to 25 y: 60 LAP patients, 60 healthy siblings (HS), and 60 unrelated healthy controls (HC). Subgingival plaque was collected from LAP sites—diseased (PD ≥5 mm with bleeding on probing) and healthy (PD ≤3 mm with no bleeding on probing)—and from healthy sites of HS and HC. Plaque DNA was extracted and analyzed by polymerase chain reaction for the detection of the JP2 and non-JP2 sequences of A. actinomycetemcomitans. Overall, 90 (50%) subjects tested positive for the JP2 sequence. Fifty (83.33%) LAP subjects were carriers of the highly leukotoxic JP2 sequence, detected in 45 (75%) diseased sites and 34 (56.67%) healthy sites. Additionally, JP2 carriage was found in 16 HS (26.67%) and 24 HC (40%; P < 0.0001, among groups). The non-JP2 sequence was detected in 26 (14.44%) total subjects: 17 (28.33%) LAP patients detected in equal amounts of diseased and healthy sites (n = 11, 18.33%), 6 (10%) HS sites, and 3 (5%) HC sites (P < 0.05, among groups). The JP2 sequence was strongly associated with LAP-diseased sites in young African Americans, significantly more so than the non-JP2 (ClinicalTrials.gov NCT01330719).
Knowledge Transfer Statement: Clinicians may use the results of this study to identify susceptible individuals to aggressive periodontitis, potentially leading to more appropriate selection of therapeutic choices.
Keywords: bacterial virulence, inflammation, juvenile periodontitis, microbiology, bacteria, children
Introduction
Localized aggressive periodontitis (LAP) is a severe inflammatory disease that affects the periodontal tissues of the first molars and incisors. Characterized by pronounced attachment loss and alveolar bone destruction, aggressive periodontitis ordinarily has an early age of onset and a rapid rate of progression and, if left untreated, results in tooth mobility and loss (Armitage 1999; Albandar 2014). Literature confirms the prevalence of LAP to be higher among individuals of African descent (6.9%) compared with Hispanics (1.02%) and Caucasians (0.14%; Löe and Brown 1991; Albandar and Tinoco 2002; Susin et al. 2014).
The anaerobic gram-negative coccobacillus Aggregatibacter actinomycetemcomitans (Aa)—a nonmotile common inhabitant of the oral microflora—expresses a number of virulence factors. Among these factors, the production of leukotoxin has been highlighted for its pathogenic role in the progression of aggressive periodontitis. This cell-specific leukotoxin induces the cytolysis of human polymorphonuclear leukocytes and monocytes, thereby disabling host immune defenses and allowing the microbe to evade destruction (Zambon et al. 1983; Fives-Taylor et al. 1999). The strong association between LAP and Aa presence has long been a topic of study. Previous research suggests this species to be an important etiologic agent in aggressive periodontitis (Slots et al. 1980; Darby and Curtis 2001; Fine et al. 2007; Haubek et al. 2008; Shaddox et al. 2012). However, the presence of Aa in periodontally compromised and periodontally healthy individuals implies varying degrees of virulence (Socransky and Haffajee 1992; Asikainen and Chen 1999; Mombelli et al. 1999).
JP2 is a highly leukotoxic serotype b strain of Aa that is reportedly associated with aggressive periodontitis (Zambon et al. 1996; Haubek et al. 2002; Ennibi et al. 2012). Analyses of the JP2 leukotoxin (ltx) operon were compared with those of minimally leukotoxic non-JP2 sequence, and the results showed that while the 2 maintain similar transcriptional organization, the JP2 ltx operon possessed a unique 530–base pair deletion sequence that sanctions an increased production of leukotoxin: about 10 to 20 times more than the non-JP2 strain (Brogan et al. 1994). Additionally, evidence shows the JP2 strain preferentially infects LAP populations of African origin, suggesting that this strain exhibits racial tropism toward this group (Haubek et al. 1997). A study reported that of the 326 Aa isolates collected across 29 countries, 38 were of the JP2 phenotype, and all were exclusively associated with adolescent LAP patients of African descent (Haubek et al. 1997). Similarly, a later study found the 530–base pair deletion in 34 of 41 (83%) young adults with LAP in Morocco (Ennibi et al. 2012).
Conversely, this sequence was not detected in 228 Greek participants with chronic or aggressive periodontitis (n = 91), nonperiodontitis (n = 77), or periodontitis with supportive periodontal treatment (n = 60; Sakellari et al. 2011). Looking at yet different populations, Mombelli et al. (1999) evaluated 185 Chinese subjects, of which 116 were identified as carrying Aa isolates, but none of which contained the 530–base pair deletion sequence.
Since there is apparent variation in the presence of the JP2 sequence among racially distinct populations, the purpose of this study is to investigate the prevalence of the Aa highly leukotoxic JP2 sequence and minimally leukotoxic non-JP2 sequence within a cohort of African Americans diagnosed with LAP, periodontally healthy siblings (HS) of LAP patients, and unrelated healthy controls (HC) in north Florida.
Materials and Methods
Participant Population
A subject group of 180 African American participants (aged 5 to 25 y) were evaluated in this study. The present cross-sectional study reports the baseline findings of an ongoing longitudinal trial, geared toward determining the microbiological and immunologic mechanisms associated with LAP children and the impact of periodontal treatment on the progression of this disease (clinical trial registration at ClinicalTrials.gov: NCT01330719). One of its aims is to evaluate the presence and stability of the JP2 and non-JP2 sequences of Aa in this cohort of LAP individuals, their HS, and unrelated controls, which is the purpose of the current study. Information regarding the investigation was verbally communicated to all participants and legal guardians of those aged <18 y. Written consent/assent was obtained from all subjects and/or legal guardians before the commencement of the study. The study protocol was reviewed and approved by the Institutional Review Board at the University of Florida (201400349).
Participants were recruited from the following locations in Florida (USA): the Leon County Health Department (Tallahassee; April 2007 to November 2015), the Dental Clinical Research Unit (Gainesville; February 2014 to March 2014), the Duval County Health Department (Jacksonville; February 2014 to March 2016), and the Jackson County Health Department (Marianna; April 2015 to April 2016). Patients with a possible diagnosis of LAP were referred to us by the attending doctors in each facility and were screened for inclusion criteria. Once a diagnosis was confirmed, parents of the LAP patient were asked to bring any siblings for screening as well. HC were selected at random and were age and race matched from each facility. The inclusion criteria for this study maintained that each participant must be 1) systemically in good overall health (absence of systemic diseases or conditions that could influence periodontal disease diagnosis and/or progression; diabetes, leukemia, Down’s syndrome, etc.) as evidenced by medical history and 2) diagnosed with LAP according to previously published classification (Armitage 1999; Albandar 2014), presenting at least 2 sites with pocket depth (PD) >4 mm with bleeding on probing (BoP) with concomitant presence of attachment loss and radiographically detected bone loss on incisors and/or first molars. HS of LAP participants and age-matched unrelated HC had no periodontal disease, as defined by the absence of PD >4 mm with BoP, no clinical attachment loss, and no radiographic bone loss. Patients were excluded if they were diagnosed with any systemic diseases or conditions that could influence the characteristics or clinical presentation of periodontal disease (e.g., diabetes or blood disorders), if they reported the use of antibiotics within 3 mo prior to first visit or the use of any medications that could influence periodontal characteristics or interfere with response to periodontal treatment (e.g., cyclosporine, phenytoin), pregnant and/or lactating women, and smokers. The patient pool for the current study was selected on the basis of having sufficient plaque samples available to conduct the polymerase chain reaction (PCR) assay.
Clinical Examination and Sampling
The following clinical periodontal parameters were measured at 6 sites per tooth for all patients: PD, clinical attachment level (CAL), dichotomous BoP, and visible plaque index (Ainamo and Bay 1975). CAL was calculated by adding PD and gingival margin position, where the latter was measured as a negative number if the margin was coronal to the cementoenamel junction. CAL was given a value of zero/undetermined in healthy sites with no attachment loss/recession, when the cementoenamel junction was not detected. Probing was performed manually with a UNC-15 probe, and all parameter data were stored with Florida Probe software (Florida Probe Corporation). Bitewing and periapical radiographs were taken to assess bone levels and confirm diagnosis. Intra- and interexaminer calibration, prior to the study, was achieved when 80% of duplicate measures of PD and CAL were within 1 mm (included diseased and healthy sites).
With sterile endodontic paper points (size, medium; Kerr Endodontics), subgingival plaque was collected from the deepest periodontally diseased site (PD ≥5 mm with BoP, attachment loss ≥2 mm, and radiographic bone loss) and from periodontally healthy sites (PD ≤3 mm with no BoP and no attachment loss/no recession) in each LAP subject. Subgingival plaque was also collected from 1 periodontally healthy site of HS and HC (PD ≤3 mm with no BoP and no attachment loss). Paper points containing samples were placed in 1.5-mL Eppendorf tubes and stored at −80 °C until processed. Bacterial DNA was isolated from the subgingival plaque samples with a DNA Purification Kit, following the manufacturer’s instructions (MasterPure EPICENTRE Biotechnologies).
Target sequences were amplified within a standard 25-μL PCR with the Phusion High-Fidelity PCR Master Mix with HF buffer (New England BioLabs Inc.) and the following primers: ltx3 (5′-GCCGACACCAAAGACAAAGTCT-3′) and ltx4 (5′-GCCCATAACCAAGCCACATAC-3′; described in Poulsen et al. 2003). PCR reaction mix and thermocycling conditions were adapted from manufacturer recommendations: temperature profile included 30 cycles of denaturation at 98 °C for 10 s, annealing at 60 °C for 30 s, and extension at 72 °C for 30 s. PCR products were electrophoresed on an ethidium bromide–stained 1.5% TBE agarose gel, and the presence of JP2 and non-JP2 sequences was confirmed by 0.7- and 1.2-kb amplicons, respectively (Poulsen et al. 2003).
Statistical Analysis
Based on previous reports on the prevalence of the JP2 clone, our power analysis indicated that 50 subjects per group would be enough to provide a difference of up to 30% in the proportion of this species between LAP and HC with a power of at least 80%. Statistical analyses on the presence of JP2 and non-JP2 sequences of Aa sequence and correlations with clinical parameters of the sampled site were conducted with statistical software (GraphPad Prism, version 5.00). Chi-square and Fisher’s exact tests were used to compare the prevalence of positive sequence (JP2 and non-JP2) among groups. A Shapiro-Wilk test was run for all data to determine normality. Analysis of variance and Kruskal-Wallis with Dunn’s multiple comparisons were used to compare clinical and demographic parameters among groups. Mann-Whitney test was used to compare 2 groups when indicated. Significance was determined at P value <0.05.
Results
JP2 and Non-JP2 Bacterial Sequences Detection
The present study evaluated 180 young African Americans living in northern Florida. This population included 60 LAP patients, 60 HS of LAP patients, and 60 HC. No significant differences were found among the mean ages of each clinical group. Information regarding demographics and clinical parameters are detailed in the Table. A total of 240 subgingival plaque samples were analyzed by PCR; this included 2 samples from each LAP patient—1 diseased site (LAPd) and 1 healthy site (LAPh)—and 1 sample from HS and HC.
Table.
Demographics and Clinical Parameters.
| LAP |
Healthy |
|||
|---|---|---|---|---|
| Diseased (n = 60) | Healthy (n = 60) | Siblings (n = 60) | Controls (n = 60) | |
| Age, y | 13.25 ± 4.12 | 11.45 ± 3.61 | 14.07 ± 6.46 | |
| Sex, female:male | 29:31 | 32:28 | 35:25 | |
| Dentition,a prim:perm | 17:43 | 9:51 | 9:51 | 11:49 |
| Mean PD site,b mm | 6.55 ± 1.78c | 2.65 ± 0.68 | 2.75 ± 0.68 | 2.73 ± 0.52 |
| Mean CAL site,b mm | 3.98 ± 1.85 | NDd | NDd | NDd |
| PD ≥4 mm, e % | 16.07 ± 10.74c | 3.78 ± 5.58 | 4.71 ± 5.82 | |
| Affected sites,e,f n (%) | 15.51 ± 12.30 (10.01 ± 8.81)c | 0 | 0 | |
| Plaque index,e % | 36.57 ± 23.87 | 37.02 ± 21.30 | 37.15 ± 25.18 | |
| BoP, % | 20.28 ± 13.45 | 10 ± 11.28g | 16.07 ± 25.86h | |
All participants were African Americans. Note that 2 samples were analyzed from each LAP patient (1 diseased, 1 healthy; n = 120). Values are presented as mean ± SD or n. Significance was determined by Kruskal-Wallis with Dunn’s multiple comparisons among groups.
BoP, bleeding on probing; CAL, clinical attachment level; LAP, localized aggressive periodontitis; ND, not determined; perm, permanent; PD, pocket depth; prim, primary.
Dentition type refers to the tooth from which the sample was collected (permanent or primary).
PD and CAL were calculated on sampled affected/diseased sites only.
P < 0.0001, compared with other groups.
The cementoenamel junction in healthy sites with no recession cannot be detected; thus, CAL is either zero (as we consider it) or cannot be reliably measured.
PD >4 mm (pockets >4 mm), affected sites, and plaque (sites with visible plaque) were whole-mouth parameters.
Affected site: PD >4 mm with concomitant BoP, CAL ≥ 2, and radiographically detected bone loss.
P < 0.001, compared with other groups.
P < 0.05, compared with other groups.
The highly leukotoxic JP2 sequence was identified in 90 of the 180 participants in this study (50.00%), out of which 50 (83.33%) were LAP patients who carried this Aa sequence in a diseased and/or healthy site, 16 (26.67%) were HS, and 24 (40.00%) were HC. The JP2 sequence was more prevalent in LAPd sites (n = 45, 75%) than in the LAPh sites (n = 34, 56.67%) and healthy individual sites (P < 0.001, among all groups; Fig. 1a). Further comparisons by Fisher’s exact test showed a trend (P = 0.054) for higher carriage of JP2 in LAPd than in LAPh sites but a highly significant difference of JP2 carriage in LAPd versus HS (P < 0.001) and HC (P < 0.001; Fig. 1a). The minimally leukotoxic non-JP2 sequence was detected in 14.44% (n = 26) of this study’s population: 28.33% (n = 17) were LAP patients carrying the non-JP2 sequence in 18.33% (n = 11) of both LAPd and LAPh sites; 10.00% (n = 6) were HS; and 5% (n = 3) were HC (P < 0.05 between LAP sites and HC; Fig. 1b).
Figure 1.

Presence of JP2 and non-JP2 among groups. (a) Presence of JP2 sequence among groups, P < 0.0001 (chi-square exact test). Fisher’s exact test shows a trend difference between LAPd and LAPh sites (+P = 0.054) and a significant difference between LAPd sites and HS (****P < 0.0001) and HC (***P < 0.001). (b) Presence of Aggregatibacter actinomycetemcomitans minimally leukotoxic sequence among groups. Fisher’s exact test shows difference between HC and LAPd sites (*P < 0.05) and LAPh sites (*P < 0.05). HC, healthy unrelated controls; HS, healthy siblings; LAP, localized aggressive periodontitis (d, diseased sites; h, healthy siblings).
Figure 2 describes the distribution of the JP2 and non-JP2 sequences among all LAP sites (diseased and healthy) versus all healthy sites (HS and HC). The JP2 sequence was identified in 66.00% (n = 79) of all LAP sites and 33.00% (n = 40) of all healthy sites (P < 0.001), while the non-JP2 sequence was found in 18% of all LAP sites and 8% of all healthy sites (P < 0.05).
Figure 2.
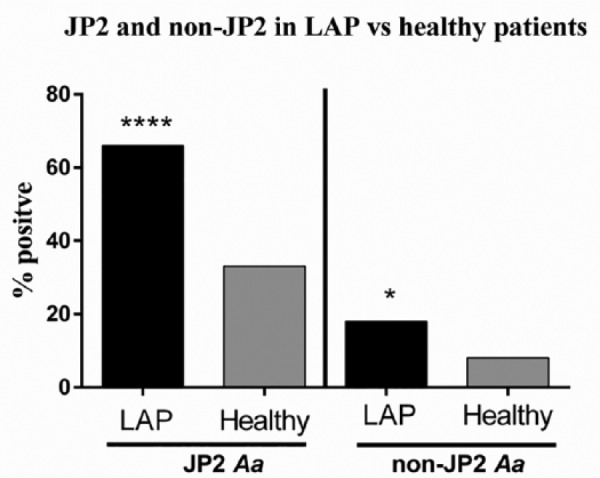
Localized aggressive periodontitis (LAP) versus healthy participant sites: JP2, ****P < 0.0001; non-JP2, *P < 0.05 (Fisher’s exact test). Aa, Aggregatibacter actinomycetemcomitans.
JP2 and Non-JP2 Sequence Detection and Disease Severity
No significant difference was found when comparing the mean PD of JP2-positive LAP-diseased sites with the mean PD of JP2-negative LAP-diseased sites (Fig. 3a). However, a significant difference was found when comparing the mean PD of all LAP sites positive for JP2 with all LAP sites negative for JP2 (P = 0.034; Fig. 3b). Comparisons between the mean CAL of JP2-positive diseased sites (4.11 mm) and JP2-negative diseased sites (3.6 mm) were also nonsignificant (Fig. 3c). However, in terms of all LAP sites, the Mann-Whitney test showed a significant difference between the mean CAL of the JP2-positive sites and the mean CAL of the JP2-negative sites: 2.34 mm and 1.32 mm, respectively (P = 0.019; Fig. 3d). Among the JP2-negative sites, there were only 2 instances where both the LAP-diseased site and the LAP healthy site carried a non-JP2 Aa sequence. All the remaining JP2-negative sites were negative for all Aa.
Figure 3.

Comparison of pocket depth (PD) and clinical attachment level (CAL) in diseased sites within localized aggressive periodontitis (LAP; a, c) and in all healthy and diseased sites in LAP (*P < 0.05; b, d). Mann-Whitney test showed significance for PD difference between JP2-positive and JP2-negative sites (P = 0.034; b) and a significant difference in mean CAL values between those sites (P = 0.019; d). Boxes indicate means; whiskers, SEM.
In the LAP group approximately 10% of the sites positive for JP2 were also simultaneously positive for the non-JP2 strain. To observe any differences in the clinical presentation of LAP under the influence of JP2 alone versus the influence of the JP2 and non-JP2 sequences, the PD and CAL means of sites positive for only JP2 were compared with sites positive for both sequences and to sites negative for JP2. Comparison of PD from LAP-diseased sites that are coinfected with both sequences (5.78 mm), infected with JP2 alone (6.61 mm), and not infected with JP2 at all (6.87 mm) was nonsignificant (P = 0.131; Fig. 4a). Furthermore, no significance was found among the mean PDs of these groups when considering all LAP sites (P = 0.074; Fig. 4b). However, a significant difference was found in terms of the PD of sites with no JP2 versus JP2 (P = 0.020; Fig. 4b). CAL comparisons with prevalence of both sequences, JP2 alone, or no JP2 were also nonsignificant (4.22, 4.08, and 3.6 mm, respectively; P = 0.537; Fig. 4c). However, Kruskal-Wallis with Dunn’s multiple comparisons showed significance between CAL means for all LAP sites infected with JP2 only and those not infected with JP2 (P = 0.040; Fig. 4d).
Figure 4.

Comparisons of pocket depth (PD), clinical attachment level (CAL), and JP2 presence alone or with non-JP2, in terms of localized aggressive periodontitis (LAP diseased sites only (LAPd; a, c) or all diseased and healthy sites in LAP patients (b, d). No difference was found on PD by analysis of variance among the 3 categories of JP2 (b), but Mann-Whitney test comparing only presence versus absence of JP2 was significant (*P < 0.05). Kruskal-Wallis with Dunn’s multiple comparisons test showed a difference in mean CAL between JP2-positive and JP2-negative sites in all LAP sites (d; *P < 0.05), specifically between sites negative for JP2 and sites with only JP2.
Discussion
Despite a low prevalence in North America, studies have shown that LAP may affect children of African descent at almost 3 times the prevalence than that of Caucasians (Susin et al. 2014). This disease severely compromises the periodontal structures of the first molar and incisors and can lead to tooth loss at an early age if left untreated. Much like the epidemiology of LAP, the JP2 sequence of Aa is found in higher numbers within this ethnic group (Haubek et al. 1997; Bueno et al. 1998). Additionally, the JP2 sequence has been recently associated with the etiology of LAP initiation (Darby and Curtis 2001; Fine et al. 2007; Haubek et al. 2008).
The present cross-sectional study discusses the prevalence of the highly leukotoxic JP2 and the minimally leukotoxic non-JP2 sequence of Aa within a cohort of 180 young African Americans, with and without LAP. The results of this study describe a strong association between LAP and JP2 presence, as 83.33% of LAP subjects harbored the JP2 sequence, which was also more prevalent in the LAP-diseased sites (75.00%) than in the LAP healthy sites (56.67%). The minimally leukotoxic non-JP2 sequence appears to be less associated with LAP, with detection in only 28.33% of LAP subjects. Unlike the JP2 sequence, the non-JP2 sequence does not appear to preferentially infect diseased sites and was equally distributed between diseased and healthy sites of LAP patients, at 18.33%. These findings were consistent with a number of previous reports that examined populations of similar ancestral backgrounds. For instance, a recent study evaluated 25 patients with aggressive periodontitis, of whom 52% carried the highly leukotoxic strain and 11% carried the minimally leukotoxic strain of Aa (Cortelli et al. 2005). Additionally, a population of 82 African Americans, 44 Caucasians, 13 Hispanics, and 7 Asian Americans were examined in separate study, where 54 African Americans, 8 Caucasians, and 9 Hispanics had LAP. Regarding only the LAP population, 31 (57%) African Americans, 7 (78%) Hispanics, and 1 (2%) Caucasian carried the highly leukotoxic Aa strain (Haraszthy et al. 2000).
Since the JP2 sequence presented so frequently within the present study population, the clinical impacts of this sequence were analyzed by correlating its presence with severity of disease (PD and CAL). Within the LAP-diseased sites only, the PD of sites positive for JP2 (alone or with non-JP2) was not significantly different from the PD of JP2-negative sites (6.44 vs. 6.87 mm, respectively; P > 0.05). Similar findings were observed for mean CAL of JP2-positive sites (4.11 mm), which was slightly greater than the mean CAL of the JP2-negative sites (3.60 mm), however not significant (P > 0.05). Fine et al. (2007) support our findings, as their data analysis of PDs and CALs did not show any significant correlation between the JP2 phenotype and an increased severity of LAP symptoms. Only when healthy sites along with diseased sites were taken into consideration within the LAP group did we observe significant differences for CAL in sites with JP2 only. This lack of difference when examining only diseased sites within LAP could be due to the small number of LAP-diseased sites that were indeed negative for the JP2 clone.
To determine any differences between the influences of JP2 alone versus JP2 in combination with non-JP2 on LAP severity, diseased pockets infected with both sequences were grouped and compared with diseased pockets with JP2 alone and diseased pockets negative for JP2. The JP2/non-JP2 sites generally presented lower PD and CAL values than the JP2-only group, although no significance was detected there. This could suggest that colonization by non-JP2 sequences may offer a form of indirect protection against the pathogenicity of the JP2 sequence within a site, possibly due to competition. A recent study reported that the relative risk of developing attachment loss was lower in sites coinfected with the JP2 and non-JP2 sequences (relative risk: 12.4) than in those infected with only the JP2 sequence (relative risk: 18.0; Haubek et al. 2008). A subsequent study by the same group discusses the concept of competitive exclusion among Aa sequences within a common niche and further surmised that colonization by the non-JP2 sequence could reduce the “infectious dose” of JP2 or “the number of samplings positive for JP2,” thereby offering a possible explanation for the reduced relative risk of attachment loss in JP2 + non-JP2 coinfected sites (Haubek et al. 2009).
Due to the familial nature of LAP, siblings of LAP patients often present with the disease. Given this, along with the well-recognized concept of intrafamilial dissemination of the JP2 sequence, we hypothesized that the JP2 sequence would present more frequently within the HS group (Bueno et al. 1998; Haubek et al. 2007). Interestingly, this was not the case in the present investigation, as our healthy unrelated controls had a higher prevalence of JP2 (40.00%) than HS (26.67%), although this difference was not statistically significant. One of these previous studies (Bueno et al. 1998) showed that 53.5% of subjects (6 of 11) within families where LAP disease (juvenile periodontitis as reported by authors) was initiated presented the JP2 clone and only 15.4% (2 of 13) within families without conversion to disease presented the JP2 clone. Although these differences were not statistically significant, this paper discussed the possibility of JP2 being more present in individuals or families of individuals that convert to disease and a possible risk indicator for future disease. Although lower percentages were found in LAP siblings on the present investigation, only 2 of these 60 siblings have developed the disease so far (roughly 5 y of follow-up), and both of those were negative for JP2.
In a further look at the periodontally healthy sites, we observed that LAP patients’ healthy sites carried the highest proportion of JP2 detection of all the healthy sites at 56.67%. This further confirms the affinity of the JP2 sequence for LAP subjects in general; however, it does not confirm that JP2 is associated with an increased periodontal destruction in these patients. In fact, the relatively high proportion of the JP2 sequence in healthy unrelated controls sites (40.00%) makes us wonder about the actual interaction of this strain with an individual host defense to possibly explain its association with LAP disease. In corroboration with this idea, a “hyperresponsive” phenotype, characterized by a heighted inflammatory response, has been reported in LAP subjects, possibly explaining these individuals’ susceptibility to severe host tissue destruction upon contact with bacterial toxins (Shaddox et al. 2010). In fact, a recent study showed a differentiated host response and a distinct microbial profile in LAP patients with sites harboring Aa compared with those not harboring this bacteria (Gonçalves et al. 2013). Thus, not only the presence of a specific strain may be important here, but also how the host responds to these bacteria and a specific microbial flora along with it becomes essential for the disease development. These aspects were not evaluated here and thus require further investigation.
An interesting finding of the present study is the relatively high frequency of the JP2 detection within our healthy population; this potentially supports the claim that the JP2 strain may be more of an opportunistic pathogen that overgrows due to homeostatic imbalance in the oral microbiota and thus increases the risk of disease development (Asikainen and Chen 1999; Bartold and Van Dyke 2013). This concept is in accordance with the previously mentioned “infectious dose” argument (Haubek et al. 2009). Alternatively, the JP2 sequence may work with and/or inadvertently create an environment that incites the pathogenic nature of other microorganisms. Previous data indicate differences in the composition of the microbial communities associated with low/nondetectable Aa levels versus >104 Aa levels (Gonçalves et al. 2013). A separate study, in agreement with this theory, discusses the association of Aa with Streptococcus parasanguinis and Filifactor alocis as having a higher likelihood of causing bone loss when compared with Aa alone (Fine et al. 2013). As mentioned, the absence of a complete analysis of the microbiological composition of the subgingival environment is a limitation in this study. A prior study on this population reported on a distinct microbial profile in the presence of Aa (Gonçalves et al. 2013), however not specifically accounting for the JP2 sequence. Thus, without these data, possible variations within the microbial communities associated with the JP2 and non-JP2 sequences and no Aa may occur as reported before (Gonçalves et al. 2013); thus, the varying impacts of these communities cannot be properly accounted for here. Another limitation of this study is the lack of quantification of copy numbers of JP2 in the different groups. Thus, higher levels of this organism could be present in diseased sites versus low levels in healthy sites. PCR is very sensitive and hence could explain the high detection rate in the healthy control sites, even if it presented a few copies of the target sequence. Therefore, further studies looking into quantification of this species would be helpful to fully understand JP2’s role in the pathogenesis of LAP.
In summary, the findings reported herein provide additional evidence of the strong association of the Aa JP2 strain with young African American LAP patients and a possible, however yet to be confirmed, protective role for the non-JP2 strain. However, further studies are required to decipher the true role of this bacterial strain in the pathogenesis of LAP.
Author Contributions
D. Burgess, contributed to conception, design, data analysis, and interpretation, drafted and critically revised the manuscript; H. Huang, contributed to design and data analysis, critically revised the manuscript; P. Harrison, contributed to conception, design, data analysis, and interpretation, critically revised the manuscript; I. Aukhil, contributed to conception, design, and data acquisition, critically revised the manuscript; L. Shaddox, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Footnotes
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
We thank the National Institutes of Health / National Institute of Dental and Craniofacial Research (R01DE019456) for the financial support provided for this research, as well as the doctors and staff at the Leon, Duval, and Jackson county health department dental centers for their assistance with coordination of our visits to their clinics, patient referrals, and dental care.
References
- Ainamo J, Bay I. 1975. Problems and proposals for recording gingivitis and plaque. Int Dent J. 25(4):229–235. [PubMed] [Google Scholar]
- Albandar JM. 2014. Aggressive periodontitis: case definition and diagnostic criteria. Periodontol 2000. 65(1):13–26. [DOI] [PubMed] [Google Scholar]
- Albandar JM, Tinoco EM. 2002. Global epidemiology of periodontal diseases in children and young persons. Periodontol 2000. 29:153–176. [DOI] [PubMed] [Google Scholar]
- Armitage GC. 1999. Development of a classification system for periodontal diseases and conditions. Ann Periodontol. 4(1):1–6. [DOI] [PubMed] [Google Scholar]
- Asikainen S, Chen C. 1999. Oral ecology and person-to-person transmission of Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Periodontol 2000. 20:65–81. [DOI] [PubMed] [Google Scholar]
- Bartold PM, Van Dyke TE. 2013. Periodontitis: a host-mediated disruption of microbial homeostasis. Unlearning learned concepts. Periodontol 2000. 62(1):203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogan JM, Lally ET, Poulsen K, Kilian M, Demuth DR. 1994. Regulation of Actinobacillus actinomycetemcomitans leukotoxin expression: analysis of the promoter regions of leukotoxic and minimally leukotoxic strains. Infect Immun. 62(2):501–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno LC, Mayer MP, DiRienzo JM. 1998. Relationship between conversion of localized juvenile periodontitis-susceptible children from health to disease and Actinobacillus actinomycetemcomitans leukotoxin promoter structure. J Periodontol. 69(9):998–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortelli JR, Cortelli SC, Jordan S, Haraszthy VI, Zambon JJ. 2005. Prevalence of periodontal pathogens in Brazilians with aggressive or chronic periodontitis. J Clin Periodontol. 32(8):860–866. [DOI] [PubMed] [Google Scholar]
- Darby I, Curtis M. 2001. Microbiology of periodontal disease in children and young adults. Periodontol 2000. 26:33–53. [DOI] [PubMed] [Google Scholar]
- Ennibi OK, Benrachadi L, Bouziane A, Haubek D, Poulsen K. 2012. The highly leukotoxic JP2 clone of Aggregatibacter actinomycetemcomitans in localized and generalized forms of aggressive periodontitis. Acta Odontol Scand. 70(4):318–322. [DOI] [PubMed] [Google Scholar]
- Fine DH, Markowitz K, Fairlie K, Tischio-Bereski D, Ferrendiz J, Furgang D, Paster BJ, Dewhirst FE. 2013. A consortium of Aggregatibacter actinomycetemcomitans, Streptococcus parasanguinis, and Filifactor alocis is present in sites prior to bone loss in a longitudinal study of localized aggressive periodontitis. J Clin Microbiol. 51(9):2850–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine DH, Markowitz K, Furgang D, Fairlie K, Ferrandiz J, Nasri C, McKiernan M, Gunsolley J. 2007. Aggregatibacter actinomycetemcomitans and its relationship to initiation of localized aggressive periodontitis: longitudinal cohort study of initially healthy adolescents. J Clin Microbiol. 45(12):3859–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fives-Taylor PM, Meyer DH, Mintz KP, Brissette C. 1999. Virulence factors of Actinobacillus actinomycetemcomitans. Periodontol 2000. 20:136–167. [DOI] [PubMed] [Google Scholar]
- Gonçalves PF, Klepac-Ceraj V, Huang H, Paster BJ, Aukhil I, Wallet SM, Shaddox LM. 2013. Correlation of Aggregatibacter actinomycetemcomitans detection with clinical/immunoinflammatory profile of localized aggressive periodontitis using a 16S rRNA microarray method: a cross-sectional study. PLoS One. 8(12):e85066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraszthy VI, Hariharan G, Tinoco EM, Cortelli JR, Lally ET, Davis E, Zambon JJ. 2000. Evidence for the role of highly leukotoxic Actinobacillus actinomycetemcomitans in the pathogenesis of localized juvenile and other forms of early-onset periodontitis. J Periodontol. 71(6):912–922. [DOI] [PubMed] [Google Scholar]
- Haubek D, Dirienzo JM, Tinoco EM, Westergaard J, López NJ, Chung CP, Poulsen K, Kilian M. 1997. Racial tropism of a highly toxic clone of Actinobacillus actinomycetemcomitans associated with juvenile periodontitis. J Clin Microbiol. 35(12):3037–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubek D, Ennibi OK, Abdellaoui L, Benzarti N, Poulsen S. 2002. Attachment loss in Moroccan early onset periodontitis patients and infection with the JP2-type of Actinobacillus actinomycetemcomitans. J Clin Periodontol. 29(7):657–660. [DOI] [PubMed] [Google Scholar]
- Haubek D, Ennibi OK, Poulsen K, Vaeth M, Poulsen S, Kilian M. 2008. Risk of aggressive periodontitis in adolescent carriers of the JP2 clone of Aggregatibacter (Actinobacillus) actinomycetemcomitans in Morocco: a prospective longitudinal cohort study. Lancet. 371(9608):237–242. [DOI] [PubMed] [Google Scholar]
- Haubek D, Ennibi OK, Vaeth M, Poulsen S, Poulsen K. 2009. Stability of the JP2 clone of Aggregatibacter actinomycetemcomitans. J Dent Res. 88(9):856–860. [DOI] [PubMed] [Google Scholar]
- Haubek D, Poulsen K, Kilian M. 2007. Microevolution and patterns of dissemination of the JP2 clone of Aggregatibacter (Actinobacillus) actinomycetemcomitans. Infect Immun. 75(6):3080–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löe H, Brown LJ. 1991. Early onset periodontitis in the United States of America. J Periodontol. 62(10):608–616. [DOI] [PubMed] [Google Scholar]
- Mombelli A, Gmür R, Lang NP, Corbert E, Frey J. 1999. Actinobacillus actinomycetemcomitans in Chinese adults: serotype distribution and analysis of the leukotoxin gene promoter locus. J Clin Periodontol. 26(8):505–510. [DOI] [PubMed] [Google Scholar]
- Poulsen K, Ennibi OK, Haubek D. 2003. Improved PCR for detection of the highly leukotoxic JP2 clone of Actinobacillus actinomycetemcomitans in subgingival plaque samples. J Clin Microbiol. 41(10):4829–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakellari D, Katsikari A, Slini T, Ioannidis I, Konstantinidis A, Arsenakis M. 2011. Prevalence and distribution of Aggregatibacter actinomycetemcomitans serotypes and the JP2 clone in a Greek population. J Clin Periodontol. 38(2):108–114. [DOI] [PubMed] [Google Scholar]
- Shaddox L, Wiedey J, Bimstein E, Magnuson I, Clare-Salzler M, Aukhil I, Wallet SM. 2010. Hyper-responsive phenotype in localized aggressive periodontitis. J Dent Res. 89(2):143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaddox LM, Huang H, Lin T, Hou W, Harrison PL, Aukhil I, Walker CB, Klepac-Ceraj V, Paster BJ. 2012. Microbiological characterization in children with aggressive periodontitis. J Dent Res. 91(10):927–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slots J, Reynolds HS, Genco RJ. 1980. Actinobacillus actinomycetemcomitans in human periodontal disease: a cross-sectional microbiological investigation. Infect Immun. 29(3):1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socransky SS, Haffajee AD. 1992. The bacterial etiology of destructive periodontal disease: current concepts. J Periodontol. 63(4 Suppl):322–331. [DOI] [PubMed] [Google Scholar]
- Susin C, Haas AN, Albandar JM. 2014. Epidemiology and demographics of aggressive periodontitis. Periodontol 2000. 65(1):27–45. [DOI] [PubMed] [Google Scholar]
- Zambon JJ, Christersson LA, Slots J. 1983. Actinobacillus actinomycetemcomitans in human periodontal disease: prevalence in patient groups and distribution of biotypes and serotypes within families. J Periodontol. 54(12):707–711. [DOI] [PubMed] [Google Scholar]
- Zambon JJ, Haraszthy V, Hariharan G, Lally ET, Demuth DR. 1996. The microbiology of early-onset periodontitis: association of highly toxic Actinobacillus actinomycetemcomitans strains with localized juvenile periodontitis. J Periodontol. 67(3s):282–290. [DOI] [PubMed] [Google Scholar]