

Abstract
To increase the levels of pulmonary gene transfer by nonviral vectors, we have adopted electroporation protocols for use in the lung. A volume of 100–200 μl of purified plasmid DNA suspended in saline was instilled into the lungs of anesthetized mice. Plasmids expressed luciferase, or β-galactosidase under control of the CMV immediate-early promoter and enhancer. Immediately following delivery, a series of eight square wave electric pulses of 10 ms duration each at an optimal field strength of 200 V/cm were administered to the animals using 10 mm Tweezertrodes (Genetronics, San Diego, CA, USA). The electrodes were placed on either side of the chest, which had been wetted with 70% ethanol. The animals recovered and survived with no apparent trauma until the experiments were terminated at the desired times, between 1 and 7 days post-treatment. Gene expression was detected by 1 day postelectroporation and peaked between 2 and 5 days. By 7 days, expression was back to baseline. By contrast, essentially no gene expression was detected in the absence of electric pulses. Using a β-galactosidase-expressing plasmid, the distribution of gene expression appeared to be concentrated in the periphery of the lung, but was also present throughout the parenchyma. The primary cell types expressing gene product include alveolar type I and type II epithelial cells. No inflammation or lung injury was detected histologically or by cytokine measurements in lungs at either 1 or 24 h following electroporation treatment. These results provide evidence that electroporation is a safe and effective means for introducing naked DNA into the lung and form the basis for future studies on targeted pulmonary gene therapy.
Keywords: transfection, electroporation, pulmonary gene therapy, alveolar epithelial cells, plasmid, nonviral vectors
Introduction
Numerous viral and nonviral approaches for gene delivery to the lung have been developed, but most have serious limitations. Inflammation, immunological responses, and nonspecificity of cell targeting are a few of the problems associated with viral vectors. By contrast, the major drawbacks to current nonviral methods have been inefficiency of gene transfer and subsequent low-level gene expression. Over the past 10 years, the main approach for nonviral gene delivery to the lung has used liposomes complexed with plasmid DNA. Although lipoplex has proven safe in clinical trials of pulmonary gene transfer, the levels of gene expression obtained using liposomes remain well below those seen with adenoviral vectors and at the limit of that required for therapeutic efficacy in animal models.1,2 Polyplex delivery systems, including polyethyleneimine (PEI), have shown promise for use in pulmonary gene transfer in vitro and in vivo, but again, the resulting levels of gene expression in vivo remain lower than desired.3,4
One characteristic of the respiratory tract that makes it amenable to gene transfer is that it can be targeted from both the vascular surface and the epithelial surface. It has been widely demonstrated that systemic administration of liposome–DNA complexes by intravenous injection, either as complexes or sequentially, results in deposition and high-level uptake in the pulmonary microvasculature.5–9 Such delivery yields endothelial cell expression, but relatively little expression in other cell types owing to low vascular permeability. By contrast, delivery of lipoplex, polyplex, and viruses via the airways results in gene transfer primarily to cells of the epithelium and resident alveolar macrophages; tight junctions between epithelial cells prevent vector access to other cell types.9–11 Thus, when desired, a limited degree of cell-specific targeting can be accommodated by delivery route. However, gene delivery to all cell types within the lung remains very difficult to achieve by current methods. Further, both of these access routes have considerable physical and biological barriers to vector delivery that limit access as well as destroy the vector, including mucins and surfactants in the lung and proteases, DNAses, and opsins in the circulation.12
Electroporation uses electrical fields to create transient pores in the cell membrane that allow the entry of normally impermeable macromolecules into the cytoplasm.13. Such molecules can include DNA, RNA, and proteins. Although this technique is routinely used to transfer DNA to bacteria, yeast, and mammalian cells in culture,14 it has only recently been applied to living animals. To date, electroporation has been adapted for use in skeletal muscle,15–17 liver,18,19 cardiac tissue,20 skin,21 the vasculature,22,23 cornea,24,25 and kidney.26 In most studies, electroporation causes a 100- to 1000-fold increase in gene expression compared to DNA injection alone.16,22,25 In all of these tissues, the procedure is rapid, reproducible, and requires relatively low doses of plasmid DNA that can be produced inexpensively. Further, at the appropriate field strengths, electroporation has proven to be a safe and effective method. One advantage of electroporation is that it mediates the transfer of DNA to multiple cell types and cell layers within a tissue. For example, in the vasculature, we have shown that when added from the adventitial surface, electroporation results in gene transfer to and expression in cells of the adventitia, medial and intimal smooth muscle layers, and the endothelium.22 Thus, the use of electroporation for DNA transfer appears to be able to circumvent tight junctions and other physical barriers that limit gene transfer using other types of vectors. In the present study, we investigated whether electroporation could be used as an effective gene delivery method for the lung in living animals.
Materials and methods
Plasmids
The plasmids pCMV-lux-DTS and pCMV-lacZ-DTS express fire fly luciferase and β-galactosidase, respectively, from the CMV immediate-early promoter/enhancer.27,28 Plasmids were propagated in Escherichia coli and purified using Qiagen Giga-prep kits, as described by the manufacturer (Qiagen, Chatsworth, CA, USA). For electroporation, plasmids were suspended to the desired concentration in 10 mM Tris, pH 8.0, 1 mM EDTA, and 140 mM NaCl. Agarose gel electrophoretic analysis demonstrated that greater than 80% of the purified DNA was present in the supercoiled form, and no RNA was detected.
Ex vivo gene transfer to the lung
Female Balb/c mice (15–18 g) were euthanized with sodium pentobarbital (260 mg/kg body weight) followed by cervical dislocation. The chest and neck were opened by a midline incision, the ribs retracted, and the lungs exposed. A volume of 200 μl of plasmid at the indicated concentration suspended in 10 mM Tris, pH 8.0, 1 mM EDTA, and 140 mM NaCl was injected into the lungs via the trachea. Immediately following DNA delivery, 10 mm Tweezertrodes (Genetronics, San Diego, CA, USA) were placed on either side of individual lobes and a series of eight 10 ms pulses at a field strength of 200 V/cm were administered at 1 s intervals using an ECM830 square wave electroporator (Genetronics). After electroporation, the lungs were removed and placed into DMEM containing 10% fetal bovine serum and 1× antibiotic/antimycotic solution (Invitrogen) and incubated at 37°C in a humidified incubator with 5% CO2. After 24 h the lungs were removed from the medium, rinsed in PBS, and snap frozen in liquid nitrogen for extract preparation.
In vivo gene transfer to the lung
Female Balb/c mice (15–18 g) were anesthetized with sodium pentobarbital (50 mg/kg body weight) and placed in the supine position. A 1 cm incision was made in the skin of the neck, exposing the sternhyoid muscles above the trachea. The muscle layer was teased apart and a 30-gauge needle was inserted between cartilage rings into the trachea at a 45° angle toward the lungs. A solution of 100–200 μl of plasmid in 10 mM Tris, pH 8.0, 1 mM EDTA, and 140 mM NaCl was administered between breaths over a 2 s period, the needle was removed, and the animal was allowed to recover breathing for 30–60 s. Immediately following this, a series of eight square wave electric pulses of 10 ms duration each were administered to the animals using 10 mm Tweezertrodes and an ECM830 electroporator. The electrodes were placed externally on either side of the chest, which had been wetted with 70% ethanol. Unless otherwise stated, a field strength of 200 V/cm was applied. The incision was closed with 2–3 resorbable sutures. The animals were allowed to recover from anesthesia and were returned to the vivarium. After 1–7 days, the mice were anesthetized and euthanized by pentobarbital overdose and cervical dislocation. The lungs were removed and either snap frozen immediately in liquid nitrogen for preparation of cell extracts or perfused with saline and fixed in 4% paraformaldehyde followed by 10% buffered formalin for histological analysis. All experiments were conducted in accordance with institutional guidelines in compliance with the recommendations of the Guide for Care and Use of Laboratory Animals.
Histological analysis
Immediately following euthanasia, lungs were inflated to total lung capacity with cold 4% paraformaldehyde in PBS and placed in a container of the same fixative for 4 h at 4°C. The lungs were rinsed with PBS, and stained with X-gal for 24 h at 37°C, after which they were rinsed with PBS and fixed in 10% buffered formalin for paraffin embedding and thin section preparation. For immunohistochemistry, 6 μm thin sections were cut, reacted with antibodies directed against β-galactosidase (Chemicon, Temecula, CA, USA), and stained with an alkaline phosphatase Vector ABC system and Vector Blue reagent (Vector Laboratories, Burlingame, CA, USA). Sections were counterstained with eosin. For pathological analysis, hemotoxylin- and eosin-stained sections were blinded and reviewed by a pathologist for lung injury. Four criteria were scored (vascular congestion, polymorphonuclear cell infiltrates, interstitial infiltrates, and hyaline membranes) on a five-point scale.
Measurement of reporter gene expression
Lungs were frozen in liquid nitrogen immediately after removal, and extracts were prepared using a Bio-Pulverizer (Biospec Products, Bartlesville, OK, USA). After the tissue was made into a powder, it was suspended in 400–800 μl of lysis buffer (Promega, Madison, WI, USA) and the samples were thawed at room temperature and vortexed for 15 sec. The samples were refrozen in liquid nitrogen and three freeze/thaw cycles were performed by alternating between liquid nitrogen and a room temperature water bath. Debris was removed by centrifugation. Luciferase activity was measured in duplicate using the Luciferase Assay System (Promega) in a Turner luminometer. Purified recombinant luciferase (Promega) was used to produce a standard curve for each experiment.
IL-6 measurements
Lobes or entire lungs were removed and snap frozen in liquid nitrogen. Lysates were prepared exactly as described for the measurement of gene expression. IL-6 levels were determined in duplicate by ELISA (R & D Systems, Minneapolis, MN, USA) using recombinant IL-6 to produce a standard curve for each experiment.
Statistical analysis
Nonparametric, Mann–Whitney U-tests were performed to determine statistical significance using Instat 2.03 software (GraphPad Software, San Diego, CA, USA).
Results
Use of electroporation for ex vivo plasmid delivery and expression in isolated lungs
We previously have used electroporation successfully to transfer genes to skeletal muscle, the cornea, and the vasculature. In all of these tissues, electrodes could be placed directly on the target tissue for efficient delivery of the electric field. To determine whether electroporation could also be used in the lung, we first tested whether genes could be delivered ex vivo to the tissue, by similarly placing flat electrodes on either side of isolated lung lobes from mice. Animals were euthanized, their chests opened, and 200 μl of a solution of plasmid DNA in 10 mM Tris, pH 8.0, 1 mM EDTA, and 140 mM NaCl was administered into the trachea. Within 30 s of DNA delivery, 10 mm diameter Tweezertrodes were placed on either side of each lobe and an electric field of 200 V/cm was applied in a series of eight 10 ms pulses at a frequency of 1 pulse/s. These field parameters were chosen based on our previous findings that these were the optimal conditions for gene transfer to a number of different tissues, including the vasculature, skeletal muscle, and cornea. After electroporation of one lobe, the electrodes were moved and successive lobes were treated similarly. Upon completion of electroporation, the lobes were separated, placed in growth medium, and incubated overnight at 37°C. Over 5 ng of luciferase per gram wet weight was expressed when only 20 μg of DNA was delivered to the lungs using electroporation (Figure 1). By contrast, approximately 10 pg of luciferase per gram wet weight was expressed in lungs receiving 20 μg of DNA when no electric field was applied.
Figure 1.

Gene transfer by electroporation in the explanted lung. A quantity of 20 μg of pCMV-lux-DTS in 200 μl of 10 mM Tris, pH 8, containing 1 mM EDTA and 140 mM NaCl was injected into the trachea of euthanized mice. Immediately following DNA delivery, eight square wave pulses of 10 ms duration each were administered at a field strength of 200 V/cm via Tweezertrode electrodes placed on either side of individual lobes, which had been exposed by opening the chest. Lungs received no DNA (control), DNA without electroporation (DNA only), or DNA and electroporation (n=4 per condition). Following DNA delivery, the lungs were placed in DMEM containing 10% FBS and antibiotics for 24 h, at which time luciferase expression was measured. Mean expression levels were calculated and standard error of the mean (s.e.m) was determined using Instat software (GraphPad Inc., San Diego, CA, USA).
In vivo gene transfer using electroporation
Based on the ability of electroporation to transfer genes successfully to the ex vivo lung, we next determined whether electroporation could be used in the living animal to mediate pulmonary gene transfer. A volume of 100–200 μl of a plasmid solution was delivered to the lungs via injection into the trachea. To do this, a small incision was made in the neck, the muscle layer was teased apart to reveal the trachea, and the DNA was injected into the trachea between cartilage rings between breaths (Figure 2). The animals were placed on a platform and held at a 60° incline with their heads up to facilitate delivery and even distribution of the bolus to the lungs. Within 1 min of DNA delivery, the chest was wetted with 70% ethanol and electrodes were placed on either side of the chest. A series of eight pulses of 10 msec duration each were administered at the indicated field strengths. Following delivery of the electric field, the animals were allowed to recover and gene expression was assayed at later times, as indicated. In these experiments, no trauma or mortality was detected in any of the animals as a result of the electroporation procedure. In total, 88% of the treated animals survived and the only cases of mortality were due to the delivery of the fluid into the lungs.
Figure 2.

Cartoon of in vivo electroporation procedure for murine lungs. A volume of 100–200 μl of a DNA solution was injected into the trachea, which was exposed through a small incision in the neck, and allowed to distribute throughout the lungs by holding the animal upright. The chest was wetted with 70% ethanol and within 30–60 s of DNA delivery, electrodes were placed on either side of the chest and the animal was electroporated. Optimal conditions used eight square wave pulses of 10 ms duration each at a field strength of 200 V/cm.
When 20 μg of plasmid was delivered to the lungs in the absence of an electric field, essentially no gene expression above the limit of detection was detected at 2 days after DNA delivery (Figure 3). Similarly, when field strengths of 50 or 100 V/cm were used, no increase in gene expression was detected. By contrast, when a field strength of 200 V/cm was delivered to the chest, we detected almost 60 pg of gene product per gram wet weight of lung. As the field strength was raised to 400 V/cm, gene expression decreased, but was still well above background, similar to the profile of gene delivery and expression seen in the vasculature and cornea. Consequently, in all subsequent experiments, electroporations were carried out at a field strength of 200 V/cm. Electroporation-mediated gene delivery and expression were also dose-dependent (Figure 4). Very little gene expression was detected at doses of DNA below 20 μg, but significant levels were obtained at both 20 and 100 μg of DNA. In all cases, the same volume of DNA solution was delivered to the animals; only the concentration differed. When 100 μg of DNA was delivered by electroporation to the animals, almost 10 ng of gene product per gram wet weight of lung was obtained. For a typical mouse, this corresponds to almost 150 pg per animal.
Figure 3.
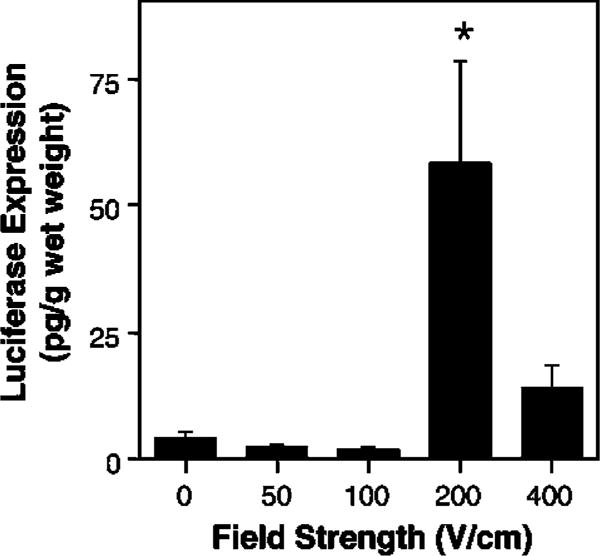
Voltage dependency of gene transfer. A total quantity of 20 μg of pCMV-Lux-DTS in 200 μl was injected intratracheally and varying field strengths were applied to the chest. In all cases, eight pulses of 10 ms duration each were used. The levels of gene expression were measured at 2 days post-treatment. The mean expression is shown±s.e.m. (n = 4 animals/point). *P<0.02 versus 0 V/cm by Mann-Whitney U-test.
Figure 4.
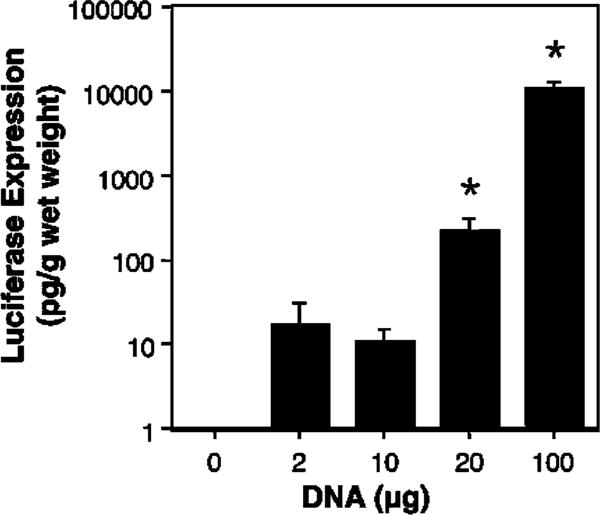
Dose curve for electroporation-mediated gene transfer. Mice were injected intratracheally with varying concentrations of pCMV-Lux-DTS (200 μl) and electroporated at 200 V/cm as described in Figure 2 (n = 4 animals per concentration). After 2 days, luciferase activities were measured as described in the section materials and methods. Mann–Whitney U-tests were performed to determine statistical significance. *P<0.001 compared to no DNA.
A time course of expression was performed after delivery and electroporation of DNA (Figure 5). As has been seen in other tissues, gene expression is detected by 1 day postelectroporation and is maximal between 2 and 5 days. By 7 days postdelivery, essentially no gene expression was detected over background levels. Thus, this technique is very effective for short-term gene expression.
Figure 5.

Time course of gene expression in the mouse lung. A total quantity of 20 μg of pCMV-Lux-DTS in 200 μl was injected intratracheally and the chest was electroporated at 200 V/cm. At the indicated times, the animals were euthanized and luciferase activity was measured in cell extracts prepared from the excised lungs. The mean expression is shown±s.e.m. (n=4 animals per time). P<0.02 versus 0 days by Mann–Whitney U-test.
Localization of gene expression in the lung
To determine where gene expression is occurring following DNA delivery by electroporation, we transferred a plasmid expressing β-galactosidase to the lung and stained for enzyme activity using X-gal (Figure 6). The left lobe of lungs that received 20 μg of the lacZ plasmid but were not electroporated showed no β-galactosidase activity (Figure 6a). Similarly, lungs that did not receive plasmid showed no blue staining (not shown). By contrast, when the same amount of plasmid was delivered and electroporated, there was a significant amount of β-galactosidase activity as shown in idividual lobes (Figure 6b) or in entire lungs (Figure 6c and d). At higher magnification, it could be seen that β-galactosidase activity was concentrated within small “dots” that may correspond to alveolar sacs. This was confirmed when thin sections were cut from the paraffin-embedded lungs (Figure 6e–g). Most of the X-gal-reactive β-galactosidase activity appeared to be localized to the periphery of the lungs, and there was nonuniform, spotty activity in parenchymal tissue. As can be seen, intense and light-blue staining can be seen in all cells of the alveoli (Figure 6e–g). Thus, both alveolar type I and type II cells appear to receive and express the transgene. Immunohistochemical analysis using antibodies against β-galactosidase confirmed that gene expression is focused in peripheral alveoli with some limited expression in airways and blood vessels in focal areas (not shown). These results suggest that electroporation can be used for high-level gene transfer and expression to the alveolar epithelium throughout the lung.
Figure 6.

Distribution of gene expression in electroporated mouse lungs. A quantity of 20 μg of pCMV-lacZ-DTS in 100 μl was injected intratracheally into animals that were either not electroporated (a) or electroporated at 200 V/cm (b–d). After 2 days, lungs were perfused, fixed, and stained with X-gal for β-galactosidase expression. Panels (a) and (b) show the left lobes and panels (c) and (d) show the entire lungs of four different animals. Panels (e–g) show areas of gene expression in lungs electroporated with pCMV-lacZ-DTS at higher magnification in 6 μm thin sections. Bars=1 mm (a and b), 5 mm (c and d), 100 μm (e and f), and 50 μm (g).
Determination of inflammatory response and procedure safety
In order to assess the safety of the delivery and electroporation procedure on lung tissue, sections of treated and naïve lungs were examined for histological changes due to the procedure at 1 h and 24 h following treatment (Figure 7). Mice received no fluid instillation or pCMV-lux-DTS with or without electroporation. Lungs were inflated to total lung capacity with formalin prior to paraffin embedding and sectioning. Blinded pathological examination revealed no apparent histological differences between untreated lungs, those receiving DNA without electroporation, those that were electroporated without DNA addition, and those that had been electroporated with plasmid at 200 V/cm. Using a five point scale for lung injury and scoring four criteria (vascular congestion, hyaline membranes, polymorphonuclear cell infiltrates, and interstitial infiltrates), it was impossible to detect any lung injury or to distinguish between the blinded samples (Table 1). There was no evidence of infiltrating cells, no thickening of alveolar or airway walls, and no hemorrhaging. To ensure that the procedure caused no inflammatory response, IL-6 levels were also measured in these lungs at 24 h post-treatment (Tabel 1). As seen in the pathological analysis, neither DNA delivery nor electroporation caused any increases in IL-6 levels. Further, the expression of the foreign transgene did not affect any parameters. Based on these results and the health of the animals, the procedure seems well-tolerated and safe.
Figure 7.

Histological analysis of electroporated and untreated lungs. Lungs from naïve animals (a and e), animals that received 100 μl of plasmid only (no electroporation, b and f), animals that received no DNA but were electroporated (c and g), or animals that received 100 μl of plasmid and were electroporated (d and h) were removed 1 h (a–d) or 24 h (e–h) post-treatment, inflated, paraffin-sectioned, and stained with hematoxylin and eosin.
Table 1.
Lung Injury and Inflammation
| Treatment |
Vascular congestion
|
IL-6 (pg/mg protein) | Luciferase (pg/g wet weight) | |
|---|---|---|---|---|
| 1 h | 24 h | |||
| Naive | 0 | 1 | 24.4±14.4 | 1.1±0.3 |
| Electroporation only | 1 | 0 | 9.9±0.7 | 2.0±0.8 |
| DNA only | 1 | 0 | 18.6±8.0 | 118±47 |
| DNA+electroporation | 0 | 0 | 13.8±5.5 | 9568±2140 |
Mice (n=2) were either untreated (naïve), given 100 μl of plasmid only (no electroporation), electroporated without DNA addition (electroporation only), or given 100 μl of plasmid (100 μg) and electroporated as described in the section materials and methods. At 1 h after treatment, the lungs were inflated, paraffin-sectioned, and stained with hematoxylin and eosin. At 24 h after treatment, the left lobe was tied off and removed for cytokine and luciferase measurements prior to inflation and fixation. Samples were blinded and scored on a five point scale for lung injury (0=healthy, 5=severe lung injury). Scoring for hyaline membranes, polymorphonuclear infiltrates, and interstitial infiltrates gave a score of 0 (completely healthy) for all sections and are not shown. IL-6 and luciferase levels at 24 h post-treatment were determined by ELISA and luciferase assays, respectively (mean±s.e.m.; an additional lung was included for the 24 h point, n=3).
Discussion
In this study, we have shown that delivery of naked DNA using electroporation is a simple and reproducible method for gene transfer to the lungs. By delivering relatively low levels of purified plasmid to the lungs of mice via an intratracheal injection, and then applying an electric field across the chest of the animal using eight square wave pulses of 10 ms duration each, up to nanogram levels of gene product per gram wet weight could be detected by 2 days postelectroporation. Maximal gene expression is obtained at a field strength of 200 V/cm. By contrast, with no applied electric field, essentially no gene transfer and expression is detected. Histological analysis of treated lungs revealed that gene transfer is seen throughout the periphery with limited distribution in the parenchyma, resulting in gene expression primarily in alveolar type I and type II epithelial cells. In aggregate, this technique may have wide appeal for pulmonary gene transfer owing to its ease, efficacy, and safety.
Apart from exposing the trachea through an incision and the instillation of the DNA solution, the current procedure results in little trauma. Others have demonstrated success delivering similar volumes of fluid to the lungs of mice using nasal inhalation or an endotracheal tube.9,29,30 By employing similar methods, we may be able to reduce the length of the procedure or potential complications due to minor surgery, making lung electroporation an even more attractive method. The optimal field strength for gene transfer is 200 V/cm, a value similar to that found by us in a number of other tissues in both mice and rats, including the vasculature, cornea, and skeletal muscle.22,25,31 This field strength corresponds to less than 1 J of energy (W s) and is well below that used to defibrillate the heart (200–360 J). Indeed, we have seen no trauma or damage induced by the electroporation procedure, either macro- or microscopically. IL-6 levels were unchanged in treated lungs and histological analysis of the lungs showed no inflammatory infiltrates or other pathological changes induced by electroporation. Thus, the procedure is well tolerated by the animals and is safe.
When compared to the level of gene expression obtained after ex vivo DNA delivery and electroporation, the amount of expression seen resulting from in vivo delivery of the same amount of DNA was approximately 20-fold less. The most likely reason for this is differences in the distribution and relative strength of the electric field at the lung between the two procedures. When the electrodes are placed directly on the pleural surface of the lung in the ex vivo setting, the full field strength (200 V/cm) is delivered to the lung. By contrast, by applying the electrodes to the surface of the chest, the field must travel through skin, fat, muscle, bone, cartilage, and interstitial fluid prior to reaching the lungs. Since the field distribution within the chest has not been characterized, it is not clear whether the lungs actually see a field strength of 200 V/cm, but it is likely that they see only a fraction of the field due to the electrical parameters of the other tissue components.32 Our laboratory is currently exploring this.
The amount of gene expression obtained in the mouse lung using electroporation is on the order of 10–100 pg product per μg of administered DNA per gram of tissue at high doses of DNA (100 μg per animal). This corresponds to approximately 20–200 pg luciferase per μg of administered DNA per mg protein. Although it is difficult to directly compare these expression levels to those using other delivery techniques, electroporation-mediated DNA delivery to the lung appears to be a very desirable method for delivery. For example, studies using aerosol delivery of polyethyleneimine:DNA complexes in mice gave roughly 2 pg of gene product per gram tissue per μg of DNA,4 whereas intravenous delivery of cationic lipid–protamine–DNA complexes in mice yielded 400 pg luciferase/μg DNA/mg protein.6 Thus, electroporation yields levels that are comparable to those obtained previously.
High-level expression was seen by day 2 and peaked at day 5 postelectroporation. However, by 7 days postdelivery, the levels of gene expression had fallen back to baseline. Similar time courses of gene expression have been observed by us using plasmids delivered by electroporation in the vasculature and cornea.22,25 Similarly, Liu and colleagues, using intravenous delivery of lipoplex, found that gene expression in the lung peaked at 1 day and then decreased by 2- to 3-logs by 4 days postdelivery.7 In all of these cases, the promoters used to drive gene expression have been strong promoter/enhancer combinations from viruses, most notably the CMViep. By contrast, Yew et al33,34 have reported that the use of a human ubiquitin B promoter or a hybrid CMV–ubiquitin B promoter can increase the duration of expression up to 3 months over the typical 7-day expression pattern seen with the CMViep alone. Thus, it appears that the short duration of gene expression seen in our experiment is due to the choice of promoter, and that use of alternative promoters may increase both the duration and amount of gene expression in electroporated lungs. However, even with such short durations of gene expression, this is a valuable approach for delivery of genes where expression for limited times is desired.
One major advantage of this method for gene delivery to the lung is that it efficiently transfers genes to multiple cell types. Perhaps most exciting is its ability to mediate gene delivery and expression in alveolar type I and type II cells, which are typically very difficult to transfect, either in vivo or in vitro. Viruses and most nonviral DNA delivery systems typically deliver genes to only the cell layer with which they come into contact. Thus, when delivered systemically, gene transfer is seen primarily to the pulmonary vascular endothelial cells,6,7,9,35,36 whereas airway delivery results in transfer and expression in the epithelium.9,10,35,37,38 One exception to this appears to be PEI, where it has been reported that intravenous delivery of PEI–DNA complexes can in fact lead to expression in alveolar epithelial type II cells.3,39 By contrast, electroporation results in gene transfer to alveolar epithelial cells, as well as cells beyond the epithelial permeability barrier, including some smooth muscle and endothelial cells in focal areas of the lung. How the DNA is traversing the epithelium remains unclear, but it is possible that transient holes are opened up between cells by the electric field, thus allowing DNA to move to lower cell layers. Alternatively, it is also possible that the DNA is moving through the cells themselves during the eight pulses. Similar movement of DNA through multiple cell layers has also been seen in the vasculature where DNA added to the adventitial surface of the blood vessel is delivered to and expresses in the medial and intimal smooth muscle layer as well as the endothelium.22
As seen from the lacZ expression studies, the distribution of gene expression is heterogenous and was focused primarily in the alveolar epithelium at the periphery (Figure 6). Most surprising was the fact that there was very little detectable gene transfer and expression in airway epithelial cells. There are several possible reasons for this. First, it is likely that we are not getting uniform distribution of the delivered plasmid solution. It has been demonstrated that much more homogenous distribution of fluid throughout the lung is obtained when the liquid forms a plug in the trachea before being carried through the airways by inspired air than when liquid is delivered into the trachea and moves by gravity into the airways.40 Based on our method of injection, it is likely that we are relying on gravity to deliver the fluid, and, thus, less homogeneity is possible. In the future, the formation of a liquid plug and/or the use of surfactant may aid in better distribution of the DNA to the parenchyma prior to electroporation and hence more uniform expression. Another possibility is that the applied electric field is not uniform throughout the lung and that regions receiving a lower field strength did not show expression. The electrodes used in the present study were 10 mm in diameter, placed on either side of the chest. It is possible that the use of larger electrodes could increase the area of high-level gene expression. Future studies will test whether the combination of better fluid delivery techniques and electrodes can increase the distribution to provide gene transfer and expression to all cells in the lung.
Acknowledgments
We thank Joseph N Benoit (University of North Dakota), and G.R. Scott Budinger, Joshua Meeks, and Chris Capaccio (Northwestern University) for insightful discussions. This work was supported in part by grants from the Crane Asthma Fund and the Sandler Program for Asthma Research.
References
- 1.Albelda SM, Wiewrodt R, Zuckerman JB. Gene therapy for lung disease: hype or hope? Ann Intern Med. 2000;132:649–660. doi: 10.7326/0003-4819-132-8-200004180-00008. [DOI] [PubMed] [Google Scholar]
- 2.Chadwick SL, et al. Safety of a single aerosol administration of escalating doses of the cationic lipid GL-67/DOPE/DMPE-PEG5000 formulation to the lungs of normal volunteers. Gene Therapy. 1997;4:937–942. doi: 10.1038/sj.gt.3300481. [DOI] [PubMed] [Google Scholar]
- 3.Bragonzi A, et al. Comparison between cationic polymers and lipids in mediating systemic gene delivery to the lungs. Gene Therapy. 1999;6:1995–2000. doi: 10.1038/sj.gt.3301039. [DOI] [PubMed] [Google Scholar]
- 4.Gautam Densmore CL, Xu B, Waldrep JC. Enhanced gene expression in mouse lung after PEI–DNA aerosol delivery. Mol Ther. 2000;2:63–70. doi: 10.1006/mthe.2000.0087. [DOI] [PubMed] [Google Scholar]
- 5.Barron L, Uyechi L, Szoka FC. Cationic lipids are essential for gene delivery mediated by intravenous administration of lipoplexes. Gene Therapy. 1999;6:1179–1183. doi: 10.1038/sj.gt.3300929. [DOI] [PubMed] [Google Scholar]
- 6.Li S, Huang L. In vivo gene transfer via intravenous administration of cationic lipid–protamine–DNA (LPD) complexes. Gene Therapy. 1997;4:891–900. doi: 10.1038/sj.gt.3300482. [DOI] [PubMed] [Google Scholar]
- 7.Song YK, Liu F, Chu S, Liu D. Characterization of cationic liposome-mediated gene transfer in vivo by intravenous administration. Hum Gene Ther. 1997;8:1585–1594. doi: 10.1089/hum.1997.8.13-1585. [DOI] [PubMed] [Google Scholar]
- 8.Tan Y, et al. Sequential injection of cationic liposome and plasmid DNA effectively transfects the lung with minimal inflammatory toxicity. Mol Ther. 2001;3:673–682. doi: 10.1006/mthe.2001.0311. [DOI] [PubMed] [Google Scholar]
- 9.Uyechi LS, Gagne L, Thurston G, Szoka FC., Jr Mechanism of lipoplex gene delivery in mouse lung: binding and internalization of fluorescent lipid and DNA components. Gene Therapy. 2001;8:828–836. doi: 10.1038/sj.gt.3301461. [DOI] [PubMed] [Google Scholar]
- 10.Yonemitsu Y, et al. Efficient gene transfer to airway epithelium using recombinant Sendai virus. Nat Biotechnol. 2000;18:970–973. doi: 10.1038/79463. [DOI] [PubMed] [Google Scholar]
- 11.Stern M, et al. The effects of jet nebulisation on cationic liposome-mediated gene transfer in vitro. Gene Therapy. 1998;5:583–593. doi: 10.1038/sj.gt.3300629. [DOI] [PubMed] [Google Scholar]
- 12.Weiss D. Delivery of gene transfer vectors to lung: obstacles and the role of adjunct techniques for airway administration. Mol Therapy. 2002;6:148. doi: 10.1006/mthe.2002.0662. [DOI] [PubMed] [Google Scholar]
- 13.Somiari S, et al. Theory and in vivo application of electroporative gene delivery. Mol Therapy. 2000;2:178–187. doi: 10.1006/mthe.2000.0124. [DOI] [PubMed] [Google Scholar]
- 14.Ausubel FM, et al. Current Protocols in Molecular Biology. John Wiley & Sons; New York: 1994. [Google Scholar]
- 15.Mathiesen I. Electropermeabilization of skeletal muscle enhances gene transfer in vivo. Gene Therapy. 1999;6:508–514. doi: 10.1038/sj.gt.3300847. [DOI] [PubMed] [Google Scholar]
- 16.Mir LM, et al. High-efficiency gene transfer into skeletal muscle mediated by electric pulses. Proc Natl Acad Sci USA. 1999;96:4262–4267. doi: 10.1073/pnas.96.8.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aihara H, Miyazaki J. Gene transfer into muscle by electroporation in vivo. Nat Biotechnol. 1998;16:867–870. doi: 10.1038/nbt0998-867. [DOI] [PubMed] [Google Scholar]
- 18.Heller R, et al. In vivo gene electroinjection and expression in rat liver. FEBS Lett. 1996;389:225–228. doi: 10.1016/0014-5793(96)00590-x. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki T, et al. Direct gene transfer into rat liver cells by in vivo electroporation. FEBS Lett. 1998;425:436–440. doi: 10.1016/s0014-5793(98)00284-1. [DOI] [PubMed] [Google Scholar]
- 20.Harrison RL, Byrne BJ, Tung L. Electroporation-mediated gene transfer in cardiac tissue. FEBS Lett. 1998;435:1–5. doi: 10.1016/s0014-5793(98)00987-9. [DOI] [PubMed] [Google Scholar]
- 21.Dujardin N, et al. In vivo assessment of skin electroporation using square wave pulses. J Control Release. 2002;79:219–227. doi: 10.1016/s0168-3659(01)00548-x. [DOI] [PubMed] [Google Scholar]
- 22.Martin JB, Young JL, Benoit JN, Dean DA. Gene transfer to intact mesenteric arteries by electroporation. J Vasc Res. 2000;37:372–380. doi: 10.1159/000025753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsumoto T, et al. Successful and optimized in vivo gene transfer to rabbit carotid artery mediated by electronic pulse. Gene Therapy. 2001;8:1174–1179. doi: 10.1038/sj.gt.3301502. [DOI] [PubMed] [Google Scholar]
- 24.Oshima Y, et al. Targeted gene transfer to corneal endothelium in vivo by electric pulse. Gene Therapy. 1998;5:1347–1354. doi: 10.1038/sj.gt.3300725. [DOI] [PubMed] [Google Scholar]
- 25.Blair-Parks K, Weston BC, Dean DA. Gene delivery to the cornea by plasmid injection and electroporation. J Gene Med. 2002;4:92–100. doi: 10.1002/jgm.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsujie M, et al. Electroporation-mediated gene transfer that targets glomeruli. J Am Soc Nephrol. 2001;12:949–954. doi: 10.1681/ASN.V125949. [DOI] [PubMed] [Google Scholar]
- 27.Dean BS, Byrd JN, Jr, Dean DA. Nuclear targeting of plasmid DNA in human corneal cells. Cur Eye Res. 1999;19:66–75. doi: 10.1076/ceyr.19.1.66.5344. [DOI] [PubMed] [Google Scholar]
- 28.Vacik J, Dean BS, Zimmer WE, Dean DA. Cell-specific nuclear import of plasmid DNA. Gene Therapy. 1999;6:1006–1014. doi: 10.1038/sj.gt.3300924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wheeler CJ, et al. A novel cationic lipid greatly enhances plasmid DNA delivery and expression in mouse lung. Proc Natl Acad Sci USA. 1996;93:11454–11459. doi: 10.1073/pnas.93.21.11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stern M, et al. Pretreatment with cationic lipid-mediated transfer of the Na+ K+-ATPase pump in a mouse model in vivo augments resolution of high permeability pulmonary oedema. Gene Therapy. 2001;7:960–966. doi: 10.1038/sj.gt.3301193. [DOI] [PubMed] [Google Scholar]
- 31.Li S, et al. Muscle-specific enhancement of gene expression by incorporation of the SV40 enhancer in the expression plasmid. Gene Therapy. 2001;8:494–497. doi: 10.1038/sj.gt.3301419. [DOI] [PubMed] [Google Scholar]
- 32.Gabriel C, Lau RW, Gabriel S. The dielectric properties of biological tissues: III. Parametric models for the dielectric spectrum of tissues. Phys Med Biol. 1996;41:2271–2293. doi: 10.1088/0031-9155/41/11/003. [DOI] [PubMed] [Google Scholar]
- 33.Yew NS, et al. Optimization of plasmid vectors for high-level expression in lung epithelial cells. Hum Gene Ther. 1997;8:575–584. doi: 10.1089/hum.1997.8.5-575. [DOI] [PubMed] [Google Scholar]
- 34.Yew NS, et al. High and sustained transgene expression in vivo from plasmid vectors containing a hybrid ubiquitin promoter. Mol Therapy. 2001;4:75–82. doi: 10.1006/mthe.2001.0415. [DOI] [PubMed] [Google Scholar]
- 35.Canonico AE, Conary JT, Meyrick BO, Brigham KL. Aerosol and intravenous transfection of human alpha 1-antitrypsin gene to lungs of rabbits. Am J Respir Cell Mol Biol. 1994;10:24–29. doi: 10.1165/ajrcmb.10.1.8292378. [DOI] [PubMed] [Google Scholar]
- 36.Reynolds PN, et al. A targetable, injectable adenoviral vector for selective gene delivery to pulmonary endothelium in vivo. Mol Therapy. 2000;2:562–578. doi: 10.1006/mthe.2000.0205. [DOI] [PubMed] [Google Scholar]
- 37.Dumasius V, et al. b2-Adrenergic receptor overexpression increases alveolar fluid clearance and responsiveness to endogenous catecholamines in rats. Circ Res. 2001;89:907–914. doi: 10.1161/hh2201.100204. [DOI] [PubMed] [Google Scholar]
- 38.Weiss DJ, Bonneau L, Liggitt D. Use of perfluorochemical liquid allows earlier detection of gene expression and use of less vector in normal lung and enhances gene expression in acutely injured lung. Mol Therapy. 2001;3:734–745. doi: 10.1006/mthe.2001.0321. [DOI] [PubMed] [Google Scholar]
- 39.Goula D, et al. Rapid crossing of the pulmonary endothelial barrier by polyethylenimine/DNA complexes. Gene Therapy. 2000;7:499–504. doi: 10.1038/sj.gt.3301113. [DOI] [PubMed] [Google Scholar]
- 40.Cassidy KJ, et al. A rat lung model of instilled liquid transport in the pulmonary airways. J Appl Physiol. 2001;90:1955–1967. doi: 10.1152/jappl.2001.90.5.1955. [DOI] [PubMed] [Google Scholar]