

Abstract
The genomes of cancers deficient in mismatch repair (MMR) contain exceptionally high numbers of somatic mutations. In a proof-of-concept study, we previously showed that colorectal cancers with MMR deficiency were sensitive to immune checkpoint blockade with anti-PD-1 antibodies. We have expanded this study to now evaluate efficacy of PD-1 blockade in patients with advanced MMR-deficient cancers across 12 different tumor types. Objective radiographic responses were observed in 53% of patients and complete responses were achieved in 21% of patients. Responses were durable with median progression-free and overall survival still not reached. Functional analysis in a responding patient demonstrated rapid in vivo expansion of neoantigen-specific T cell clones that were reactive to mutant neopeptides found in the tumor. These data support the hypothesis that the large proportion of mutant neoantigens in MMR-deficient cancers make them sensitive to immune checkpoint blockade, regardless of the cancers’ tissue of origin.
Therapy with immune checkpoint inhibitors has uncovered a subset of tumors that are highly responsive to an endogenous adaptive immune response (1). When the interaction between the checkpoint ligands and their cognate receptors on the effector cells is blocked, a potent and durable anti-tumor response can be observed and on occasion this response can be accompanied by severe autoimmunity (2–5). These findings support the notion that many cancer patients contain in their immune system the capacity to react selectively to their tumors, ostensibly through recognition of tumor-specific antigens.
The molecular determinants that define this subset of tumors is still unclear, however several markers, including PD-L1 expression, RNA expression signatures, mutational burden and lymphocytic infiltrates have been evaluated in specific tumor types (6–10). Though such markers appear to be helpful in predicting response in specific tumor types, none of them have been evaluated prospectively as a pan-tumor biomarker. Another potential determinant of response is mutation-associated neoantigens (MANAs) that are encoded by cancers (11–14). As previously described, mismatch-repair deficient cancers are predicted to have a very large number of MANAs that might be recognized by the immune system (15–18). This prediction led us to conduct a small phase II study, focused on eleven patients with colorectal cancers, in which it was demonstrated that PD-1 blockade was an effective treatment for many patients with these tumors (19). Since the initiation of that trial, other studies have shown that the number of mutations in MMR-proficient cancers correlates with the response to PD-1 blockade, providing further support for a relationship between mutation burden and treatment response (20).
The genomes of mismatch repair-deficient tumors all harbor hundreds to thousands of somatic mutations, regardless of their cell of origin. We therefore sought to investigate the effects of PD-1 blockade (using the anti-PD-1 antibody pembrolizumab) in mismatch-repair tumors independent of the tissue of origin. In the current study, we prospectively evaluated the efficacy of PD-1 blockade in a range of different subtypes of mismatch repair-deficient cancers (Clinical-Trials.gov number, NCT01876511).
Eighty-six consecutive patients were enrolled between September 2013 and September 2016 (table S1). The data cutoff was December 19, 2016. All patients received at least one prior therapy and had evidence of progressive disease prior to enrollment. Twelve different cancer types were enrolled in the study (Fig. 1). All enrolled patients had evidence of mismatch repair-deficiency as assessed by either polymerase chain reaction or immunohistochemistry. For most cases, germline sequencing of MSH2, MSH6, PMS2 and MLH1 was performed to determine if the mismatch repair-deficiencies were associated with a germline change in one of these genes (i.e., whether the patients had Lynch Syndrome) (table S2). Germline sequence changes diagnostic of Lynch syndrome were noted in 32 cases (48%), with MSH2 being the most commonly mutated gene. In seven additional cases where germ line testing was not performed, the patient reported a family history consistent with a diagnosis of Lynch syndrome.
Fig. 1. Patient survival and clinical response to Pembrolizumab across 12 different tumor types with mismatch repair deficiency.
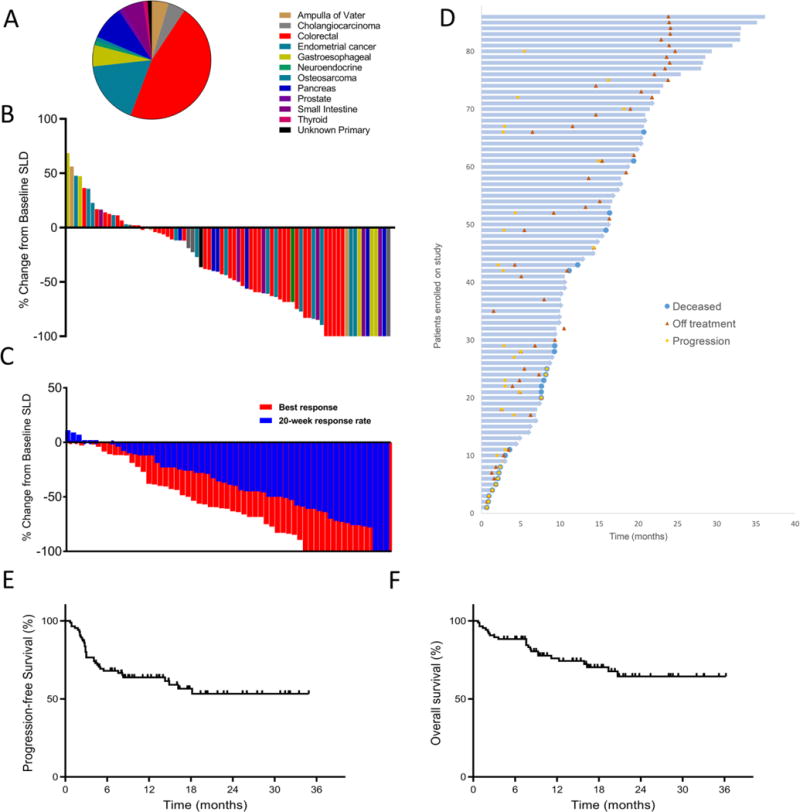
(A) Tumor types across 86 patients. (B) Waterfall plot of all radiographic responses across 12 different tumor types at 20 weeks. Tumor responses were measured at regular intervals and values show the best fractional change of the sum of longest diameters (SLD) from the baseline measurements of each measurable tumor. (C) Confirmed radiographic objective responses at 20 weeks in blue compared to the best radiographic responses in the same patients in red. The mean time to the best radiographic response was 28 weeks. (D) Swimmer plot showing survival for each patient with mismatch repair deficient tumors, indicating death, progression and time off therapy. (E) Kaplan-Meier estimates of progression-free survival and (F) overall patient survival.
The adverse events to treatment were manageable, and similar to those found in other clinical studies employing pembrolizumab, as shown in table S3. While 74% of patients experienced an adverse effect, most were low grade. Endocrine disorders, mostly hypothyroidism, occurred in 21% of patients and was easily managed with thyroid hormone replacement.
Seventy-eight patients had disease that could be evaluated by Response Evaluation Criteria In Solid Tumors (RECIST) (Table 1). Objective radiographic responses were noted in 53% of patients (46 of 86 patients; 95% CI, 42–64%), with 21% (n = 18) achieving a complete radiographic response. Disease control (measured as partial response + complete response + stable disease) was achieved in 66 (77%) of the 86 patients (95% CI, 66–85%). Radiographic responses could be separated into two classes. First, in twelve cases, scans at 20 weeks showed stable disease, which eventually converted to an objective response (measured as tumor size reduction in response to therapy, according to RECIST criteria). Second, in eleven additional cases, we observed an initial partial response or stable disease at the 20-week scan that later converted to complete responses while treatment was continued. The average time to any response was 21 weeks and the average time to complete response was 42 weeks (Fig. 1). Of note, the objective response rate was similar between colorectal cancer versus other cancer subtypes. Specifically, objective responses were observed in 52% (95% CI 36 to 68%) of patients with colorectal cancers and 54% (95% CI 39 to 69%) of the patients with cancers originating in other organs (tables S4 and S5). There was also no significant difference in the objective response rate between Lynch and non-Lynch syndrome-associated tumors; 46% (95% CI of 30 to 63%) versus 59% (95% CI of 41 to 76%), respectively (p = 0.27).
Table 1. Summary of therapeutic response to pembrolizumab (anti-PD-1) treatment.
Radiographic responses, progression-free survival (PFS) and overall survival (OS) estimates were measured using the Response Evaluation Criteria In Solid Tumors (RECIST v1.1) guidelines. Patients were considered not evaluable if they did not undergo a 12-week scan due to clinical progression. The rate of disease control was defined as the percentage of patients who had a complete response, partial response, or stable disease for 12 weeks or more. NR, not reached.
| Type of response | Patients (n = 86) |
|---|---|
| Complete response | 18 (21%) |
| Partial response | 28 (33%) |
| Stable disease | 20 (23%) |
| Progressive disease | 12 (14%) |
| Not evaluable | 8 (9%) |
| Objective response rate 95% CI |
53% 42% to 64% |
| Disease control rate 95% CI |
77% 66% to 85% |
| Median progression-free survival time 95% CI |
NR 14.8 months to NR |
| 2-year progression-free survival rate 95% CI |
53% 42% to 68% |
| Median overall survival time 95% CI |
NR NR to NR |
| 2-year overall survival rate 95% CI |
64% 53% to 78% |
Neither median progression-free survival (PFS) nor median overall survival (OS) has yet been reached (median follow-up time of 12.5 months; Fig. 1) and the study is ongoing. However the estimates of PFS at 1- and 2-years were 64% and 53%, respectively. The estimates of OS at 1- and 2-years were 76% and 64%, respectively, which is markedly higher than expected based on the advanced state of disease in this cohort (21). The PFS and OS was not significantly different in patients with colorectal cancers as compared to those with other cancer types (fig. S1). Neither PFS (HR 1.2; 95% CI of 0.582 to 2.512, p = 0.61) or OS (HR 1.71; 95% CI of 0.697 to 4.196; p = 0.24) were influenced by tumors associated with Lynch Syndrome.
Eleven patients achieved a complete response and were taken off therapy after two years of treatment. No evidence of cancer recurrence has been observed in those patients with a median time off therapy of 8.3 months. Seven other patients had residual disease by imaging, but pembrolizumab was discontinued after reaching the 2-year milestone or because of intolerance to therapy. To date, the average time off therapy for this group was 7.6 months. As of the data cutoff, none of these patients has shown evidence of progression since discontinuation of pembrolizumab.
Twenty patients with measurable radiographic disease underwent percutaneous biopsies between 1 month and 5 months after the initiation of therapy. Twelve of these biopsies demonstrated no evidence of tumor cells and were shown to have varying degrees of inflammation, fibrosis and mucin, consistent with an ongoing immune response (fig. S2). The other eight cases showed residual tumor cells. The absence of cancer cells in post-treatment biopsies was a strong predictor of progression free survival (HR for PFS was 0.189, 95% CI 0.046 to 0.767, p = 0.012) with median PFS of 25.9 months versus 2.9 months for biopsies with evidence of residual tumor. While there was no significant difference in OS between patients whose biopsies were positive or negative for tumor cells, median OS has not yet been reached in patients with negative biopsies (table S6).
Primary clinical resistance to initial therapy with pembrolizumab (as measured by progressive radiographic disease on the first study scan) was noted in twelve (14%) patients (Table 1). We determined the exomic sequences of tumor and matched normal DNA from three of these patients and compared them to the exomes of 15 primary tumors from patients that had achieved objective responses to the therapy (table S7). The three therapy-insensitive tumors harbored an average of 1,413 non-synonymous mutations, not significantly different from the number in patients with objective responses (1,644 non-synonymous mutations; p = 0.67, Student’s t test). The gene (B2M) encoding β2-microglobulin, a protein required for antigen presentation (22), was not mutated in any of the primary tumors from the resistant group (table S8).
Only five cases of acquired resistance were noted, where patients developed progressive disease after an initial objective response to pembrolizumab. Three of these cases were atypical in that the tumors emerged in occult sites such as the brain (two cases) or bone (one case). All three cases were treated with local therapy (radiation or surgery) and patients survived and continued treatment with pembrolizumab. However, in accordance with study design, these three patients are listed in Fig. 1 as having progressive disease.
We performed exome sequencing of brain metastases biopsies from two patients and compared the results with those of their primary tumors (fig. S3 and table S7). In the first case, the primary duodenal tumor and brain metastasis shared 397 non-synonymous somatic mutations, providing unequivocal evidence that the metastasis was derived from the primary duodenal tumor rather than from an independent tumor. Moreover, the metastasis harbored 1,010 non-synonymous new mutations not present in the primary tumor, while the primary tumor harbored 964 mutations not present in the metastasis (table S9). In the second case, the primary colorectal tumor and brain metastasis shared 848 non-synonymous somatic mutations, similarly providing unequivocal evidence of a genetic relationship between the two lesions. The brain metastasis harbored 221 non-synonymous mutations not present in the primary colorectal tumor, while the primary tumor harbored 100 mutations not present in the metastasis (table S10). Of note, the brain metastases from both these patients contained mutations in the B2M gene. In the patient with the duodenal tumor, a truncating mutation (L15Ffs*41) in the B2M gene was identified in the metastasis but not in the primary tumor. The primary colorectal tumor harbored a truncating mutation in beta-2-microglobulin (V69Wfs*34) while the metastasis retained this mutation and acquired a second B2M mutation (12L>P; table S7).
We also evaluated the exomes of three primary tumors from patients that originally had stable disease by RECIST criteria at 20 weeks, but disease progressed within 8 months of initiating therapy. The average mutational burden was 1,647 for this group, similar to those of the other patients described above. Interestingly, two of these three tumors harbored mutations of B2M (table S7).
We next sought to directly test the hypothesis that checkpoint blockade induces peripheral expansion of tumor-specific T cells and that mismatch repair deficient tumors harbor functional MANA-specific T cells. Deep sequencing of TCR CDR3 regions (TCRseq) has emerged as a valuable technique to evaluate T cell clonal representation in both tumors and peripheral blood. We performed TCRseq on tumors from three responding patients (obtained from archival surgical resections) and identified intratumoral clones that were selectively expanded in the periphery (Fig. 2A). These clones were present at very low frequency (often undetectable) in the peripheral blood before pembrolizumab treatment, but many rapidly increased after treatment initiation, followed by a contraction that generally occurred before radiologic responses were observed. To characterize functional T cell clones’ specific for mutant peptides, we obtained peripheral blood from one of the patients (subject 19). We tested the patients post-treatment peripheral blood for reactivity against the 15 top candidate MANAs as identified via a neoantigen prediction algorithm (specified by the patient’s HLA class I alleles – see supplemental materials) with an IFNγ ELISpot assay. Based on either spot-forming cells (SFC) or a cytokine activity analysis, T cell responses were observed against 7/15 peptides (Fig. 2, B and C). We next interrogated the expanded lymphocyte populations against these 7 peptides with TCRseq. Clonal T cell expansion was noted in response to 3/7 peptides (Fig. 2D), with specificity demonstrated by a lack of significant expansion in response to any other peptide tested (fig. S4). In the peripheral blood, T cell expansion to these 3 mutant peptides resulted in 142 unique TCR sequences, seven of which were found in the tumor sample (2 from MANA1, 3 from MANA2 and 2 from MANA4) (Fig. 2D). Of note, the mutant peptides that scored positive all resulted from frameshift mutations -the type of mutation that is most characteristic of MMR-deficient cancers.
Fig. 2. TCR clonal dynamics and mutation associated neoantigen recognition in patients responding to PD-1 blockade.
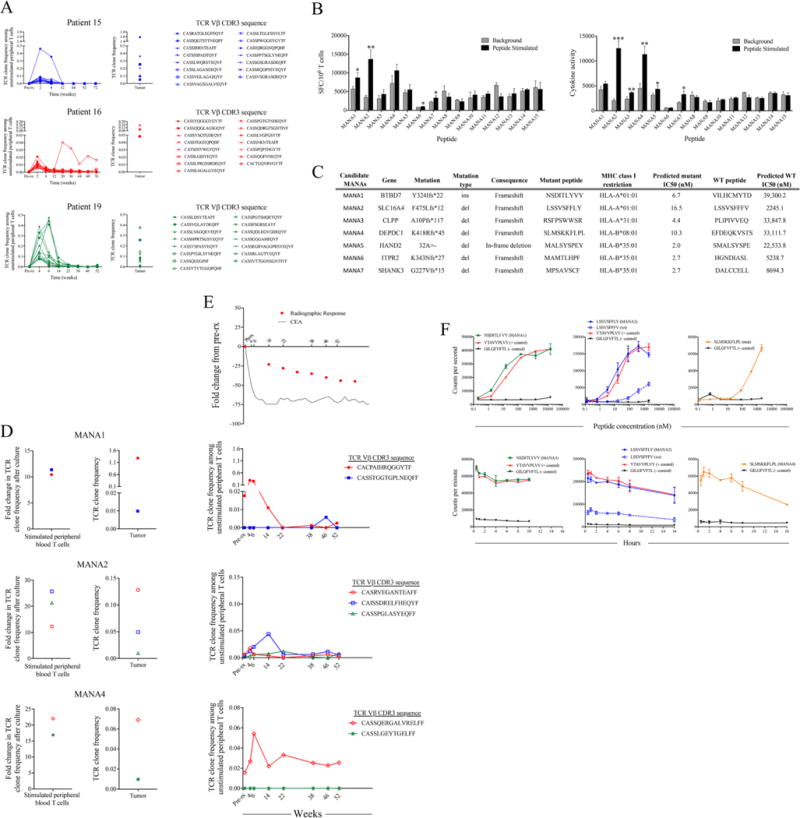
(A) T cell receptor (TCR) sequencing was performed on serial peripheral T cell samples obtained before and after PD-1 blockade. Tumor tissue with mismatch repair deficiency was obtained from three responding patients. Figures show 15 TCR clones with the highest fold change in frequency after treatment (left panels) that was also found in the original tumor (right panels). (B) Whole exome sequencing was performed on tumor and matched normal tissue from patient 19. Somatic alterations were analyzed using a neo-antigen prediction pipeline to identify putative mutation associated neoantigens (MANAs). Reactivity to 15 candidate MANAs was tested in a 10 day cultured IFNγ ELISpot assay. Data are shown as the mean number of spot forming cells (SFC) per 106 T cells (top) or mean cytokine activity (bottom) of triplicate wells +/− SD. (C) Seven candidate MANAs were selected for TCR analysis based on ELISpot reactivity (D) MANA-specific T cell responses were identified against 3/7 candidate MANAs (MANA1, MANA2 and MANA4) after a 10-day in vitro stimulation (left panels). MANA specific clones were identified by significant expansion in response to the relevant peptide and no significant expansion in response to any other peptide tested (fig. S3). Data are shown as the fold change in TCR clone frequency compared to the frequency of that clone after identical culture without peptide. These T cell clones were also found in the original tumor biopsy (right panels). (E) Frequency of MANA-specific clones, carcinoembryonic antigen (CEA) and radiographic response in the tumor [from (D)] were tracked in the peripheral blood before treatment, and at various times after pembrolizomab treatment. Time is shown in weeks after first pembrolizumab dose. (F) In vitro binding and stability assays demonstrate the affinity kinetics of each relevant MANA and the corresponding WT peptide (when applicable) for their restricting HLA I allele. The A*02:01-restricted Influenza M GILGFVTL epitope was used as a negative control for each assay and known HLA-matched epitopes were used as positive controls when available. Data are shown as counts per second with increasing peptide concentration for binding assays (top panel) or counts per minute over time for stability assays (bottom panel). Data points indicate the mean of two independent experiments +/− SD. *p < 0.05, **p < 0.01, ***p < 0.001.
All seven of the MANA-reactive TCRs were detectable in peripheral blood at very low frequency (less than 0.02%) before treatment. However, four of the clones rapidly increased in frequency in the peripheral blood following anti-PD-1 treatment (Fig. 2E). Similar to results from the three patients analyzed above, frequencies of these functionally validated MANA-specific T cell clones peaked soon after treatment and corresponded with normalization of the systemic tumor marker and predated objective radiographic response by several weeks. This peak in T-cell clonal expansion was followed by significant decreases in frequency, reminiscent of T cell responses to acute viral infections (Fig. 2E). Because all the MANAs were from frameshift mutations, only MANA2 had a similar wild-type counterpart (differing in the two carboxy-terminal amino acids). The corresponding wild-type peptide bound to HLA with >100 fold lower affinity than the mutant peptide counterpart (Fig. 2F), consistent with the mutation conferring enhanced HLA binding.
To estimate the fraction of cancer patients to which the results of this study might be applicable, we evaluated 12,019 cancers representing 32 distinct tumor types for MMR-deficiency using an NGS based approach (Fig. 3). In accordance with a recent independent estimate using a different approach (23), we found that >5% of adenocarcinomas of the endometrium, stomach, small Intestine, colon and rectum, cervix, prostate bile duct and liver, as well as neuroendocrine tumors, non-epithelial ovarian cancers and uterine sarcomas, were MMR deficient. Across these 11 tumor types, 8% of stage I to stage III cancers and 4% of stage IV cancers were MMR-deficient. This represents roughly 40,000 annual stage I–III and 20,000 stage IV diagnoses in the United States alone. Because genetic and immunohistochemical tests for MMR-deficiency are already widely available, these results tie immunity, cancer genetics, and therapeutics together in a manner that will likely establish a new standard-of-care and in the future, testing for MMR-deficiency in patients’ refractory to other treatments might be considered in order to identify those who may benefit from PD-1 pathway blockade, regardless of tumor type.
Fig. 3. Mismatch repair deficiency across 12,019 tumors.
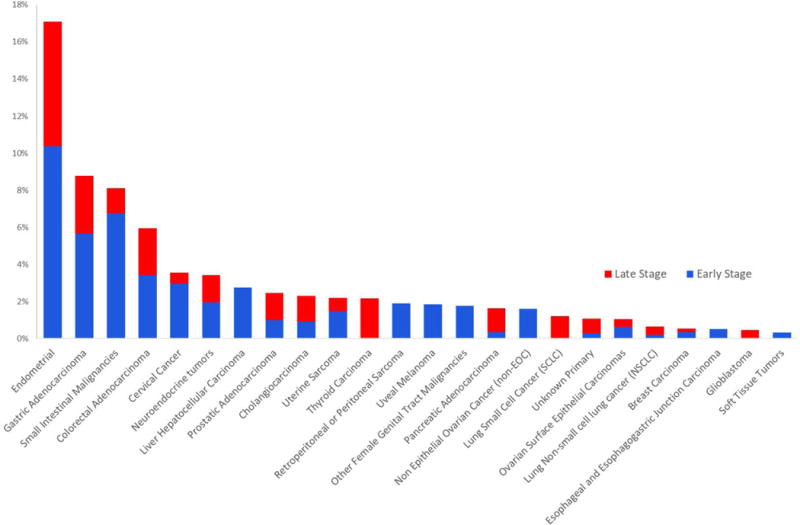
Proportion of tumors deficient in mismatch repair in each cancer subtype, expressed as a percentage. Mismatch repair deficient tumors were identified in 24 out of 32 tumor subtypes tested, more often in early stage (defined as stage < IV) disease.
Supplementary Material
Acknowledgments
The data reported are tabulated in the main text and supplemental materials. The raw TCR RNA sequence data have been deposited into the ImmuneACCESS project repository of the Adaptive Biotech database, under the following link: https://clients.adaptivebiotech.com/pub/diaz-2017-science. We would like to acknowledge K. Helwig for administrative support and C. Blair for outstanding technical assistance. We also want to thank E. H. Rubin, R. Dansey and R. Permutter at Merck & Co., Inc., Kenilworth, NJ USA for supporting this research. This work was funded by The Swim Across America Laboratory at Johns Hopkins, The Ludwig Center for Cancer Genetics and Therapeutics, The Howard Hughes Medical Institutes, The Bloomberg-Kimmel Institute for Cancer Immunotherapy at Johns Hopkins, The 2017 Stand Up To Cancer Colon Cancer Dream Team, The Commonwealth Fund, The Banyan Gate Foundation, The Lustgarten Foundation for Pancreatic Cancer Research, The Bloomberg Foundation, The Sol Goldman Pancreatic Cancer Research Center, Merck & Co., Inc., Kenilworth, NJ USA, Gastrointestinal SPORE grant P50CA062924 and NIH grants P30CA006973, CA163672, CA43460, CA203891, CA67941, CA16058 and CA57345. LD, DL, BV, NP and KWK are inventors on patent application (PCT/US2015/060331 or WO 2016077553 A1) submitted by Johns Hopkins University that covers checkpoint blockade and microsatellite instability. LD, BV, NP and KWK are founders of PapGene and Personal Genome Diagnostics (PGDx). LD is a consultant for Merck, Illumina, PGDx and Cell Design Labs. PGDx and PapGene, as well as other companies, have licensed technologies from Johns Hopkins University, on which LD, BV, NP and KWK are inventors. Some of these licenses and relationships are associated with equity or royalty payments.
Footnotes
The terms of these arrangements are being managed by Johns Hopkins and Memorial Sloan Kettering in accordance with its conflict of interest policies.
SUPPLEMENTARY MATERIALS
www.sciencemag.org/cgi/content/full/science.aan6733/DC1
Materials and Methods
References (24–36)
REFERENCES AND NOTES
- 1.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McDermott DF, Drake CG, Sznol M, Choueiri TK, Powderly JD, Smith DC, Brahmer JR, Carvajal RD, Hammers HJ, Puzanov I, Hodi FS, Kluger HM, Topalian SL, Pardoll DM, Wigginton JM, Kollia GD, Gupta A, McDonald D, Sankar V, Sosman JA, Atkins MB. Survival, durable response, and long-term safety in patients with previously treated advanced renal cell carcinoma receiving nivolumab. J Clin Oncol. 2015;33:2013–2020. doi: 10.1200/JCO.2014.58.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD, Leming PD, Lipson EJ, Puzanov I, Smith DC, Taube JM, Wigginton JM, Kollia GD, Gupta A, Pardoll DM, Sosman JA, Hodi FS. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020–1030. doi: 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gettinger SN, Horn L, Gandhi L, Spigel DR, Antonia SJ, Rizvi NA, Powderly JD, Heist RS, Carvajal RD, Jackman DM, Sequist LV, Smith DC, Leming P, Carbone DP, Pinder-Schenck MC, Topalian SL, Hodi FS, Sosman JA, Sznol M, McDermott DF, Pardoll DM, Sankar V, Ahlers CM, Salvati M, Wigginton JM, Hellmann MD, Kollia GD, Gupta AK, Brahmer JR. Overall survival and long-term safety of nivolumab (anti-programmed death 1 antibody, BMS-936558, ONO-4538) in patients with previously treated advanced non-small-cell lung cancer. J Clin Oncol. 2015;33:2004–2012. doi: 10.1200/JCO.2014.58.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Chen L, Pardoll DM, Topalian SL, Anders RA. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20:5064–5074. doi: 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS, Zhang M, Papadopoulos N, Kinzler KW, Vogelstein B, Sears CL, Anders RA, Pardoll DM, Housseau F. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5:43–51. doi: 10.1158/2159-8290.CD-14-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, Kohrt HE, Horn L, Lawrence DP, Rost S, Leabman M, Xiao Y, Mokatrin A, Koeppen H, Hegde PS, Mellman I, Chen DS, Hodi FS. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, Seja E, Lomeli S, Kong X, Kelley MC, Sosman JA, Johnson DB, Ribas A, Lo RS. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2017;168:542. doi: 10.1016/j.cell.2017.01.010. [DOI] [PubMed] [Google Scholar]
- 11.Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, Vogelstein B, Allison JP. Epitope landscape in breast and colorectal cancer. Cancer Res. 2008;68:889–892. doi: 10.1158/0008-5472.CAN-07-3095. [DOI] [PubMed] [Google Scholar]
- 12.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, Mulder GE, Toebes M, Vesely MD, Lam SS, Korman AJ, Allison JP, Freeman GJ, Sharpe AH, Pearce EL, Schumacher TN, Aebersold R, Rammensee HG, Melief CJ, Mardis ER, Gillanders WE, Artyomov MN, Schreiber RD. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 14.Ward JP, Gubin MM, Schreiber RD. The role of neoantigens in naturally occurring and therapeutically induced immune responses to cancer. Adv Immunol. 2016;130:25–74. doi: 10.1016/bs.ai.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 16.Kim H, Jen J, Vogelstein B, Hamilton SR. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol. 1994;145:148–156. [PMC free article] [PubMed] [Google Scholar]
- 17.Smyrk TC, Watson P, Kaul K, Lynch HT. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer. 2001;91:2417–2422. doi: 10.1002/1097-01422001061591:12<2417:AID-CNCR1276>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 18.Dolcetti R, Viel A, Doglioni C, Russo A, Guidoboni M, Capozzi E, Vecchiato N, Macrì E, Fornasarig M, Boiocchi M. High prevalence of activated intraepithelial cytotoxic T lymphocytes and increased neoplastic cell apoptosis in colorectal carcinomas with microsatellite instability. Am J Pathol. 1999;154:1805–1813. doi: 10.1016/S0002-94401065436-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Duffy SM, Goldberg RM, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban RH, Wood LD, Cuka N, Pardoll DM, Papadopoulos N, Kinzler KW, Zhou S, Cornish TC, Taube JM, Anders RA, Eshleman JR, Vogelstein B, Diaz LA., Jr PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Overman M, Lonardi S, Leone F, McDermott RS, Morse MA, Wong KYM, Neyns B, Leach JL, Garcia Alfonso P, Lee JJ, Hill A, Lenz H-J, Desai J, Moss RA, Cao ZA, Ledeine J-M, Tang H, Kopetz S, Andre T. Nivolumab in patients with DNA mismatch repair deficient/microsatellite instability high metastatic colorectal cancer: Update from CheckMate 142. J Clin Oncol. 2017;35(suppl):519. doi: 10.1200/JCO.2017.35.4_suppl.519. [DOI] [Google Scholar]
- 21.Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, Humblet Y, Bouché O, Mineur L, Barone C, Adenis A, Tabernero J, Yoshino T, Lenz HJ, Goldberg RM, Sargent DJ, Cihon F, Cupit L, Wagner A, Laurent D. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:303–312. doi: 10.1016/S0140-67361261900-X. [DOI] [PubMed] [Google Scholar]
- 22.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, Saco J, Homet Moreno B, Mezzadra R, Chmielowski B, Ruchalski K, Shintaku IP, Sanchez PJ, Puig-Saus C, Cherry G, Seja E, Kong X, Pang J, Berent-Maoz B, Comin-Anduix B, Graeber TG, Tumeh PC, Schumacher TN, Lo RS, Ribas A. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hause RJ, Pritchard CC, Shendure J, Salipante SJ. Classification and characterization of microsatellite instability across 18 cancer types. Nat Med. 2016;22:1342–1350. doi: 10.1038/nm.4191. [DOI] [PubMed] [Google Scholar]
- 24.Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, Zhang T, Adleff V, Phallen J, Wali N, Hruban C, Guthrie VB, Rodgers K, Naidoo J, Kang H, Sharfman W, Georgiades C, Verde F, Illei P, Li QK, Gabrielson E, Brock MV, Zahnow CA, Baylin SB, Scharpf RB, Brahmer JR, Karchin R, Pardoll DM, Velculescu VE. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. 2017;7:264–276. doi: 10.1158/2159-8290.CD-16-0828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carlson CS, Emerson RO, Sherwood AM, Desmarais C, Chung MW, Parsons JM, Steen MS, LaMadrid-Herrmannsfeldt MA, Williamson DW, Livingston RJ, Wu D, Wood BL, Rieder MJ, Robins H. Using synthetic templates to design an unbiased multiplex PCR assay. Nat Commun. 2013;4:2680. doi: 10.1038/ncomms3680. [DOI] [PubMed] [Google Scholar]
- 26.Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, Riddell SR, Warren EH, Carlson CS. Comprehensive assessment of T-cell receptor β-chain diversity in αβ T cells. Blood. 2009;114:4099–4107. doi: 10.1182/blood-2009-04-217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bacher JW, Flanagan LA, Smalley RL, Nassif NA, Burgart LJ, Halberg RB, Megid WM, Thibodeau SN. Development of a fluorescent multiplex assay for detection of MSI-high tumors. Dis Markers. 2004;20:237–250. doi: 10.1155/2004/136734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murphy KM, Zhang S, Geiger T, Hafez MJ, Bacher J, Berg KD, Eshleman JR. Comparison of the microsatellite instability analysis system and the Bethesda panel for the determination of microsatellite instability in colorectal cancers. J Mol Diagn. 2006;8:305–311. doi: 10.2353/jmoldx.2006.050092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolchok JD, Hoos A, O’Day S, Weber JS, Hamid O, Lebbé C, Maio M, Binder M, Bohnsack O, Nichol G, Humphrey R, Hodi FS. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin Cancer Res. 2009;15:7412–7420. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 30.Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS, Zhang M, Papadopoulos N, Kinzler KW, Vogelstein B, Sears CL, Anders RA, Pardoll DM, Housseau F. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5:43–51. doi: 10.1158/2159-8290.CD-14-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, Chen L. Colocalization of inflammatory response with B7-H1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:127ra37. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cuka N, Hempel H, Sfanos K, De Marzo A, Cornish T. PIP: An open source framework for multithreaded image analysis of whole slide images. Lab Invest. 2014;94:398A. [Google Scholar]
- 33.Cupitt J, Martinez K. Electronic Imaging: Science & Technology. International Society for Optics and Photonics; 1996. pp. 19–28. [Google Scholar]
- 34.Jones S, Anagnostou V, Lytle K, Parpart-Li S, Nesselbush M, Riley DR, Shukla M, Chesnick B, Kadan M, Papp E, Galens KG, Murphy D, Zhang T, Kann L, Sausen M, Angiuoli SV, Diaz LA, Jr, Velculescu VE. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci Transl Med. 2015;7:283ra53. doi: 10.1126/scitranslmed.aaa7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sausen M, Leary RJ, Jones S, Wu J, Reynolds CP, Liu X, Blackford A, Parmigiani G, Diaz LA, Jr, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE, Hogarty MD. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet. 2013;45:12–17. doi: 10.1038/ng.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970;48:443–453. doi: 10.1016/0022-28367090057-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.