

Abstract
Our research on concussion-induced axonal injury may lead to identification of biomarkers that enable noninvasive diagnosis and treatment.
The interchangeable terms concussion and mild traumatic brain injury (mTBI) denote the phenomenon of a change in brain function associated with blunt trauma to the head or body. Beyond the fact that concussion can occur without the victim losing consciousness, researchers are only just beginning to uncover some of the essential aspects of this major health issue.
Initiated by a biomechanical event, concussion is unique among central nervous system disorders, and humans, more than other species, are particularly prone to brain damage from head impact. For example, if a human and a pig undergo the same head rotational acceleration, the human will fare far worse, as shown by animal and human studies. The approximately 1500 g human brain endures substantially greater mass effects than the 100 g pig brain under similar mechanical loading, where regions of the brain push and pull against each other as the brain is rapidly deformed (Browne et al. 2011; Meaney and Smith 2011; Smith and Meaney 2000; Smith et al. 2003b).
An important finding in animal and human studies of concussion is that selective damage to axons, known as diffuse axonal injury (DAI), is a key anatomic phenomenon of mTBI (Johnson et al. 2013; Meaney and Smith 2011; Smith and Meaney 2000; Smith et al. 2013a). Moreover, although in most cases many of the mental function deficits associated with concussion completely resolve, up to 20 percent of individuals with a single mTBI have persisting cognitive dysfunction (Hanten et al. 2013; McCauley et al. 2014). There is also growing evidence and concern that one or more concussions can trigger long-term neurodegenerative changes in the brain (Hay et al. 2016; Smith et al. 2013b).
Axonal Injury from Concussion
While the whole brain suffers dynamic tissue deformation during concussion, the white matter is at greatest risk of damage, possibly because of its highly organized, highly directional structure (anisotropy). Axon tracts, the long transmission lines of the white matter, suffer damage in several unique ways from the tensile and shear forces induced by rotational head acceleration. In particular, the classic viscoelastic nature of axons appears to be a major contributing factor (Meaney and Smith 2015; Smith et al. 1999b).
Under normal daily mechanical loading conditions, axons can easily stretch to at least twice their resting length and relax back unharmed to their prestretch straight geometry (Tang-Schomer et al. 2010). However, under dynamic loading with rapid stretch, the axonal cytoskeleton can physically break, evidenced by an undulating appearance upon relaxation immediately afterward (figure 1). These axonal undulations have been observed in an in vitro model of dynamic stretch injury of axons, in preclinical TBI models, and in human TBI (Smith et al. 1999b; Tang-Schomer et al. 2010, 2012).
FIGURE 1.

Undulations of neurons in the brain shown for three cases after blunt trauma. (A) 3-hour survival in adult swine traumatic brain injury (TBI). (B) 18-year-old human male, 10-hour survival. (C) 18-year-old human female, 22-hour survival. Image (B) is reprinted with permission from Tang-Schomer et al. (2012).
Using a customized in vitro axonal stretch injury model, we found that the stiffest components of the axon, microtubules, are the structures that most overtly display physical breaking at the time of trauma (Tang-Schomer et al. 2010). The break sites along the micro-tubule lattice appear to account for the undulating course of injured axons, by impeding the sliding of adjacent microtubules to return back to the relaxed straight geometry after injury.
Because microtubules essentially serve as the anatomical tracks for protein transport, proteins pile up at points of individual micro-tubule disconnection, resulting in varicose swellings distributed periodically along the injured axon (Johnson et al. 2013; Tang-Schomer et al. 2012) as well as partial transport interruption (Tang-Schomer et al. 2012) rather than complete transport failure in an entire region of the axon. In this scenario, some protein transport can continue through areas of swelling along remaining intact microtubules, but may be derailed farther along the axon because of microtubule disruption there.
Association between Concussion and Neurodegeneration
There is evidence that one or repeated head blows can switch the brains of some individuals from a normal aging track to an accelerated neurodegenerative path, commonly referred to as chronic traumatic encephalopathy (CTE) (Hay et al. 2016; Johnson et al. 2010; Smith et al. 2013b).
The initiating source of postconcussive neurodegenerative changes has yet to be identified, but DAI is a leading candidate (Johnson et al. 2013). CTE histopathological occurrences of neurofibrillary tau protein tangles and amyloid-beta plaques found to a greater extent in individuals with histories of multiple concussions (compared with age-matched controls) are similar to tissue accumulations in other neurodegenerative diseases (Hay et al. 2016; Johnson et al. 2010, 2012; Smith et al. 2013b).
Curiously, tau protein and amyloid precursor protein (APP), the parent protein of amyloid-beta, are normally most abundant in axons (Chen et al. 2004, 2009; Johnson et al. 2009; Smith et al. 2003a; Uryu et al. 2007). Therefore, it has been suggested that axonopathy in concussion may lead to the aberrant production and/or aggregation of tau and amyloid-beta (Johnson et al. 2010, 2012).
Role of Tau Protein
Using a new computational model of traumatic axonal injury, we identified a potential viscoelastic spring underlying microtubule breaking, the tau protein (Ahmadzadeh et al. 2014, 2015).
Tau proteins crosslink and stabilize the parallel arrangement of microtubules along the axon. As the axon is stretched under normal loading conditions, adjacent microtubules slide past each other, stretching the stabilizing tau proteins, which must unfold from their resting conformation. We propose that this tau extension includes the breaking of hydrogen bonds along the tau protein. This model predicts that the thermodynamics of hydrogen bond breaking along tau proteins during dynamic axon stretch injury cannot keep up with the rapid microtubule sliding. Thus, the pulling of tau proteins against the microtubules induces enough mechanical strain to rupture the microtubule (Ahmadzadeh et al. 2014, 2015).
While axons themselves rarely disconnect at the time of trauma, large swellings from transport interruption can cause a secondary disconnection of axons (Johnson et al. 2013; Smith and Meaney 2000). There is a broad range in the rate and morphology of axonal swellings identified with undulated axons, varicose swellings, and disconnected axonal bulbs.
The gold standard for postmortem clinical diagnosis of these types of DAI is immunohistochemistry using anti-APP antibodies, which can identify axonal swellings in white matter within hours of injury (Johnson et al. 2013). Although APP+ swollen axonal profiles are indicative of DAI, the vast majority of axons in white matter tracts appear relatively normal after TBI, even in severe cases (Johnson et al. 2013; Smith et al. 2003c). TBI must therefore involve injury beyond axon transport interruption and swelling.
Role of Sodium and Calcium Channels
Ionic concentration disturbances in traumatic axonal injury may play an important role in dysfunction (Iwata et al. 2004; Smith and Meaney 2000; Smith et al. 2003b; Wolf et al. 2001). Our in vitro model revealed that stretch injury impaired regulation of axonal sodium channels, where the channel inactivation gate is disabled and can no longer block excessive sodium influx (figure 2). In turn, the sodium-calcium exchangers are reversed and the high-voltage state maintains open calcium channels, thereby progressively increasing intra-axonal calcium concentrations after injury (figure 3; Wolf et al. 2001). The cascade can continue to the point where high calcium levels activate calcium-dependent proteases, inducing secondary cytoskeletal disruption (Iwata et al. 2004; von Reyn et al. 2012; Yuen et al. 2009).
FIGURE 2.
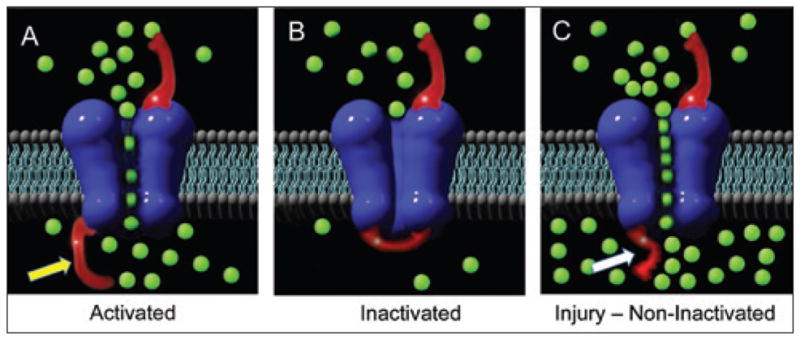
Diagram of the axonal membrane showing (A) sodium channel (blue) assemblage with the inactivation gate or “flapper valve” (shown with yellow arrow) with normal sodium influx (green balls) that create an action potential; (B) normal inactivation closes the gate to allow efflux of sodium; (C) upon injury, the inactivation gate is rendered dysfunctional (white arrow), allowing unmitigated influx of sodium ions.
FIGURE 3.
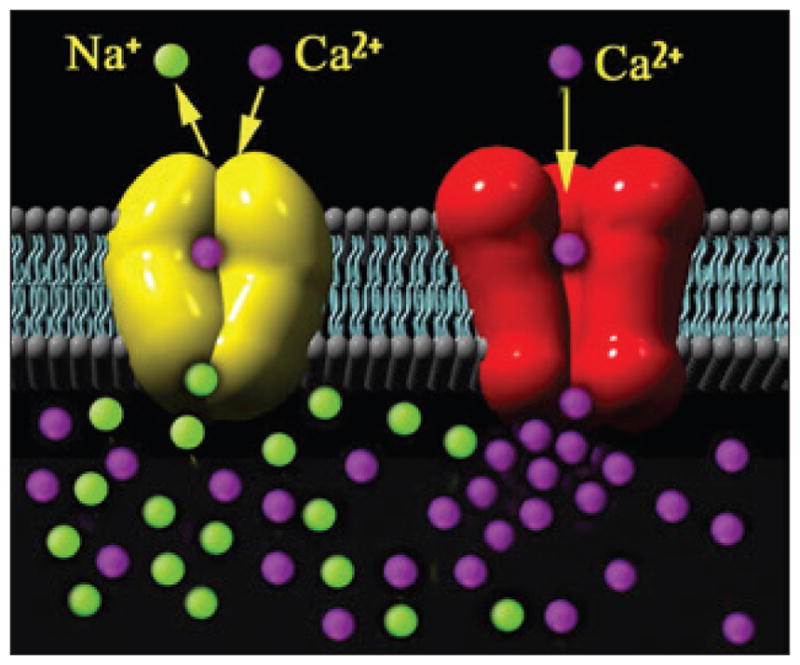
After traumatic axonal injury, there is uncontrolled influx of sodium (Na+) ions (green balls), as illustrated in figure 2, triggering reversal of the sodium-calcium (Ca2+) exchanger (yellow) and sustained opening of the voltage-gated calcium channels (red), resulting in progressive increases of intra-axonal calcium ions (purple). Adapted from Wolf et al. (2001).
We propose that the loss of function of axonal sodium channels throughout the brain network underlies common neurocognitive symptoms of concussion, such as loss of consciousness, decreased processing speed, and memory dysfunction. With too much sodium entering the axon, the capacity to generate action potentials (the rapid exchange of ions across the neural membrane) is lost or dysfunctional, interrupting or slowing signaling in the axon and throughout the neural network of the brain (Johnson et al. 2013).
Our in vitro model revealed that for some injured axons, sodium channels may remain dysfunctional, resulting in an increase in the number of sodium channels over time (Yuen et al. 2009). This may be a temporary adaptive change to compensate for the dysfunctional channels.
However, this “fix” may come at a cost. With even mild stretch injury of axons in vitro inducing very little sodium or calcium influx, the axons added more sodium channels. With a second very mild stretch injury a day later, massive sodium and calcium influx occurred. If this is also shown to be the case for concussion, it raises the intriguing possibility that sodium channelopathy plays a role in the suggested “period of vulnerability” after concussion.
Potential Biomarkers
In concert with the in vitro model, a swine mTBI model of head rotational acceleration has been developed and scaled to match the brain tissue deformation of human concussion (Browne et at. 2011; Kimura et al. 1996; Meaney et al. 1995; Smith et al. 1997, 2000). With this model, extensive APP+ axonal pathology is produced throughout the white matter with an appearance identical to human DAI (Johnson et al. 2015, 2016; McGowan et al. 1999; Smith et al. 1999a).
The use of this mTBI model in conjunction with examination of human moderate-to-severe TBI has revealed additional axonal injury not seen with APP+ analysis (Johnson et al. 2015, 2016), using an immunostain for the calpain-cleaved spectrin N-terminal fragment (SNTF). The appearance of SNTF generated exclusively in axons implies that the injury induced substantial increases in intra-axonal calcium concentrations, resulting in calpain activation and cleavage of axonal spectrin (Johnson et al. 2016). These observations corroborate the dysregulation of calcium concentrations in axon trauma observed in vitro (figure 3).
Notably, we found that SNTF appears in the blood shortly after concussion in individuals who were later found to have persistent cognitive dysfunction (Siman et al. 2013); its presence thus signals permanent brain damage through the degeneration of axons (Johnson et al. 2016). SNTF and tau protein were also found in the blood of professional ice hockey players after concussions and could remain at elevated levels for days (Siman et al. 2015; Zetterberg et al. 2013).
These studies suggest that SNTF and/or other axonal proteins may serve as blood biomarkers to evaluate the severity of concussion and to specifically diagnose DAI. The identification and validation of a biomarker would be a valuable tool for managing concussed patients as it could provide an objective measure of the effectiveness of treatment. Indeed, biomarkers may allow for the stratification of mTBI patients according to severity, thereby increasing the power of treatment trials.
Conclusion
While substantial progress has been made in understanding the pathophysiology of concussion far beyond this brief overview of our efforts, it is nonetheless certain that the field remains in its infancy. There is no evidence-based practice or medicine for treating concussion, leaving many patients and their families frustrated with their care. And in the absence of noninvasive tests to identify patients at risk, there is little information to provide a long-term prognosis for concussed individuals (Levin and Smith 2013; Smith et al. 2013b).
Research priorities for concussion should include efforts to identify therapeutic targets for DAI (Smith et al. 2013a), characterize the period of vulnerability after concussion for patient management and return to play decisions (Meany and Smith 2011), and uncover the trigger for biochemical events that result in progressive neurodegenerative changes.
References
- Ahmadzadeh H, Smith DH, Shenoy VB. Viscoelasticity of tau proteins leads to strain rate-dependent breaking of microtubules during axonal stretch injury: Predictions from a mathematical model. Biophysical Journal. 2014;106(5):1123–1133. doi: 10.1016/j.bpj.2014.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadzadeh H, Freedman BR, Connizzo BK, Soslowsky LJ, Shenoy VB. Micromechanical poroelastic finite element and shear-lag models of tendon predict large strain dependent Poisson’s ratios and fluid expulsion under tensile loading. Acta Biomaterialia. 2015;22:83–91. doi: 10.1016/j.actbio.2015.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne KD, Chen XH, Meaney DF, Smith DH. Mild traumatic brain injury and diffuse axonal injury in swine. Journal of Neurotrauma. 2011;28(9):1747–1755. doi: 10.1089/neu.2011.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. American Journal of Pathology. 2004;165(2):357–371. doi: 10.1016/s0002-9440(10)63303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathology. 2009;19(2):214–223. doi: 10.1111/j.1750-3639.2008.00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanten G, Li X, Ibarra A, Wilde EA, Barnes A, McCauley SR, McCarthy J, Hoxhaj S, Mendez D, Hunter JV, Levin HS, Smith DH. Updating memory after mild traumatic brain injury and orthopedic injuries. Journal of Neurotrauma. 2013;30(8):618–624. doi: 10.1089/neu.2012.2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay J, Hunter JV, Smith DH, Stewart W. Chronic traumatic encephalopathy: The neuropathological legacy of traumatic brain injury. Pathology: Mechanisms of Disease. 2016:11. doi: 10.1146/annurev-pathol-012615-044116. (epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata A, Stys PK, Wolf JA, Chen XH, Taylor AG, Meaney DF, Smith DH. Traumatic axonal injury induces proteolytic cleavage of the voltage-gated sodium channels modulated by tetrodotoxin and protease inhibitors. Journal of Neuroscience. 2004;24(19):4605–4613. doi: 10.1523/JNEUROSCI.0515-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Graham DI, Stewart JE, Praestgaard AH, Smith DH. A neprilysin polymorphism and amyloid-beta plaques following traumatic brain injury. Journal of Neurotrauma. 2009;26(8):1197–1202. doi: 10.1089/neu.2008.0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: A link to Alzheimer’s disease? Nature Reviews Neuroscience. 2010;11(5):361–370. doi: 10.1038/nrn2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathology. 2012;22(2):142–149. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Experimental Neurology. 2013;246:35–43. doi: 10.1016/j.expneurol.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Meaney DF, Cullen DK, Smith DH. Animal models of traumatic brain injury. Handbook of Clinical Neurology. 2015;127:115–128. doi: 10.1016/B978-0-444-52892-6.00008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Weber MT, Cullen DK, Siman R, Smith DH. SNTF immunostaining reveals previously undetected axonal pathology in traumatic brain injury. Acta Neuropathologica. 2016;131(1):115–135. doi: 10.1007/s00401-015-1506-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Meaney DF, McGowan JC, Grossman RI, Lenkinski RE, Ross DT, McIntosh TK, Gennarelli TA, Smith DH. Magnetization transfer imaging of diffuse axonal injury following experimental brain injury in the pig: Characterization by magnetization transfer ratio with histopathologic correlation. Journal of Computer Assisted Tomography. 1996;20(4):540–546. doi: 10.1097/00004728-199607000-00007. [DOI] [PubMed] [Google Scholar]
- Levin H, Smith D. Traumatic brain injury: Networks and neuropathology. Lancet Neurology. 2013;12(10):15–16. doi: 10.1016/S1474-4422(12)70300-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauley SR, Wilde EA, Barnes A, Hanten G, Hunter JV, Levin HS, Smith DH. Patterns of early emotional and neuropsychological sequelae after mild traumatic brain injury. Journal of Neurotrauma. 2014;31(10):914–925. doi: 10.1089/neu.2012.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan JC, McCormack TM, Grossman RI, Mendonça R, Chen XH, Berlin JA, Meaney DF, Xu BN, Cecil KM, McIntosh TK, Smith DH. Diffuse axonal pathology detected with magnetization transfer imaging following brain trauma in the pig. Magnetic Resonance in Medicine. 1999;41(4):727–733. doi: 10.1002/(sici)1522-2594(199904)41:4<727::aid-mrm11>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Meaney DF, Smith DH, Shreiber DI, Bain AC, Miller RT, Ross DT, Gennarelli TA. Biomechanical analysis of experimental diffuse axonal injury. Journal of Neurotrauma. 1995;12(4):689–694. doi: 10.1089/neu.1995.12.689. [DOI] [PubMed] [Google Scholar]
- Meaney DF, Margulies SS, Smith DH. Diffuse axonal injury. Journal of Neurosurgery. 2011;95(6):1108–1110. doi: 10.3171/jns.2001.95.6.1108. [DOI] [PubMed] [Google Scholar]
- Meaney DF, Smith DH. Biomechanics of concussion. Clinics in Sports Medicine. 2011;30(1):19–23. doi: 10.1016/j.csm.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney DF, Smith DH. Cellular biomechanics of central nervous system injury. Handbook of Clinical Neurology. 2015;127:105–114. doi: 10.1016/B978-0-444-52892-6.00007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siman R, Giovannone N, Hanten G, Wilde EA, McCauley SR, Hunter JV, Li X, Levin HS, Smith DH. Evidence that the blood biomarker SNTF predicts brain imaging changes and persistent cognitive dysfunction in mild TBI patients. Frontiers in Neurology. 2013;4:190. doi: 10.3389/fneur.2013.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siman R, Shahim P, Tegner Y, Blennow K, Zetterberg H, Smith DH. Serum SNTF increases in concussed professional ice hockey players and relates to the severity of postconcussion symptoms. Journal of Neurotrauma. 2015;32(17):1294–1300. doi: 10.1089/neu.2014.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Meaney DF. Axonal damage in traumatic brain injury. Neuroscientist. 2000;6(6):483–495. [Google Scholar]
- Smith DH, Chen XH, Xu BN, McIntosh TK, Gennarelli TA, Meaney DF. Characterization of diffuse axonal pathology and selective hippocampal damage following inertial brain trauma in the pig. Journal of Neuropathology and Experimental Neurology. 1997;56(7):822–834. [PubMed] [Google Scholar]
- Smith DH, Wolf JA, Lusardi TA, Lee VM, Meaney DF. High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. Journal of Neuroscience. 1999b;19(11):4263–4269. doi: 10.1523/JNEUROSCI.19-11-04263.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, Saatman KE, Leoni MJ, Xu BN, Wolf JA, Meaney DF. Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. Journal of Neuropathology and Experimental Neurology. 1999a;58(9):982–992. doi: 10.1097/00005072-199909000-00008. [DOI] [PubMed] [Google Scholar]
- Smith DH, Nonaka M, Miller R, Leoni M, Chen XH, Alsop D, Meaney DF. Immediate coma following inertial brain injury dependent on axonal damage in the brainstem. Journal of Neurosurgery. 2000;93(2):315–322. doi: 10.3171/jns.2000.93.2.0315. [DOI] [PubMed] [Google Scholar]
- Smith DH, Chen XH, Iwata A, Graham DI. Amyloid beta accumulation in axons after traumatic brain injury in humans. Journal of Neurosurgery. 2003a;98(5):1072–1077. doi: 10.3171/jns.2003.98.5.1072. [DOI] [PubMed] [Google Scholar]
- Smith DH, Meaney DF, Shull WH. Diffuse axonal injury in head trauma. Journal of Head Trauma Rehabilitation. 2003b;18(4):307–316. doi: 10.1097/00001199-200307000-00003. [DOI] [PubMed] [Google Scholar]
- Smith DH, Uryu K, Saatman KE, Trojanowski JQ, McIntosh TK. Protein accumulation in traumatic brain injury. Neuromolecular Medicine. 2003c;4(1–2):59–72. doi: 10.1385/NMM:4:1-2:59. [DOI] [PubMed] [Google Scholar]
- Smith DH, Hicks R, Povlishock JT. Therapy development for diffuse axonal injury. Journal of Neurotrauma. 2013a;30(5):307–323. doi: 10.1089/neu.2012.2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Johnson VE, Stewart W. Chronic neuropathologies of single and repetitive TBI: Substrates of dementia? Nature Reviews Neurology. 2013b;9(4):211–221. doi: 10.1038/nrneurol.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang-Schomer MD, Patel AR, Baas PW, Smith DH. Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, micro-tubule disassembly, and axon degeneration. FASEB Journal. 2010;24(5):1401–1410. doi: 10.1096/fj.09-142844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang-Schomer MD, Johnson VE, Baas PW, Stewart W, Smith DH. Partial interruption of axonal transport due to microtubule breakage accounts for the formation of periodic varicosities after traumatic axonal injury. Experimental Neurology. 2012;233(1):364–372. doi: 10.1016/j.expneurol.2011.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uryu K, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI, Lee VM, Trojanowski JQ, Smith DH. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Experimental Neurology. 2007;208(2):185–192. doi: 10.1016/j.expneurol.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Reyn CR, Mott RE, Siman R, Smith DH, Meaney DF. Mechanisms of calpain mediated proteolysis of voltage gated sodium channel alpha-subunits following in vitro dynamic stretch injury. Journal of Neurochemistry. 2012;121(5):793–805. doi: 10.1111/j.1471-4159.2012.07735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. Journal of Neuroscience. 2001;21(6):1923–1930. doi: 10.1523/JNEUROSCI.21-06-01923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen TJ, Browne KD, Iwata A, Smith DH. Sodium channelopathy induced by mild axonal trauma worsens outcome after a repeat injury. Journal of Neuroscience Research. 2009;87(16):3620–3625. doi: 10.1002/jnr.22161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterberg H, Smith DH, Blennow K. Biomarkers of mild traumatic brain injury in cerebrospinal fluid and blood. Nature Reviews Neurology. 2013;9(4):201–210. doi: 10.1038/nrneurol.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]