

Abstract
System xc− is a sodium-independent electroneutral transporter, comprising a catalytic subunit xCT (SLC7A11), which is involved in importing cystine. Certain cancers such as gliomas upregulate the expression of system xc−, which confers a survival advantage against the detrimental effects of reactive oxygen species (ROS) by increasing generation of the antioxidant glutathione. However, ROS have also been shown to function as targeted, intracellular second messengers in an array of physiological processes such as proliferation. Several studies have implicated ROS in important cancer features such as migration, invasion, and contribution to a cancer stem cell (CSC)-like phenotype. The role of system xc− in regulating these ROS-sensitive processes in glioblastoma multiforme (GBM), the most aggressive malignant primary brain tumor in adults, remains unknown. Stable SLC7A11 knockdown and overexpressing U251 glioma cells were generated and characterized to understand the role of redox and system xc− in glioma progression. SLC7A11 knockdown resulted in higher endogenous ROS levels and enhanced invasive properties. On the contrary, overexpression of SLC7A11 resulted in decreased endogenous ROS levels as well as decreased migration and invasion. However, SLC7A11-overexpressing cells displayed actin cytoskeleton changes reminiscent of epithelial-like cells and exhibited an increased CSC-like phenotype. The enhanced CSC-like phenotype may contribute to increased chemoresistance and suggests that overexpression of SLC7A11 in the context of GBM may contribute to tumor progression. These findings have important implications for cancer management where targeting system xC− in combination with other chemotherapeutics can reduce cancer resistance and recurrence and improve GBM patient survival.
Keywords: : SLC7A11, glioma, redox, glutathione, cancer stem cells
Introduction
Glioblastoma multiforme (GBM) is the most common malignant primary adult brain tumor and newly diagnosed patients have a life expectancy of less than a year [1,2]. Despite major advances in understanding solid tumor biology, neuroimaging, improved surgical techniques, and chemo/radiation therapies, GBM patients inevitably progress or relapse an average of only 6.9 months after treatment [3] with 80% of tumors found within 3 cm of the initial resection site [4]. The origin and development of GBM are complex and not fully understood, but involve various genetic alterations that confer growth advantages, including, but not limited to, rapid cell proliferation and aggressive invasion of the surrounding normal brain tissues. GBM cells are able to escape the tumor mass and migrate large distances through the brain parenchyma, and this infiltrative pattern makes tumor cells difficult to target. Thus, understanding the mechanisms that promote migration and invasion of glioma cells could lead to the development of more effective therapeutic strategies to increase survival of GBM patients.
System xc− is a sodium-independent membrane transporter involved in the efflux of glutamate along with the influx of extracellular cystine that is reduced to cysteine, the rate-limiting precursor for the major antioxidant glutathione (GSH) [5]. Expression of system xc− is upregulated in gliomas and has been shown to promote redox balance by maintaining high levels of GSH, which can scavenge or neutralize reactive oxygen species (ROS), as well as conjugate to xenobiotic agents that are then exported out of the cell [6,7]. In addition, the exported glutamate has been shown to be involved in glioma invasion by stimulating cell motility. The released glutamate activates the Ca2+-permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor and induces intracellular Ca2+ oscillations that promote cell migration [8]. Decreasing glutamate release through pharmacological inhibition of system xC− was shown to decrease chemotactic invasion and migration of the glioma, which could be rescued with addition of exogenous glutamate. Thus, it has been hypothesized that system xc− is involved in promoting survival of glioma under oxidative stress through increased GSH production as well as promoting tumor cell infiltration into the normal brain through released glutamate that acts as an autocrine/paracrine mitogen (migration-promoting signal) for cell motility and invasion.
ROS, including the superoxide anion (O2•−), hydrogen peroxide (H2O2), and the hydroxyl radical (OH•−), serve as secondary messengers of signal transduction pathways that regulate cell proliferation, survival, and apoptosis [9]. There is accumulating evidence that ROS can also contribute to tumor progression by activating pathways for tumor invasion and migration [9–11]. Chiu et al. showed that U87 glioma treated with 12-O-tetradecanoylphorbol-13-acetate (TPA) increased ROS generation that induced invasion/migration through activation of mitogen-activated protein kinase (MAPK), matrix metalloproteinase (MMP)-9, and cyclooxygenase-2/prostaglandin E2, whereas inhibition of ROS with exogenous antioxidant treatment blocked the TPA-induced migration/invasion [9].
These findings based on how ROS levels increase migration/invasion are conflicting with the published data on how system xc− plays a role in tumor proliferation, invasion, and migration. Most of the previous studies showing the contribution of system xc− to glioma growth, survival, and migration were performed with pharmacological inhibitors such as sulfasalazine, which have numerous off-target effects, and indirectly link system xc− to cellular functions. Therefore, it is difficult to draw conclusions as to the role system xc− plays in glioma progression. The contribution of system xc− to tumor invasion/migration in the context of system xc− expression in glioma cells has not been investigated. We circumvented this problem by genetically modifying the expression of SLC7A11, the catalytic subunit of system xc−, to generate stable overexpressing and knockdown U251 glioma cell lines.
Recently, studies have suggested that a small population of stem-like cells, termed cancer stem cells (CSCs), or tumor-initiating cells, is responsible for GBM invasiveness. These CSCs, enriched with the stem cell marker CD133, showed greater migratory and invasive properties compared with matched CD133-negative cells derived from GBM [12]. The CSCs have been identified on the ability to self-renew, initiate tumorigenesis, and are resistant to chemotherapeutic agents [13]. However, CSCs, similar to normal stem cells, are quiescent slow-cycling cells with low endogenous ROS levels, which account for their self-renewal capacity. The low ROS levels in CSCs can be attributed to decreased ROS generation, enhanced ROS-scavenging systems, or a combination of both. CSCs have also been shown to differentially express various solute carriers (SLCs) [14], but SLC7A11 expression in CSCs has not yet been thoroughly investigated.
In this study, we show by RNA sequence analysis that several genes involved in cell adhesion, migration, and morphogenesis are downregulated in the SLC7A11-overexpressing U251 glioma. We found that the SLC7A11-overexpressing cells indeed displayed altered cell morphology and cytoskeletal changes. These cells were more cuboidal with large spherical bodies and more diffuse filamentous actin, and this correlated with decreased migratory and invasive properties. Conversely, the SLC7A11 knockdown cells displayed more pronounced filamentous actin at the plasma membrane and increased invasion. Overexpression of SLC7A11 in U251 glioma cells also resulted in upregulation of the CSC-like phenotype. These results suggest that high expression of system xc− is correlated with an increased CSC-like phenotype that may promote tumor recurrence, but not necessarily tumor metastasis/migration.
Materials and Methods
Cell culture
Human glioma cell lines (U251) were purchased from American Type Culture Collection and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM GlutaMAX (Gibco), penicillin (100 U/mL), and streptomycin (100 μg/mL). All cell cultures were incubated (6% CO2, 37°C) in a humidified chamber. For chemoresistance studies, cells were treated 24 h after plating with 300 mM Temozolimide (TMZ; Sigma-Aldrich) for 72 h. For sphere formation studies, cells were cultured in DMEM F-12, 50:50 (MediaTech, Inc.), 1 M HEPES, B-27 supplement (Gibco); 0.420 U heparin (American Pharmaceutical Partners), 2 mM GlutaMAX, penicillin (100 U/mL), and streptomycin (100 μg/mL) supplemented with 20 ng/mL epidermal growth factor (EGF; R&D Systems) and 20 ng/mL basic fibroblast growth factor (bFGF; R&D Systems) every 3 days. Sphere-like cells were subcultured using Accutase (Innovative Cell Technologies, Inc.) to break clusters into single-cell suspension.
Production of shSLC7A11 and SLC7A11 U251 glioma cell lines
Lentivirus particles were produced by transfection of HEK 293T cells with 15 μg of human TRC-pLKO.1-SLC7A11 shRNA (TRCN0000043126; Sigma-Aldrich), 15 μg of pLK01-nontargeting shRNA (Mission shRNA; Sigma-Aldrich), or 15 μg of a human SLC7A11-pLX304 plasmid (DNASU Plasmid Repository) using calcium phosphate coprecipitation. The culture medium was replaced with fresh 10% FBS in 1 × DMEM after 8 h and supernatant was collected 48 h after transfection. After determination of viral titers, U251 cells were incubated with a viral vector containing the appropriate overexpressing RNA, shRNA, or control shRNA, using a multiplicity of infection of 0.5. Blasticidin (1.0 μg/mL; Sigma-Aldrich) or puromycin (10 μg/mL; Sigma-Aldrich) selection was used to obtain stable recombinant SLC7A11-overexpressing and shSLC7A11 knockdown U251 cells, respectively. Parental U251 cells served as controls for SLC7A11-overexpressing cells, while cells transduced with an empty vector served as controls for the SLC7A11 knockdown cells.
RNA sequence data generation and analysis
Sequencing libraries were prepared with the TruSeq RNA Sample Preparation Kit V2 (Illumina, San Diego) according to the manufacturer's protocol with minor modifications, as previously described [15]. Briefly, 500 ng of total RNA from each sample was used for polyadenylated RNA enrichment with oligo dT magnetic beads, and the poly(A) RNA was fragmented with divalent cations under elevated temperature. First-strand cDNA synthesis produced single-stranded DNA copies from the fragmented RNA by reverse transcription. After second-strand cDNA synthesis, the double-stranded DNA underwent end repair, and the 3′ ends were adenylated. Finally, universal adapters were ligated to the cDNA fragments, and 10 cycles of PCR were performed to produce the final sequencing library. Library templates were prepared for sequencing using the cBot cluster generation system (Illumina) with TruSeq SR Cluster V3 Kit. Sequencing run was performed in single-read mode of 51 cycles of read1 and 7 cycles of index read using the Illumina HiSeq 2500 platform with TruSeq SBS V3 Kits. Real-time analysis software was used to process image analysis and base calling. Sequencing runs generated ∼40 million single reads for each sample. The refSeq annotation for the hg19 version of the human genome was used to create a transcriptome Bowtie [16] index (version 0.12.7), to which reads were aligned with the following settings: -v 3-a. Gene expression levels were estimated using eXpress [17] (version 1.4.1), and DESeq [18] was used for evaluating differential expression.
Immunofluorescence microscopy for F-actin
Cells (200,000 cells/well) were plated (12-well plate), incubated (24 h, 4°C), and then fixed [15 min, 4% paraformaldehyde in phosphate-buffered saline (PBS)]. Cells were then permeabilized (0.1% Triton X-100; 15 min, room temperature). After washes in PBS, cells were incubated with Alexa Fluor 488 phalloidin (A12379; Molecular Probes; 1:40) for 1 h. After three 5-min washes, cells were mounted in Dako fluorescent mounting medium and imaged on an LSM 510 Meta inverted 2-photon confocal microscope.
Boyden chamber cell migration assay
In vitro cell migration assays were performed using 8-μm pore Millicell cell culture inserts (Millipore; P18P01250). A total of 2.0 × 104 cells/0.5 mL medium [5% bovine serum albumin (BSA) in DMEM] were placed in the top chamber of the insert and 10% FBS was placed in the bottom wells to serve as a chemoattractant. As a negative control, 5% BSA was added to the bottom of the Transwell. After 6 h of incubation, cells that had migrated through the pores to the bottom of the insert were detached using Accutase and counted using a Guava EasyCyte flow cytometer. Additionally, any cells that fell to the bottom of the well were also counted.
Cell invasion assay
In vitro cell invasion assays were performed using BD BioCoat Matrigel Invasion Chambers (354480; BD Biosciences). The top surface of each Transwell chamber was coated with Matrigel matrix to block noninvasive cells from migrating through 8-μm membrane pores. A total of 2.5 × 104 cells/0.5 mL DMEM were placed in the top chamber and bottom wells were filled with 10% FBS, which served as a chemoattractant. The cells were incubated at 37°C, 6% CO2, for 24 h, after which invasive cells on the bottom surface of the insert were detached using Accutase and counted using a Guava EasyCyte flow cytometer.
Adhesion assay
To demonstrate cell attachment, 96-well plates were coated with either 5 μg/mL fibronectin (BD Biosciences, NJ) or 5 μg/mL type I collagen (Advanced BioMatrix, San Diego, CA) in 100 μL PBS overnight at 4°C. The plates were blocked with 2.5 mg/mL BSA for 2 h in DMEM at 37°C. Cells were trypsinized and 2 × 104 cells were seeded in each well for 60 min at 37°C. The culture surfaces were washed with Dulbecco's phosphate-buffered saline and fixed with 10% trichloroacetic acid at 4°C for 1 h. Wells were then washed with dH2O and allowed to air dry, after which 100 μL of sulforhodamine B solution 0.4% in 1% acetic acid was added. After 15 min of incubation, plates were washed in 1% acetic acid, allowed to air dry, and 200 μL of 10 mM Tris base was added to each well. Absorbance was read at 570 nm using a SpectraMax M3.
Tumorsphere formation assay and limiting dilution assay
Cells were cultured for at least 10 days in sphere-forming medium. Sphere-like cells were subcultured into single-cell suspension, and cells of ≥90% viability were used for sphere formation studies. Cells were serially diluted from 500 to 2 cells per well of 96-well plates, as previously described [19]. Cells were cultured in DMEM F-12, 50:50, supplemented with HEPES, B-27 supplement, pen-strep antibiotics, and GlutaMAX. Every 3 days, fresh EGF and bFGF were added to the media, and cultures were analyzed for spheres. Limiting dilution assay data were analyzed using the extreme limiting dilution algorithm [20].
Flow cytometry
Flow cytometry was performed on cells stained with Mushasi-1, SOX2, Nestin, and Nanog antibodies (Santa Cruz). Secondary antibodies used were coupled to either Alexa Fluor 647 or Alexa Fluor 488 (Invitrogen). Intracellular staining was performed after fixing single-cell suspensions with 2% paraformaldehyde, permeabilizing with ice-cold 100% methanol, blocking with PBS/1% BSA for 1 h at 4°C, and incubating (30–45 min, room temperature) with the appropriate antibody (1:50) in PBS/1% BSA. Stained single-cell suspensions were analyzed using an LSR Fortessa cytometer (BD Biosciences); data were analyzed using FlowJo 7.6.1.
Cell viability assay
Cell counting kit-8 (Dojindo Molecular Technologies) was used to measure cell viability according to the manufacturer's protocol. Absorbance was measured at 450 nm using a SpectraMax M3.
Statistical analysis
Experiments were performed in at least triplicate and repeated at least three independent times. Statistical analysis was calculated using a two-sided Student's t-test. Differences are considered statistically significant when P < 0.05.
Results
Differential gene expression in SLC7A11-overexpressing and knockdown glioma cells involved in adhesion and migration
RNA sequence analysis was performed to assess gene expression changes upon SLC7A11 overexpression and knockdown in U251 glioma. Differential expression analysis revealed over 4,000 differential genes between SLC7A11 overexpression and respective control cells and over 3,000 differential genes when SLC7A11 is knocked down in U251 cells [15]. Gene Ontology (GO) enrichment analysis identified the number of GO terms that are underrepresented in the SLC7A11-overexpressing glioma and in the SLC7A11 knockdown glioma compared with respective controls. Representative GO terms indicate that several genes involved in regulation of adhesion, migration, and extracellular matrix are downregulated in the SLC7A11-overexpressing glioma compared with control cells (Fig. 1A). GO enrichment analysis of the SLC7A11 knockdown glioma compared with control cells revealed that genes involved in the cell leading edge were downregulated as well as a number of genes involved in proliferation such as those functioning in mitosis, chromosome organization, and nuclear division were downregulated (Fig. 1B).
FIG. 1.

Gene expression changes upon SLC7A11 overexpression and knockdown in U251 cells. (A) Representative GO terms enriched in genes downregulated upon SLC7A11 overexpression. (B) Representative GO terms enriched in genes downregulated upon SLC7A11 knockdown. GO enrichment analysis was carried out using FuncAssociate [42]. GO, gene ontology.
SLC7A11 modification in U251 glioma alters migration, invasion, and cell adhesion
To assess whether the modulation of genes involved in migration and adhesion had any functional consequences, the SLC7A11-modified U251 cells were examined for their migratory, invasive, and adhesive properties. Cell migration requires forward movement of the plasma membrane at the cell leading edge, and despite a downregulation of genes involved in this process, the SLC7A11 knockdown cells showed no difference in migration compared with respective control cells (Fig. 2A). While there was no difference in migration, there was a significant increase in invasion in the knockdown cells (Fig. 2B). On the other hand, the SLC7A11-overexpressing cells exhibited lower migration (Fig. 2A) and invasion (Fig. 2B) compared with their respective control cells. This was consistent with the downregulation of genes in the SLC7A11-overexpressing cells involved in cell-substrate adhesion, actin and fibronectin binding, epithelial cell migration, and regulation of chemotaxis. To assess the impact of downregulation of genes involved in adhesion and extracellular matrix, cell attachment was analyzed. Surprisingly, SLC7A11-overexpressing cells had significantly increased cell attachment after 1 h compared with respective control cells. On the other hand, SLC7A11 knockdown cells had significantly decreased (P value = 5.44 × 10−7) cell attachment compared with control cells (Fig. 2C). Of note, it was observed that the SLC7A11-overexpressing cells, at or near 100% confluency, released from the cell culture plate in large sheets indicating that the adhesive properties of the cells were dynamic.
FIG. 2.

Modification of SLC7A11 in U251 glioma is correlated with altered cell migratory, invasive, and adhesive properties. (A) Boyden chamber migration assay, (B) Matrigel invasion assay, and (C) adhesion assay in SLC7A11-overexpressing and knockdown gliomas compared with respective control cells. Error bars indicate SD; ***P < 0.001, NS = not significant. SD, standard deviation.
SLC7A11 overexpression is correlated with altered cell morphology and cytoskeletal changes
To further assess their adhesive/migratory properties, morphology of the cells was examined. While the SLC7A11 knockdown cells retained similar astrocytoid morphology compared with the control cells with well-developed, long, branching cytoplasmic extensions, the SLC7A11-overexpressing cells had a cuboidal-like morphology with large spherical bodies and central nuclei (Fig. 3A). Since actin is critical to cell adhesion and migration, fluorescent phalloidin staining was performed to visualize filamentous actin (F-actin). Whereas both control cells displayed visible, stretched actin stress fibers with an elongated polarized shape, both SLC7A11-modified cells had actin cytoskeleton changes (Fig. 3B). The SLC7A11 knockdown cells displayed more pronounced F-actin staining at the plasma membrane, while some cells displayed an absence of elongated stress fibers and staining only at the plasma membrane. On the other hand, the SLC7A11-overexpressing cells displayed more diffuse, cytoplasmic F-actin staining (Fig. 3B). The redistribution of F-actin from the cell membrane to the cytoplasm correlated with SLC7A11-overexpressing cells exhibiting lower migration (Fig. 4A) and invasion (Fig. 4B).
FIG. 3.
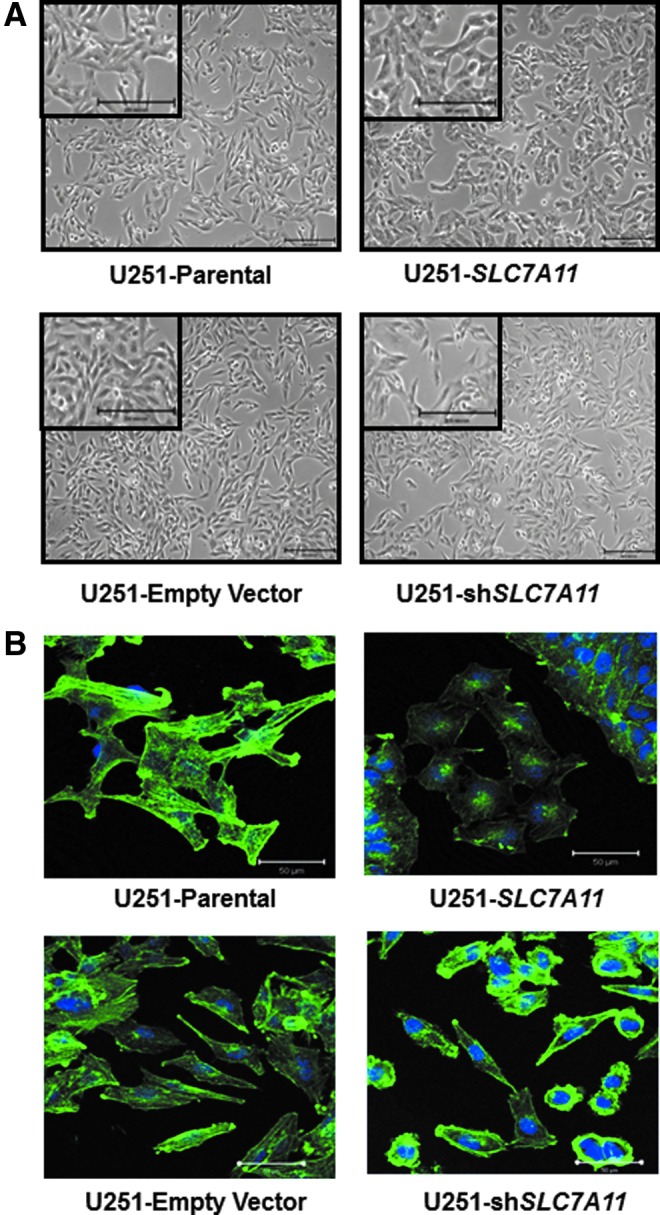
SLC7A11 overexpression in U251 glioma is correlated with altered cell morphology. (A) Representative bright-field images showing the cell morphology of SLC7A11-modified U251 glioma compared with respective control cells. Scale bars: 200 μm. (B) Representative fluorescent images of phalloidin staining to selectively label F-actin in SLC7A11-overexpressing, knockdown, and respective control cells. Scale bars: 50 μm.
FIG. 4.

SLC7A11 overexpression is correlated with increased tumorsphere formation. (A) Photomicrographs of SLC7A11-modified U251 and control cells after 10 days of culture in CSC-enriching or normal media, scale bars, 300 and 100 μm, respectively. (B) SLC7A11-modified U251 and control cells were cultured for a total of 12 days in CSC-enriching media. Numbers of tumorspheres formed at indicated time points were calculated. Data shown are means from a representative experiment. (C) Limiting dilution analysis of frequencies of sphere-forming cells present after 14 days of culture for SLC7A11-modified U251 and control cells. (D) SLC7A11-modified U251 and control cells were cultured for a total of 12 days in CSC-enriching media. Number of tumorspheres was counted before single-cell dissociation and replating for another 12 days of enrichment in CSC-enriching media for secondary tumorsphere cultures and again for tertiary cell cultures. Data shown are mean ± SD from a representative experiment. **P < 0.01; ***P < 0.001. CSC, cancer stem cell.
SLC7A11-overexpressing U251 cells develop a more CSC-like phenotype
The growth of the SLC7A11-overexpressing glioma as tight cuboidal cells in a sheet resembled epithelial-like cells, while the elongated astrocytoid morphology of SLC7A11 knockdown cells resembled the more invasive mesenchymal cells. It has been hypothesized that as cancer cells undergo this epithelial–mesenchymal transition (EMT), which includes loss of cell–cell junctions and restructuring of the cytoskeleton, there is enhanced tumorigenicity and a correlation with increased CSC-like properties [2]. To test whether the SLC7A11-modified cells have an altered differentiation state, we cultured the parental SLC7A11 overexpression and SLC7A11 knockdown U251 cell lines in CSC-enriching media. Within 7–10 days, the SLC7A11-overexpressing cells formed large (∼300 μm in diameter) tumorspheres, indicative of the presence of cancer stem/progenitor cells. In contrast, SLC7A11 knockdown, parental, and empty vector cells formed either much smaller spheres (≤50 μm) or cell aggregates (Fig. 4A).
Additional analysis by tumorsphere formation assay showed that SLC7A11-overexpressing cells formed significantly more tumorspheres than did the SLC7A11 knockdown, parental, and empty vector cells as early as 4 days, and the number of tumorspheres formed by the SLC7A11-overexpressing cells drastically increased by day 12 (Fig. 4B). A limiting dilution assay revealed an increased frequency of cancer stem/progenitor cells in the SLC7A11-overexpressing cell line (Fig. 4C). A secondary and tertiary tumorsphere formation assay was performed to obtain a more highly enriched stem/progenitor cell population versus a more heterogeneous population in the primary spheres [21]. The SLC7A11-overexpressing U251 glioma cells had significantly higher numbers of tumorspheres formed at secondary and tertiary passage, which correlate with the number of multipotential stem cell/progenitor cells present in the population (Fig. 4D). The SLC7A11 knockdown and empty vector controls formed very few tumorspheres with numbers comparable with the parental controls (data not shown). To further confirm that a higher percentage of SLC7A11-overexpressing cells showed a CSC-like phenotype, we performed intracellular staining for the CSC-associated markers, Nanog, Musashi-1, Sox-2, and Nestin. Flow cytometry analysis revealed that all four CSC-associated markers were more highly expressed in the SLC7A11-overexpressing cells compared with the SLC7A11 knockdown, parental, and empty vector cells (Fig. 5).
FIG. 5.

SLC7A11 overexpression is correlated with an increase in CSC-associated markers. Flow cytometric analyses of expression levels of the CSC-associated markers, Nanog, Musashi-1, Sox-2, and Nestin, in SLC7A11-modified and control cells after culture in CSC-enriching media.
Characterization of SLC7A11-overexpressing glioma cells has also revealed that they have increased chemoresistance to TMZ due to increased GSH levels and increased mitochondrial respiration [15]. Recent studies suggest that CSCs also mediate chemoresistance in various tumors, including glioma. We hypothesized that overexpression of SLC7A11 in U251 glioma, which promotes a CSC-like phenotype, also renders the cells resistant to chemotherapy. After enriching CSCs in the parental and SLC7A11-overexpressing U251 lines, we dissociated the tumorspheres into a single-cell suspension and treated them with 300 μM TMZ. Even though the viability decreased in both the CSC-enriched parental and SLC7A11-overexpressing gliomas after TMZ treatment, the SLC7A11-overexpressing cells still maintained higher viability than control cells treated with TMZ (Fig. 6).
FIG. 6.

CSC-enriched SLC7A11-overexpressing U251 cells maintain higher viability under basal conditions and when treated with Temozolomide (TMZ) compared with control cells. Single cells were plated after 2-week enrichment for CSCs and then treated with or without 300 μM TMZ. Viability was measured after 72 h of treatment. Data shown are mean ± SD from a representative experiment. Data shown are mean ± SD from a representative experiment where *** indicates P < 0.001.
Discussion
Recurrence in GBM patients is inevitable even with maximal resection due to the highly invasive behavior of the tumor. Brain tumors have an ability to invade the surrounding healthy brain tissue. These migratory cells can also travel large distances where they can form recurrent tumors at both adjacent and distant sites in the brain. Therefore, it is necessary to understand the molecular mechanisms involved in glioma migration and invasion so that new approaches can be found to treat the cells left behind after surgical resection.
GBM utilizes highly metabolic reactions that generate O2•−, H2O2, HO•, HOCl, and 1O2 as by-products. Whereas HO• can rapidly inactivate proteins and degrade lipids and nucleic acids resulting in cell damage, O2•− and H2O2 are often implicated in cellular signaling and transcription. The role of ROS in migration, invasion, and EMT has been well established where ROS activates the MAPK signaling cascade and nuclear factor-kappaB pathway leading to activation of target genes such as urokinase plasminogen activators/MMPs and EMT genes [22]. We have shown that system xc− transporter is involved in regulating endogenous ROS levels and modulating oxidative stress [15]. In this work, we have identified SLC7A11 overexpression in glioma as a potential novel mechanism contributing to GBM dissemination and tumorigenesis.
For glioma cells to migrate, it requires a change in morphology where the cell becomes polarized, and membrane protrusions develop, including extensions at the leading edge, with subsequent cytoskeletal contractions that allow advancement forward [23]. Our data show that SLC7A11-overexpressing gliomas have a cuboidal-like morphology with large spherical bodies that are not conducive to migration, while the SLC7A11 knockdown cells retain an elongated polarized shape with more pronounced F-actin staining at the plasma membrane. It may be plausible that the high endogenous ROS levels in the SLC7A11 knockdown cells promote actin polymerization and cytoskeleton remodeling, leading to cell migration. Actin polymerization induced by Rac 1, an upstream regulator of the ROS-producing enzyme NOX, has been shown to be stimulated by increased O2•− levels, and antioxidant treatment suppressed the actin elongation [24,25]. Antioxidants such as N-acetylcysteine have also been shown to dampen the activity of coffin, an actin-binding protein that modulates lamellipoda formation [26].
The SLC7A11-overexpressing glioma cells, which exhibit low endogenous ROS levels, display significantly reduced migration and invasion, which is consistent with their morphology. Since ROS serve as signaling molecules, it may be that the cells lack sufficient signaling for stimulation of migration/invasion. ROS can oxidize critical cysteine residues in signaling molecules involved in cell migration such as in protein kinase C, an activator of receptor tyrosine kinases (RTKs), as well as in negative regulators of RTKs such as protein tyrosine phosphatases [22]. Increased ROS production activated by growth factor receptors has been shown to increase cell migration in vascular smooth muscle cells [27] and in endothelial cells [28]. Higher ROS levels in SLC7A11 knockdown glioma cells likely activate important signaling pathways involved in the increased cell invasion.
EMT has been recognized as a crucial process in progression of certain cancers such as breast cancer. During EMT, cells that are epithelial-like, which are characterized by an immobile, aligned, and tightly packed phenotype, undergo a transition to a more mesenchymal-like state characterized by mobilization and detachment, invasion and migration, and resistance to anchorage dependence [29]. After circulating/migrating cells reach secondary sites with a conducive microenvironment, they undergo a reverse transition known as mesenchymal-to-epithelial transition (MET). ROS have been shown to activate signaling pathways involved in induction of EMT [30]. It is interesting that the SLC7A11-overexpressing cells with low endogenous ROS levels exhibit a more epithelial-like phenotype, while the SLC7A11 knockdown cells with higher endogenous ROS levels exhibit a more mesenchymal-like phenotype. An important characteristic of EMT is loss of intracellular adhesion where increased ROS levels have been implicated in intracellular dissociation [31,32]. The SLC7A11-overexpressing glioma did exhibit higher adhesion, while the SLC7A11 knockdown cells showed lower adhesion compared with respective control cells. It is important to note that EMT and MET are not mutually exclusive and cancer cells exhibit epithelial–mesenchymal plasticity. Additionally, ROS induction of EMT is not absolute, but is cellular context and tissue-type dependent [33]. While EMT has been studied extensively in breast cancer, strong evidence of the existence of EMT in GBM is still lacking [34]. Thus, further investigation is warranted to examine whether SLC7A11 modification can promote EMT-like and MET-like changes in GBM.
Cancer-associated EMT contributes to the generation of cell populations with stem-like characteristics [30]. Since CSCs have been suggested to be responsible for GBM invasiveness and EMT, and ROS levels seem to play an important role in CSC self-renewal, we hypothesized that overexpression of SLC7A11 levels in glioma may promote a CSC-like phenotype. Therefore, we enriched the CSC-like population in the SLC7A11-modified and control glioma cells. Compared with SLC7A11 knockdown and control cells, the SLC7A11-overexpressing cells showed greater tumorsphere formation and a higher frequency of sphere-forming cells, both of which are indicative of an increased CSC-like phenotype. In addition, SLC7A11 overexpression correlated with increased expression of the progenitor/stem cell markers, Musashi-1, Nanog, Sox-2, and Nestin, which are also CSC-associated markers. It is possible that SLC7A11 overexpression drives the maintenance of CSCs because redox balance plays an important role in stem cell renewal [35,36]. Despite reports that CSCs exhibit EMT and increased invasive properties, we did not see a correlation between the increased CSC-like phenotype in the SLC7A11-overexpressing glioma cell and invasiveness/migration. However, Brabletz et al. proposed that CSCs can switch back and forth between a stationary/proliferative phenotype and a migratory/invasive phenotype to promote growth of the tumor core and colonization of the neighboring normal tissue [37]. This theory may explain the observations made with SLC7A11-overexpressing cells.
CSCs have been shown to express various membrane transporters of the SLC series as well as ATP-binding cassette (ABC) transporters. For instance, ABCB5 is expressed on a distinct subset of chemoresistance, stem cell phenotype-expressing tumor cells in melanoma [38]. Indeed, ABC transporters are expressed at higher levels in the distinct side population of tumor cells with stem-like characteristics that may account for the more efficient secretion of chemotherapeutics from CSCs and thus render them more resistant [39]. The quiescence of stem cells also protects them from chemo- and radiotherapy and therefore CSCs may be responsible for tumor reoccurrence in glioma. Similar to normal stem cells, certain subsets of CSCs in breast tumors, when compared with nontumorigenic cells, have lower ROS levels that contributed to their radioresistance [40]. These CSCs maintained low endogenous ROS levels due to upregulation of free radical scavenging systems, such as GSH. Furthermore, expression of one of the CD44+ variant isoforms (CD44v) in CSCs was shown to interact and stabilize the xCT subunit in gastrointestinal cancer cells, contributing to the upregulation of GSH synthesis [41]. Since several studies suggest that CSCs may mediate chemoresistance, we wondered whether overexpression of SLC7A11 may render glioma cells resistant to chemotherapy by promoting the CSC-like phenotype. Indeed, the SLC7A11-overexpressing cells enriched for the CSC-like phenotype still retained higher viability after TMZ treatment compared with control U251 cells enriched for the CSC-like phenotype and treated with TMZ. Thus, the SLC7A11-overexpressing glioma cells may have decreased chemosensitivity to TMZ not only due to increased GSH levels [15] but also due to an increased CSC-like phenotype.
ROS are known to actively participate in alterations in cell structure required for migration and cytoskeletal dynamics, in the expression of adhesion molecules, and activation of signaling processes involved in cell migration [26]. The altered cell morphology and changes in the invasive and migratory phenotype of the SLC7A11-modified cells most likely are reflective of their low endogenous ROS levels based on cystine/cystine-mediated GSH generation. While cancer metastasis involves local invasion and dissemination of cancer cells to distant sites, the outgrowth of micrometastatic cells into macroscopic metastases is associated with self-renewal, a well-defined characteristic of CSCs [2]. Previously, we have shown that generation of stable SLC7A11-overexpressing glioma cells demonstrated characteristics of higher GSH generation and lower ROS production [15]. Surprisingly, we found that overexpression of SLC7A11 levels in glioma is correlated with an increased CSC-like phenotype. This may also be reflective of low endogenous ROS levels since high ROS levels can lead to stem cell differentiation and loss of stem-like characteristics.
In conclusion, we have shown that overexpression of SLC7A11 in glioma is correlated with a less migratory CSC-like phenotype that may contribute to GBM progression by tumor cell planting and chemoresistance. Further studies are warranted to explore whether the observed results are strictly GSH mediated, involve compensatory strategies, or involve alternative pathways. The contributions of ROS to pathways that regulate cell proliferation, invasion, migration, survival, and apoptosis have only begun to be elucidated. Clearly, system xc− plays an important role in maintaining redox balance by promoting GSH generation. It will be necessary to decipher the pathways that are affected by upregulation of the transporter and determine whether targeting the transporter in cancer cells may provide improved clinical outcome in patients diagnosed with GBM.
Acknowledgments
The authors acknowledge California Institute of Regenerative Medicine (TG2-01150), the Rosalinde and Arthur Gilbert Foundation, STOP Cancer, and the National Cancer Institute Cancer Center Support Grant (P30CA033572) for funding. The authors also acknowledge the technical support of the City of Hope RNAi Core (Dr. Claudia M. Kowolik), the Light Microscopy Digital Imaging Core (Dr. Brian Armstrong and Tina Patel), and Dr. Keely L. Walker for critical reading and editing of the article.
Author Disclosure Statement
K.S.A. is a shareholder, director, and officer of TheraBiologics, Inc., a clinical-stage biopharmaceutical company focused on the development of stem cell-mediated cancer therapies. No competing financial interests exist for other authors.
References
- 1.Brandes AA, Tosoni A, Spagnolli F, Frezza G, Leonardi M, Calbucci F. and Franceschi E. (2008). Disease progression or pseudoprogression after concomitant radiochemotherapy treatment: pitfalls in neurooncology. Neuro Oncol 10:361–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scheel C. and Weinberg RA. (2012). Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol 22:396–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watkins S. and Sontheimer H. (2012). Unique biology of gliomas: challenges and opportunities. Trends Neurosci 35:546–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giese A. and Westphal M. (2001). Treatment of malignant glioma: a problem beyond the margins of resection. J Cancer Res Clin Oncol 127:217–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chung WJ, Lyons SA, Nelson GM, Hamza H, Gladson CL, Gillespie GY. and Sontheimer H. (2005). Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci 25:7101–7110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colvin OM, Friedman HS, Gamcsik MP, Fenselau C. and Hilton J. (1993). Role of glutathione in cellular resistance to alkylating agents. Adv Enzyme Regul 33:19–26 [DOI] [PubMed] [Google Scholar]
- 7.Lo M, Wang YZ. and Gout PW. (2008). The x(c)− cystine/glutamate antiporter: a potential target for therapy of cancer and other diseases. J Cell Physiol 215:593–602 [DOI] [PubMed] [Google Scholar]
- 8.Lyons SA, Chung WJ, Weaver AK, Ogunrinu T. and Sontheimer H. (2007). Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res 67:9463–9471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiu WT, Shen SC, Chow JM, Lin CW, Shia LT. and Chen YC. (2010). Contribution of reactive oxygen species to migration/invasion of human glioblastoma cells U87 via ERK-dependent COX-2/PGE(2) activation. Neurobiol Dis 37:118–129 [DOI] [PubMed] [Google Scholar]
- 10.Drukala J, Urbanska K, Wilk A, Grabacka M, Wybieralska E, Del Valle L, Madeja Z. and Reiss K. (2010). ROS accumulation and IGF-IR inhibition contribute to fenofibrate/PPARalpha-mediated inhibition of glioma cell motility in vitro. Mol Cancer 9:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsieh CH, Chang HT, Shen WC, Shyu WC. and Liu RS. (2012). Imaging the impact of Nox4 in cycling hypoxia-mediated U87 glioblastoma invasion and infiltration. Mol Imaging Biol 14:489–499 [DOI] [PubMed] [Google Scholar]
- 12.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD. and Rich JN. (2006). Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444:756–760 [DOI] [PubMed] [Google Scholar]
- 13.Eramo A, Ricci-Vitiani L, Zeuner A, Pallini R, Lotti F, Sette G, Pilozzi E, Larocca LM, Peschle C. and De Maria R. (2006). Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ 13:1238–1241 [DOI] [PubMed] [Google Scholar]
- 14.El-Gebali S, Bentz S, Hediger MA. and Anderle P. (2013). Solute carriers (SLCs) in cancer. Mol Aspects Med 34:719–734 [DOI] [PubMed] [Google Scholar]
- 15.Polewski MD, Reveron-Thornton RF, Cherryholmes GA, Marinov GK, Cassady K. and Aboody KS. (2016). Increased expression of system xc− in glioblastoma confers an altered metabolic state and temozolomide resistance. Mol Cancer Res 14:1229–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Langmead B, Trapnell C, Pop M. and Salzberg SL. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts A. and Pachter L. (2013). Streaming fragment assignment for real-time analysis of sequencing experiments. Nat Methods 10:71–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anders S. and Huber W. (2010). Differential expression analysis for sequence count data. Genome Biol 11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, Oh YT, Kim H, Rheey J, et al. (2013). Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 23:839–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu Y. and Smyth GK. (2009). ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 347:70–78 [DOI] [PubMed] [Google Scholar]
- 21.Rota LM, Lazzarino DA, Ziegler AN, LeRoith D. and Wood TL. (2012). Determining mammosphere-forming potential: application of the limiting dilution analysis. J Mammary Gland Biol Neoplasia 17:119–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tochhawng L, Deng S, Pervaiz S. and Yap CT. (2013). Redox regulation of cancer cell migration and invasion. Mitochondrion 13:246–253 [DOI] [PubMed] [Google Scholar]
- 23.Demuth T. and Berens ME. (2004). Molecular mechanisms of glioma cell migration and invasion. J Neurooncol 70:217–228 [DOI] [PubMed] [Google Scholar]
- 24.Moldovan L, Irani K, Moldovan NI, Finkel T. and Goldschmidt-Clermont PJ. (1999). The actin cytoskeleton reorganization induced by Rac1 requires the production of superoxide. Antioxid Redox Signal 1:29–43 [DOI] [PubMed] [Google Scholar]
- 25.Selvakumar B, Hess DT, Goldschmidt-Clermont PJ. and Stamler JS. (2008). Co-regulation of constitutive nitric oxide synthases and NADPH oxidase by the small GTPase Rac. FEBS Lett 582:2195–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munnamalai V. and Suter DM. (2009). Reactive oxygen species regulate F-actin dynamics in neuronal growth cones and neurite outgrowth. J Neurochem 108:644–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sundaresan M, Yu ZX, Ferrans VJ, Irani K. and Finkel T. (1995). Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 270:296–299 [DOI] [PubMed] [Google Scholar]
- 28.Ushio-Fukai M, Tang Y, Fukai T, Dikalov SI, Ma Y, Fujimoto M, Quinn MT, Pagano PJ, Johnson C. and Alexander RW. (2002). Novel role of gp91(phox)-containing NAD(P)H oxidase in vascular endothelial growth factor-induced signaling and angiogenesis. Circ Res 91:1160–1167 [DOI] [PubMed] [Google Scholar]
- 29.Ilmer M, Vykoukal J, Recio Boiles A, Coleman M. and Alt E. (2014). Two sides of the same coin: stem cells in cancer and regenerative medicine. FASEB J 28:2748–2761 [DOI] [PubMed] [Google Scholar]
- 30.Cichon MA. and Radisky DC. (2014). ROS-induced epithelial-mesenchymal transition in mammary epithelial cells is mediated by NF-kB-dependent activation of Snail. Oncotarget 5:2827–2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nigam S, Weston CE, Liu CH. and Simon EE. (1998). The actin cytoskeleton and integrin expression in the recovery of cell adhesion after oxidant stress to a proximal tubule cell line (JTC-12). J Am Soc Nephrol 9:1787–1797 [DOI] [PubMed] [Google Scholar]
- 32.Inumaru J, Nagano O, Takahashi E, Ishimoto T, Nakamura S, Suzuki Y, Niwa S, Umezawa K, Tanihara H. and Saya H. (2009). Molecular mechanisms regulating dissociation of cell-cell junction of epithelial cells by oxidative stress. Genes Cells 14:703–716 [DOI] [PubMed] [Google Scholar]
- 33.Cannito S, Novo E, di Bonzo LV, Busletta C, Colombatto S. and Parola M. (2010). Epithelial-mesenchymal transition: from molecular mechanisms, redox regulation to implications in human health and disease. Antioxid Redox Signal 12:1383–1430 [DOI] [PubMed] [Google Scholar]
- 34.Ortensi B, Setti M, Osti D. and Pelicci G. (2013). Cancer stem cell contribution to glioblastoma invasiveness. Stem Cell Res Ther 4:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi X, Zhang Y, Zheng J. and Pan J. (2012). Reactive oxygen species in cancer stem cells. Antioxid Redox Signal 16:1215–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ding S, Li C, Cheng N, Cui X, Xu X. and Zhou G. (2015). Redox regulation in cancer stem cells. Oxid Med Cell Longev 3:23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brabletz T, Jung A, Spaderna S, Hlubek F. and Kirchner T. (2005). Opinion: migrating cancer stem cells—an integrated concept of malignant tumour progression. Nat Rev Cancer 5:744–749 [DOI] [PubMed] [Google Scholar]
- 38.Frank NY, Margaryan A, Huang Y, Schatton T, Waaga-Gasser AM, Gasser M, Sayegh MH, Sadee W. and Frank MH. (2005). ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res 65:4320–4333 [DOI] [PubMed] [Google Scholar]
- 39.Dean M. (2009). ABC transporters, drug resistance, and cancer stem cells. J Mammary Gland Biol Neoplasia 14:3–9 [DOI] [PubMed] [Google Scholar]
- 40.Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, et al. (2009). Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458:780–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, Oshima M, Ikeda T, Asaba R, et al. (2011). CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(−) and thereby promotes tumor growth. Cancer Cell 19:387–400 [DOI] [PubMed] [Google Scholar]
- 42.Berriz GF, Beaver JE, Cenik C, Tasan M. and Roth FP. (2009). Next generation software for functional trend analysis. Bioinformat 25:3043–3044 [DOI] [PMC free article] [PubMed] [Google Scholar]